 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lead(II) chloride ionic liquids and organic/inorganic hybrid materials – a study of chloroplumbate(II) speciation†
Fergal
Coleman
,
Guo
Feng
,
Richard W.
Murphy
,
Peter
Nockemann
,
Kenneth R.
Seddon
and
Małgorzata
Swadźba-Kwaśny
*
QUILL, The Queen's University of Belfast, Belfast, BT9 5AG, UK. E-mail: m.swadzba-kwasny@qub.ac.uk
First published on 17th January 2013
Abstract
A range of chloroplumbate(II) organic salts, based on the two cations, 1-ethyl-3-methylimidazolium and trihexyl(tetradecyl)phosphonium, was prepared by ionothermal synthesis. Depending on the structure of the organic cation and on the molar ratio of PbCl2 in the product, χPbCl2, the salts were room-temperature ionic liquids or crystalline organic/inorganic hybrid materials. The solids were studied using Raman spectroscopy; the crystal structure of [C2mim]{PbCl3} was determined and shown to contain 1D infinite chloroplumbate(II) strands formed by edge-sharing tetragonal pyramids of pentacoordinate (PbCl5) units. The liquids were analysed using 207Pb NMR and Raman spectroscopies, as well as viscometry. Phase diagrams were constructed based on differential scanning calorimetry (DSC) measurements. Discrete anions: [PbCl4]2− and [PbCl3]−, were detected in the liquid state. The trichloroplumbate(II) anion was shown to have a flexible structure due to the presence of a stereochemically-active lone pair. The relationship between the liquid phase anionic speciation and the structure of the corresponding crystalline products of ionothermal syntheses was discussed, and the data were compared with analogous tin(II) systems.
Introduction
Salts comprising of organic cations and halometallate anions are of interest in several fields of material sciences. For example, crystalline chloroplumbate(II) systems may be considered as organic/inorganic hybrid compounds, with a tendency to form 2D perovskites and low-dimensional crystalline materials with electro-optic characteristics (photoluminescence, electroluminescence and nonlinear optical properties).1 Lower melting organic halometallates have been investigated as soft materials (ionic liquid crystals2 or ionic liquids).3 Both solids and liquids have been studied quite extensively by solid-state and ionic liquid chemists, but each group adopts their own research methodology and approaches. In the solid state, the focus is on crystal structure and material properties. Chlorometallate ionic liquids, on the other hand, have been used for electrochemistry,4 catalysis5 and separations,6 their anionic speciation has been thoroughly investigated,7–10 but they have rarely been used for inorganic syntheses,11 and never for the systematic study on the ionothermal preparation of organic/inorganic hybrid materials (i.e. direct crystallisation from the molten ionic liquid). It would be interesting to adopt a more holistic approach: to prepare a set of halometallates based on the same metal halide, and (through changes of the cation and the reactant ratios) access both groups of materials: ionic liquids and crystalline organic/inorganic hybrid materials. In this study, chloroplumbate(II) salts were chosen as a model system, due to their flexibility in structural arrangements, with varying coordination numbers and stereochemistry.12,13Solid state perspective
The structural organisation of homoleptic chloroplumbate(II) anions with organic cations is governed by: (a) the very flexible coordination sphere of lead(II), influenced by the ‘inert electron pair’ and relativistic effects,14 (b) the flexible coordination environments of halide anions, and (c) steric and hydrogen bonding effects deriving from the cation. This has given rise to a plethora of chloroplumbate(II) structures, from rare discrete anions,15,16 to much more common 1D strands13,17 and 2D sheets of polyanions, often forming perovskite-type materials.13In order to tune the electro-optic properties of these materials, attempts have been made to modify the solid-state structures of chloroplumbate(II) materials in a controlled manner. This can be achieved by using deliberately selected cations, able to form strong hydrogen bonds or to induce specific steric effects. For example, amine-functionalised cations (e.g. 2-methylpentane-1,5-diammonium or N,N′-dimethylpiperazinium) were demonstrated to direct the crystal packing by hydrogen bonding,12,13,17 while the combination of hydrogen bonding and steric effects deriving from cations has been shown to induce the formation of discrete chloroplumbate(II) anions, e.g. [PbCl5]3−,15 [PbCl6]4− or [Pb2Cl10]6−,16 or extraordinary arrangements of the chloroplumbate(II) units, such as channel polymers.18 Besides the influence of cation, the choice of halide (Cl, Br, or I) was reported to have some effect on the structural arrangement.19 Noteworthy, the molar ratio of the reactants (expressed as mole fraction of lead(II) halide, χPbX2) was not considered as a key variable in the syntheses.
Chloroplumbate(II) organic/inorganic hybrid salts are typically prepared by crystallisation from acidic aqueous solutions.12,13 Ionothermal syntheses from ionic liquids20 have been limited to two publications,21,22 reporting jointly five structures based on 1-alkyl-3-methylimidazolium bromoplumbates(II), but none based on chloroplumbates(II). From this perspective, it appeared interesting to make the first attempt to prepare chloroplumbate(II) organic/inorganic hybrid materials via the ionothermal route. Furthermore, it seemed worthwhile to analyse the influence of χPbCl2 of the melt on the structure of the crystalline products.
Ionic liquid perspective
Ionic liquids based on halometallate anions are characterised (in their liquid phase) by dynamic equilibria between several anionic species.7–10 The presence and concentration of each species is known to depend on the metal, and on the mole fraction of metal halide, χMXx, whereas ‘standard’, i.e. non-functionalised, organic cations were typically found not to affect the liquid-state anionic speciation (viz. for example abundant work on chloroaluminate(III) systems based on various cations).23 Consequently, the variables taken into account when studying halometallate ionic liquids differ from those investigated routinely by solid-state chemists. Importantly, in the liquid state, only discrete chlorometallate anions (monomers, dimers, or sometimes trimers and tetramers) may be present; the formation of polyanionic structures naturally results in the crystallisation of a solid.9There have been no room-temperature chloroplumbate(II) ionic liquids reported to date. This study presents an exciting opportunity to prepare and investigate a new group of metal-based ionic liquids.
Approach and methodology in this work
In this paper, a range of chloroplumbate(II) organic salts, based on two cations: trihexyl(tetradecyl)phosphonium, [P6 6 6 14]+ and 1-ethyl-3-methylimidazolium, [C2mim]+, was prepared via the ionothermal route. The variables taken into account were: the mole fraction, χPbCl2, and the organic cation structure. Depending on their melting point, the products were studied as ionic liquids, as crystalline materials and, where possible, in both states. The relationship between the speciation of chloroplumbate(II) anions in the liquid state, and the structure of crystalline materials synthesised via the ionothermal route, was studied.Experimental
Lead(II) chloride, (ex BDH Chemicals Ltd., 99%), was dried under high vacuum (120 °C, 3 d, 10−2 mbar) and stored in a dinitrogen-filled glovebox (MBraun LabMaster dp; <0.1 ppm O2 and H2O). Trihexyl(tetradecyl)phosphonium chloride (97.7% by 31P NMR spectroscopy) was provided by Cytec Industries, Inc., benzyl(triphenyl)phosphonium chloride was purchased from Sigma-Aldrich. 1-Ethyl-3-methylimidazolium chloride24 was prepared as described elsewhere, dried under high vacuum (75 °C, 7 d, 10−2 mbar) and stored in the glovebox.Synthesis of chloroplumbate(II) systems
Chloroplumbate(II) systems based on [P6 6 6 14]+ and [C2mim]+ were prepared for a wide range of compositions (0.05 ≤ χPbCl2 ≤ 0.75).Syntheses were carried out in the glovebox. Typically, an appropriate amount of [cation]Cl was weighed into a sample vial (10 cm3) containing a PTFE-coated stirring bar. An appropriate amount of lead(II) chloride was then carefully added to achieve the desired ratio of the reactants. Subsequently, the sample vial was closed with a cap, placed in a multi-well heater-stirrer, and stirred vigorously (80–150 °C, overnight). The samples were stored in the glovebox prior to study. Accurate masses of reactants are given in Table 1.
| χ PbCl2 | [C2mim]Cl–PbCl2 | [P6 6 6 14]Cl–PbCl2 | χ PbCl2 | ||
|---|---|---|---|---|---|
| m [C2mim]Cl | m PbCl2 | m [P6 6 6 14]Cl | m PbCl2 | ||
| 0.05 | 2.2734 | 0.2270 | 0.10 | 2.8313 | 0.1683 |
| 0.10 | 2.0623 | 0.4346 | 0.20 | 2.6458 | 0.3555 |
| 0.20 | 1.6961 | 0.8043 | 0.25 | 2.5422 | 0.4556 |
| 0.25 | 1.5320 | 0.9686 | 0.30 | 2.4434 | 0.6503 |
| 0.30 | 1.3774 | 1.1197 | 0.28 | 1.6304 | 0.3386 |
| 0.33 | 1.2929 | 1.2079 | 0.33 | 2.3933 | 0.6309 |
| 0.40 | 1.1033 | 1.3952 | 0.37 | 1.5342 | 0.4827 |
| 0.45 | 0.9795 | 1.5201 | 0.40 | 2.2206 | 0.7927 |
| 0.50 | 0.8636 | 1.6381 | 0.45 | 2.0894 | 0.9151 |
| 0.55 | 0.7529 | 1.7455 | 0.50 | 1.9624 | 1.0501 |
| 0.60 | 0.6515 | 1.8537 | 0.55 | 1.8043 | 1.1806 |
| 0.65 | 0.5581 | 1.9660 | 0.60 | 1.6699 | 1.3459 |
| 0.67 | 0.5148 | 1.9825 | 0.65 | 1.5176 | 1.5102 |
| 0.75 | 0.3736 | 2.1259 | 0.67 | 1.4386 | 1.5640 |
| — | — | — | 0.75 | 1.1465 | 1.8422 |
Analytical methods
[P6 6 6 14]Cl–PbCl2, was stirred overnight with a small amount of activated charcoal (80 °C, overnight) to remove fluorescent impurities. The charcoal was allowed to partially settle (but was not removed) prior to the Raman measurements; its presence did not interfere with the measurements.
This system and a solid [C2mim]Cl–PbCl2 system were measured at ambient temperature. To obtain Raman spectra of molten samples of the [C2mim]Cl–PbCl2 system, the samples (sealed in quartz cuvettes) were heated with a heat gun above the melting point (150 °C), and two 4-second scans were immediately recorded. The temperature drop measured within this time (for a sample outside of spectrometer) was 4 °C, hence the temperature of the experiment is assumed to be 148 ± 2 °C. For comparison, several samples of the [P6 6 6 14]Cl–PbCl2 system were also tested under these conditions.
All spectra were measured at 80 °C, using a Bruker DRX 500 spectrometer. The 207Pb NMR spectra of the [P6 6 6 14]Cl–PbCl2 system were recorded at 104.8 MHz. Lead(II) nitrate in D2O (1.0 M solution) was used as an external reference (δ = −1963 ppm). 207Pb is a spin 1/2 nucleus, characterised by medium sensitivity (2.00 × 10−3 relative to 1H natural abundance).25
For [P6 6 6 14]Cl–PbCl2, the temperature was ramped four times between −90 and 80 °C, at 5 °C min−1, each time stabilised for 5 min at minimum and maximum temperature. All scans for the liquid samples were modulated. For [C2mim]Cl–PbCl2, the temperature was ramped three times between −90 to 200 °C, at 5 °C min−1, each time stabilised for 5 min at minimum and maximum temperature.
Crystal data for [C2mim]{PbCl3} were collected using a Bruker Nonius KappaCCD diffractometer with a FR591 rotating anode and a molybdenum target at ca. 120 K in a dinitrogen stream.26 Lorentz and polarisation corrections were applied. The structure was solved by direct methods. Hydrogen-atom positions were located from difference Fourier maps and a riding model with fixed thermal parameters (Uij = 1.2Ueq for the atom to which they are bonded, 1.5 for methyl), was used for subsequent refinements. The function minimised was Σ[ω(|Fo|2 − |Fc|2)] with reflection weights ω1 = [σ2 |Fo|2 + (g1P)2 + (g2P)] where P = [max|Fo|2 + 2|Fc|2]/3. The SHELXTL package and OLEX2 were used for structure solution and refinement.27,28
Results and discussion
Synthesis
The [C2mim]Cl–PbCl2 system was chosen to study because the [C2mim]+ cation is a popular motif in design of ionic liquids and, at the same time, the [C2mim]Br–PbBr2 system is known to give organic/inorganic hybrid materials which exhibit non-linear optical behaviour.21 Although analogous materials based on the [C2mim]Cl–PbCl2 system are not known, this system was expected to yield materials of similar properties.The [P6 6 6 14]Cl–PbCl2 system, incorporating a bulky cation with long, flexible alkyl chains, was designed to give room-temperature ionic liquids, facilitating liquid-state speciation studies.
A range of compositions of both systems was prepared. In the course of ionothermal synthesis (i.e. at 80–150 °C) homogenous melts were obtained for χPbCl2 ≤ 0.45. For χPbCl2 > 0.45, samples contained a white powder, identified as unreacted lead(II) chloride by Raman spectroscopy.
At ambient temperature, all samples of [C2mim]Cl–PbCl2 were crystalline solids, whilst all compositions of [P6 6 6 14]Cl–PbCl2 were viscous liquids (see Fig. 1), apart from the lead(II) chloride suspended in χPbCl2 > 0.45. These are the first room-temperature ionic liquids based on chloroplumbate(II) anions reported to date.
![Photographs of the room temperature ionic liquids of the selected samples of the [P6 6 6 14]Cl–PbCl2 system; (a) χPbCl2 = 0.28 and (b) χPbCl2 = 0.37.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f1.gif) | ||
| Fig. 1 Photographs of the room temperature ionic liquids of the selected samples of the [P6 6 6 14]Cl–PbCl2 system; (a) χPbCl2 = 0.28 and (b) χPbCl2 = 0.37. | ||
[C2mim]Cl–PbCl2 system
![Phase diagram of the [C2mim]Cl–PbCl2 system; A – [C2mim]Cl, B – [C2mim]2{PbCl4}, C – [C2mim]{PbCl3}, D – lead(ii) chloride and L – liquid.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f2.gif) | ||
| Fig. 2 Phase diagram of the [C2mim]Cl–PbCl2 system; A – [C2mim]Cl, B – [C2mim]2{PbCl4}, C – [C2mim]{PbCl3}, D – lead(II) chloride and L – liquid. | ||
Analysing the phase diagram alone, the crystal structures of phases B and C are expected to be polymeric, rather than contain discrete anions. This is supported by relatively high melting points, as well as by incongruent melting of both phases. Based on the stoichiometry, general formulas of [C2mim]2{PbCl4} and [C2mim]{PbCl3} could be tentatively suggested for phases B and C, respectively.
As described in the introduction, chloroplumbate(II) structures based on organic cations feature a plethora of various structural arrangements. The known compounds are characterised by various χPbCl2 values, and their structure depends strongly on the cation. Unfortunately, in the literature, there are no chloroplumbate(II) structures based on the imidazolium cation. Six bromoplumbate(II) crystalline materials based on imidazolium cations, five synthesised ionothermally21,22 and one grown from an aqueous solution,29 are listed in Table 2.
| Cation | Product chracteristics | Ref | |
|---|---|---|---|
| χ PbBr2 | Bromoplumbate(II) structure | ||
| [Cnmim]+ – 1-alkyl-3-methylimidazolium cation, where n is the carbon number in the alkyl chain. | |||
| [C4mim]+ | 0.33 | Trimers, [Pb3Br12]6− | 21 |
| [C2mim]+ | 0.50 | Strands of face-sharing octahedral (PbBr6) | 28 |
| [C2mim]+ | 0.50 | Strands of edge-sharing (PbBr5) | 21 |
| [C3mim]+ | 0.50 | Strands of edge-sharing (PbBr5) | 22 |
| [Callylmim]+ | 0.50 | Strands of edge-sharing (PbBr5) | 22 |
| [C6mim]+ | 0.50 | Strands of edge-sharing (PbBr5) | 22 |
Four of them were isostructural, containing 1D chains of pentacoordinate (PbBr5) units in the form of bridging basal chlorine atoms from tetragonal pyramids, effectively giving [Cnmim]{PbBr3} stoichiometric compounds (χPbBr2 = 0.50), as shown in Fig. 3a. Another structure28 of [C2mim]{PbBr3} formula and χPbBr2 = 0.50 stoichiometry formed 1D strands of face-sharing octahedra (PbBr6), as shown in Fig. 3b. Finally, a structure of a different stoichiometry (χPbBr2 = 0.33), featuring a trinuclear [Pb3Br12]6− anion depicted in Fig. 3c, was determined for [C4mim]6[Pb3Br12].21 In all structures, there was a dense network of hydrogen bonds between the bromoplumbate(II) strands and the surrounding cations.
![Connectivity in bromoplumbate(ii) oligomeric or polymeric chains found in bromoplumbate(ii) organic/inorganic hybrid materials based on the [Cnmim]+ cation.21,22,28](/image/article/2013/DT/c3dt32742f/c3dt32742f-f3.gif) | ||
| Fig. 3 Connectivity in bromoplumbate(II) oligomeric or polymeric chains found in bromoplumbate(II) organic/inorganic hybrid materials based on the [Cnmim]+ cation.21,22,28 | ||
In analogy, it may be expected that phases B and C in the [C2mim]–PbCl2 system (Fig. 2) contain polymeric 1D strands: [C2mim]2{PbCl4} and [C2mim]{PbCl3}, respectively.
To determine the precise structure of the anion, a single crystal was grown directly from the molten χPbCl2 = 0.40 composition. Its structure contained infinite chloroplumbate(II) strands formed by edge-sharing tetragonal pyramids of pentacoordinate (PbCl5) units (Fig. 4), with an overall stoichiometry of [C2mim]{PbCl3}, χPbCl2 = 0.50. This confirms that phase C (Fig. 2) is isostructural with the analogous bromoplumbate(II) salt,21 shown in Table 2 and Fig. 3a. In particular, the mean Clap–Pb–Cleq at 88(6)° is the same as that of the analogous Brap–Pb–Breq at 89(5)°, and both structures show strong evidence for the existence of a stereochemically active lone pair, not interacting with any of the cationic hydrogen-bond donor sites.
![A section of the {PbCl3}n strands along the a-axis in the crystal structure of [C2mim]{PbCl3} showing the hydrogen bonding interactions with the surrounding [C2mim]+ cations.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f4.gif) | ||
| Fig. 4 A section of the {PbCl3}n strands along the a-axis in the crystal structure of [C2mim]{PbCl3} showing the hydrogen bonding interactions with the surrounding [C2mim]+ cations. | ||
The asymmetric unit contains one Pb atom, three different Cl− anions and one imidazolium cation. The Pb atoms form three shorter bonds to chloride anions ranging from 2.629(1) to 2.778(1) Å, and have two longer contacts at 3.074(1) and 3.108(1) Å (Fig. 5). Four of the five chloride anions are μ2-bridging to the neighbouring Pb atoms and link the five-coordinate, edge-sharing, distorted tetragonal pyramids into strands along the a-axis. The sixth position of the ‘octahedron’ is vacant in the crystal structure, and is presumably occupied by a lone pair of electrons from the lead(II).
![Coordination geometry of a Pb atom in a distorted tetragonal pyramid in the crystal structure of [C2mim]{PbCl3}.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f5.gif) | ||
| Fig. 5 Coordination geometry of a Pb atom in a distorted tetragonal pyramid in the crystal structure of [C2mim]{PbCl3}. | ||
Two of the three ring protons of the [C2mim]+ cations are involved in C–H⋯Cl hydrogen bonding (see Fig. 6), with the C2–H and C5–H protons showing relatively weak interactions at 2.906(1) and 2.985(1) Å, respectively. Shorter C–H⋯Cl contacts to the {PbCl3}n strands are found for the N-methyl and terminal CH3 group of the N-ethyl group, ranging from 2.697(7) to 2.885(7) Å, respectively.
![The C–H⋯Cl hydrogen bonding interactions of the [C2mim]+ cations in [C2mim]{PbCl3}.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f6.gif) | ||
| Fig. 6 The C–H⋯Cl hydrogen bonding interactions of the [C2mim]+ cations in [C2mim]{PbCl3}. | ||
Each imidazolium cation is hydrogen bonded (via both the ring protons and two N-methyl protons) to the adjacent {PbCl3}n strands, resulting in a three-dimensional C–H⋯Cl network, shown in Fig. 7. The crystallographic data are summarised in Table 3.
![Packing in the crystal structure of [C2mim]{PbCl3}.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f7.gif) | ||
| Fig. 7 Packing in the crystal structure of [C2mim]{PbCl3}. | ||
| [C2mim]{PbCl3} | |
|---|---|
| Formula | C6H11Cl3N2Pb |
| M w/g mol−1 | 424.71 |
| Dimensions/mm | 0.12 × 0.06 × 0.02 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| a/Å | 8.1267(5) |
| b/Å | 9.1916(6) |
| c/Å | 14.8385(10) |
| α/° | 90.00 |
| β/° | 90.00 |
| γ/° | 90.00 |
| V/Å3 | 1108.40(12) |
| Z | 4 |
| D calc/g cm−3 | 2.480 |
| Crystal shape | block |
| Crystal colour | Colourless |
| μ/mm−1 | 15.894 |
| F(000) | 733 |
| Meas. reflections | 8412 |
| Unique reflections | 2832 |
| Parameters refined | 111 |
| Goof on F2 | 0.829 |
| R 1 | 0.0140 |
| wR2 | 0.0319 |
| R 1(all data) | 0.0149 |
| wR2(all data) | 0.0322 |
| Flack parameter | 0.007(4) |
| CCDC | xxxxx |
To confirm the identification of phase B, a single crystal was grown (directly from the melt) from χPbCl2 = 0.33. Unfortunately, the crystalline material was extremely hygroscopic and decomposed prior to measurement. Therefore, to gain a better insight into the possible structure of phase B, Raman spectra were recorded for a range of [C2mim]Cl–PbCl2 compositions.
In [C2mim]{PbCl3} (higher χPbCl2 values, phase C), lead(II) is pentacoordinate, with shorter Pb–Cl bonds and therefore blue-shifted vibrational frequencies are to be expected. In [C2mim]2{PbCl4} (lower χPbCl2 values), lead is anticipated to be hexacoordinate, with longer Pb–Cl bonds, and therefore the vibrational frequencies are expected to be red-shifted.36
Bands characteristic of the Pb–Clt (terminal) stretching frequencies fall between ca. 200 and 300 cm−1.34 The ratio of a bridging metal–halogen stretching frequency to a terminal metal–halogen stretching frequency, ζ, is typically between 0.60 and 0.85.30 The relevant Raman bands recorded for the [C2mim]Cl–PbCl2 system are detailed in Fig. 8 and Table 4.
![Raman spectra of the [C2mim]Cl–PbCl2 system (ambient temperature, solid state), compared to the Raman spectrum of lead(ii) chloride (main band indicated in black). The bands indicated in red originate from [C2mim]2{PbCl4} and those indicated in blue from [C2mim]{PbCl3}.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f8.gif) | ||
| Fig. 8 Raman spectra of the [C2mim]Cl–PbCl2 system (ambient temperature, solid state), compared to the Raman spectrum of lead(II) chloride (main band indicated in black). The bands indicated in red originate from [C2mim]2{PbCl4} and those indicated in blue from [C2mim]{PbCl3}. | ||
| χ PbCl2 | [C2mim]2{PbCl4} | [C2mim]{PbCl3} | PbCl2 |
|---|---|---|---|
| a The intensities of the peaks are represented in parentheses (s = strong, m = medium, and sh = shoulder). | |||
| 0.20 | 116(s), 163(m), 217(m) | ||
| 0.30 | 117(s), 162(m), 216(m) | ||
| 0.33 | 117(s), 163(m), 215(m) | ||
| 0.40 | 117(s), 163(m), 218(m) | 174(sh), 221(sh) | |
| 0.45 | 117(s), 161(sh), 220(h) | 172(m), 225(sh) | |
| 0.50 | 172(m), 228(s) | ||
| 0.55 | 173(sh), 226(s) | 158(s) | |
| 0.65 | 229(m) | 127(s), 157(s), 176(sh) | |
| 1.00 | 133(sh), 159(s), 179(sh) | ||
| 1.0034 | 131(m), 157(s), 179(m) | ||
For higher χPbCl2 (phase C), a band for Pb–Clt is found around 226 cm−1, and a band for Pb–Clb (bridging) around 173 cm−1, with ζ = 0.75. For lower χPbCl2 values (phase B), both Pb–Cl stretching frequencies are red-shifted to around 218 cm−1 and 163 cm−1, respectively, with ζ = 0.77. Significantly, Raman spectroscopy is less sensitive than DSC for the detection of small amounts of new phases, and hence they are detected at lower χPbCl2 values in Fig. 2 than in Fig. 8.
Phase C, present for χPbCl2 > 0.33, was unambiguously identified as [C2mim]{PbCl3}, with pentacoordinate lead(II) strands (Fig. 3–7). Raman spectra for phase B, present predominantly for χPbCl2 < 0.33, strongly suggest that lead(II) is hexacoordinate (viz. red-shifted bands, indicating longer Pb–Cl bonds). The proposed structure of [C2mim]2{PbCl4} contains most likely one of the two structural motifs: 2D sheets (Fig. 9a) or 1D strands (Fig. 9b). Perovskite-like, 2D chloroplumbate(II) layers shown in Fig. 9a are the dominant structural arrangements in chloroplumbate(II) organic/inorganic hybrid materials with χPbCl2 = 0.33 stoichiometry.12,13,16b Alternatively, the presence of strongly hydrogen-bonding [C2mim]+ cation may direct the formation of infinite 1D chloroplumbate(II) chains with edge-sharing octahedra (Fig. 9b), in analogy to bromoplumbate cations; however, such arrangements are very rare in chloroplumbate(II) systems.31
![Proposed structure of the anion in solid [C2mim]2{PbCl4}.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f9.gif) | ||
| Fig. 9 Proposed structure of the anion in solid [C2mim]2{PbCl4}. | ||
Raman spectra of three molten samples of the [C2mim]Cl–PbCl2 system (Fig. 10) were recorded at 148 ± 2 °C. The absence of vibrations in the region associated with Pb–Clb (200–150 cm−1) strongly suggests the presence of only mononuclear anions in the liquid state.
![Raman spectra (148 ± 2 °C, liquid state) of the [C2mim]Cl–PbCl2 system. The bands indicated in red originate from [PbCl4]2− and those indicated in blue from [PbCl3]−.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f10.gif) | ||
| Fig. 10 Raman spectra (148 ± 2 °C, liquid state) of the [C2mim]Cl–PbCl2 system. The bands indicated in red originate from [PbCl4]2− and those indicated in blue from [PbCl3]−. | ||
There are a number of publications on the liquid state speciation of molten chloroplumbate(II) inorganic salts; the studies are typically carried out at temperatures above 500 °C.32–35 For various systems, the existence of one or more of the following chloroplumbate(II) species was suggested: [PbCl]+, [Pb2Cl5]−, [PbCl3]−, [PbCl4]2−, [PbCl5]3− and [PbCl6]4−.31 The Pb–Cl Raman stretching frequencies for liquid-phase chloroplumbate(II) systems reported in the literature34,36 are as follows: ca. 201 cm−1 for hexacoordinate lead(II) in molten lead(II) chloride, 205 cm−1 for [PbCl6]4− in molten CsCl–PbCl2, ca. 232 cm−1 for [PbCl4]2− in molten CsCl–PbCl2, and 249 cm−1 for [PbCl3]− in the aqueous solution. As expected, with decreasing coordination number the average Pb–Cl bond length decreases, and therefore the blue shift is observed in the relevant stretching frequencies. Noteworthy, the Pb–Cl stretching frequency for [PbCl3]− was much lower in molten CsCl–PbCl2 (χPbCl2 = 0.50) than in the aqueous solution. This was attributed either to pseudo-tetrahedral (C3v) geometry of [PbCl3]− in the CsCl–PbCl2 system, with a sterically active lone pair, or to the presence of equilibrated polyhedra clusters, [PbxCl3x]x− (x = 1–4), but no definitive conclusion was reached.34
The observed Pb–Cl stretching frequencies were around 224 cm−1 for χPbCl2 = 0.20 and 0.33, and around 237 cm−1 for χPbCl2 = 0.45. In both cases, Pb–Cl vibrations in the molten state (224 and 237 cm−1) were blue-shifted compared to the Pb–Clt vibrations for the same compositions in the solid state (218 and 226 cm−1), suggesting lower coordination numbers of chloroplumbate(II) anions in the melt.36 Considering both the literature data and the stoichiometry of the samples (limited chloride availability at higher χPbCl2 values), it may be assumed that [PbCl4]2− is the predominant species for χPbCl2 = 0.20 and 0.33, and [PbCl3]− dominates at χPbCl2 = 0.45. This is also logical, as upon crystallisation, these monomeric units would naturally assemble into {PbCl4}− and {PbCl3}− polymeric chains, further proving a close relationship between the solid and the liquid phase.
This Raman study gave an insight into the liquid-phase anionic speciation of chloroplumbate(II) systems, but the distribution of the anions as a function of composition was not resolved. To investigate this, different techniques, such as multinuclear NMR spectroscopy, should be used. Therefore, room temperature ionic liquid system based on the [P6 6 6 14]+ cation was prepared.
[P6 6 6 14]Cl–PbCl2 system
The [P6 6 6 14]+ cation was selected to frustrate crystallisation; DSC measurements revealed remarkably low first-order phase transitions (−65 to −70 °C; 1 to 6 kJ mol−1) for all compositions.† The partial phase diagram is shown in Fig. 11. A single first-order found for the lower χPbCl2 values were tentatively assumed to correspond to the formation of [P6 6 6 14]2[PbCl4], lowering the melting point of [P6 6 6 14]Cl. For χPbCl2 ≥ 0.40, a second peak was found at higher temperatures, possibly corresponding to [P6 6 6 14][PbCl3]. The relatively high phase transition for an ionic liquid based on the singly charged [PbCl3]− might be attributable to the presence of a trigonal planar (D3h), rather than a pseudo-tetrahedral (C3v) anion.![Phase diagram of the [P6 6 6 14]Cl–PbCl2 system.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f11.gif) | ||
| Fig. 11 Phase diagram of the [P6 6 6 14]Cl–PbCl2 system. | ||
207Pb NMR spectroscopy (which has a normal chemical shift range from −5500 to 6000 ppm)36 was used to study trends in the liquid state speciation as a function of composition. Spectra acquired for [P6 6 6 14]Cl–PbCl2 (at 80 °C to lower the viscosity) are shown in Fig. 12. Single peaks were found for each composition, indicating the existence of exchange between the chloroplumbate(II) species, rapid on the NMR time scale. The 207Pb NMR signal is shifted within the range 960 to 1360 ppm; peak width also changes noticeably.
![207Pb NMR spectra (104.8 MHz, 80 °C, neat liquid) of the [C8mim]Cl–PbCl2 system, referenced to 1.0 M solution of Pb(NO3)2 in D2O, δref = −1963 ppm.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f12.gif) | ||
| Fig. 12 207Pb NMR spectra (104.8 MHz, 80 °C, neat liquid) of the [C8mim]Cl–PbCl2 system, referenced to 1.0 M solution of Pb(NO3)2 in D2O, δref = −1963 ppm. | ||
To facilitate quantitative analysis of the spectroscopic data, the chemical shift, δ, and the peak width at half-height, Δν1/2, were plotted as a function of composition, χPbCl2 (Fig. 13).
![Plots of: (●) chemical shifts, δ (ppm), and (▲) peak widths at half-height, Δν1/2 (Hz), of the 207Pb NMR signals, as well as () viscosities (mPa s) measured for the [P6 6 6 14]Cl–PbCl2 system at 80 °C, and plotted as functions of composition, χPbCl2. Dashed trend lines and dotted lines at χPbCl2 = 0.33 and 0.45 are only visual guidelines.](/image/article/2013/DT/c3dt32742f/c3dt32742f-f13.gif) | ||
Fig. 13 Plots of: (●) chemical shifts, δ (ppm), and (▲) peak widths at half-height, Δν1/2 (Hz), of the 207Pb NMR signals, as well as ( ) viscosities (mPa s) measured for the [P6 6 6 14]Cl–PbCl2 system at 80 °C, and plotted as functions of composition, χPbCl2. Dashed trend lines and dotted lines at χPbCl2 = 0.33 and 0.45 are only visual guidelines. ) viscosities (mPa s) measured for the [P6 6 6 14]Cl–PbCl2 system at 80 °C, and plotted as functions of composition, χPbCl2. Dashed trend lines and dotted lines at χPbCl2 = 0.33 and 0.45 are only visual guidelines. | ||
The δ value depends on the electron density on the 207Pb nuclei, which in this study is related directly to the coordination number of chloroplumbate(II) complexes. The peak width, Δν1/2, is influenced by the electronic environment of the nuclei and by the viscosity of the medium. The electronic environment is related to the geometry of the anion, which depends on coordination number and on the stereochemical activity of the lone pair. To identify the contribution of the electronic environment, the viscosity of the samples at 80 °C was plotted as a function of temperature (Fig. 13).
The δ values increase from the starting plateau between χPbCl2 = 0.05 and 0.15 to another plateau between χPbCl2 = 0.45 and 0.55 (see Fig. 13, upper, with three ‘speciation zones’: A, B and C). This indicates the existence of two chloroplumbate(II) species in equilibrium with each other, one predominant for low χPbCl2 (A), the other predominant for high χPbCl2 (C), and both present in significant concentrations for the intermediate compositions (B), where they must be in dynamic equilibrium. The χPbCl2 < 0.30 compositions (A) contain high concentrations of chloride; as expected, the 207Pb NMR signal is shifted upfield, indicating higher electron density on the metal nuclei (i.e. relatively high coordination number). In contrast, the χPbCl2 > 0.45 compositions (C) contain lower chloride concentrations, leading to lower coordination number of lead(II), and hence a downfield shift in the 207Pb NMR signal.
Considering the viscosity of neat ionic liquids, signals for χPbCl2 < 0.30 (A) are relatively narrow (<100 Hz), as shown in Fig. 12. This indicates high symmetry species, such as octahedral [PbCl6]4− or tetrahedral [PbCl4]2−. The increase in the Δν1/2 values for these compositions is paralleled by the increase in viscosity, as expected when replacing the small negative chloride ion with the larger, doubly or quadruply charged chloroplumbate(II) anions (cf. ref. 9 and 10).
The peak width for χPbCl2 = 0.30 is much greater than that for lower χPbCl2 values (A). Furthermore, for χPbCl2 lying between 0.30 and 0.45 (B), the Δν1/2 values increase as the viscosity of the samples decrease. This is indicative of the presence of a new species, characterised by lower charge inducing the viscosity decrease. The signal broadening derives from the dynamic exchange between two chloroplumbate(II) anions (vide supra), and also may contain a contribution from the lower symmetry species.
Signals corresponding to χPbCl2 > 0.45 (C) are broad, with the Δν1/2 values reaching a plateau for the χPbCl2 > 0.50 compositions. A plateau reached by both δ and Δν1/2 may suggest the existence of one predominant chloroplumbate(II) anion of low symmetry, but more likely it points towards an equilibrium state involving two chloroplumbate(II) anions, [PbCl4]2− and [PbCl3]−; since PbCl2 precipitates for all χPbCl2 > 0.45 compositions, the composition containing exclusively [PbCl3]− anions is never formed.
There is a paucity of 207Pb NMR data for chloroplumbate(II) ionic liquids. Based on the evidence presented here, the most plausible suggestion for the identity of chloroplumbate(II) species would be: [PbCl4]2− in A, predominance of [PbCl3]− equilibrated with [PbCl4]2− in C, and a dynamic exchange between the two anions in B. The [PbCl3]− anion is of the lowest symmetry, D3h, and its single charge would result in a viscosity decrease. [PbCl4]2− would produce more viscous ionic liquids, with signals corresponding to higher symmetry.
To confirm this speciation, Raman spectra of the [P6 6 6 14]Cl–PbCl2 system were recorded at ambient temperature. Spectra of freshly prepared compositions were of very poor quality due to fluorescence, deriving from impurities which originate from the organic chloride precursor, [P6 6 6 14]Cl. In most cases, this could be solved by recrystallisation of [cation]Cl prior to the syntheses of chloroplumbate(II) systems, but this is not practical in the case of [P6 6 6 14]Cl, which is a commercial room temperature ionic liquid (Cyphos 101). To tackle this problem, the prepared compositions were stirred overnight with activated charcoal. The presence of small amounts of suspended charcoal did not interfere with the measurements; acquired spectra were of acceptable quality and were used in this work.
The regions of Raman spectra characteristic of Pb–Cl vibrations are shown in Fig. 14. Two chloroplumbate(II) anions were detected, one present in all compositions (band at ca. 245 cm−1), and another found for χPbCl2 ≥ 0.33 (band at ca. 267 cm−1). The vibrational frequencies are higher than typical for Pb–Cl bonds; this indicates low coordination numbers and very short bonds, supporting the presence of [PbCl4]2− in all compositions and [PbCl3]− for χPbCl2 ≥ 0.33.
![Raman spectra (ambient temperature, liquid state) of the [P6 6 6 14]Cl–PbCl2 system, compared to Raman spectrum of lead(ii) chloride (χPbCl2 = 1.00). The bands indicated in red originate from [P6 6 6 14]2[PbCl4] and those indicated in blue from [P6 6 6 14][PbCl3].](/image/article/2013/DT/c3dt32742f/c3dt32742f-f14.gif) | ||
| Fig. 14 Raman spectra (ambient temperature, liquid state) of the [P6 6 6 14]Cl–PbCl2 system, compared to Raman spectrum of lead(II) chloride (χPbCl2 = 1.00). The bands indicated in red originate from [P6 6 6 14]2[PbCl4] and those indicated in blue from [P6 6 6 14][PbCl3]. | ||
In order to assess the temperature effect, several spectra for the [P6 6 6 14]Cl–PbCl2 system were recorded also at 148 ± 2 °C, but the changes in the stretching frequencies were negligible (within 2 cm−1).
It is noteworthy that PbCl2 precipitates for compositions where χPbCl2 > 0.45, although [P6 6 6 14][PbCl3] might be expected to form stoichiometrically at χPbCl2 = 0.50. This suggests that the equilibrium concentrations of both chloroplumbate(II) anions (see the equilibria shown in eqn (1)) are shifted quite strongly to the left by the precipitation of lead(II) chloride.
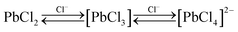 | (1) |
Comparison of two ionic liquid systems
It is striking, that the Pb–Cl stretching frequencies for [PbCl4]2− and [PbCl3]− in the liquid [P6 6 6 14]Cl–PbCl2 system (245 and 267 cm−1, respectively) are strongly blue-shifted compared to the same vibrations in molten [C2mim]Cl–PbCl2 (224 and 237 cm−1). This shows that the structures of chloroplumbate(II) anions are very flexible, as is commonly found for complexes containing a stereochemically active lone pair. For example, the stretching frequency for [PbCl3]− was found to be 237 cm−1 in molten [C2mim]Cl–PbCl2, 249 cm−1 in aqueous solution37 and 267 cm−1 in [P6 6 6 14]Cl–PbCl2 (liquid). As already pointed out by Dracopoulos et al.,34 the trigonal planar structure of [PbCl3]− is expected to have shorter Pb–Cl bonds than the pseudo-tetrahedral one. Consequently, in the hydrophobic environment of [P6 6 6 14]Cl–PbCl2, the structure of [PbCl3]− appears to be very close to planar, with short lead–chlorine bonds. In an aqueous solution, the anion distorts towards C3v symmetry. Finally, in hydrophilic [C2mim]Cl–PbCl2, with well-documented tendency of the cation to form strong hydrogen bonds with halometallate anions,10 a pseudo-tetrahedral structure of the [PbCl3]− anion is expected, with well-pronounced influence of the free electron pair (for comparison, viz. the analogous crystal structure of [C2mim][SnCl3],10 with a dense network of hydrogen bonds and pseudo-tetrahedral anion). However, in contrast to the tin(II) system, the lead(II) anion forms infinite 1D chains upon crystallisation.Conclusions
The first examples of chloroplumbate(II) room temperature ionic liquids are reported. Furthermore, the first organic/inorganic chloroplumbate(II) materials prepared via the ionothermal route are described.At ambient temperature, the imidazolium system is in the solid state, whereas the phosphonium system is in the liquid state, giving the interesting opportunity to compare and contrast these systems. Based on the phase diagrams and Raman spectra for both systems, on 207Pb NMR spectroscopy of the [P6 6 6 14]Cl–PbCl2 system, and single crystal X-ray crystallography of the [C2mim]Cl–PbCl2 system, it was established that anionic speciation regions in the solid and liquid states mirror each other, with major speciation changes at χPbCl2 = 0.30 and 0.45. This indicates, that the solid-state anionic structure of chloroplumbate(II) polymeric strands is derived directly from the liquid-state structure of chloroplumbate(II) monomeric anions.
In the liquid phase, the geometry of chloroplumbate(II) anions depends strongly on the cation. As shown by Raman spectroscopy, in the hydrophobic environment of tetraalkylphosphonium cations, [PbCl3]− is close to trigonal planar, with extremely short Pb–Cl bonds, whilst in the presence of strongly hydrogen-bonding [C2mim]+ cations, pseudo-tetrahedral geometry, with well-pronounced influence of the free electron pair, is favoured.
Acknowledgements
The authors would like to thank Prof. D. R. MacFarlane (Monash University) and Dr J. D. Holbrey for very useful comments on phase diagrams. The EPSRC UK National Crystallography Service (NCS) is acknowledged for crystal data collection. Moreover, the authors acknowledge QUILL and its Industrial Advisory Board for support. P.N. thanks the EPSRC for a RCUK fellowship.Notes and references
- D. G. Billing and A. Lemmerer, CrystEngComm, 2009, 11, 1549 RSC , and references therein.
- (a) K. Binnemans, Y. G. Galyametdinov, R. Van Deun, D. W. Bruce, S. R. Collinson, A. P. Polishchuk, I. Bikchantaev, W. Haase, A. V. Prosvirin, L. Tinchurina, I. Litvinov, A. Gubajdullin, A. Rakhmatullin, K. Uytterhoeven and L. Van Meervelt, J. Am. Chem. Soc., 2000, 122, 4335 CrossRef CAS; (b) J. D. Martin, C. L. Keary, T. A. Thornton, M. P. Novotnak, J. W. Knutson and J. C. W. Folmer, Nat. Mater., 2006, 5, 271 CrossRef CAS.
- (a) M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS; (b) Y. Yoshida and G. Saito, Phys. Chem. Chem. Phys., 2010, 12, 1675 RSC.
- (a) A. Taubert, Angew. Chem., Int. Ed., 2004, 43, 5380 CrossRef CAS; (b) J. Estager, P. Nockemann, K. R. Seddon, G. Srinivasan and M. Swadźba-Kwaśny, ChemSusChem, 2012, 5, 117 CrossRef CAS.
- (a) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1 CrossRef CAS; (b) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- (a) A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471 RSC; (b) X. Chen, D. Song, C. Asumana and G. Yu, J. Mol. Catal. A: Chem., 2012, 359, 8 CrossRef CAS.
- H. Øye, M. Jagtoyen, T. Oksefjell and J. S. Wilkes, Mater. Sci. Forum, 1991, 73–75, 183 CrossRef.
- (a) C. Hardacre, R. W. Murphy, K. R. Seddon, G. Srinivasan and M. Swadźba-Kwaśny, Aust. J. Chem., 2010, 63, 845 CrossRef CAS; (b) D. C. Apperley, C. Hardacre, P. Licence, R. W. Murphy, N. V. Plechkova, K. R. Seddon, G. Srinivasan, M. Swadźba-Kwaśny and I. J. Villar-Garcia, Dalton Trans., 2010, 39, 8679 RSC.
- J. Estager, P. Nockemann, K. R. Seddon, M. Swadźba-Kwaśny and S. Tyrrell, Inorg. Chem., 2011, 50, 5258 CrossRef CAS.
- M. Currie, J. Estager, P. Licence, S. Men, P. Nockemann, K. R. Seddon, M. Swadźba-Kwaśny and C. Terrade, Inorg. Chem., 2012 DOI:10.1021/ic300241p.
- (a) D. Freudenmann and C. Feldmann, Dalton Trans., 2010, 40, 452 RSC; (b) K. Biswas, Q. Zhang, I. Chung, J.-H. Song, J. Androulakis, A. J. Freeman and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 14760 CrossRef CAS; (c) P. Mahjoor and S. E. Latturner, Cryst. Growth Des., 2009, 9, 1385 CrossRef CAS.
- A. B. Corradi, A. M. Ferrari, G. C. Pellacani, A. Saccani, F. Sandrolini and P. Sgarabotto, Inorg. Chem., 1999, 38, 716 CrossRef CAS.
- A. B. Corradi, A. M. Ferrari, L. Righi and P. Sgarabotto, Inorg. Chem., 2001, 40, 218 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier Science, Oxford, 1997 Search PubMed.
- I. Kalf and U. Englert, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m129 Search PubMed.
- (a) S. A. Bourne and Z. Mangombo, CrystEngComm, 2004, 6, 437 RSC; (b) L. Dobrzycki and K. Woźniak, CrystEngComm, 2008, 10, 577 RSC; (c) L. Dobrzycki and K. Woźniak, J. Mol. Struct., 2009, 921, 18 CrossRef CAS; (d) M. Geselle and H. Fuess, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 51, 242 CrossRef.
- A. B. Corradi, S. Bruni, F. Cariati, A. M. Ferrari, A. Saccani, F. Sandrolini and P. Sgarabotto, Inorg. Chim. Acta, 1997, 254, 137 CrossRef.
- M. C. Aragoni, M. Arca, C. Caltagirone, F. A. Devillanova, F. Demartin, A. Garau, F. Isaia and V. Lippolis, CrystEngComm, 2005, 7, 544 RSC.
- (a) G. A. Mousdis, V. Gionis, G. C. Papavassiliou, C. P. Raptopoulou and A. Terzis, J. Mater. Chem., 1998, 8, 2259 RSC; (b) A. B. Corradi, S. Bruni, F. Cariati, A. M. Ferrari, A. Saccani, F. Sandrolini and P. Sgarabotto, Inorg. Chim. Acta, 1997, 254, 137 CrossRef.
- R. E. Morris, Chem. Commun., 2009, 2990 RSC.
- A. Thirumurugan and C. N. R. Rao, Cryst. Growth Des., 2008, 8, 1640 CAS.
- J.-P. Niu, Q.-G. Zhai, J.-H. Luo, S.-N. Li, Y.-C. Jiang and M.-C. Hu, INOCHE, 2011, 14, 663 CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- J. S. Wilkes, J. A. Levisky, C. L. Hussey and M. L. Druelinger, Proc. Int. Symp. Molten Salts, 1980, 81–89, 245 Search PubMed.
- E. S. Claudio, M. A. Horst, C. E. Forde, C. L. Stern, M. K. Zart and H. A. Godwin, Inorg. Chem., 2000, 39, 1391 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- T. Chen, Z. Sun, J.-P. Niu, Q.-G. Zhai, C. Jin, S. Wang, L. Li, Y. Wang, J. Luo and M. Hong, J. Cryst. Growth, 2011, 325, 55 CrossRef CAS.
- J. R. Ferraro, Low Frequency Vibrations of Inorganic and Co-ordination Compounds, Plenum Press, New York, 1971, 173 Search PubMed.
- I. F. Burshtein and A. L. Poznyak, Kristallografiya (Russ.) (Crystallogr. Rep.), 2002, 47, 62 Search PubMed.
- F. G. McCarty and O. J. Kleppa, J. Phys. Chem., 1964, 68, 3846 CrossRef CAS.
- V. A. Maroni, J. Chem. Phys., 1971, 54, 4126 CrossRef CAS.
- V. I. Posypaiko and E. A. Alekseeva, Phase Equilibria in Binary Halides, IFI/Plenum Media Company, New York, 1987 Search PubMed.
- V. Dracopoulos, D. T. Kastrissios and G. N. Papatheodorou, Polyhedron, 2005, 24, 619 CrossRef CAS.
- H. Chr. Marsmann and F. Uhlig, Further advances in germanium, tin and lead NMR, in Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd., New York, 2009 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds (Part A), Wiley, New York, 5th edn, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 919679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt32742f |
| This journal is © The Royal Society of Chemistry 2013 |