 Open Access Article
Open Access ArticleMorphology controlled NH4V3O8 microcrystals by hydrothermal synthesis
G. S. Zakharova*abc, Ch. Täschnerb, T. Kolbc, C. Jähnec, A. Leonhardtb, B. Büchnerb and R. Klingelerc
aInstitute of Solid State Chemistry, Ural Division, Russian Academy of Sciences, Pervomaiskaya ul. 91, Yekaterinburg, 620990 Russia
bLeibniz Institut für Festkörper- und Werkstoffforschung IFW Dresden, Helmholtzstr. 20, 01069 Dresden, Germany
cKirchhoff Institute for Physics, INF 227, University of Heidelberg, 69120 Heidelberg, Germany. E-mail: ruediger.klingeler@kip.uni-heidelberg.de
First published on 14th January 2013
Abstract
Single crystalline ammonium trivanadate NH4V3O8 with variable morphologies, including shuttles, flowers, belts, and plates, was synthesized by the hydrothermal treatment of NH4VO3 with acetic acid. The crystals optimally grow under gentle conditions of 140 °C for 48 h. The resulting NH4V3O8 microcrystals were characterized by means of X-ray diffraction, scanning electron microscopy, infrared and Raman spectroscopy, static magnetization studies, and thermal analysis. The key factors to control the size and morphology of the crystals are the pH value and the vanadium concentration. A tentative microscopic growth mechanism is proposed and it is demonstrated how shape and morphology of the resulting microcrystalline material can be tuned by appropriate synthesis parameters.
1. Introduction
Microcrystals are a subject of intensive research not only because of the possible novel properties associated with individual particles with reduced dimensions but also owing to new collective properties and advanced tunable functions arising from particle ensembles. Much attention has been hence paid to the self-assembly and transformation of individual particles into highly ordered superstructures with controllable architectures.1 Such microcrystal ensembles are characterized by high crystallinity, high porosity, and subunit alignment, rendering them promising materials for many applications such as catalysis, sensing, or energy storage.2 The controlled synthesis of such hierarchical structures is however demanding and it remains a great challenge to transfer microcrystal ensembles into applications because their formation processes are still poorly understood.There has been renewed interest in different ammonium vanadates owing to their new application as cathode materials in rechargeable lithium batteries.3–5 Ammonium trivanadate NH4V3O8 as a representative of the trivanadate family has attractive electrochemical characteristics (high specific energy, good rate capacity, low cost, and long cycle life) comparable with LiV3O8 (see, e.g., ref. 6 and 7). The structure of NH4V3O8 consists of VO5 square pyramids and VO6 octahedrons with NH4+ cations between the vanadium-oxide layers. Each pair of VO5 square pyramids is bridged by an edge to form a double V2O8 group. The V2O8 groups are connected by sharing corners to produce a sinusoidal zigzag chain along the b axis. These zigzag chains are held together through VO6 octahedrons to form a layer structure parallel to the (00l) plane.8
It is well known that the preparation method has significant influences on the properties, structure, and morphology of the compound. Traditionally, ammonium trivanadate NH4V3O8 has been prepared by the classical acidification of an NH4VO3 solution with hydrochloric or sulfuric acid up to pH = 1.5 and a subsequent treatment under hydrothermal conditions.9 This method yields NH4V3O8·0.75H2O with a belt-like shape. Wang et al.10,11 have synthesized NH4V3O8·nH2O by the surfactant (sodium dodecyl sulfonate or sodium dodecyl benzene sulfonate) assisted hydrothermal method. The morphology of the product and the water content depend on the type of the surfactant used, i.e., an NH4V3O8·0.2H2O flake-like powder was synthesized using H2SO4 to adjust the pH of the solution at 2–3 and sodium dodecyl sulfonate (SDS) as a soft template.10 The proper use of SDS promotes the agglomeration of larger trivanadate ammonium particles. In contrast, in the presence of sodium dodecyl benzene sulfonate (SDBS) and hydrochloric acid (pH = 4.0) NH4V3O8·0.37H2O nanorod-shaped crystallites are produced, which display promising electrochemical properties with a maximum specific discharge capacity of up to 327.1 mA h g−1 at 30 mA g−1 and good cycling stability.11 In comparison, the product synthesized without SDBS is composed of NH4V3O8·0.42H2O and exhibits a flake-like morphology. According to the electrochemical performance,11 the lithium ion insertion/extraction was greatly improved owing to the insertion of the surfactant and the nanorod morphology. This is also affected by doping as shown for F-ions introduced into ammonium trivanadate, (NH4)0.9V3O7.9F0.1·0.9H2O, which was prepared by precipitation from aqueous HF-acidified ammonium vanadate solutions.12 The discharge capacity is strongly influenced by the cation type, and the lithium capacity of this ammonium vanadate type is 35% greater than that of the corresponding potassium vanadate. The homogeneous precipitation method using urea is an effective way of obtaining NH4V3O8·nH2O nanorods with a diameter of 60 nm and a length of several microns.13 Ammonium trivanadate can be formed by the interaction of V2O5 with NH3 and an admixture of NH4Cl under hydrothermal conditions.14
Herein, we report a facile method for a scalable synthesis of NH4V3O8 with different morphologies based on the hydrothermal strategy without sophisticated techniques and surfactants.
2. Experimental
All the starting materials, NH4VO3 and acetic acid were analytically pure and used directly without any purification. At first, 0.2 g NH4VO3 was dissolved in 12 ml deionized water to form a light-yellow solution. Then an appropriate amount of acetic acid with the concentration of 100% was added drop-wise under stirring to adjust the desired pH value of the solution (from 2.5 up to 6.0). The clear-orange solution was transferred into a 15 ml teflon-lined stainless steel autoclave. The autoclave was heated at 140 °C for 48 h and then cooled to room temperature naturally. The obtained product was filtered, washed with deionized water and dried in air under ambient conditions.The product was characterized by powder X-ray diffraction (XRD) by means of a Shimadzu XRD-7000 S using Cu Kα radiation, λ = 1.5418 Å, with a step size of Δ2Θ = 0.02°. The morphology of the powder was determined by scanning electron microscopy (SEM, Nano-SEM; FEI). Elemental analysis was carried out using an energy-dispersive X-ray spectrometer (EDX). Fourier transform infrared (FT-IR) spectra were recorded using a Spectrum One B (Perkin-Elmer) with an automatic diffuse reflectance accessory. A thoroughly ground sample was applied as a thin layer to a purpose-designed holder plate. The spectra were measured at least three times, and the scatter in peak positions was within ±1 cm−1. Raman spectra were recorded on a Thermo Scientific DXR SmartRaman spectrometer using a 532 nm laser at a laser output power of 8 mW. Differential scanning calorimetry (DSC) was carried out using an SDT Q600 from TA Instruments. The powdery sample was heated with a rate of 10 K min−1 from room temperature up to 800 °C in a nitrogen atmosphere. Magnetization studies were done using a Quantum Design MPMS XL5 SQUID magnetometer.
3. Results and discussion
XRD patterns of the as-prepared products using different pH values are shown in Fig. 1. All diffraction peaks are indexed to a monoclinic NH4V3O8 crystal phase with the lattice constants a = 4.9993 Å, b = 8.4230 Å, c = 7.8490 Å, β = 96.4260° (JCPDS No. 088-1473). No peaks from other phases are detected indicating that the as-prepared samples are composed of ammonium trivanadate exclusively. For all pH values applied in the synthesis procedure, the data imply the same crystallographic structure, i.e. there is no change of the crystal structure in pH-dependent experiments. There are, however, subtle changes in the spectra with respect to the peak widths and intensities. Upon increasing the pH value the intensity of the (001) peak becomes stronger which indicates that the degree of crystallinity increases. The data imply that the particles size of NH4V3O8 depends on the pH value and therefore on the acidity of the solution. The dimension in the direction along the (001) plane, corresponding to layers of the VOn polyhedra of NH4V3O8 prepared at pH = 2.5, is the smallest in this series. This implies that among all products this sample features the shortest diffusion path for any insertion ion (for example, Li+) between the planes.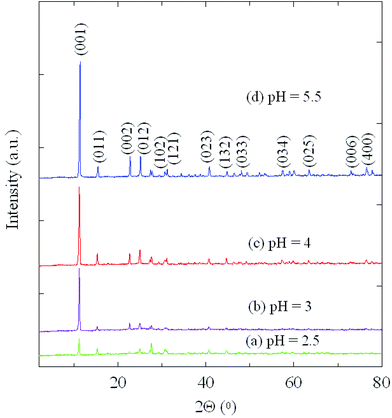 | ||
| Fig. 1 XRD patterns of NH4V3O8 obtained under different pH conditions: (a) pH = 2.5, (b) pH = 3, (c) pH = 4, (d) pH = 5.5. | ||
There are several parameters affecting the morphology characteristics of NH4V3O8 crystals such as the reaction time, the treatment temperature, the vanadium concentration, and the pH-value of the solution. In the synthesis series the reaction time was prolonged from 24 h till 56 h, and the treatment temperature changed between 120, 140 and 180 °C. While there is no obvious influence of the reaction time and temperature on the general morphology of the product the size of the resulting particles becomes smaller with increasing temperature and time of the treatment.
A main result of our study concerns the relevance of the pH value and the vanadium concentration on the crystallization and shape control of ammonium trivanadate. Ammonium trivanadate was synthesized by varying the pH value (2.5–6.0) and the vanadium concentration (CV5+ = 0.03 ÷ 0.13 mol l−1). Vanadium in the highest oxidation state of V5+ is known to exhibit various isopolyvanadate forms in aqueous solutions at different pH values and concentrations.15 In the mentioned range of the acidity and the concentration, vanadium forms vanadate-ions with the maximum of condensation degree: V10O26(OH)24−, V10O27(OH)5−, and V10O286− ions. In order to elucidate these effects, both the pH value of the solution and the vanadium concentration were varied. Fig. 2 shows SEM images of the resulting ammonium trivanadate produced at different pH values and vanadium concentrations. The figures in general clearly show that the morphology of the end products can be nicely tuned under appropriate synthesis conditions. We note that this observation is in good agreement with previous experimental results15,16 on V5+ species in aqueous solutions.
 | ||
| Fig. 2 SEM images of NH4V3O8 obtained under different acidity: (a, b) pH = 2.5, (c) pH = 3, (d) pH = 4, (e) pH = 4.5 ÷ 5.5, (f) pH = 6. | ||
NH4V3O8 microcrystals obtained at the pH value equal to 2.5 and CV5+ = 0.03 mol l−1 (Fig. 2a and b) consist of a large quantity of relatively uniform 3D rose-like crystals. The single particle size varies from 23 to 11 μm (Fig. 2b). The 3D rose-like crystals are formed by superimposed nanoplatelets with a width of 2–6 μm and a thickness of 500–800 nm. If the vanadium concentration vs. the pH value phase diagram is considered, the NH4V3O8 sample with the rose-like shape is found around the equilibrium region where V10O26(OH)24− ions and V2O5 exist.15 The shuttle-like NH4V3O8 microcrystals displayed in Fig. 2c are formed at pH = 3 and CV5+ = 0.13 mol l−1. In this region of the phase diagram (pH and CV5+), the aqueous solutions exhibit the vanadium(V) precursors as V10O26(OH)24− ions. The shuttle-like crystals have two flat tips and six symmetrically grown edges. They are 29–34 μm in length, 13–16 μm in width and 1.2–1.5 μm in thickness. In contrast to the abovementioned conditions, NH4V3O8 prepared at pH = 3.5 and CV5+ = 0.10 mol l−1 does not result in a unique and well-defined morphology. Under these conditions, irregular microplates, microflakes, and nanoparticles are revealed by SEM investigation. Vanadium(V) species in a ratio of V10O26(OH)24− and V10O27(OH)5− roughly equal to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 are present in this solution. When the pH value is set at around 4 and CV5+ equals to 0.01–0.1 mol l−1, perfect single-crystal NH4V3O8 microbelts are obtained (Fig. 2d). They have an average length of about 25–45 μm, a width of 2–15 μm, and a thickness of 600–900 nm. Smooth and clean surfaces are observed. According to the equilibrium diagram of vanadium(V) species, only V10O27(OH)5− ions can be found in aqueous solution. The NH4V3O8 samples prepared at pH = 4.5 ÷ 5.5 (Fig. 2e) show a plate-like morphology. They have typical lengths of about 60–80 μm, widths of 30–60 μm, and thicknesses ranging from 1.1 to 2.3 μm. It is quite possible that the NH4V3O8 microplates are composed of coalesced single-crystal NH4V3O8 microbelts. As discussed above, only vanadium(V)-ions in the form of V10O27(OH)5− are present in the aqueous solution. When the pH value of the solution is adjusted to about 6, the obtained product possesses plate- and filaments-like morphologies (Fig. 2f). The vanadium concentration ranges within 0.05–0.1 mol l−1. Vanadium(V) species can be found as well as V10O27(OH)5− and V10O286− ions, and the ratio between both is 1. Further increase of the pH is not possible because the acidity of the pure NH4VO3 solution is around 6.2. To summarize, tuning the pH value during the solution process can be used to control the morphology of NH4V3O8 material from 3D rose-like microcrystals up to single-crystal microbelts and microplates. The acidity of the solution obviously plays a key role among the synthesis parameters for controlling the morphology of NH4V3O8 architectures.
:1 are present in this solution. When the pH value is set at around 4 and CV5+ equals to 0.01–0.1 mol l−1, perfect single-crystal NH4V3O8 microbelts are obtained (Fig. 2d). They have an average length of about 25–45 μm, a width of 2–15 μm, and a thickness of 600–900 nm. Smooth and clean surfaces are observed. According to the equilibrium diagram of vanadium(V) species, only V10O27(OH)5− ions can be found in aqueous solution. The NH4V3O8 samples prepared at pH = 4.5 ÷ 5.5 (Fig. 2e) show a plate-like morphology. They have typical lengths of about 60–80 μm, widths of 30–60 μm, and thicknesses ranging from 1.1 to 2.3 μm. It is quite possible that the NH4V3O8 microplates are composed of coalesced single-crystal NH4V3O8 microbelts. As discussed above, only vanadium(V)-ions in the form of V10O27(OH)5− are present in the aqueous solution. When the pH value of the solution is adjusted to about 6, the obtained product possesses plate- and filaments-like morphologies (Fig. 2f). The vanadium concentration ranges within 0.05–0.1 mol l−1. Vanadium(V) species can be found as well as V10O27(OH)5− and V10O286− ions, and the ratio between both is 1. Further increase of the pH is not possible because the acidity of the pure NH4VO3 solution is around 6.2. To summarize, tuning the pH value during the solution process can be used to control the morphology of NH4V3O8 material from 3D rose-like microcrystals up to single-crystal microbelts and microplates. The acidity of the solution obviously plays a key role among the synthesis parameters for controlling the morphology of NH4V3O8 architectures.
In order to discuss the microscopic origin of the pH-dependence of the crystallite morphology the reaction mechanism should be considered in detail. The basic reaction for the synthesis of ammonium trivanadate microcrystals can be formally formulated as follows:
| 3NH4VO3 + CH3COOH ⇆ NH4V3O8 + NH3 + CH3COONH4 + H2O. |
During the dissolving in water, vanadium polyanions are surrounded by dipolar water molecules giving [V(OH)h(OH2)6−h](5−h)+ solvated species. The hydrolysis ratio h depends on the pH value leading to the formation of different forms of vanadium precursors. The six-fold coordinated neutral precursor [VO(OH)3(OH2)2]0 is formed within the range 3 < pH < 6.15 The condensation of the vanadium precursor goes via oxalation as well as preferably via the faster olation reaction along the H2O–V–OH direction (cf.Fig. 3a) and leads to the formation of a 1D-vanadium oxide network within the xy plane.17 This is schematically visualized in Fig. 3c. Belt- or shuttle-like particles are generated in this case. In the z direction (H2O–V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) no V–OH groups are present. Therefore no condensation reaction can be started. At a lower acidity of about 2.5 (pH < 3), the positive precursor [VO(OH)2(OH2)3]+ is observed.15 Likewise, the condensation occurs predominantly via olation along the H2O–V–OH directions. Besides, both x and y directions are equivalent regarding condensation and 2D-NH4V3O8 crystallites (platelets) are formed (cf.Fig. 3b), which is confirmed by our results.
O) no V–OH groups are present. Therefore no condensation reaction can be started. At a lower acidity of about 2.5 (pH < 3), the positive precursor [VO(OH)2(OH2)3]+ is observed.15 Likewise, the condensation occurs predominantly via olation along the H2O–V–OH directions. Besides, both x and y directions are equivalent regarding condensation and 2D-NH4V3O8 crystallites (platelets) are formed (cf.Fig. 3b), which is confirmed by our results.
 | ||
| Fig. 3 Schematic sketch of precursor condensation forming low-dimensional NH4V3O8 networks. The simplified olation and oxolation processes are shown in (a). Considering predominant fast olation reactions, positively charged (b) and neutral (c) precursors yield the formation of 2D and 1D networks, respectively. | ||
Further information on the valence of the V-ions in the materials can be obtained from static magnetization measurements displayed in Fig. 4. In the case of V4+-ions, i.e. electronic configuration [Ar]3d1, spin S = 1/2 is expected which is associated with a saturation moment of about 1 μB depending on the g-factor which usually is very close to 2 in V4+-compounds. Instead, V5+-ions ([Ar]3d0) are essentially non-magnetic. Fig. 4 shows the static magnetization of the single-crystalline NH4V3O8 microbelts synthesized at pH 4 (see Fig. 2d). The temperature dependence of the static magnetic susceptibility χ = M/B exhibits a Curie-like behavior which is expected for quasi-free paramagnetic moments. Fitting the data by means of a Curie–Weiss law χ(T) = χ0 + C/(T − Θ), with the Curie constant C, the Weiss temperature Θ, and a temperature-independent contribution χ0 yields χ0 ≈ 8 × 10−5 emu mol−1, Θ ≈ 0. From the Curie constant we infer the effective magnetic moment peff = 0.07 μB. This value is much smaller than the value of peff = 1.73 μB which would be expected for a system with S = 1/2. Quantitatively, our results imply that nearly all V-ions are non-magnetic, i.e. V5+, while only ∼0.16% of magnetic species with S = 1/2 are observed, which can be attributed to V4+-ions. This very low number of magnetic species in the material is confirmed by the field dependence of the magnetization in Fig. 4b, which implies a very low magnetization at T = 2 K. Summarizing, the analysis of the static magnetization data implies that about 99.9% of the V ions are in the 5+ state.
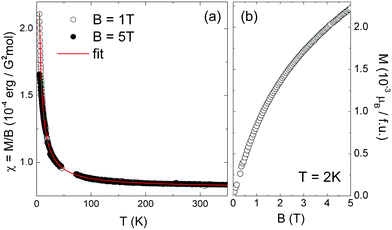 | ||
| Fig. 4 (a) Temperature dependence of the static magnetic susceptibility χ = M/B of NH4V3O8 microbelts (prepared at pH = 4) at B = 1 T and 5 T, and (b) magnetic field dependence of the magnetization at T = 2 K. The line in (a) is a Curie–Weiss fit to the data (see the text). | ||
All samples show similar FT-IR curves (Fig. 5). There are several absorption bands at 3232, 1403, 1006, 962, 727, and 522 cm−1, respectively, which can be assigned to the different excitations of the ammonium trivanadate structure. The line at 1006 cm−1 corresponds to the stretching vibration of a VO terminal bond with the length of 1.597(4) Å of the distorted octahedron. The peak at 962 cm−1 is associated with a stretching vibration of the VO terminal bond length of 1.602(3) Å in the distorted square pyramids. The band at 727 cm−1 is assigned to an asymmetric stretching vibration of the V–O–V bridges with bond distances of 1.80–1.88 Å. The absorption band centered around 522 cm−1 is due to stretching vibrations of the groups incorporating a bridging oxygen coordinated to three or four vanadium atoms with bond distances of 1.9–2.9 Å.18 These groups are located in double chains of distorted octahedrons and square pyramids forming the V–O framework in many vanadium oxide compounds with a significant degree of polymerization. The band at 1403 cm−1 is attributed to a symmetric bending vibration of NH4+ groups. The corresponding asymmetric stretching vibrations show up as the peak at 3232 cm−1.19 The strong broadening of this stretching band is due to the presence of a dense network of hydrogen bonds. In addition, crystal water is identified by a characteristic band at 1644 cm−1.20 The shift of the bending vibration into the region of higher wavenumbers may be described by the formation of hydrogen bonds between water and ammonium molecules.
 | ||
| Fig. 5 FTIR spectrum of NH4V3O8 prepared at pH = 4. | ||
The results of Raman spectroscopy are presented in Fig. 6 and show ammonium trivanadate NH4V3O8 in comparison with the NH4VO3 precursor. The Raman spectrum of NH4VO3 (Fig. 6a) consists of a strong band at 926 cm−1, medium peaks at 895, 645 cm−1 and weak bands at 493, 259 and 209 cm−1. This spectrum is in good agreement with literature data21 and indicates a chain-like structure of NH4VO3 formed by connected tetrahedral basic units. The Raman spectrum of NH4V3O8 (Fig. 6b) in the wavelength range of 60–1200 cm−1 is dominated by the peaks at 109, 150, 182, 210, 240, 260, 372, 427, 500, 514, 556, 673, 813, 963, 993 cm−1. The bands at 963 and 993 cm−1 are assigned to VO stretching modes of distorted octahedral and distorted square pyramids, respectively. The band at 813 cm−1 can be assigned to V–O stretching bonds.22 The bands at 514, 556, 673 cm−1 can be attributed to V–O–V stretching modes. A very strong band at 673 cm−1 confirms predominately the existence of a framework than the chain-like structure similar to NH4VO3.23 Other bands in the frequency range below 400 cm−1 can be assigned to V–O and N–H bending modes.24
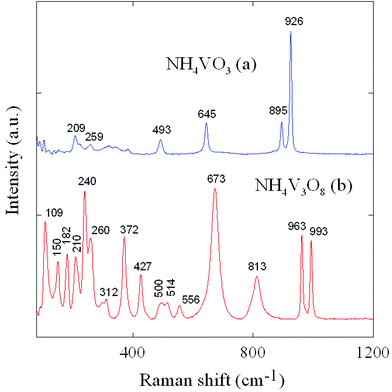 | ||
| Fig. 6 Raman spectra of (a) NH4VO3 and (b) NH4V3O8. | ||
In order to determine the thermal stability of ammonium trivanadate and the amount of crystal water in the sample, TG and DSC studies of the as-prepared materials were carried out (Fig. 7). There is an exothermal peak at 344 °C, with an integrated weight loss of about 10.6%, which can be attributed to the decomposition of ammonium trivanadate according to the following eqn:
| 2NH4V3O8·xH2O → 3V2O5 + 2NH3↑ + (1 + 2x)H2O. |
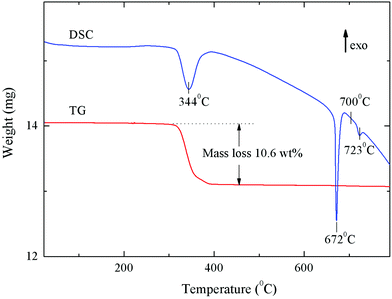 | ||
| Fig. 7 Thermogravimetric analysis of NH4V3O8 under an N2 atmosphere. | ||
The measured mass loss allows estimating the amount of crystal water in the as-prepared material. The analysis yields a water content of 0.35H2O per formula unit (NH4V3O8·0.35H2O). These data are in good agreement with ref. 10, implying the similarity of the as-prepared materials with a small amount of crystal water (about 0.2 per unit in ref. 10) and the water-free NH4V3O8. The sharp endothermic effect at 672 °C visible in the DSC data demonstrates the melting process of vanadium pentoxide.
In summary, a facile hydrothermal method was developed to create ammonium trivanadate NH4V3O8 with various morphologies. The influence of the pH value was studied in detail. In addition, the optical properties of NH4V3O8 have been examined by Raman and IR spectra. A possible growth mechanism is proposed. The phase morphology of ammonium trivanadate is governed by the actual V5+ species available in the aqueous solution, which mainly depends on the pH value. This facile pH-different hydrothermal route can be significant for the production of NH4V3O8 microstructures with controlled shape and morphology which can be used for the preparation of high performance battery materials.
Acknowledgements
Technical support by S. Pichl, M. Gierth, and G. Kreutzer is gratefully acknowledged. This work was supported by the Deutsche Forschungsgemeinschaft through grant KL 1824/4, by the BMBF via LIB 2015 (project 03SF0397) and by the Presidium of the Russian Academy of Sciences (program Novel Processes for the Preparation of Chemical Substances and Fabrication of Advanced Materials).References
- (a) J. H. Fendler, Chem. Mater., 1996, 8, 1616–1624 CrossRef CAS; (b) Y. F. Shi, Y. Wan and D. Y. Zhao, Chem. Soc. Rev., 2011, 40, 3854–3878 RSC; (c) H. C. Zeng, J. Mater. Chem., 2011, 21, 7511–7526 RSC; (d) V. Polshettiwar, B. Baruwati and R. S. Varma, ACS Nano, 2009, 3, 728–736 CrossRef CAS.
- (a) P. X. Gao, P. Shimpi, H. Y. Gao, C. H. Liu, Y. B. Guo, W. J. Cai, K. T. Liao, G. Wrobel, Z. H. Zhang, Z. Ren and H. J. Lin, Int. J. Mol. Sci., 2012, 13, 7393–7423 CrossRef CAS; (b) M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC; (c) R. Z. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef CAS; (d) S. D. Zhang, Y. M. Li, C. Z. Wu, F. Zheng and Y. Xie, J. Phys. Chem. C, 2009, 113, 15058–15067 CrossRef CAS.
- K.-F. Zhang, G.-Q. Zhang, X. Liu, Z.-X. Su and H.-L. Li, J. Power Sources, 2006, 157, 528–532 CrossRef CAS.
- H.-L. Fei, Z.-R. Shen, J.-G. Wang, H.-J. Zhou, D.-T. Ding and T.-H. Chen, Electrochem. Commun., 2008, 10, 1541–1544 CrossRef CAS.
- H. Fei, Z. Shen, J. Wang, H. Zhou, D. Ding and T. Chen, J. Power Sources, 2009, 189, 1164–1166 CrossRef CAS.
- Y. Q. Qiao, X. L. Wang, J. P. Zhou, J. Zhang, C. D. Gu and J. P. Tu, J. Power Sources, 2012, 198, 287–293 CrossRef CAS.
- H. Liu, Y. Wang, W. Yang and H. Zhou, Electrochim. Acta, 2011, 56, 1392–1398 CrossRef CAS.
- B.-Z. Lin and S.-X. Liu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1961–1963 Search PubMed.
- L. Q. Mai, C. S. Lao, B. Hu, J. Zhou, Y. Y. Qi, W. Chen, E. D. Gu and Z. L. Wang, J. Phys. Chem. B, 2006, 110, 18138–18141 CrossRef CAS.
- H. Wang, K. Huang, S. Liu, C. Huang, W. Wang and Y. Ren, J. Power Sources, 2011, 196, 788–792 CrossRef CAS.
- H. Wang, Y. Ren, W. Wang, X. Huang, K. Huang and Y. Wang, J. Power Sources, 2012, 199, 315–321 CrossRef CAS.
- C. C. Torardi and C. R. Miao, Chem. Mater., 2002, 14, 4430–4433 CrossRef CAS.
- H.-K. Park and G. Kim, Solid State Ionics, 2010, 181, 311–314 CrossRef CAS.
- S. D. Huang and Y. Shan, Chem. Commun., 1998, 1069–1070 RSC.
- J. Livage, Coord. Chem. Rev., 1998, 178, 999–1018 CrossRef.
- H. L. Fei, H. J. Zhou, J. G. Wang, P. C. Sun, D. T. Ding and T. H. Chen, Solid State Sci., 2009, 11, 102–107 CrossRef CAS.
- G. S. Zakharova, V. L. Volkov, Ch. Täschner, I. Hellmann, A. Leonhardt, R. Klingeler and B. Büchner, Solid State Commun., 2009, 149, 814–817 CrossRef CAS.
- A. Jin, W. Chen, Q. Zhu, Y. Yang, V. L. Volkov and G. S. Zakharova, Thin Solid Films, 2009, 517, 2023–2028 CrossRef CAS.
- C. Zheng, X. Zhang, Z. Qiao and D. Lei, J. Solid State Chem., 2001, 159, 181–185 CrossRef CAS.
- G. S. Zakharova, V. L. Volkov, C. Täschner, I. Hellmann, A. Leonhardt, R. Klingeler and B. Büchner, Mater. Lett., 2011, 65, 579–582 CrossRef CAS.
- S. Onodera and Y. Ikegami, Inorg. Chem., 1980, 19, 615–618 CrossRef CAS.
- L. V. Kristallov, O. V. Koryakova, L. A. Perelyaeva and M. T. Tsvetkova, Russ. J. Inorg. Chem., 1987, 32, 1811–1816 CAS.
- A. Shimizu, T. Watanabe and M. Inagaki, J. Mater. Chem., 1994, 4, 1475–1478 RSC.
- J. Twu, C.-F. Shih, T.-H. Guo and K.-H. Chen, J. Mater. Chem., 1997, 7, 2273–2277 RSC.
| This journal is © The Royal Society of Chemistry 2013 |