 Open Access Article
Open Access ArticleA different route to functional polyolefins: olefin–carbene copolymerisation†
Nicole M. G.
Franssen
ab,
Joost N. H.
Reek
a and
Bas
de Bruin
*a
aVan't Hoff Institute for Molecular Sciences (HIMS), Department of Homogeneous and Supramolecular Catalysis, Universiteit van Amsterdam, P.O. Box 94720, 1090 GS Amsterdam, The Netherlands. E-mail: b.debruin@uva.nl; Fax: +31 20 525 5604; Tel: +31 20 525 6495
bDutch Polymer Institute DPI, P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 16th January 2013
Abstract
Copolymerisation of carbenes and olefins (ethene), mediated by Rh-based catalyst precursors, is presented as a new, proof-of-concept methodology for the controlled synthesis of functional polymers. The reactions studied show that olefin–carbene polymerisation reactions provide a viable alternative to more traditional olefin polymerization techniques. RhIII-catalyst precursors, while active in the homopolymerisation of either olefins or carbenes, proved to be virtually inactive in olefin–carbene copolymerization. Conversely, the use of RhI(cod) catalyst precursors allows the synthesis of high molecular-weight, highly functionalized copolymers. The reactions yield a mixture of copolymers and some carbene homopolymers, which proved to be difficult to separate. Polyethylene was not formed under the applied reaction conditions. The average ethene content in this mixture could be increased up to 11%, although analysis of the mixture revealed that the ethene content in fractions of the copolymer mixture can be as high as 70%. Attempts to increase the ethene content by increasing the ethene pressure unexpectedly led to lower average ethene contents, which is most likely due to changes in the ratio of copolymers vs. carbene homopolymer. This behaviour is most likely a result of the reactivity difference of different active Rh-species formed under the applied reaction conditions. Apparently, higher ethene concentrations slow down the copolymerisation process (mediated by yet unidentified Rh-species) compared to the formation of homopolymers (mediated by different Rh-catalysts; most likely (allyl)RhIII-alkyl species), thereby changing the product ratio in favour of the homopolymer. The average ethene content in the copolymer mixture therefore decreases, while the ethene content within the copolymer fraction has likely increased at higher ethene concentrations (but simply less copolymer is formed). The obtained copolymers exhibit a blocky microstructure, with the functional blocks being highly stereoregular. Branching does occur and the functional groups are present in the polymer backbone as well as at the branches. Formation of copolymers was confirmed by Maldi-ToF analysis, which revealed incorporation of several ethene units into the copolymers.
Introduction
Non-functionalised polyolefins (i.e. polyethene and polypropene) have found their way in many (commodity) applications due to their outstanding properties, such as solvent resistance and thermal stability. Nowadays, these materials can easily be obtained in large scales and at low cost with very high precision of the polymer microstructures. However, due to the lack of functional groups in these polymers, they perform badly when surface chemistry is involved.1–3 Incorporation of only a small amount of functionalised monomers, randomly placed in the polymer backbone, has a large effect on the surface properties of the resulting polymers, while the beneficial properties of the original non-functionalised polyolefins are retained.1,4 The properties of the resulting materials can be further tuned by varying the structure of the functional groups, the amount of incorporation and the distribution of the polar functionalities along the polymer chain.Despite the existence of various different approaches to incorporate functionalities into polyolefin chains,3,5 synthetic methods that allow controlled incorporation of tunable amounts of polar functionalities into an otherwise non-functionalised polyolefin are rather scarce. The most widely-applied commercial approach to obtain functionalised polyolefins is via post-functionalisation reactions on existing polyolefin chains, although this often requires harsh conditions and is accompanied by many undesired side reactions, thus allowing very limited control.2,6 More promising approaches to incorporate functionalities in a controlled way are based on either acyclic diene metathesis polycondensation (ADMET) or ring-opening metathesis polymerisation (ROMP), and in this way polyolefins bearing different functional groups have been prepared exhibiting precise sequence distributions.7–9 However, the required use of specially designed monomers is a significant drawback of these approaches, both in terms of cost-effectiveness and synthetic limitations.
Obtaining functionalised polymers via direct copolymerisation of non-functionalised olefins and polar vinyl monomers is of particular interest, since control over the amount of polar monomers and their distribution along the chain could in principle be achieved by exploiting the reactivity differences of both monomers.3,10–12 Large scale applications of this approach are mainly based on radical copolymerisations of olefins and predominantly acrylates, although the general low reactivity of alkenes (especially ethene) towards radical polymerisation requires the use of quite harsh polymerisation conditions.13,14 The resulting copolymers are highly branched and contain large amounts of acrylates. Radical polymerisation is therefore not suitable for the synthesis of copolymers with a well-defined (blocky) microstructure, especially if stereoregularity of the functionalised block is required.15 Such materials are expected to exhibit interesting material properties resulting from phase separation on the microscopic level.16 Recently, Monteil and coworkers have shown that these materials might be accessible by applying radical polymerisation in combination with coordination–insertion polymerisation using a nickel catalyst, although there is still limited control over the stereoselectivity and the incorporation of polar monomers is moderate (<35%).17 Further research is necessary to exploit this technique to its full potential.
Development of suitable transition-metal (TM) catalysed processes that allow such copolymerisations via a coordination–insertion mechanism (Scheme 1) is expected to give better (stereo)control in polymerisation, and is in that respect advantageous over radical polymerisations.18
 | ||
| Scheme 1 Schematic representation of TM-catalysed coordination–insertion polymerisation. | ||
Advances in the field of lanthanide metallocene complexes and group 4 early transition metal metallocenes might enable the synthesis of such desirable copolymers in the near future. These systems are active in the controlled synthesis of syndiotactic and isotactic (rich) homopolymers of a variety of polar vinyl monomers, obtained via living coordination–addition mechanisms (metal-controlled anionic polymerisation).19–22 However, so far these systems only allow the synthesis of di- or triblock copolymers upon combination with coordination–insertion polymerisation of ethene. Furthermore, catalytic synthesis (instead of ‘stoichiometric’ living polymerisation) cannot be achieved with these systems yet.
Another promising route towards the synthesis of well-defined copolymers bearing polar functionalities is late transition metal (LTM) catalysed insertion polymerisation of alkenes and polar vinyl monomers.3,11,12 Recent advances in the field of catalyst development have recently been reviewed by Takeuchi.23 In the early 1990s Brookhart and coworkers developed Ni and Pd catalysts bearing diimine ligands, and with these systems highly branched copolymers could be obtained in which the polar monomers were mainly present at the branches.24–26 Synthesis of linear copolymers has been achieved by using (phosphino-sulfonato)Pd catalysts that even allow consecutive insertions of polar monomers, albeit limited to 3 or 4 units inserted subsequently in a row.27,28 A drawback of these systems is that the incorporation of polar monomers is generally much less than 50%, with the Mw of the resulting polymers decreasing drastically (to as low as 3 kDa) with increasing polar content. Aside from this, stereospecific copolymerisation of polar and non-polar monomers cannot be achieved with these systems. In fact, to the best of our knowledge, there are no reported examples of any LTM catalysts capable of stereospecific (co)polymerisation of polar vinyl monomers.19
The above-described examples clearly demonstrate the recent success in the field of TM-catalysed copolymerisation of non-polar olefins and polar vinyl monomers, but also emphasise the remaining challenges, in particular with respect to consecutive insertions of polar and non-polar monomers, leading to the synthesis of well-defined block copolymers, and stereoregularity. Therefore, the development of alternative pathways to introduce functionalities directly into the growing polymer chain via a transition-metal catalysed process, based on monomers other than polar vinyl compounds, is of growing importance.
Polymerisation of so-called C1 monomers (building-up a polymer chain with one backbone-carbon unit at the time) as an alternative to traditional vinyl monomers (C2 monomers; extending the growing polymer chain by two carbon units at each insertion step) might help to overcome some of these challenges.29,30 The use of functionalised carbenes (i.e. C1 monomers) as ‘functional-group carriers’ in coordination–insertion polymerisation has proven to be successful, especially if densely functionalised, highly stereoregular polymers are desired.29–42 Highly stereospecific (co)polymerisation of diazoesters (N2CHCOOR) can be achieved in good yields and with high Mw in a Rh catalysed process.30,43–51 This leads to the formation of syndiotactic polymers bearing an ester functionality at every single carbon atom of the polymer main chain.
Combining polymerisation of functionalised carbenes with coordination–insertion polymerisation of non-functionalised olefins (in particular ethene) (Scheme 2) might lead to the development of new materials that are inaccessible via traditional copolymerisation techniques. Recently we have shown that copolymers of functionalised and non-functionalised carbenes (thus exhibiting similar structures to copolymers of carbenes and ethene) showed indeed unique material properties, thereby emphasizing the potential of this approach.52,53 However, these copolymerisations required the use of diazomethane or dimethylsulfoxonium ylides, which limits the application of this process significantly. In this respect, extending this concept to the use of ethene would be beneficial. Previous attempts to achieve these copolymerisations with Pd catalysts proved to be unsuccessful, due to clear differences in the nature of the active species responsible for both homopolymerisation reactions.42
 | ||
| Scheme 2 Proposed copolymerisation of olefins and functionalised carbenes. | ||
We decided to study these copolymerisations in more detail in the presence of Rh as a catalyst due to the activity of Rh towards both the polymerisation of functionalised carbenes (vide supra) as well as ethene polymerisation.54–56 We anticipated that these two processes are potentially compatible, since ethene polymerisation is catalysed by RhIII complexes55,56 and recently we demonstrated that the active species for carbene polymerisation is based on RhIII as well.49 In this paper we report our findings and demonstrate the potential of this approach for the controlled synthesis of functional polyolefins.
Results and discussion
The activity of different well-defined homogeneous Rh catalyst precursors (Fig. 1) was studied in the copolymerisation of ethene and ethyl diazoacetate (EDA) (Fig. 2 and Scheme 3). These Rh complexes are chosen because of their known activity in either carbene polymerisation (complexes 1–3) or ethene polymerisation (complexes 4 and 5). Complexes 1 and 2 are RhI(cod) catalyst precursors, but mechanistic studies on the homopolymerisation of EDA revealed that they are converted in situ to (a mixture of different) active RhIII(allyl)-type species, which are responsible for the actual carbene polymerisation event.49 | ||
| Fig. 1 Rh catalysts tested in the copolymerisation of ethene and ethyl diazoacetate (EDA). | ||
 | ||
| Fig. 2 Schematic representation of Rh catalysed copolymerisation of ethene and ethyl diazoacetate (EDA). | ||
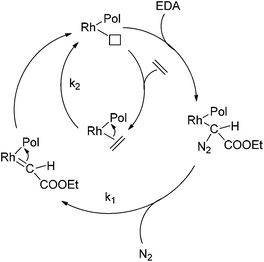 | ||
| Scheme 3 Schematic representation of the mechanistic steps leading to ethene–EDA copolymers catalysed by an Rh catalyst. The results described in this paper imply that k1vs. k2 differs for the various active species present during the reaction. | ||
1. Olefin-carbene copolymerizations
The copolymerisation experiments were performed by first incubating a solution of the catalyst in DCM with ethene at a pressure of 2 bar for 2 h, after which EDA was added to the reaction mixture and the reaction mixture was allowed to stir overnight.Under these conditions, catalyst precursors 1–3 show activity in ethene–carbene copolymerisation (Fig. 2 and Scheme 3), giving rise to high Mw polymers with polydispersity indices (PDI) between 1.7 and 4.6 (Table 1, entries 1–3). Incorporation of ethene can be achieved up to 11 mol%, depending on the applied catalyst precursor. The highest ethene incorporation could be achieved with catalyst precursors 1 and 2. Catalyst precursor 3 gave rise to the highest polymer yield, but a lower amount of ethene is incorporated into the polymers (entry 3) than in the reaction with complexes 1 and 2.
| Entry | Catalyst | Yield (%) |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (kDa) b (kDa) |
PDIb | Mol% ethenec |
|---|---|---|---|---|---|
| a Reaction conditions: DCM (5 mL); Rh catalyst (0.04 mmol); Pethene 2 bar; EDA (2.0 mmol) added after 2 h; 16 h at room temperature. b Determined by size exclusion chromatography (GPC) in DCM at 35 °C vs. polystyrene. c Based on 1H NMR integration. d MAO (500 eq. w.r.t. Rh) was added as a cocatalyst. | |||||
| 1 | 1 | 14 | 182 | 4.6 | 7 |
| 2 | 2 | 50 | 1022 | 3.5 | 11 |
| 3 | 3 | 68 | 270 | 1.7 | 2 |
| 4 | 4 | 0 | — | — | — |
| 5d | 5 | 0 | — | — | — |
Catalysts 4 and 5 did not show any significant (co)polymerisation activity under the applied reaction conditions (entries 4 and 5). No product formation was observed at all employing catalyst 4, and mainly unreacted EDA was recovered after the reaction. Apparently, the presence of EDA in the reaction mixture blocks the activity of this complex towards ethene homopolymerisation, probably due to strong coordination of EDA to the Rh center. This is in agreement with the lack of activity of this complex in the homopolymerisation of EDA.48 In a separate experiment, complex 5 was activated in situ by applying methyl aluminoxane (MAO) as a cocatalyst, prior to addition of EDA and ethene to the reaction mixture. Neither copolymers nor polyethene or polyEDA homopolymers were formed under these conditions. As was observed for catalyst 4, EDA seems to deactivate the (ethene) polymerization activity of these catalysts. The reaction is further hampered by side reactions of MAO with EDA, and as a result the catalyst system is not active towards polymerisation.
The behaviour of precatalysts 1–3 in this ethene–EDA copolymerisation was studied in more detail.
Analysis of the products obtained in the copolymerisation reaction revealed in all cases the formation of an inhomogeneous polymer mixture consisting of copolymers and homopolymers of EDA (vide infra).57 The ethene incorporation listed in Table 1 is therefore a measure for the average content of ethene in the polymer mixture, rather than the ethene content of the copolymer fractions. Attempts to separate the copolymers from EDA homopolymers57 were not successful due to their very similar properties (e.g. solubility and Mw as determined by size-exclusion chromatography (SEC), see ESI†).
Homopolymers of ethene are not formed under the applied conditions. Activation of the catalyst precursors with one equivalent of EDA was not sufficient to achieve ethene homopolymerisation, indicating that the presence of excess EDA in the reaction mixture is required in order to achieve (co)polymerisation activity.
For all three catalysts (1–3), the Mw of polymers obtained in the copolymerisation reactions is higher than the Mw of the homopolymers obtained in reactions without ethene (e.g. 182 kDa versus 120 kDa for complex 143), indicating that the presence of ethene slows down chain termination reactions. This is in agreement with previously-described chain transfer/termination pathways.50,58 The polymer yield drops upon incorporation of ethene as compared to the yields obtained in the carbene homopolymerisation reactions. Previous studies on the EDA homopolymerisation revealed that chain transfer occurs only to a very limited extent. Therefore, this lower yield, in combination with the increased Mw, points to a lower initiation efficiency in the presence of ethene.
Remarkably, increasing the ethene pressure did not lead to a higher ethene incorporation (Table 2). For both catalysts 1 and 2 increasing the pressure from 2 bar to 6 bar actually led to a slightly lower ethene content (Table 2, entries 1 and 2), although the decrease is minimal. The yield does not change significantly. Also the Mw of the copolymers decreases only slightly, emphasizing that chain termination is not significantly enhanced upon increasing the ethene pressure. The unexpected lower average incorporation of ethene at higher ethene pressures seems to be related to the increased ethene concentration with respect to EDA, as is emphasised by dilution experiments. Upon increasing the solvent volume (and thus increasing the absolute amount of ethene) while keeping the ethene pressure at 2 bar, the overall incorporation of ethene into the copolymers decreased as well (entry 3 vs. 4). Decreasing the ethene pressure to 0.75 bar, on the other hand, did not lead to an increased ethene incorporation (entry 5).
| Entry | Catalyst | P ethylene (bar) | Yield (%) |
M
wb (kDa) |
PDIb | Mol% ethenec |
|---|---|---|---|---|---|---|
| a Reaction conditions: DCM (5 mL); Rh catalyst (0.04 mmol); Pethene; EDA (2.0 mmol) added after 2 h; 16 h at room temperature; b Determined by size exclusion chromatography (GPC) in DCM at 35 °C vs. polystyrene. c Based on 1H NMR integration. d CHCl3 (5 mL) was used as a solvent. e CHCl3 (200 mL) was used as a solvent. | ||||||
| 1 | 1 | 6 | 14 | 145 | 3.5 | 4 |
| 2 | 2 | 6 | 60 | 742 | 2.3 | 9 |
| 3d | 1 | 2 | 24 | 125 | 2.7 | 6 |
| 4e | 1 | 2 | 23 | 96 | 2.9 | 1 |
| 5 | 1 | 0.75 | 17 | 173 | 2.7 | 1 |
Kinetic studies on the copolymerisation reaction catalysed by complex 1 at an ethene pressure of 2 bar revealed a similar kinetic profile to that observed in the homopolymerisation of EDA, although some additional side products are formed during the reaction (for more details about the kinetic profile and these side products, see Fig. S2, ESI†).45
Mechanistic studies on the homopolymerisation of EDA revealed that all three catalyst precursors are in situ activated for carbene polymerisation upon reaction with EDA. Complexes 1 and 2 give rise to multiple active species in this process that exhibit different reactivity towards carbenes, leading to the formation of a mixture of dimers, oligomers and polymers in the carbene homopolymerisation reaction (an overview of these proposed catalyst-activation steps is given in Fig. S3†).48 Recently we reported that (one of) the active species responsible for the formation of high-Mw polymers from carbenes is the RhIII(allyl)(alkyl) species 6 depicted in Fig. 3.49 Complex 3 is closer in structure to this active RhIII(allyl)(alkyl) species and thus provides a more effective (and most likely also a better defined) route to the formation of this active species under the applied reaction conditions, which is reflected in almost selective formation of polymers in the carbene homopolymerisation process.
 | ||
| Fig. 3 Proposed active species for the Rh-catalysed stereoselective polymerisation of diazoesters, depicted with a growing polymer chain attached to the metal centre. | ||
Since catalyst precursor 3 gives rise to rather low incorporation of ethene in the copolymerisation reaction, we can conclude that this RhIII(allyl)(alkyl) species 6 most active in the homopolymerisation of carbenes is poorly reactive to ethene, thus giving rise to very low ethene incorporation (Table 1, entry 3). Since active species 6 is very active towards EDA homopolymerisation, the amount of PEA homopolymers in the copolymer mixture obtained with catalyst 3 is rather large, resulting in a rather low average ethene content. Possibly, species 6 is not reactive towards ethene at all, and the formation of a small amount of a different active species from complex 3 under the applied reaction conditions could be responsible for the observed ethene incorporation with pre-catalyst 3.
In situ activation of RhI(cod) precursors 1 and 2 under the applied reaction conditions apparently leads to more effective formation of the active species responsible for the copolymerisation reaction, thus giving rise to different ratios of the species that do show activity towards ethene vs. the ones that do not show activity towards ethene. This leads to a different composition of the polymer mixture, and higher average ethene contents in the reactions with complexes 1 and 2 compared to 3.
Differences in the structures of the active species could also explain the counter-intuitive effect of the ethene concentration on the observed average ethene incorporation into the copolymer mixture. A higher concentration of ethene with respect to EDA enhances coordination of ethene to the metal centre and thus slows down the polymerisation by competition with EDA for a vacant site (vide supra). Higher amounts of ethene incorporation are expected under such conditions, but lower amounts are actually observed. Most likely, the rate of consecutive EDA insertions at the Rh-species responsible for the formation of EDA homopolymers is less affected by increasing the ethene pressure than the overall rate of the copolymerisation reaction, which is catalysed by a different active species. As a result of a different pressure dependence of the reaction rates of both polymerisation reactions, the product ratio produced in the reaction mixture changes in favour of the homopolymer. The average ethene content in the copolymer mixture therefore decreases, while the ethene content within the copolymer fraction has likely increased at higher ethene pressures/concentrations (but simply less copolymer is formed). At lower ethene concentrations (i.e., 0.75 bar vs. 2 bar) the chances for ethene incorporation in the copolymerisation reaction decrease in favour of EDA insertions. This leads to a decreased ethene content in the copolymeric chains, which also leads to a lower average ethene content.
Alternatively, ethene might have an influence on the catalyst activation process, possibly affecting the relative amounts of active Rh species formed. This could well influence the ratio of species responsible for EDA homopolymerisation versus species responsible for ethene–EDA copolymerisation, thus causing a similar effect on the ratio of EDA homopolymers versus ethene–EDA copolymers as described above. To rule out this possibility, we first incubated the catalyst with EDA for 15 min to allow the formation of the active species in an ethene-free environment, after which the system was pressurized with ethene (both 2 bar and 6 bar). In these cases, the amount of ethene incorporated into the polymers and their yields and Mw were similar to those reported in Table 2, indicating that the catalyst activation process is not significantly affected by the presence of ethene. Most likely, the pressure dependence of the average ethene content is therefore a result of the above-described structural differences between the active species responsible for homopolymerization of EDA and those leading to EDA–ethene copolymerization.
The exact structure of the active species responsible for ethene–carbene copolymerisation remains elusive. As stated above, excess EDA is necessary to achieve polymerisation activity. This, and the formation of multiple oligomer/polymer products (associated with vigorous N2 gas formation) right after the start of the reaction, hampered detailed NMR studies of the catalyst activation process.
2. Characterisation by NMR
All copolymers were analysed by NMR in CDCl3 at room temperature. All 1H NMR spectra reveal incorporation of both monomers. Resonances of the carbene units are observed at 4.2 ppm (CH2CH3), 3.15 ppm (backbone CH) and 1.2 ppm (CH2CH3), while ethene units give rise to a peak at 1.3 ppm, which overlaps with the CH2CH3 signal of the ester groups. The composition of the copolymers in terms of ethene content has already been discussed above, and the values were obtained by comparing the integral value of the backbone of the ester block at 3.1 ppm and the integral value at 1.2 ppm corrected for the contribution of the ester CH3 groups. The additional value of the integral at 1.2 ppm is ascribed to –(CH2–CH2)n– blocks. These two integral values give the molar ratio of CH(COOEt) units and CH2–CH2 units, which was further transformed into the molar composition in terms of percentage as listed in the corresponding tables. Additional signals (e.g., CH2 units at the end of a block or at the branches, vide infra) were not taken into account. NMR analysis confirmed that the reactions yield a mixture of EDA homopolymers57 and ethene–EDA copolymers. Spectra of different samples from the same copolymer batch varied in ethene content (Fig. 4), showing that the thus formed polymeric material is inhomogeneous in nature. These fractions were obtained by subjecting the copolymer samples to CHCl3 for a short time, allowing a rough qualitative separation based on small solubility differences. The bottom spectrum reveals that part of the copolymers contain a very high ethene content (up to 70%).59 All fractions were soluble in CDCl3 at room temperature, thereby excluding enhancement of the signal at 1.3 ppm by polyethene homopolymers and thus confirming ethene incorporation into the copolymers. The fact that these copolymers have very similar properties to carbene homopolymers and, as a result, are very difficult to separate from PEA indicates that they might well have desirable properties to function as blending agents.53 This underlines the potential of this new copolymerisation approach.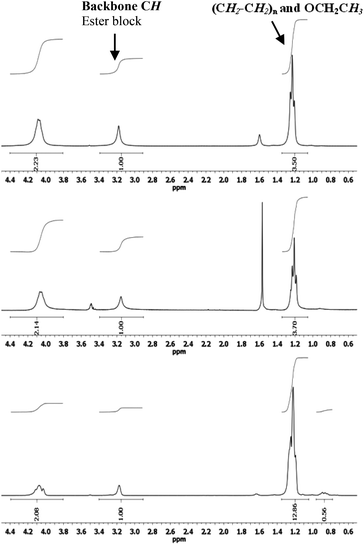 | ||
| Fig. 4 1H NMR spectra of three fractions of the same copolymer sample with increasing ethene content (top to bottom) emphasizing the inhomogeneous nature of the sample. | ||
While the EDA homopolymers are obtained as linear chains, the copolymers are branched. As a representative example, Fig. 5 displays the 1H NMR spectrum of copolymers obtained with catalyst 1 containing 9% ethene. The spectrum reveals that the copolymers have a blocky microstructure, indicated by the presence of a –(CH(COOEt)n– block (4.2 ppm (CH2CH3), 3.15 ppm (backbone CH) and 1.2 ppm (CH2CH3)) and a –(CH2–CH2)n– block. The signal of the backbone of the ester block is relatively sharp, indicating that a syndiotactic block is formed. The signals of the groups at the block interface were observed at 2.9 ppm (CH(COOEt)–(CH2–CH2)n) and 1.6 ppm ((CH(COOEt))n–CH2). Random incorporation of ester units (or CH2–CH2 units) most likely takes place as well. This is most probably responsible for the somewhat low integral value observed for the backbone signal of the ester block with respect to that of the ester CH2 units (i.e., 1:∼2.15 instead of 1:2). However, additional signals for these groups were not observed (likely due to signal overlap). The branched nature of the copolymers was emphasised by the observation of a signal at 2.1 ppm, which corresponds to the backbone signal of CH(branch) moieties. Both alkyl branches and branches containing ester groups are present in the copolymer. The latter give rise to signals at 2.3 ppm due to the presence of CH2COOEt groups at the end of a branch. The neighbouring CH2 signals were observed at 1.6 ppm and their correlation was confirmed by COSY experiments. Based on integration, the average degree of branching is one per 100 backbone carbon units, but this value is underestimated due to the presence of EDA homopolymers in the sample, which contribute to a higher integral value for the backbone signal.
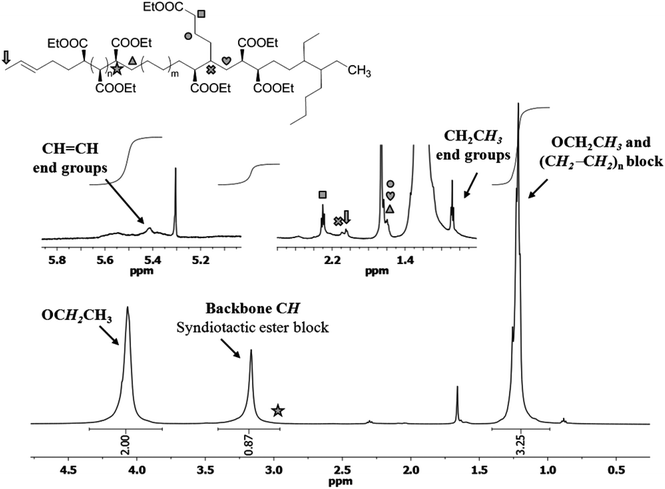 | ||
| Fig. 5 1H NMR spectrum of a copolymer sample with 9% ethene in CDCl3 at room temperature. | ||
End-groups were observed as CH2CH3 signals at 0.9 ppm, and in some cases signals were observed at 5.4 ppm (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) and 2.05 ppm (CHCH3) indicative of internal olefinic end groups formed by β-hydride elimination. To study the origin of this branching, we performed a computational study, the results of which are described in the ESI.†
CH) and 2.05 ppm (CHCH3) indicative of internal olefinic end groups formed by β-hydride elimination. To study the origin of this branching, we performed a computational study, the results of which are described in the ESI.†
13C NMR analysis confirms the presence of blocks and as a representative example the spectrum of a copolymer sample with 9% ethene is depicted in Fig. 6. The –(CH(COOEt))n– blocks give rise to signals at 171 ppm (CO), 60 ppm (OCH2CH3), 45 ppm (backbone CH, syndiotactic block) and 14 ppm (OCH2CH3), while the –(CH2–CH2)n– blocks are observed at 29.7 ppm. Detailed analysis of the branching pattern was hampered by the small average amount of ethene that is incorporated into the copolymers and the fact that they exist as a mixture with EDA homopolymers. However, with the help of proton-carbon correlation spectroscopy (HMQC) additional carbon signals could be assigned. The CH2 unit next to an ester block is observed at 31ppm, and in the HMQC spectrum a low-intensity signal at 34 ppm was observed, corresponding to the CH2COOEt groups at the end of a branch. The next CH2 group in this branch was observed at 24 ppm, although this signal might as well stem from the backbone carbon of a CH2 group in-between a branch point and an ester block. The signal of the CH backbone of the ester unit next to a –(CH2–CH2)n– block is expected to appear at 45 ppm, but cannot be observed due to overlap with the CH backbone signals of the other units in the ester block. The CH3 end group signal is not observed separately for similar reasons. Additional signals were not observed.
 | ||
| Fig. 6 13C NMR spectrum of a copolymer sample with 9% ethene in CDCl3 at room temperature. | ||
1H NMR analysis of the oligomeric fraction revealed that part of the oligomers are formed with syndiotactic blocks, while the majority is formed as atactic material. The formation of syndiotactic oligomers is specific for these copolymerisation reactions and is not observed in the homopolymerisation of EDA. The atactic oligomers dissolve readily in MeOH, but the syndiotactic material is slightly less soluble, thereby facilitating separation by precipitation with MeOH. Thus, an atactic fraction and a syndiotactic-rich fraction containing 64% syndiotactic material and 36% atactic oligomers were isolated. Representative 1H NMR spectra of these fractions are depicted in Fig. S4.† Signals similar to the high-Mw copolymers are observed. The ethene content in both fractions turned out to be 4%, present as –(CH2–CH2)n– blocks. Random incorporation or branching could not be detected for these oligomers, due to overlap with the broad signal of the backbone of the atactic material. This indicates that the different active species responsible for both oligomer fractions reveal similar activity towards ethene, and both fractions are most likely a mixture of homo- and copolymers as is the case for the high-Mw polymers.
3. Mass analysis
Incorporation of both ethene and EDA units into the polymer chain was confirmed by mass analysis. Since the Mw of the polymers obtained in the reactions of ethene and EDA is too high for decent mass analysis, we analysed the oligomeric fraction obtained in the reaction catalysed by complex 1 by Maldi-ToF (Fig. 7). Two major series of signals are observed, both representing repeating carbene (CHC(OOEt)) units (Δm/z = 86). The series are separated by a mass difference of 28, corresponding to the mass of repeating ethene units. The oligomer chains bear a hydroxyl functionality on one end, and a vinylic end group on the other end, giving rise to a general structure of [HO–[(CH(COOEt))m–(CH2–CH2)n]–CHCH2]H+ (e.g. m = 10 and n = 3 for m/z = 989). This indicates that chain initiation occurs in the same way as for carbene homopolymerisation and most likely the chain starts with insertions of carbene units. This is in agreement with the above-described lack of activity of these catalysts towards ethene homopolymerisation. β-Hydride elimination is observed as the main chain-termination pathway. In contrast, carbene homopolymerisation terminates mainly via protonation, while β-hydride elimination has only a minor contribution to chain termination.59 Apparently, incorporation of ethene units enhances termination via β-hydride elimination and this is in agreement with the above-described observation of branches in the copolymers. The ethene content in the oligomer fraction is higher than the average ethene content of the polymeric fraction (e.g. 28.5% for m/z = 989). The possibility of incorporation of dinitrogen units stemming from the diazo compounds (Δm/z = 28, similar to ethene units) was ruled out by elemental analysis, revealing a low nitrogen content (<0.8%). The carbon and hydrogen content are increased compared to that of oligomers obtained by homopolymerisation of EDA (Table 3), emphasizing the incorporation of ethene units.
 | ||
| Fig. 7 MALDI-ToF spectrum of the oligomeric fraction obtained in the copolymerisation of ethene and EDA. | ||
| %C | %H | %N | |
|---|---|---|---|
| Homo-oligomers of EDA | 51.22 | 6.36 | 1.19 |
| Copolymers | 55.37 | 7.01 | 0.76 |
4. Thermal properties
Thermal analysis by differential scanning calorimetry (DSC) revealed that the melting and crystallisation properties of the ethene–EDA copolymer samples depend highly on the average ethene content (Table 4 and Fig. S5†). The copolymer samples obtained with catalyst 1 reveal three melting transitions, observed at 109 °C, 64 °C and 58 °C. The broad melting transition at 109 °C is a combination of two distinct melting transitions of the –(CH(COOEt))n– blocks in the copolymers and in the EDA homopolymers. Incorporation of CH2–CH2 units in the copolymers shifts the melting transition to slightly higher temperatures but this peak overlaps with that of the homopolymers. The two melting peaks at lower temperatures represent the fraction of branched copolymers, since the presence of branches is known to affect the crystallisation behaviour and typically leads to lower values for Tm, Tc and ΔH. An additional melting peak around 130 °C caused by the –(CH2–CH2)n– block could not be observed separately. Contribution of the different (co)polymers present in the sample to different transitions leads to an underestimation of the values for ΔH, since they are based on the total mass of the sample instead of the mass of the separate fractions. Upon cooling only one crystallisation peak was observed at 99 °C, with a relatively sharp onset and a long tail towards lower temperatures. This crystallisation temperature is clearly shifted towards higher temperatures compared to PEA homopolymers as a result of incorporation of ethene, which is in agreement with the thermal behaviour of similar copolymers prepared from EDA and diazomethane.52| Entry | Catalyst | Mol% ethene | T m (°C) | T c (°C) | ΔH |
|---|---|---|---|---|---|
| a 30 °C–180 °C or 30 °C–200 °C, 10 °C min−1; Values are taken from the 2nd cycle. b PEA homopolymers with a Mw of 120 kDa. c Copolymers obtained at an ethene pressure of 6 bar. d PEA homopolymers with a Mw of 800 kDa. e PEA homopolymers with a Mw of 164 kDa. | |||||
| 1 | 1 | 9 | 110 | 99 | 17 |
| 64 | 0.5 | ||||
| 58 | 0.2 | ||||
| 2b | 1 | — | 105 | 77 | 21 |
| 3c | 1 | 4 | 103 | 78 | 13 |
| 4 | 2 | 11 | 125 | 84 | 16 |
| 5d | 2 | — | 123 | ||
| 6 | 3 | 2 | 122 | 84 | 17 |
| 7e | 3 | — | 122 | 100 | 9 |
| 88 | 2 | ||||
Copolymer samples having 4% ethene incorporation obtained with catalyst 1 at an ethene pressure of 6 bar reveal only one melting and crystallisation peak, very similar to PEA homopolymers (entry 3). This is in agreement with the larger amount of PEA homopolymers present in this sample as is discussed in section 1, and apparently they dominate the thermal properties of the sample. However, the value for heat change is lower than that of PEA homopolymers, due to the presence of branched copolymers that affect the crystal packing. Additional thermal transitions of these copolymers were not observed.
Copolymers made by catalyst 2 show higher values for Tm and Tc than the copolymers made by complex 1, as a consequence of their increased Mw. Similar effects of the thermal behaviour on changes in the molecular weights are observed for PEA homopolymers (entry 2 vs. 5), although the Tm and Tc of the copolymers appear at slightly higher values due to the incorporation of CH2–CH2 units (entry 4). Additional peaks were not observed.
The copolymers made by catalyst 3 show a similar thermal behaviour to that observed for the corresponding homopolymers, as is expected, since the incorporation of ethene into these copolymers is very small and the effect is therefore negligible (entries 6 and 7). The slight bimodal molecular-weight distribution (see ESI† for a discussion on the SEC data) is reflected in the observation of two crystallisation peaks, which appear at slightly higher temperatures than the crystallisation of the corresponding homopolymers due to their increased Mw (entry 6 vs. 7).
Conclusions
In this paper we show as a proof-of-concept that it is possible to copolymerise carbenes with ethene using late transition metal catalysts. High-Mw, highly functionalised copolymers were obtained upon reaction of ethyl diazoacetate (EDA) and ethene in the presence of different Rh catalyst precursors. These pre-catalysts are converted in situ to several catalytically active species, which exhibit a different reactivity to ethene. As a result, the reactions yield a mixture of carbene homopolymers (PEA) and random PE-PEA copolymers. Ethene incorporation could be achieved up to a level of 11 mol% averaged over the whole copolymer mixture. Analysis of different fractions of the same copolymer batch revealed inhomogeneous polymer mixtures in which some copolymer fractions contain ethene up to 70%. The properties of these copolymers are similar to those of the EDA homopolymers, thereby hampering their separation. This behaviour emphasizes that ethene–EDA copolymers have desirable properties, which make them potentially suitable as blending agents. Since the active species responsible for the formation of carbene homopolymers are believed to be (allyl)RhIII(alkyl) species, we think that applying slight changes to the ligand structure of such (allyl)RhIII(alkyl) species could well lead to the formation of more selective catalysts for the formation of ethene–EDA copolymers, thus avoiding the formation of copolymer mixtures.Attempts to increase the ethene content by increasing the ethene pressure unexpectedly resulted in a lower average incorporation. This is likely a result of the reactivity difference of the different active Rh-species formed under the applied reaction conditions. Higher ethene concentrations slow down the copolymerisation process (mediated by yet unidentified Rh-species) compared to the formation of homopolymers (mediated by different Rh-catalysts; most likely (allyl)RhIII-alkyl species), thereby changing the product ratio in favour of the homopolymer. The average ethene content in the copolymer mixture therefore decreases. Similar results were obtained upon diluting the reaction mixture and thus increasing the (dissolved) ethene content relative to EDA, emphasizing that this concentration dependence is general.
Consecutive insertions of both carbene units and ethene take place, giving rise to copolymers with a blocky –(CH2–CH2)n–(CHC(OOEt))n– microstructure of which the ester block is highly stereoregular (syndiotactic). Branching does occur, leading to the formation of both alkyl branches and branches containing ester functionalities. Incorporation of both monomers was confirmed by MALDI-ToF analysis. The thermal behaviour of the copolymers depends on the ethene content, leading to higher values for Tm and Tc. The branched nature is reflected in the observation of additional melting transitions at lower temperatures, as a result of less favourable crystallisation. The obtained materials are therefore unique and differ from their linear analogues obtained by copolymerisation of functional carbenes with diazomethane or dimethylsulfoxonium ylides.
Experimental section
The syntheses of (1,5-cyclooctadiene)Rh(L-pro) (1),43 (1,5-dimethyl-1,5-cyclooctadiene)Rh(L-prolinate) (2),46 [(1,5-cyclooctadiene)RhCl]2 (3),60 [Rh(PhN3Ph)(HO-C8H11)] (4),61 [(Cn)RhMe](BF4)2 (5)53 and (S3-ethano-9)RhCl362 have been reported previously. Complex 2 was ‘aged’ prior to use by exposure to air according to procedures described previously.44,46 Dichloromethane was dried using an M-Braun SPS (Solvent Purification System). All other solvents were used directly from the bottle, without further purification steps. Other chemicals were purchased from chemical suppliers and used as received without further purification. NMR analyses were carried out on a Varian Mercury 300 spectrometer (300 MHz for 1H and 75 MHz for 13C, respectively), a Bruker DRX 300 spectrometer (300 MHz for 1H and 75 MHz for 13C, respectively) and a Varian IMC 500 spectrometer (500 MHz for 1H and 125 MHz for 13C, respectively). Molecular mass distributions were measured using size exclusion chromatography (SEC) on a Shimadzu LC-20AD system with two PLgel 5 μm MIXED-C columns (Polymer Laboratories) in series and a Shimadzu RID-10A refractive index detector, using dichloromethane as mobile phase at 1 mL min−1 and T = 35 °C. Polystyrene standards in the range of 760–1880000 g mol−1 (Aldrich) were used for calibration of the SEC column. IR analysis was carried out on a Shimadzu FTIR-8400S Fourier transform infrared spectrophotometer, equipped with an MKII Golden Gate single reflection ATR system. DSC was measured using a Perkin Elmer Jade DSC with closed aluminium cups under an N2 atmosphere (flow 5 mL min−1). Heating and cooling rates of 10 °C were used over a temperature range of 30–200 °C.
General procedure for ethene–EDA copolymerisations
A solution of catalyst in DCM (5 mL) was pressurised with ethene (2 bar, unless stated otherwise) and the system was stirred for 2 hours at room temperature while the ethene pressure was kept at 2 bar. EDA (2 mmol) was added to the system and the mixture was allowed to stir overnight at constant ethene pressure. After venting the system, the volatiles were removed in vacuo and MeOH (ca. 30 mL) was added to the remaining yellow oil. The white polymeric precipitate was removed by centrifugation and washed three times with fresh MeOH. The combined supernatant fractions were evacuated to remove the solvent, yielding the crude oligomer mixture as a yellow oil.Acknowledgements
We kindly thank the Dutch Polymer Institute DPI, PO Box 902, 5600 AX Eindhoven, The Netherlands, (project #646 and #647) and the European Research Council (ERC, EU, 7th framework program, grant agreement 202886-CatCIR) for financial support.Notes and references
- A. O. Patil, Chem. Innovation, 2000, 30, 19 Search PubMed.
- T. C. Chung, Functionalization of Polyolefins, Academic Press, London, 2002 Search PubMed.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS.
- I. Novak, E. Borsig, L. Hrckova, A. Fiedlerova, A. Kleinova and V. Pollak, Polym. Eng. Sci., 2007, 47, 1207 Search PubMed.
- J.-Y. Dong and Y. Hu, Coord. Chem. Rev., 2006, 250, 47 CrossRef CAS.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267 RSC.
- M. R. Buchmeiser, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH Verlag GMBH, 2009, p. 197 Search PubMed.
- A. Leitgeb and J. Wappel, Polymer, 2010, 51, 2927 CrossRef CAS.
- H. Mutlu, L. Montero de Espinosa and M. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC.
- A. R. Padwa, Prog. Polym. Sci., 1989, 14, 811 CrossRef CAS.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- H. F. Mark, N. M. Bikales, C. G. Overberger and G. Mendes, in Encyclopedia of Polymer Science and Engineering, Wiley, New York, 1986, vol. 13 Search PubMed.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120 CrossRef CAS.
- H.-C. Kim, S. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS.
- A. Leblanc, E. Grau, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Macromolecules, 2011, 44, 3293 Search PubMed.
- A. Berkefeld and S. Mecking, Angew. Chem., Int. Ed., 2008, 47, 2538 CrossRef CAS.
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS.
- S. Collins and D. G. Ward, J. Am. Chem. Soc., 1992, 114, 5460 CrossRef CAS.
- A. D. Bolig and E. Y.-X. Chen, J. Am. Chem. Soc., 2001, 123, 7943 CrossRef CAS.
- H. Deng, T. Shiono and K. Soga, Macromolecules, 1995, 28, 3067 CrossRef CAS.
- D. Takeuchi, Dalton Trans., 2010, 39, 311 RSC.
- L. K. Johnson, A. Bennet, K. Dobbs, E. Hauptman, A. Ionkin, S. D. Ittel, E. F. McCord, S. J. McLain, C. Radzewich, Z. Yin, L. Wang, Y. Wang and M. Brookhart, PMSE Prepr., 2002, 86, 319 Search PubMed.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS.
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS.
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 744 RSC.
- D. Guironnet, P. Roesle, T. Rünzi, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2009, 131, 422 CrossRef CAS.
- E. Ihara, Adv. Polym. Sci., 2010, 231, 191 Search PubMed.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706 RSC.
- N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. de Bruin, Catal. Sci. Technol., 2011, 1, 153 RSC.
- E. Ihara, N. Haida, M. Iio and K. Inoue, Macromolecules, 2003, 36, 36 CrossRef CAS.
- E. Ihara, M. Fujioka, N. Haida, T. Itoh and K. Inoue, Macromolecules, 2005, 38, 2101 CrossRef CAS.
- E. Ihara, M. Kida, M. Fujioka, N. Haida, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1536 CrossRef CAS.
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, Polym. J., 2008, 40, 1094 CrossRef CAS.
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1638 CrossRef CAS.
- E. Ihara, Y. Ishiguro, N. Yoshida, T. Hiraren, T. Itoh and K. Inoue, Macromolecules, 2009, 42, 8608 CrossRef CAS.
- E. Ihara, Y. Goto, T. Itoh and K. Inoue, Polym. J., 2009, 41, 1117 Search PubMed.
- E. Ihara, K. Saiki, Y. Goto, T. Itoh and K. Inoue, Macromolecules, 2010, 43, 4589 Search PubMed.
- E. Ihara, H. Takahashi, M. Akazawa, T. Itoh and K. Inoue, Macromolecules, 2011, 44, 3287 Search PubMed.
- E. Ihara, Y. Hara, T. Itoh and K. Inoue, Macromolecules, 2011, 44, 5955 Search PubMed.
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Polym. Chem., 2011, 2, 422 RSC.
- D. G. H. Hetterscheid, C. Hendriksen, W. I. Dzik, J. M. M. Smits, E. R. H. van Eck, A. E. Rowan, V. Busico, M. Vacatello, V. Van Axel Castelli, A. Segre, E. Jellema, T. G. Bloemberg and B. de Bruin, J. Am. Chem. Soc., 2006, 128, 9746 CrossRef CAS.
- E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631 CrossRef CAS.
- M. Rubio, E. Jellema, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Dalton Trans., 2009, 8970 RSC.
- E. Jellema, A. L. Jongerius, A. J. C. Walters, J. M. M. Smits, J. N. H. Reek and B. de Bruin, Organometallics, 2010, 29, 2823 CrossRef CAS.
- E. Jellema, A. L. Jongerius, G. Alberda van Ekenstein, S. D. Mookhoek, T. J. Dingemans, E. M. Reingruber, A. Chojnacka, P. J. Schoenmakers, R. Sprenkels, E. R. H. van Eck, J. N. H. Reek and B. de Bruin, Macromolecules, 2010, 43, 8892 CrossRef CAS.
- A. J. C. Walters, E. Jellema, M. Finger, P. Aarnoutse, J. M. M. Smits, J. N. H. Reek and B. de Bruin, ACS Catal., 2012, 2, 246 Search PubMed.
- A. J. C. Walters, O. Troeppner, I. Ivanović-Burmazović, C. Tejel, M. P. del Río, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2012, 51, 5157 Search PubMed.
- M. Finger, M. Lutz, J. N. H. Reek and B. de Bruin, Eur. J. Inorg. Chem., 2012, 9, 1437 Search PubMed.
- B. Bantu, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2007, 692, 5272 CrossRef CAS.
- N. M. G. Franssen, K. Remerie, T. Macko, J. N. H. Reek and B. de Bruin, Macromolecules, 2012, 45, 3711 Search PubMed.
- A. I. Olivos-Suarez, M. P. del Río, K. Remerie, J. N. H. Reek and B. de Bruin, ACS Catal., 2012, 2, 2046 Search PubMed.
- L. Wang, C. Wang, R. Bau and T. C. Flood, Organometallics, 1996, 15, 491 CrossRef CAS.
- L. Wang and T. C. Flood, J. Am. Chem. Soc., 1992, 114, 3169 CrossRef CAS.
- L. Wang, R. S. Lu, R. Bau and T. C. Flood, J. Am. Chem. Soc., 1993, 115, 6999 CrossRef CAS.
- Instead of a pure EDA homopolymer, the mixture could of course contain EDA–ethene copolymers with a very low ethene content (which is almost indistinguishable from a pure EDA homopolymer).
- M. Finger, J. N. H. Reek and B. de Bruin, Organometallics, 2011, 30, 1094 Search PubMed.
- The fact that copolymer fractions containing 70% ethene are soluble in CHCl3 at room temperature indicates that they have a rather low molecular weight. This is confirmed by integration of the 1H NMR signals belonging to the polymer end groups and those of the main chain. Since size exclusion chromatography revealed that the Mw of the copolymer sample is rather high, this implies that these low-Mw copolymers containing very high values of ethene make up only a small fraction of the total copolymer sample.
- G. Giodani and R. H. Crabtree, Inorg. Synth., 1990, 28, 88 CAS.
- C. Tejel, M. Ciriano, E. Sola, M. P. del Río, G. Ríos-Moreno, F. J. Lahoz and L. A. Oro, Angew. Chem., Int. Ed., 2005, 44, 3267 CrossRef CAS.
- S. Timonen, T. T. Pakkanen and T. A. Pakkanen, J. Mol. Catal. A: Chem., 1996, 111, 267 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular-weight distributions, chain growth–time profiles, detailed overview of thermal behaviour. See DOI: 10.1039/c3dt32941k |
| This journal is © The Royal Society of Chemistry 2013 |