DOI:
10.1039/C3DT32916J
(Paper)
Dalton Trans., 2013,
42, 10587-10596
Synthesis and properties of a novel quarternerized imidazolium [α-PW12O40]3− salt as a recoverable photo-polymerization catalyst†
Received
5th December 2012
, Accepted 13th May 2013
First published on 14th May 2013
Abstract
A recoverable photo-polymerization catalyst based on an imidazolium and a polyoxometalate, viz., (BMIm)2(DMIm)PW12O40 (where, BMIm = 1-butyl-3-methylimidazolium, DMIm = 3,3′-dimethyl-1,1′-diimidazolium) is reported. It catalyzes photo-polymerization of several industrially important monomers like styrene, methyl methacrylate, butyl methacrylate and vinyl acetate. The catalyst is recoverable and hence can be reused. The molecular weight and polydispersity index can be controlled reasonably and a reaction pathway is proposed. The synthesis of this novel catalyst is reported and the catalyst has been characterized using standard techniques such as single crystal X-ray diffraction studies, cyclic voltammetry and various spectroscopic methods such as Fourier transform infrared spectroscopy, 1H NMR spectroscopy, EPR spectroscopy and UV-Vis spectroscopy. DFT calculations have been used to explain the catalyst's photo-chemical activity.
Introduction
The fast receding reserve of resources and growing demand of ‘green’ plastics need an immediate synthetic technique that can provide plastics but has a low impact on the reserve of resources. Although photo-polymerization is a step forward in that direction, however the condition of repeated addition of initiator often limits its viability for large scale production. Hence, development of a versatile yet recoverable photo-polymerization initiator catalyst would be a welcome green step forward in that direction. Here we report such a photo-catalyst. Logically, the design of such a catalyst should incorporate a photo-active radical or a cation generating component which will facilitate and catalyze the initiation step of the polymerization reaction, a polymerization propagating component, an oligomer stabilizing hydrophobic component and a catalyst regenerating component. Our catalyst design is based on hybrid materials and has two components, namely: (i) an inorganic [α-PW12O40]3− polyoxometalate and (ii) the organic moieties of 1-butyl-3-methylimidazolium (BMIm) and 3,3′-dimethyl-1,1′-diimidazolium (DMIm). We now explain the rationale of this design.
Until recently a class of metal–oxide based clusters, polyoxometalates, have gained significant interest in various environmental and materials science based applications, primarily due to their photo-catalytic activity. Well documented photo-catalytic activity and extreme high stability of one such polyoxometalate (POM) viz., PW12O403− prompted its choice as a photo-active radical generating component of the catalyst,1–7 whereas certain interesting structural and electronic features of the BMIm based ionic liquids, such as: (i) ability to provide the reaction system with a radical or cation propagating source (the imidazolium moiety); (ii) as in ionic liquids, ability to form extended hydrophobic channel like compartments which can stabilize the emerging oligomers8,9 led to the choice of BMIm+/DMIm+ as its second component.8–10 Moreover the POM's ability of redox regeneration was also exploited. In short, our design strategy of the catalyst exploits: (i) the photo-activity of POM coupled with the presence of imidazolium to catalyze radical/cationic polymerization initiation; (ii) combines it with the advantages of the imidazolium moiety; (iii) the stabilization of the emerging oligomeric chains in the extended hydrophobic compartments by the dangling butyl chains, thus rendering the catalyst functional and (iv) redox recoverability of POM under the stipulated reaction conditions. It is now apt to review the recent literature to explain further the rationale of our choice of polyoxometalate and an imidazolium component for the design of our catalyst. In the matter of recoverable polyoxometalate catalysts, several strategies have been adopted. Broadly, recoverability achieved can be grouped into three classes as following: (i) recoverability exploiting photo-catalytic redox behaviour of the polyoxometalates.1–15 (ii) Recoverability by heterogenization,16–26 by amphiphilic27,28 or dendritic encapsulation29,30 and heterogenization to facilitate magnetic separation31,32 (iii) recovery by membrane separation of the reacted catalyst.33 Moreover different classes of reactions that have been catalyzed using the above recoverable techniques can further be broadly grouped into categories like oxidation,27,29,32,34–40 acid mediated reactions including but not limited to, hydrolysis, esterification, trans-esterification, acid catalysis, hydration, dehydration28,41–46 and a few other allied reactions like the Mannich reaction and benzylation.31,47 The above observations led us to analyze the trends in recent catalyst design in the matter of recoverable catalysts based on polyoxometalates. This analysis reveals that polyoxometalate based catalysts have been exploited as recoverable acid catalysts in cationic polymerization. They have further the potential alone to act as photo-active redox recoverable catalysts in free-radical reactions as well.1–4 These observations immediately warrant the use of polyoxometalates as a reactive and recoverable cationic radical photo-polymerization catalyst which has not been explored to date to the best of our knowledge. Of the polyoxometalates, PW12O403− has been shown to be extremely catalytically and photo-catalytically active in the above aspects and is also extremely stable. Our past and recent endeavours have shown the cationic polymerization in vinylic bonds using related polyoxometalates.48–50 Here we take the next step. To act as a recoverable versatile photo-polymerization catalyst for both cationic and radical polymerizations, there is a need for a common radical cation source that could act in synergy with polyoxometalate component, and a simple hydrogen free radical or a proton does not suffice for this purpose, hence a simple H3PW12O40 would not have sufficed as a catalyst. On the other hand, versatile polymerization using ionic liquids is a well-known phenomenon.51 A closer look at the literature reveals that the imidazolium moiety in the ionic liquids is responsible for this versatility, due partly to its ability to form both cations and radicals.51,52 Furthermore, it has been shown in the matter of construction of organic electronics that polyoxometalates can stabilize transient organic cation radicals, conducive to our design of a highly reactive, recoverable cationic radical photo-polymerization catalyst.53 Hence we chose substituted imidazolium component as a counter-ion for the design of our catalyst. Moreover, it is also worth noting that such ionic liquid based substituted imidazolium moieties are known to form localized hydrophobic pockets in liquid state by retaining their channel structure.76–86 This substitution in imidazolium by hydrophobic butyl and methyl moiety was, therefore, aimed to address the aspect of generation of hydrophobic compartment for stabilization of nascent oligomeric components. Thus hydrophobic butyl chains were introduced in the imidazolium component of the catalyst.
We now describe in the following sections the synthesis of this catalyst, its characterization techniques; along with the details of polymerization reactions, recovery of the catalyst, loading of the catalyst, reaction kinetics of polymerization, effect of addition of inhibitors and details of spectroscopic and density functional theoretical (DFT) calculations and propose a pathway for its action.
Experimental
Monomers used
All the chemicals used in these experiments were of reagent grade and were used without further purification. The ionic liquid 1-butyl-3-methylimidazolium (BMIm) chloride used in the experiments was prepared according to the method as available in the literature using available precursors.54
In a quartz tube, 0.2 g of the catalyst, (BMIm)2(DMIm)PW12O40 was mixed with ca. 3–5 ml of the monomer (styrene, methyl methacrylate, butyl methacrylate, vinyl acetate). The mixture was dissolved in 20 ml acetone. The mixture was bubbled with nitrogen for 15 minutes and the tube was closed tightly for isolation from air. The tube was then placed in a XPA-1 photo-reactor, operating with a Hg lamp at 300 W and λ = 365 nm for 2–3 hours. Within a few minutes, a blue hue appeared in the reaction tube. The reaction was terminated by adding an excess of acidified methanol. Solvents were evaporated under vacuum which leaves behind a mixture of polymer and the catalyst. The polymer was then separated by dissolving it in THF and was characterized further by GPC. The catalyst, which is insoluble in THF, was separated by centrifugation and washed 2–3 times with n-hexane and THF before drying under vacuum at room temperature. The catalyst, upon drying, underwent several changes of colour (like, from blue to yellow to dirty yellow) till a colourless white powder was formed, upon exposure to air. The catalyst was thus recovered. This recovered catalyst was then reused for the next polymerization cycle, recovered back and reused again. The catalytic activity of the catalyst was tested for 10 such polymerization cycles. The different steps of the reaction are shown in Fig. 1.
 |
| | Fig. 1 Different steps of the reaction. (a) Initial mixture of reactants in solvent acetone with catalyst (colourless, slightly turbid). (b) Reactant tube inside photo-reactor at t = 0 min. (c) After reaction, a blue hue appears in the reactant tube, at t = 30 min. (d)–(g) Drying of the solvent and successive change of color of the catalyst in the course of drying. (h) Quantitative recovery of the catalyst after polymerization. For comparison the catalyst before polymerization is shown. The slight bluish hue in the recovered catalyst disappears when left in the open air for some time. | |
Synthesis of the catalyst (BMIm)2(DMIm)PW12O40
To a solution of the starting polyoxometalate (H3PW12O40·xH2O) (2.88 g, 0.01 mol) in 20 ml water, the ionic liquid of BMImCl (17.45 g, 0.1 mol), was dropped slowly with vigorous shaking (note: the molar ratio of polyoxometalate to BMImCl is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) to form a milky white viscous dispersion. The mixture was kept at 30 °C for 24 hours until the reaction was complete. The dispersion was then allowed to stand and the white solid precipitate so formed was collected by vacuum filtration. After drying the precipitate completely under low pressure, the product was recrystallized from an equal volume mixture of acetone and acetonitrile. Upon standing at room temperature for 7 days, chunks of colourless crystals were formed, which were used for single crystal X-ray diffraction analysis. Note: for the polymerization experiments, both the crystals as well as the powder are equally effective.
:10) to form a milky white viscous dispersion. The mixture was kept at 30 °C for 24 hours until the reaction was complete. The dispersion was then allowed to stand and the white solid precipitate so formed was collected by vacuum filtration. After drying the precipitate completely under low pressure, the product was recrystallized from an equal volume mixture of acetone and acetonitrile. Upon standing at room temperature for 7 days, chunks of colourless crystals were formed, which were used for single crystal X-ray diffraction analysis. Note: for the polymerization experiments, both the crystals as well as the powder are equally effective.
Characterization of the catalyst
Crystal data for the catalyst C24H45N6O40PW12.
The crystal structure of the grown crystal of the catalyst was determined from the single crystal X-ray diffraction data. The intensity data were collected on a BRUKER SMART APEX CCD diffractometer with Mo Kα (λ = 0.71073 Å) at 291 K. The structure was solved by direct methods and refined using full-matrix least squares calculations with anisotropic displacement parameters for all non-hydrogen atoms. All hydrogen atoms were geometrically fixed to allow riding on the parent atoms to which they are attached and refined with individual isotropic displacement parameters. Unit cell parameters: a = 17.895(5) Å; b = 18.391(5) Å; c = 16.676(4) Å. Space group: Pbcn.
Details of spectroscopic methods employed
FTIR spectra were recorded as KBr pellets with a Nicolet 380.
FT-IR spectrometer in the range of 4000–500 cm−1. X-band EPR spectra were recorded on a Bruker ESP 300 spectrometer. 1H NMR spectra were recorded on a 400 MHz Bruker Avance Spectrometer. UV-Vis spectra were recorded in a solution of the catalyst in the range 200–1000 nm with a Shimadzu UV-160A spectrophotometer and evaluated with a program associated with the spectrometer.
Density functional theory (DFT) calculation details for the catalyst
The structural geometry of the catalyst was fully optimized (assuming C1 symmetry) with BP8655,56 generalized gradient approximations and VWN57 local density functional, triple-ζ plus polarization Slater type orbital basis sets (TZP), and the numerical integration parameter 6.0, as implemented in the Amsterdam density functional (ADF) package.58–60 The core shells [O: (1s)2, P: (1s2s2p)10, W: (1s2s2p3s3p3d4s4p4d)46] were kept frozen and were described by means of single Slater functions. The zero-order regular approximation was adopted in all of the calculations to account for the scalar relativistic effects.61–63 The solvent effects were employed in the calculations of geometry optimization by using a conductor-like screening model (COSMO)64 of solvation with the solvent-excluding-surface.65 The solute dielectric constant was set to 20.7 (acetone). The van der Waals radii for the POM atoms, which actually define the cavity in the COSMO, are 1.40, 1.92, and 2.10 Å for O, P, and W, respectively.66–73
Electrochemical characterization of the catalyst
The electrochemistry measurements were performed with a 69 CHI 660 electrochemistry workstation. Three-electrodes were employed for the electrochemical studies, viz., gold (Au) as working electrode, modified with the complex in acetone solution to emulate the working conditions of the catalyst. Secondly, platinum (Pt) disk was used as another counter electrode, and the third reference electrode used was a standard saturated calomel reference electrode (SCE). The oxidized gold working electrode surface was created using standard protocol i.e., by applying 2.0 V vs. SCE for 90 seconds in 2 M NaOH solution.
Determination of recovery of the catalyst
The recovery of the catalyst was tested exhaustively by gravimetric analysis. The weight and FTIR spectra of the recovered catalyst and its activity were recorded and compared after every polymerization cycle. Before any measurements, the catalyst was dried in open air overnight and then kept in an oven for 30 minutes at 50 °C.
Variation of catalyst loading
Polymerization reactions were carried out by increasing the relative concentration of an equimolar mixture of BMIm+ and DMIm+ with respect to the catalyst, thereby changing the effective loading of the catalyst. (Note in these cases chloride was used as counter-ion and the effect does not change with other halides.) This variation of loading was carried out at 5 equi-spaced mole-ratios of BMIm+ and DMIm+ to the catalyst ranging from 1 to 5.
To the polymerization reaction mixture, o-, m- and p-dihydroxy phenols were added in increasing ratio with respect to the catalyst for a fixed monomer concentration. It is well-known in the literature that polyoxometalates scavenge the dihydroxy phenols under photo-catalytic conditions used here.1–7,11–15 Hence the above di-hydroxy phenols were chosen as inhibitors in this study to investigate the role of the polyoxometalate component in the catalyst.
Gel permeation chromatography (GPC) for determining molecular weight (Mn) and polydispersity index (PDI)
Molecular weight and molecular weight distribution of the polymers were measured by PL GPC 50 at 25 °C using THF as eluent against polystyrene standards, flow rate: 1 mL min−1, sample concentration: 1 mg mL−1.
Results and discussion
The photo-polymerization catalyst reported here is found to polymerize a series of industrially important monomers. We here present the results of catalytic photo-polymerization of styrene, methyl methacrylate, vinyl acetate and butyl methacrylate. It is worth noting that the catalyst can photo-polymerize in the absence of any externally added co-initiators. The (BMIm)2(DMIm)PW12O40 salt itself acted as an initiator-catalyst. Polymerization reactions of the monomers with this catalyst take place readily under a 300 W, 365 nm light with an irradiation time of 2–3 hours at room temperature in acetone. Within a few minutes of irradiation, the contents of the tube develop a blue color due to the reduction of the POM moiety of the photo-catalyst. Time dependent UV-Vis spectrum of the reaction mixture presents two absorption bands with maxima at 470 nm and 750 nm (Fig. 2), which is characteristic of one-electron-reduced POM, viz. [PW12O40]4−.88,89
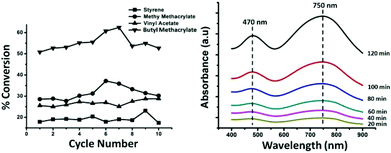 |
| | Fig. 2 Left: conversion of the monomers over 10 successive polymerization cycles; right: time dependent UV-Vis spectrum of the reaction mixture at different time steps. | |
It can be clearly seen from the spectrum that the extent of reduction increases with time, clearly indicating the participation of POM in photo-polymerization. This is discussed in detail later. The percentage of POM photo-reduced at the end of the reaction was estimated to be 39% from the reported molar extinction coefficient of [PW12O40]4− at 750 nm (ε750 = 2000 M−1 cm−1).86 The extent of conversion of the polymer obtained over 10 successive cycles of polymerization for 4 monomers, viz. styrene, methyl methacrylate, butyl acrylate and vinyl acetate are recorded (Fig. 2). It shows that for all 4 monomers the conversions vary from 20% to 60%. The optimization of the reaction conditions was achieved by varying two main operational parameters, viz. the catalyst loading and the irradiation time. It was ensured that the reaction temperature is maintained at room temperature, in order to meet two needs: (i) reduction of chain transfer and (ii) making the process less energy consuming. The control in Mn and PDI is attained by usual variation of the monomer concentration, as expected for any controlled polymerization.
Effects of catalyst and inhibitor loading on Mn of the polymers and its implications on the polymerization pathway
Now we find the effect of the catalyst loading on the Mn and PDI of the synthesized polymers. We do so to understand the pathway of this polymerization reaction. For this purpose, polymerization reactions were carried out by systematically increasing the relative concentrations of an equimolar mixture of BMIm+ and DMIm+ with respect to the catalyst. Specifically, we varied the concentration ratio [BMIm + DMIm]/[initiator] from 1 to 5 and analysed its effect on the Mn and PDI of the resulting polymer. In all these cases chloride was the counter-ion and the effect does not change if other halides are used. We observe that on increasing the concentration of BMIm+ and DMIm+, the Mn of the synthesized polymers increases, while the PDI decreases (Fig. 3). It only means that with increasing BMIm+ and DMIm+ concentrations, the relative concentration of the radicals responsible for polymerization reduces.74 The question arises: are the radicals responsible for polymerization coming from a source directly related to the polyoxometalate part of the catalyst? To test this, we added to the reaction mixture o-, m- and p-dihydroxy phenols as inhibitors as they can quench the polyoxometalate radicals. We added these di-hydroxy phenols in increasing ratio with respect to the catalyst, as these di-hydroxy phenols will scavenge the photo-excited POM and alter the Mn of the resulting polymer accordingly. We indeed observe that as the loading of these dihydroxy phenols (o-, m- or p-) increases relative to the polyoxometalate, the Mn of the polymers increase almost monotonically (Fig. 4). This shows that with an increase in dihydroxy phenol loading, lesser polyoxometalate radicals are available for polymerizing the monomer, since they are quenched by the phenols under our experimental conditions and are consumed in the process. Hence, lesser radicals are available and the Mn of the polymers obtained increases. These results, in turn, also show that the participation of polyoxometalate radicals plays a key role in photo-polymerization.
 |
| | Fig. 3 Increasing Mn (■) and decreasing PDI (●) with increasing BMIm and DMIm concentration to a fixed catalyst concentration during butyl methacrylate polymerization. (Same trend is obtained for other monomers.) | |
 |
| | Fig. 4 Increasing Mn with increasing loading of 3 different dihydroxy phenols for different monomers. | |
On the photo-catalytic activity of the catalyst and the reaction pathway
We now explain the photo-catalytic activity of the catalyst. To do so we take recourse to DFT calculations. The DFT calculations reveal a band-gap energy of 2.729 eV between the HOMO (Fig. 5a) and LUMO (Fig. 5b) of the polyoxometalate part of the catalyst.
![(a) HOMO and (b) LUMO of [α-PW12O40]3−, (c) molecular electrostatic potentials for [α-PW12O40]3−. Red regions identify nucleophilic areas whereas blue regions represent electrophilic areas, (d) EPR spectra of a reaction mixture initiated by (BMIm)2(DMIm)PW12O40 after 10 minutes of reaction in acetone.](/image/article/2013/DT/c3dt32916j/c3dt32916j-f5.gif) |
| | Fig. 5 (a) HOMO and (b) LUMO of [α-PW12O40]3−, (c) molecular electrostatic potentials for [α-PW12O40]3−. Red regions identify nucleophilic areas whereas blue regions represent electrophilic areas, (d) EPR spectra of a reaction mixture initiated by (BMIm)2(DMIm)PW12O40 after 10 minutes of reaction in acetone. | |
It hence justifies the photo-excitation of the anionic polyoxometalate part of the catalyst to form a radical upon irradiation. Hence, initially upon irradiation, photo-activation of the inorganic part of the catalyst leads to the formation of POM in excited state (POM*). POM* in turn generates an imidazolium radical cation by abstraction of hydrogen free radical from imidazolium and thereby itself gets reduced to HOM (reduced POM). The imidazolium cation radical so generated is responsible for effectively initiating free-radical or cationic photo-polymerization, rendering the catalyst its versatility. On the other hand, HOM on exposure to air regenerates the active and oxidized POM. The successive change of colour of the used catalyst during drying also supports such step-wise oxidation of the HOM to POM. Time resolved EPR spectroscopy also shows the free-radical participation in initiating the photo-polymerization reaction. The generation of a paramagnetic radical (S = 1/2) upon irradiation is shown clearly by the EPR spectrum (Fig. 5d). The spectrum also shows a second peak at g = 1.93, which perhaps corresponds to the imidazolium cation radical that is bound to POM. DFT calculations furthermore show that the anionic part of the catalyst, the POM cluster surface, has a high degree of electrophilicity (Fig. 5c). The electrophilic C3 centres of the cluster surface act as docking sites for the transfer of H˙ of imidazolium moiety to form HOM during polymerization cycles.
Structure of the crystal-lattice of the catalyst and its role in polymerization catalysis
We now address the issue of stabilization of emerging hydrophobic polymers by the catalyst. We observe that the photo-polymerization reaction with the catalyst reported here is much more facile as compared to the polymerization initiated by phosphotungstic acid alone. We therefore ask: does the catalyst exert a stabilizing role on emerging oligomeric chains? The single crystal X-ray diffraction pattern shows continuous elliptical hydrophobic channels with a diameter of 7 Å (Fig. 6a). These channels are defined by the dangling butyl groups of the imidazolium cations of the catalyst close to the POM core (Fig. 6b). Moreover, the ionic liquids are known to retain their channel framework of solid state in solution.76–85 It is postulated that the ionic liquid solutions “may not be homogenous, but have to be considered as nano-structured with polar and non-polar regions”.86 Neutron diffraction analysis yields the complementary liquid structure information. In the case of the dialkylimidazolium systems, which are similar to our reported photo-catalyst, a close relationship between the crystal structure and the liquid structure was found,87 emphasizing once again the retention of the channel framework of the solid state in solution. Specifically, nanometer-scale ordering in ionic liquids belonging to the 1-alkyl-3-methylimidazolium family is observed using molecular simulation with an all-atom force field.85 It is also shown that for ionic liquids with alkyl side chains longer than or equal to C4, aggregation of the alkyl chains occurs to form continuous hydrophobic non-polar domains in solution.85 It is thus reasonable to think that the hydrophobic channels of the reported catalyst in the solid-state is retained in solution and acts as stabilizing hydrophobic compartments for the growing polymer chains. They, thereby facilitate formation of hydrophobic polymers with long chain length. This explains that alone PW12O403− with only protons/ammonium as counter-ions, although it can act as a catalyst, it leads to polymers with extremely poor conversions. These observations support the proposition that the butyl channels of imidazolium indeed create a stabilizing hydrophobic channel. Such channels enable successful stabilization of the emerging oligomeric chains and ensure rapid recoverable photo-polymerization by the catalyst.
 |
| | Fig. 6 (a) The elliptical pore (major axis, 0.76 nm; minor axis, 0.46 nm) defined by the alternating butyl groups form a continuous hydrophobic channel and is shown in wire frame representation; (b) the close proximity of the elliptical hydrophobic channels to the anionic PW12O403− cluster core, which helps in the abstraction of H˙ from the imidazolium cation, shown in wire-frame representation. | |
 |
| | Fig. 7 NMR spectra of (a) phosphotungstic acid; (b) 1-butyl,3-methylimidazolium chloride; (c) catalyst (BMIm)2(DMIm)(PW12O40). | |
Results of electrochemical investigations of the catalyst
Fig. 8 shows cyclic voltammograms (CVs) of the catalyst in 0.5 M buffer solution of HAc–NaAc with different scan rates. Three groups of redox peaks could be observed. While the peaks at −0.07 V (b) and −0.27 V (c) are very small, the peak at 0.57 V (a) was intense. The redox potential is observed at 1.5 (V) and 0.57 (V) when the scan rate is selected as 100 mV s−1. With increasing scan rate, the oxidation peak potential becomes more positive and the reduction potential becomes more negative, while the peak currents increase linearly with increasing of the square of the scan rate. This reveals the electrochemical behavior of the complex is a diffusion confined redox process.33
 |
| | Fig. 8 Cyclic voltammograms of the catalyst, (BMIm)2(DMIm)PW12O40 in 0.5 M buffer solution of HAc–NaAc with different scan rates. Inset showing the two small peaks at −0.07V (b) and −0.27 V (c). | |
Investigation of recoverability of the catalyst
To do so, the recovered catalyst is weighed, its FTIR spectrum is recorded and its catalytic activity is tested after every polymerization cycle. Percentage recoverability of the catalyst is calculated by gravimetric analysis as (Wn/W0) × 100; where Wn = dry catalyst weight recorded at the end of nth cycle and W0 = weight of the catalyst used for the 1st cycle. It is found that the catalyst is not only highly recoverable, but is also very efficient after recovery, indicating its structural integrity. Fig. 9 shows the variation of log(Mn) and % conversion of the polymer after every cycle with styrene as a model reaction system, for 10 such cycles and the corresponding catalyst recoverability. From the FTIR spectra we wanted to understand the recovery of the catalyst on a molecular level, qualitatively. For this, we analysed the finger-print region of the catalyst in the absorbance mode. It is known that [α-PW12O40] type polyoxometalates show the following characteristic vibrational frequencies: 804 and 894 cm−1 (νas, W–O–W), 979 cm−1 (νas, W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(t)), 1078 cm−1 (νas, P–O(br)). Likewise, peaks of BMIm+ and DMIm+ appear at 2867, 2958, while 3116 and 3147 cm−1 are assigned to (νs, CH3), (νas, CH3), [νs, HC(4)–C(5)H] and [νas, HC(4)–C(5)H] respectively.9,10,75 It is reasonable to assume that retention of the finger-print implies retention of the molecular structure of the catalyst. One such recovery detail is shown (Fig. 10).
O(t)), 1078 cm−1 (νas, P–O(br)). Likewise, peaks of BMIm+ and DMIm+ appear at 2867, 2958, while 3116 and 3147 cm−1 are assigned to (νs, CH3), (νas, CH3), [νs, HC(4)–C(5)H] and [νas, HC(4)–C(5)H] respectively.9,10,75 It is reasonable to assume that retention of the finger-print implies retention of the molecular structure of the catalyst. One such recovery detail is shown (Fig. 10).
 |
| | Fig. 9 (a) Variation of log(Mn) of polymer and % recoverability of the catalyst after every cycle; (b) variation of % conversion of polymer and % recoverability of catalyst for all cycles. | |
 |
| | Fig. 10 FT-IR spectra of the catalyst before reaction (below), after 1 cycle (middle) and after 10 cycles (above). | |
Clearly, the finger print features of the IR spectrum of the catalyst are retained even after 10 polymerization cycles, indicating the retention of the molecular framework of the catalyst and that of its active sites. The structural retention and integrity is in good agreement with the functional activity of the catalyst since the conversion of the polymer at the end of the tenth consecutive polymerization cycle (Fig. 2) does not deviate significantly from that after the first few polymerization cycles. Further analysis of the FT-IR spectrum shows some decrease in the intensity of the peaks characteristic to the imidazolium moiety of the catalyst, suggesting some loss of lattice imidazolium in the course of photo-polymerization. This observation prompts us to think that the reported catalyst might be a pre-catalyst in part as well.
On the polymerization kinetics
Experiments were also performed to investigate the polymerization kinetics with styrene taken as a model reaction system for this purpose. The reaction kinetics was studied by monitoring the conversion of the monomer with reaction time. In order to understand the efficiency of the catalyst, photo-polymerization kinetics with (BMIm)2(DMIm)PW12O40 as the photo-catalyst was compared and contrasted with that of other conventional polyoxometalates such as H3PW12O40 (Fig. 11). Because of the solubility issues of H3PW12O40 in acetone, the reactions were performed in DMSO so as to make valid comparisons and to rule out any solvent effects. Not only were the polymerization kinetics found to be much faster for the reported photo-catalyst (kBMImDMImPW12 = 0.019 min−1) than that for H3PW12040 (kPW12 = 0.013 min−1), but also the conversion of the resulting polymer was three times that of H3PW12O40 alone. This is attributed to the formation of an imidazolium radical cation from the photo-excited POM* component of the catalyst. The so formed imidazolium radical cation then initiates further cationic and/or radical polymerization. Secondly, the hydrophobic butyl chains furthermore stabilize the emerging oligomers formed in the course of photo-polymerization. As no such possibility exists in the case of H3PW12O40, the corresponding photo-polymerization kinetics and conversions are very low.
 |
| | Fig. 11 Photo-polymerization kinetics of the reported photo-catalyst in comparison to that of H3PW12O40. | |
On the probable pathway of action of the catalyst
We now put forward a probable pathway of polymerization by the catalyst. Upon UV-irradiation, the catalyst (BMIm)2(DMIm)PW12O40 generates a POM in excited state (POM*), since the HOMO–LUMO band-gap corresponds to the wavelength of irradiation. A hydrogen free radical, H˙, is then transferred from imidazolium cation to POM* (POM in excited state) which generates an imidazolium radical cation and simultaneously POM* is reduced to HOM (Fig. 12).
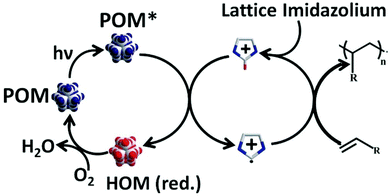 |
| | Fig. 12 Overall scheme of the polymerization reaction: generation of POM* radical center is shown which in turn generates imidazolium radicals by abstracting hydrogen. The latter is instrumental in polymerization of olefinic double bonds, the reacted imidazolium cation is replenished by the lattice DMIm+ and BMIm+ and the POM* radical center is further oxidized by aerial oxygen to give the starting POM. | |
Reason: vicinity of the acidic imidazolium hydrogen to POM* center. The reduction of the POM* centre also manifests in the change of colour of the catalyst from colourless to blue. The generated imidazolium radical cation then initiates further free-radical/cationic polymerization. This explains the high reactivity and efficiency of the catalyst in contrast to other unmodified polyoxometalates such as H3PW12O40 and renders the catalyst its versatility. Later the reacted imidazolium cation is replenished by the lattice DMIm+ and BMIm+. This is observed in the depletion of the total imidazolium content from FT-IR spectroscopic experiments. The reduced HOM on the other hand is oxidized and POM of the catalyst is regenerated in the course of solvent removal/drying. Macroscopically it is seen by successive colour changes of the catalyst solution from blue to colourless to the starting oxidized POM. The complete reaction pathway is summarized in Fig. 12.
Conclusions
In summary, the photo-catalyst, (BMIM)2(DMIM)(PW12O40), introduced in this paper can run efficiently over many cycles thereby polymerizing an array of monomers, like, styrene, methyl methacrylate, butyl methacrylate and vinyl acetate in the absence of any externally added co-initiators. These results could lead to greener ways of polymerization with well known polyoxometalates.
Notes and references
- E. Androulaki, A. Hiskia, D. Dimotikali, C. Minero, P. Calza, E. Pelizzetti and E. Papaconstantinou, Environ. Sci. Technol., 2000, 34, 2024 CrossRef CAS.
- H. Hori, E. Hayakawa, H. Einaga, S. Kutsuna, H. Kiatagawa and S. Arakawa, Environ. Sci. Technol., 2004, 38, 6118 CrossRef CAS.
- A. Hiskia, M. Ecke, A. Troupis, A. Kokorakis, H. Hennig and E. Papaconstantinou, Environ. Sci. Technol., 2001, 35, 2358 CrossRef CAS.
-
A. Hiskia, E. Androulaki, A. Mylonas, A. Troupis and E. Papaconstantinou, in Polyoxometalate Chemistry, ed. A. Mueller and M. T. Pope, Kluwer Academic Publishers, p. 417 Search PubMed.
-
M. T. Pope, in Heteropoly and isopoly oxometalates, Springer, Berlin, 1983 Search PubMed.
-
M. T. Pope and A. Mylonas, in Polyoxometalate chemistry: From topology via self-assembly to applications, ed. M. T. Pope and A. Mylonas, Kluwer Academic Publishers, Dordrecht, London, 2001 Search PubMed.
-
C. L. Hill, in Chem. Rev, ed. C. L. Hill, ACS, 1998, vol. 98(1) Search PubMed.
-
P. Wasserscheid and T. Welton, in Ionic Liquids in Synthesis, ed. P. Wasserscheid, T. Welton, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed.
-
H. Ohno, in Electrochemical aspects of ionic liquids, ed. H. Ohno, Wiley, New York, 2005 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
-
T. Kida and K. Jinnai, Jap. Pat, JP 2009108374A20090521, 2009 Search PubMed.
- Y. Guo and C. Hu, J. Mol. Catal. A: Chem., 2007, 262, 136 CrossRef CAS.
- A. Troupis, E. Gkika, A. Hiskia and E. Papaconstantinou, C. R. Chim., 2006, 9, 851 CrossRef CAS.
- A. Troupis, A. Hiskia and E. Papaconstantinou, Environ. Sci. Technol., 2002, 36, 5355 CrossRef CAS.
- A. Troupis, A. Hiskia and E. Papaconstantinou, New J. Chem., 2001, 25, 361 RSC.
- N. M. Okun, T. M. Anderson and C. L. Hill, J. Am. Chem. Soc., 2003, 125, 3194 CrossRef CAS.
- N. M. Okun, M. D. Ritorto, T. M. Anderson, R. P. Apkarian and C. L. Hill, Chem. Mater., 2004, 16, 2551 CrossRef CAS.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
-
I. V. Kozhevnikov, in Catalysts for Fine Chemicals Synthesis: Catalysis by Polyoxometalates, ed. I. V. Kozhevnikov, J. Wiley & Sons, Chichester, 2002 Search PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS.
- C. Rocchiccioli-Deltcheff, A. Aouissi, M. Bettahar, S. Launay and M. J. Fournier, J. Catal., 1996, 164, 16 CrossRef CAS.
- R. Neumann and A. M. Khenkin, Chem. Commun., 2006, 2529 RSC.
- R. G. Finke and S. Özkar, Coord. Chem. Rev., 2004, 248, 135 CrossRef CAS.
- C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS.
- A. Corma, H. Garcia and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- A. Nisar, J. Zhuang and X. Wang, Adv. Mater., 2011, 23, 1130 CrossRef CAS.
- L. H. Wee, S. R. Bajpe, N. Janssens, I. Hermans, K. Houthoofd, C. E. A. Kirschhock and J. A. Martens, Chem. Commun., 2010, 46, 8186 RSC.
- L. Plault, A. Hauseler, S. Nlate, D. Astruc, J. Ruiz, S. Gatard and R. Neumann, Angew. Chem., Int. Ed., 2004, 43, 2924 CrossRef CAS.
- S. Nlate, D. Astruc and R. Neumann, Adv. Synth. Catal., 2004, 346, 1445 CrossRef CAS.
- E. Rafiee and S. Eavani, Green Chem., 2011, 13, 2116 RSC.
- A. Nisar, Y. Lu, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2011, 50, 3187 CrossRef CAS.
- S. Roy Chowdhury, P. T. Witte, D. H. A. Blank, P. L. Alsters and J. E. ten Elshof, Chem.–Eur. J., 2006, 12, 3061 CrossRef.
- Y. Leng, J. Wang, D. Zhu, M. Zhang, P. Zhao, Z. Long and J. Huang, Green Chem., 2011, 13, 1636 RSC.
- A. G. Sathicq, G. P. Romanelli, V. Palermo, P. G. Vázquez and H. J. Thomas, Tetrahedron Lett., 2008, 49, 1441 CrossRef CAS.
-
N. Mizuno, presented in part at the 234th ACS National meeting, Boston, USA, 2007.
- S. Nlate, L. Plault and D. Astruc, Chem.–Eur. J., 2006, 12, 903 CrossRef CAS.
- A. Haimov and R. Neumann, Chem. Commun., 2002, 876 RSC.
- R. D. Gall, C. L. Hill and J. E. Walker, J. Catal., 1996, 159, 473 CrossRef CAS.
-
T. K. Shioyama and J. J. Straw, US Pat, US4419525 A 19831206, 1983 Search PubMed.
-
S. Takeshima, Jap. Pat, JP 2009291145 A 20091217, 2009 Search PubMed.
-
F.-X. Chiron, W. Fullerton, L. A. Kay, L. O'Neill and S. J. Stephens, PCT Int. Appl. WIPO, WO 2009074774 A2 20090618, 2009 Search PubMed.
-
S. Kim, D. Park, C. Son and T. Saito, Jap. Pat, JP 2007326826 A 20071220, 2007 Search PubMed.
- C. Boglio, G. Lemière, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Angew. Chem., Int. Ed., 2006, 45, 3324 CrossRef CAS.
-
E. Fuchs and F. Ohlbach, Ger. Pat, DE 19835657 A1 20000210, 2000 Search PubMed.
-
T. Sugimoto, Y. Fuuda and T. Matsuzaki, Jap. Pat, JP 11240852 A 19990907, 1999 Search PubMed.
- K. Okumura, K. Yamashita, M. Hirano and M. Niwa, J. Catal., 2005, 234, 300 CrossRef CAS.
- D. Chen, Z. Xue and Z. Su, J. Mol. Catal. A: Chem., 2003, 203, 307 CrossRef CAS.
- D. Chen, Z. Xue and Z. Su, J. Mol. Catal. A: Chem., 2004, 208, 91 CrossRef CAS.
- D. Chen, X. Wang, G. Yang, Q. Li, W. Chai, R. Yuan, S. Liu, F. Gao, H. Jin and S. Roy, J. Mol. Catal. A: Chem., 2012, 363–364, 195 CrossRef CAS.
- J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599 RSC.
-
C. Guerrero-Sanchez, F. Wiesbrock and S. Schubert Ulrich, in Ionic Liquids in Polymer Systems, ACS Symposium Series, 2005, vol. 913, pp. 37–49 Search PubMed.
- S. Triki, L. Ouahab, J. Padiou and D. Grandjean, J. Chem. Soc., Chem. Commun., 1989, 1068 RSC.
- R. S. Varma and V. V. Namboodiri, Chem. Commun., 2001, 643 RSC.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- S. D. Vosko, L. Wilk and M. Nusair, Can. J. Chem., 1980, 58, 1200 CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391 Search PubMed.
- T.C. ADF2008.01; SCM, Vrije Universiteit: Amsterdam, The Netherlands. http://www.scm.com.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- E. van Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396 CrossRef CAS.
- J. L. Pascual-ahuir, E. Silla and I. Tunon, J. Comput. Chem., 1994, 15, 1127 CrossRef CAS.
- W. Guan, L. Yan, Z. Su, S. Liu, M. Zhang and X. Wang, Inorg. Chem., 2005, 44, 100 CrossRef CAS.
- X. López, J. J. Carbó, C. Bo and J. M. Poblet, Chem. Soc. Rev., 2012, 41, 7537 RSC.
- F. Q. Zhang, H. S. Wu, D. B. Cao, X. M. Zhang, Y. W. Li and H. J. Jiao, J. Mol. Struct. (THEOCHEM), 2005, 755, 119 CrossRef CAS.
- F. Q. Zhang, H. S. Wu, Y. Y. Xu, Y. W. Li and H. J. Jiao, J. Mol. Model., 2006, 12, 551 CrossRef CAS.
- C. G. Liu, W. Guan, L. K. Yan and Z. M. Su, Dalton Trans., 2011, 40, 2967 RSC.
- X. López, J. M. Maestre, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2001, 123, 9571 CrossRef.
- J. M. Maestre, X. López, C. Bo, J. M. Poblet and N. Casan-Pastor, J. Am. Chem. Soc., 2001, 123, 3749 CrossRef CAS.
- S. M. Yue, L. K. Yan, Z. M. Su, G. H. Li, Y. G. Chen, J. F. Ma, H. B. Xu and H. J. Zhang, J. Coord. Chem., 2004, 57, 123 CrossRef CAS.
- C. L. Øpstad, T. B. Melø, H. R. Sliwka and V. Partali, Tetrahedron, 2009, 65, 7616 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- B. Jagoda-Cwiklik, P. Slavcek, L. Cwiklik, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. A, 2008, 112, 3499 CrossRef CAS.
- J. Fuller, R. T. Carlin, H. C. D. Long and D. Haworth, J. Chem. Soc., Chem. Commun., 1994, 299 RSC.
- J. Dupont, P. A. Z. Suarez, R. F. D. Souza, R. A. Burrow and J.-P. Kintzinger, Chem.–Eur. J., 2000, 6, 2377 CrossRef CAS.
- A. G. Avent, P. A. Chaloner, M. P. Day, K. R. Seddon and T. J. Welton, J. Chem. Soc., Dalton Trans., 1994, 3405 RSC.
- K. M. Dieter, C. J. Dymek, N. E. Heimer, J. W. Rovang and J. S. Wilkes, J. Am. Chem. Soc., 1988, 110, 2722 CrossRef CAS.
- A. Mele, C. D. Tran and S. H. P. Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364 CrossRef CAS.
- A. Mele, G. Romano, M. Giannone, E. Ragg, G. Fronza, G. Raos and V. Marcon, Angew. Chem., Int. Ed., 2006, 45, 1123 CrossRef CAS.
- B. G. Moutiers, A. Labet, A. E. Azzi, C. Gaillard, C. Mariet and K. Lutzenkirchen, Inorg. Chem., 2003, 42, 1726 CrossRef.
- M. G. D. Popolo and G. A. J. Voth, J. Phys. Chem. B, 2004, 108, 1744 CrossRef.
- C. Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 3330 CrossRef.
- U. Schröder, J. D. Wadhawan, R. G. Compton, F. Marken, P. A. Z. Suarez, C. S. Consorti, R. F. de Souza and J. Dupont, New J. Chem., 2000, 24, 1009 RSC.
- C. Hardacre, J. D. Holbrey, S. E. McMath, D. T. Bowron and A. K. Soper, J. Chem. Phys., 2003, 118, 273 CrossRef CAS.
- E. Papaconstantinou, Chem. Soc. Rev., 1989, 18, 1 RSC.
- S. Mandal, P. R. Selvakannan, R. Pasricha and M. Sastry, J. Am. Chem. Soc., 2003, 125, 8440 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a