 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfide ions as modulators of metal–thiolate cluster size in a plant metallothionein†
Tamara
Huber
and
Eva
Freisinger
*
Institute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: freisinger@aci.uzh.ch
First published on 2nd May 2013
Abstract
Metallothioneins are small cysteine-rich proteins coordinating various transition metal ions preferably with the electron configuration d10. They are ubiquitously present in all phyla, and next to phytochelatins they represent a successful molecular concept for high-capacity metal ion binding. Recent studies showed the incorporation of sulfide ions into the metal–thiolate cluster of metallothionein 2 from the plant Cicer arietinum (cicMT2) increasing the cadmium binding capacity and stability of the cluster. In the present work, the sulfide-induced structural changes accompanying the cluster formation and the sulfide-modulated increase in cluster size are analyzed in detail with a variety of analytical and spectroscopic techniques. Evaluation of the mechanism of sulfide containing CdII–thiolate cluster formation in cicMT2 reveals a strong dependence on the sequence of metal and sulfide additions for successful sulfide incorporation. To probe the general ability of metallothioneins to form sulfide containing larger metal–thiolate clusters, analogous experiments were performed with a mammalian metallothionein. The observation that the cadmium binding ability of rabbit liver MT2A was only slightly increased led to the development of a hypothesis in which the long cysteine-free linker regions present in certain plant metallothioneins may contribute to the accommodation of the respective larger cluster assemblies.
Introduction
Metallothioneins (MTs) comprise a diverse class of metal binding proteins and their occurrence is reported throughout the animal kingdom, in plants, several eukaryotic microorganisms, as well as in some prokaryotes.1–3 MTs are low molecular mass proteins (<10 kDa) with a high percentage of cysteine residues, which are involved in the formation of thermodynamically very stable metal–thiolate clusters with d10 metal ions, such as ZnII, CuI, CdII, and HgII. While the metal ions are relatively buried within the cluster, some of the thiolate ligands are accessible to the surrounding solvent and are more prone to oxidation, resulting in the release of a fraction of the metal ions from MTs in vitro.4,5 Accordingly, it is feasible that the cellular redox state has an influence on the availability of the coordinated metal ions and hence might indicate a function of these proteins in vivo. MTs are also reported to play a role in metal homeostasis and detoxification, as scavengers of reactive oxygen species, and in signal transduction.4,6–13 In plants and certain microorganisms, however, a family of enzymatically synthesized metal binding peptides with the general formula (γ-GluCys)nGly, the phytochelatins (PCs), are crucial for the detoxification of metal ions.7,14,15 For example, suppression of PC synthesis in the fission yeast Schizosaccharomyces pombe leads to a metal sensitive phenotype. S. pombe produces two forms of CdII–PC complexes, a dimeric sulfide-free low molecular mass complex of the composition Cd1.8[(γ-GluCys)3Gly]2 as well as a tetrameric high molecular mass complex, which contains additional sulfide ions, i.e. Cd5.4S[(γ-GluCys)3Gly]4.16,17 Acidification of the high molecular mass complex in vitro and reconstitution with CdII in the presence of Na2S further increased the CdII binding capacity, resulting in a complex with a CdII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S2−:(γ-GluCys)3Gly ratio of 1.8:1:1.17 Recruiting sulfide ions as additional ligands also leads to a higher thermodynamic stability of the PC complexes.18
:S2−:(γ-GluCys)3Gly ratio of 1.8:1:1.17 Recruiting sulfide ions as additional ligands also leads to a higher thermodynamic stability of the PC complexes.18
More recently, occurrence of sulfide ions was detected in a broad range of recombinantly expressed ZnII- and CdII-forms of GST-MT fusion proteins, including mammalian MT1 and MT4 as well as Quercus suber (cork oak) MT2.10,19–21 The only native MT-form found to contain sulfide ions so far is the CdII-loaded form of the yeast CuI-MT CUPI, which was obtained after the growth of Saccharomyces cerevisiae in CdII-supplemented media.22
The stoichiometry of the sulfide containing MT forms described so far displays a high degree of heterogeneity. It is hence not possible to establish a general rule with respect to the ratio between incorporated sulfide ions and coordinated metal ions or even between the combined content of Cys thiolate groups and sulfide ions and the metal ions. The main consensus is a S2− to metal ion ratio below one, reflecting the need to compensate the additional negative charges introduced into the cluster by the sulfide ions with additionally coordinated metal ions. Presuming that incorporation of sulfide ions has in vivo relevance for certain MTs, flexible metal ion and sulfide contents would provide a simple and fast way to adapt to changes in the physiological environment. Generally, sulfide ions take part in a variety of functions including redox and electron transfer processes, structure stabilization of proteins, enzyme regulation, iron and oxygen sensing, as well as neurotransmission and neuromodulation.23–27 Interestingly, some of these functions have also been proposed for MTs.9
The focus of this work is on the plant MT2 from Cicer arietinum (chickpea), cicMT2. The mRNA of this protein is up-regulated during epicotyls growth and shows increased expression in mature tissues.28 The amino acid sequence of cicMT2 consists of 78 amino acids and features two Cys-rich regions containing eight and six cysteine residues, respectively, separated by a 41 amino acids long linker region:
(M) ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) KMYPDMSY TEQTTSETLV
KMYPDMSY TEQTTSETLV
MGVASGKTQF EGAEMGFGAE NDG ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
cicMT2, obtained by recombinant expression in Escherichia coli, is able to coordinate five divalent metal ions such as ZnII, CdII, and CoIIin vitro.29 Previous results strongly suggest that the coordinated metal ions are arranged in a single metal–thiolate cluster formed by the combined N- and C-terminal Cys-rich regions of the protein leading to a hairpin-like structure. Recently, we evaluated the incorporation of sulfide ions into the CdII–thiolate cluster of cicMT2 in vitro.30 In the presence of initially 10 equiv. of sulfide ions a protein complex of the average composition Cd9S7cicMT2 is formed. In addition to a significantly increased binding capacity of cicMT2 for CdII, the incorporation of sulfide ions also raises the pH stability of the formed cluster by approximately 0.5 pKa units compared to Cd5cicMT2. In the present work, the sulfide-induced structural changes accompanying the cluster formation and the sulfide-modulated increase in cluster size are analyzed in more detail. Successful incorporation of sulfide ions strongly depends on the sequence of metal ion and sulfide additions, which is evaluated with UV and (magnetic) circular dichroism spectroscopy as well as with analytical methods to determine the amount of metal and sulfide ions bound to the protein. To probe the general ability of metallothioneins to form sulfide containing larger metal–thiolate clusters, analogous experiments were performed with rabbit liver MT2A, but only a marginal increase of the metal ion binding capacity was observed. We hypothesize therefore that the long cysteine-free linker region present in certain plant metallothioneins but not in mammalian MTs might be a structural prerequisite to allow hosting of larger cluster assemblies.
Materials and methods
Chemicals and solutions
The enzymes and purification systems used for plasmid construction were purchased from Promega AG (Dübendorf, Switzerland), Fermentas (Le Mont-sur-Lausanne, Switzerland), or Roche (Rotkreuz, Switzerland). All buffers and chemicals were ACS grade or comparable from Sigma-Aldrich (Buchs, Switzerland), Merck (Darmstadt, Germany), Chemie Brunschwig (Basel, Switzerland), and Roth AG (Arlesheim, Switzerland). All solutions were prepared using Millipore water. If necessary, solutions were saturated with nitrogen or argon. Whenever complete absence of oxygen was required, Millipore water was degassed by three consecutive freezing-thawing cycles under vacuum.Plasmid construction
To increase expression yields the cicMT2 coding sequence was transferred from the previously used construct in pTYB229,30 (New England Biolabs, Ipswich, USA) into the vector pGEX-4T-1 (GE Healthcare, Uppsala, Sweden) using the BamHI and XmaI restriction sites, which allows overexpression of proteins with an N-terminal glutathione S-transferase- (GST-) tag that can be proteolytically cleaved with thrombin. To increase cleavage efficiency and selectivity as well as to obtain cicMT2 with its native N-terminal Ser residue, an additional tobacco etch virus (TEV) protease cleavage site was introduced into the vector between the thrombin cleavage site and cicMT2. For this, an additional fragment consisting of two complementary oligonucleotides (ATCTTAGTGT![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) GAGAACCTTTACTTCCAATCCTGCTGTGGCGGTGCATGTCAA, restriction sites are underlined) was digested with BamHI and MfeI and inserted into the analogously digested and dephosphorylated vector. While the BamHI restriction site is part of the multiple cloning region of the vector backbone, the MfeI site is part of the cicMT2 coding sequence. Identity of the resulting plasmid pGtMT was verified by DNA sequencing (Microsynth AG, Balgach, Switzerland). For the complete vector sequence see the ESI.†
GAGAACCTTTACTTCCAATCCTGCTGTGGCGGTGCATGTCAA, restriction sites are underlined) was digested with BamHI and MfeI and inserted into the analogously digested and dephosphorylated vector. While the BamHI restriction site is part of the multiple cloning region of the vector backbone, the MfeI site is part of the cicMT2 coding sequence. Identity of the resulting plasmid pGtMT was verified by DNA sequencing (Microsynth AG, Balgach, Switzerland). For the complete vector sequence see the ESI.†
Expression and purification of Zn5cicMT2
The GST-cicMT2 fusion protein was overexpressed in the protease-deficient E. coli strain BL21(DE3) according to the GST manual (GE Healthcare). After induction with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at an optical density at 600 nm of 1, Luria-Bertani (LB) expression media were supplemented with a final concentration of 200 μM ZnCl2 and cells harvested after 6–8 h growth at 30 °C. Cells were resuspended in phosphate-buffered saline (PBS; 10 mM Na2HPO4, 1.8 mM KH2PO4, 140 mM NaCl, 2.7 mM KCl, pH 7.3), frozen at −80 °C overnight, and lysed by ultrasonication. The soluble cell extract was applied to a GST column (GSTPrep FF 16/10, GE Healthcare), equilibrated with PBS. After washing, the fusion protein was stripped from the column using 50 mM reduced glutathione and cleaved with 1 mg TEV protease per 40 mg of GST-cicMT2 fusion protein for 12 h at 4 °C in the presence of 0.5 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM dithiothreitol (DTT).31 Oxidized cicMT2 was reduced with 25 mM DTT and separated from the GST tag and TEV protease by size exclusion chromatography using a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare) in 10 mM Tris-HCl, 50 mM NaCl, pH 7.3. The protein was concentrated by ultrafiltration (Amicon, Millipore AG, Zug, Switzerland) and the oxidized cicMT2 was again reduced with 100 mM DTT. Subsequently, a slight excess of ZnII ions was added to reconstitute Zn5cicMT2 and a final purification was performed by size exclusion chromatography using the same column as above in 10 mM Tris-HCl, 10 mM NaCl, pH 7.3.TEV protease expression and purification
The plasmid coding for TEV protease (pRK793) was obtained from AddGene (Cambridge, MA, USA). Expression and purification of TEV was performed as described with slight modifications.31 The protein was expressed in the protease-deficient E. coli expression strain BL21(DE3)-RIL. After induction with 1 mM IPTG at an optical density at 600 nm of 0.5, cells were grown for 4 h at 30 °C, harvested by centrifugation, and lysed by ultrasonication. The soluble cell extract was applied to a NiII-loaded nitrilotriacetate (Ni-NTA) agarose column (Qiagen AG, Hombrechtikon, Switzerland), equilibrated in 50 mM sodium phosphate buffer pH 8.0, 200 mM NaCl, 10% glycerol, 25 mM imidazole. After washing, the TEV protease was stripped from the column using 500 mM imidazole. Eluted TEV protease was immediately applied to a HiTrap desalting column (GE Healthcare), equilibrated in 25 mM sodium phosphate buffer pH 8.0, 100 mM NaCl, 10% glycerol, to prevent precipitation of the protein in the high imidazole concentration. Average yields were 15 mg purified TEV protease per liter cell culture medium.Preparation of rabbit liver MT2A
Purified rabbit liver MT2A, isolated from its native source and purified as described,32,33 was a kind gift of Prof. Milan Vašák (Institute of Inorganic Chemistry, University of Zurich, Switzerland). For quantification of metal ions as well as potentially sulfide ions the “native” lyophilized protein was dissolved in 10 mM Tris-HCl, 10 mM NaCl, pH 8.3 and purified with size exclusion chromatography (Hiload 16/60 Superdex 75 pg column, GE Healthcare). For the reconstitution of the ZnII-form the lyophilized protein was dissolved in 10 mM Tris-HCl, 10 mM NaCl, 10 mM DTT, pH 8.3 and incubated for 12 h to ensure complete reduction of Cys-thiolate groups. Bound metal ions were removed by acidification of the protein to pH 2.0 with 1 M HCl and subsequent size exclusion chromatography (Hiload 16/60 Superdex 75 pg) in 10 mM HCl in an anaerobic chamber. 10 equiv. of ZnCl2 were added to the apo-form, the pH was increased to pH 7.3 with NaOH and the protein concentrated using an Amicon stirred cell under argon pressure.Preparation of apo-cicMT2
For the preparation of apo-cicMT2, reconstituted Zn5cicMT2 was incubated with 100 mM DTT for 1 h prior to acidification of the protein to pH 2.0 with 1 M HCl. To remove metal ions, the sample was applied to a Hiload 16/60 Superdex 75 pg size exclusion column pre-equilibrated with 10 mM HCl in an anaerobic chamber.Metal ion titrations
For the titrations with ZnII ions apo-MT was used and for titrations with CdII ions either the ZnII-loaded MT form or apo-MT. For each titration, 1 mL of a 10 μM MT solution in 10 mM Tris-HCl, 10 mM NaCl, pH 7.3 was transferred into a septum-sealed cuvette with a path length of 1 cm inside an anaerobic chamber. If required 5–30 equiv. of S2− ions were added in form of a Na2S solution and the mixture was incubated for at least 10 min. The respective metal ion solution was filled into a Hamilton glass syringe inside the anaerobic chamber. Subsequently, the metal ion solution was added to the cuvette through the septum outside of the chamber in steps of 1 equiv., and UV, CD, and/or MCD spectra were taken after incubation for approximately 2 min.To evaluate the mechanism of sulfide-containing CdII–thiolate cluster formation, three experiments were performed, in which the order of Zn5cicMT2 (10 μM), CdII (0 to ≥15 equiv.), and S2− (100 μM) addition was varied. When the protein solution was incubated with 10 equiv. of S2− ions and then titrated with increasing amounts of CdII ions, the experimental setup with one cuvette and a Hamilton glass syringe as described above was used. In the other two experiments, however, the S2− ions or the protein solution was the last component to be added and accordingly a separate sample had to be prepared for each titration step as described below.
“MT2 + xCdII + 10 S2−”: 10 μM Zn5cicMT2 was mixed with the required equiv. of CdII ions (0–19 equiv. in 1.25 equiv. steps), incubated for 2 min, and then 10 equiv. of S2− were added.
“xCdII + 10 S2− + MT2”: The required equiv. of CdII ions (0–15 equiv. in 1 equiv. steps) were mixed with 10 equiv. of S2−, incubated for 10 min, and then 10 μM Zn5cicMT2 was added.
After titration, samples were subjected to size exclusion chromatography using a Superdex 75 10/300 GL analytical column in 10 mM Tris-HCl, 10 mM NaCl, pH 7.3 to remove unbound metal and sulfide ions. The major peak corresponding to the monomeric protein species was collected and used for the determination of protein, metal, and sulfide ion concentrations.
The analogous experiment performed with apo-cicMT2 instead of Zn5cicMT2 was followed with UV spectroscopy only and is shown in the ESI.†
UV/Vis, CD, and MCD measurements
UV/Vis absorption spectra were recorded on a Cary 500 scan spectrophotometer (Agilent Technologies AG, Basel, Switzerland) using a scan speed of 600 nm min−1. CD and MCD spectra were acquired with a J-810 spectropolarimeter (Jasco Corporation, Tokyo, Japan) equipped with a 1.5 T (15 kG) magnet over a spectral range of 180–500 nm with a scan speed of 50 nm min−1 and accumulation of four spectra for each measurement. All spectra were recorded at room temperature.Determination of protein, potential cystine, sulfide, and metal ion concentrations
The protein concentration was assessed by thiol group quantification with 2,2′-dithiodipyridine (2-PDS) at pH 4.0 as described.29,34 To ensure the reduced state of cicMT2 after the different kinds of titrations the amount of disulfide bonds, i.e. cystine moieties, was determined with 5,5′-dithiobis(2-nitro-benzoic acid) (DTNB) and sodium sulfite. Disulfide bridges lead to the formation of 2-nitro-5-thio-benzoic acid, which can be quantified via its absorption at 412 nm (ε412 nm = 13600 M−1 cm−1).35,36 Results indicate that less than 5% of the Cys residues are oxidized. However, as values are within the error range of the assay, all proteins were assumed to be in the completely reduced state. The concentration of sulfide ions was determined with the methylene blue and the 2-PDS assays as described.30 The metal ion concentration was analyzed with flame atomic absorption spectroscopy (F-AAS) using an AA240FS spectrometer (Agilent Technologies AG).
Results and discussion
ZnII- and ZnII/S-forms of cicMT2
Compared to the previously used expression and purification system, i.e. pTYB2 from the IMPACT™-CN system (New England Biolabs),29,30 use of the modified GST-fusion system with an additional TEV protease cleavage site increased the yields of purified Zn5cicMT2 approximately 15 fold to 15 mg L−1 of cell culture. As an additional benefit reduction of the protein purification time by roughly 50% was achieved. The protein obtained in this way coordinates five ZnII ions as determined with F-AAS for the metal ion concentration and with the 2-PDS assay for the Cys–SH content and hence the protein concentration. The metal binding stoichiometry was confirmed by mass spectrometry at neutral pH (ESI†). The DTNB assay indicates that within the error range of the method all Cys residues of the protein are in the reduced state, which was also confirmed by ESI-MS at acidic pH (ESI†). Hence, the quality and purity of the protein are in excellent agreement with the results obtained with the IMPACT™-CN system previously used by us.29,30 In contrast to other investigations on MTs overexpressed as GST-fusion proteins in E. coli,20 cicMT2, expressed in its ZnII-form, does not contain any sulfide ions.We recently reported that titration of Zn5cicMT2 with CdII ions in the presence of 10 equivalents of sulfide ions leads to the formation of a complex with the average composition Cd9S7cicMT2 that appears to contain a single metal cluster involving the Cys residues of both the N- and C-terminal Cys-rich regions as ligands.30 To evaluate the formation of an analogous ZnII/S-form apo-cicMT2 was titrated with ZnII ions in the presence and absence of 10 equiv. of S2−. The plot of molar absorptivity of the ligand-to-metal charge transfer (LMCT) band centered around 230 nm, indicative of ZnII coordination to thiolate ligands, against the amount of ZnII ions added to the solution shows that in the absence of sulfide the apo-protein is able to coordinate up to five ZnII ions as expected (Fig. 1A). Titration in the presence of sulfide shows the evolution of an additional absorption band, or of multiple closely spaced bands, centered around 265 nm (Fig. 1B). The analogous formation of LMCT bands in the spectral range between 260 and 280 nm has been observed during the titration of a solution containing ZnII and cysteine in a ratio of 1:2 with a Na2S solution and was assigned to the formation of cysteine-capped ZnS nanocrystallites.37 A similar bathochromic shift of absorption bands indicative of sulfide incorporation into cadmium(II)–thiolate clusters has also been observed, for example, during the formation of the CdII/S-cicMT2 species, upon titration of mammalian CdII-MTs with sulfide, and for the sulfide containing PC complexes in S. pombe.21,30,38 The origin of the observed shifts of the LMCT transitions has been attributed to decreasing HOMO–LUMO gaps with increasing cadmium cluster size. A major influence is also ascribed to the transition to structures with highly coordinated sulfur atoms, e.g., μ3- or even μ4-S2− ions.39–42
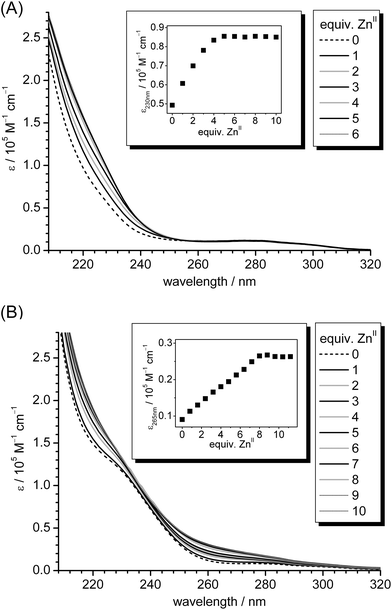 | ||
| Fig. 1 (A) UV spectra of the titration of 10 μM apo-cicMT2 in a buffer containing 10 mM Tris-HCl, 10 mM NaCl, pH 7.3 with increasing amounts of ZnII ions. The inset shows the increase of molar absorptivity of the LMCT band at 230 nm plotted against the number of ZnII ions added to the solution. (B) UV spectra of the analogous titration as shown in (A), but in the presence of 10 equiv. of S2− ions. In the inset the red-shifted LMCT band at 265 nm is plotted against the number of ZnII ions. | ||
When the absorptivity at 265 nm is plotted against the equiv. of added ZnII ions (Fig. 1, inset) an absorptivity increase up to eight ZnII ions added is observed. This indicates that the binding capacity of cicMT2 for ZnII is clearly increased in the presence of sulfide as observed for the analogous titration with CdII.30 To determine the sulfide content of the newly formed species, i.e. ZnxSycicMT2, unbound metal and sulfide ions were removed by size exclusion chromatography (SEC). Analysis of the elution peak corresponding to the monomeric protein species by a combination of F-AAS, 2-PDS assay, and the methylene blue method revealed an average stoichiometric composition of Zn7.9(6)S7(1)cicMT2. This value is similar to the results obtained for the titration of Zn5cicMT2 with CdII ions mentioned above, i.e. Cd9S7cicMT2, and contrasts previous reports in the literature that the ZnII and sulfide contents of ZnII/S-MT forms are usually around 50% lower than the CdII and sulfide contents of the respective CdII/S-MT forms.20,43 It has to be noted, however, that these literature values were for species directly isolated from E. coli and were not obtained by systematic titration experiments as described here.
Sulfide-induced changes of CdII–thiolate cluster structure
To evaluate if sulfide incorporation and the accompanied increase in cluster size leads to changes of the cluster structure, CD and MCD spectra were recorded for different points during the titration of Zn5cicMT2 with CdII ions in the presence of 10 equiv. of sulfide. Results were compared to spectra from the analogous titration without sulfide ions (Fig. 2). To complement the data also the corresponding previously published UV spectra are shown (Fig. 2A and D).29,30 The pronounced CD bands in the spectra of CdII-substituted MTs, which can be observed at higher wavelengths than the bands of the peptide backbone, are proposed to originate from peptide-induced asymmetry of the binding sites within the cluster as well as from the chirality introduced by bridging thiolate ligands.44 The cluster formation, i.e. recruitment of bridging thiolate ligands, is often, but not always, accompanied by exciton splitting as indicated by two oppositely signed CD bands with a cross-over point at the position of the absorption band around 250 nm. CD spectra of mammalian Cd7MTs typically show this feature. For Cd5cicMT2 however, only a monophasic CD profile is observed as similarly seen, for example, in the spectrum of Cd-MtnE, one of the MTs from Drosophila melanogaster.29,45 Despite this monophasic CD profile, the CdII ions in Cd5cicMT2 are clearly coordinated in the form of a metal–thiolate cluster as indicated by a red-shift of absorption bands during the titration of apo-cicMT2 with CdII or CoII ions.29 The CD spectra for the titration of Zn5cicMT2 with CdII ions (Fig. 2B) reveal a shift of the CD band, which is located around 255 nm in the ZnII-form, from 252 nm after the addition of the first equiv. of CdII to a final position at 248 nm after saturation of the protein with 5 CdII ions. In addition, a broad band with low molar ellipticity evolves around 275 nm, again very similar to the spectrum reported for D. melanogaster Cd-MtnE. The CD spectra recorded for the analogous titration in the presence of 10 equiv. of sulfide ions look distinctively different (Fig. 2E). Already after the addition of 4 equivalents of CdII, the evolution of a clearly biphasic CD profile is observed with extrema at (−)263.5 and (+)279.5 nm, the latter shifting to (+)284 nm after the addition of 10 equiv. of CdII. The cross-over point is located at 272 nm. Hence the spectrum is clearly red-shifted with respect to spectra of mammalian Cd7MTs, which show extrema located around (−)240 and (+)260 nm, but can be well compared to the spectra of two MTs from D. melanogaster, i.e. Cd-MtnA ((−)261 and (+)279.5 nm) as well as the aforementioned Cd-MtnE ((−)267.5 and (+)281 nm), both in their sulfide containing forms.45 The cross-over point at 272 nm mentioned above coincides with the position of the shoulder seen in the UV spectra (Fig. 2D), which is attributed to sulfide incorporation into cadmium–thiolate clusters as mentioned above, and hence the distinct CD profile is indicative of the presence of sulfide ions in the cluster of Cd-cicMT2.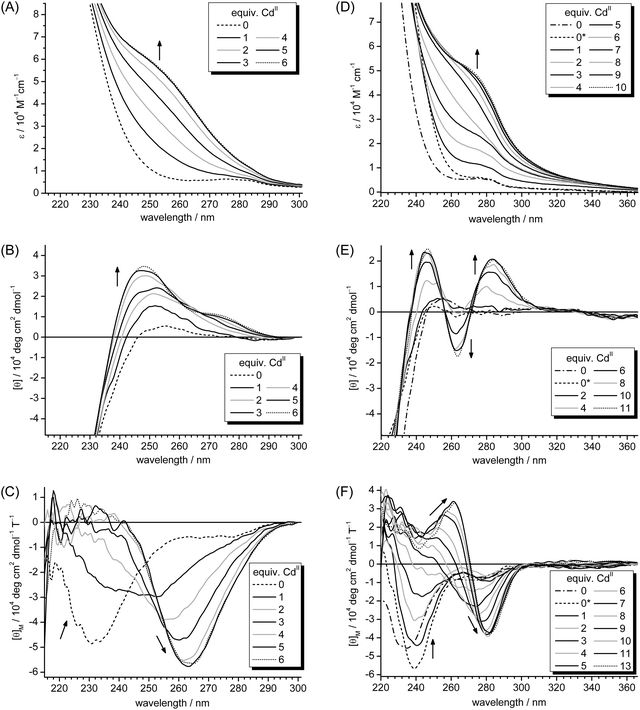 | ||
| Fig. 2 UV (A, D), CD (B, E) and MCD (C, F) spectra of the titration of Zn5cicMT2 in the absence (A, B, C) and presence of 10 equiv. of S2− (D, E, F) with CdII ions. The dash-dotted spectra (also denoted with 0) in D, E, and F correspond to Zn5cicMT2, and the dashed spectra (0*) originate from Zn5cicMT2 in the presence of 10 equiv. of S2−. The dotted spectra in all parts of the figure indicate the respective titration points at which additional CdII ions do not cause further spectral changes, i.e. when the protein is saturated with CdII ions. Arrows show the evolution or disappearance of transitions with increasing amounts of CdII ions added to the solution. A:29 Reproduced with the permission of the Royal Society of Chemistry; D: Reprinted, with permission, from ref. 30. Copyright 2013 American Chemical Society. | ||
MCD spectra can be observed when the electronic states of a chromophore are influenced by an externally applied magnetic field causing differences in absorption of the left and right circularly polarized light.44 While the MCD signal of the peptide backbone transitions is weak, a so-called A-term arises from tetrahedral CdII–thiolate units, which shows a cross-over point at the maximum of the corresponding absorption band, i.e. again around 250 nm. In the MCD spectrum of mouse Cd7MT1 this cross-over point is located at 248 nm followed by a minimum at 258 nm.44 The MCD spectra of the titration of Zn5cicMT2 with CdII ions in the absence of sulfide ions clearly show the evolution of an A-term indicative of tetrahedral CdS4 sites and additionally indicate cluster formation based on the shift of the respective cross-over points from roughly 246 nm after the addition of two CdII ions to 251 nm after five equivalents and saturation of the protein with metal ions (Fig. 2C). The corresponding minima shift from 257 to 263.5 nm. The MCD spectra for the titration of Zn5cicMT2 with CdII ions in the presence of 10 equiv. of sulfide show a biphasic profile (Fig. 2F). The negative band around (−)239 nm observed in the spectrum of Zn5cicMT2 in the presence of sulfide ions disappears after the addition of approximately four equiv. of CdII and a new positive band is formed, which reaches its highest magnetic ellipticity value after the addition of 11 equiv. of CdII at a wavelength of (+)262 nm. In parallel, a new extreme starts to evolve with the addition of four equiv. of CdII ions centered at roughly (−)272 nm, which reaches its maximum magnetic ellipticity value after the addition of nine equiv. of CdII and its final position at (−)281 nm. The cross-over point shifts from roughly 260 nm (four CdII) to 272 nm (11 CdII), again nicely coinciding with the position of the absorption band in Fig. 2D.
The distinctly different features of the UV, CD, and MCD spectra show clearly that the cluster structure in the presence of sulfide ions differs from that of Cd5cicMT2. In addition, very similarly shaped CD and MCD spectra were observed for the sulfide containing cadmium γ-glutamyl peptide complexes of the fission yeast S. pombe.46 The slight differences observed in the UV and MCD spectra of Zn5cicMT2 in the presence and absence of sulfide ions (Fig. 2D and F) are due to spectral contributions of the Na2S solution (ESI†). Accordingly, when a mixture of Zn5cicMT2 and sulfide ions is separated with SEC, analysis of the monomeric protein peak shows that still 5 ZnII ions are coordinated to the protein but no sulfide ions are detected. The CD spectrum of the Na2S solution is featureless in the spectral range used in this study.
Sulfide ions as modulators of the CdII/S-cluster size
Already 30 years ago it was shown that the PC complexes of S. pombe can contain variable amounts of sulfide ions, and that the CdII-binding capacity of the PC complexes depends on the concentration of sulfide ions in the reaction system.16–18,47,48 It is thus of interest to evaluate if also plant MTs, e.g. cicMT2, have the ability to host different sizes of metal–thiolate clusters depending on the metal ion concentration and the cellular supply of sulfide ions. To analyse this possibility in vitro, Zn5cicMT2 was titrated with CdII ions in the presence of 0, 5, 10, 20, and 30 equiv. of sulfide ions and the evolution of the LMCT bands was monitored with UV spectroscopy (Fig. 3A). As the S2− ions contribute to the overall absorption below 270 nm (dotted lines), the maximum CdII binding capacity of the protein was assessed by plotting the molar absorptivity at a higher wavelength, i.e. 275 nm, against the equiv. of CdII added to the solution. As expected, in the absence of S2− ions a Cd5cicMT2 species is formed. When S2− ions are provided, more CdII ions can be bound resulting in 15 equiv. of CdII in the presence of 30 equiv. of S2− (Table 1, column 2). To verify that the observed changes of the absorption spectra indeed originate from protein bound additional CdII and S2− ions and not from, e.g., CdS aggregates in solution, the final titration mixtures were applied to an analytical SEC column. The elution profiles are dominated by a peak at 13.5 mL corresponding to the monomeric protein species (Fig. 3B). This suggests that the structure of the protein can be expanded to accommodate the growing cluster without a major alteration of the overall protein size, or more precisely its hydrodynamic radius. With increasing amounts of sulfide ions present in the initial titration solution, peaks with lower elution volumes rise in intensity corresponding to protein dimers, trimers, and higher aggregates as well as CdS aggregates. The respective peak at 13.5 mL was collected and analyzed for protein, metal ion and sulfide ion content by colorimetric assays, i.e. 2-PDS and methylene blue assay, and F-AAS (Table 1, columns 3 and 4, Fig. 3C). The CdII contents in the purified protein fractions agree very well with the values obtained from the titration experiments. Hence it can be concluded that all of the CdII–S bonds that contributed to the detected absorption at 275 nm indeed belonged to the monomeric protein complex formed and did not originate from CdS aggregates in solution. In addition the amounts of bound CdII and S2− ions show a nearly linear dependence on the number of sulfide equivalents initially supplied. Additional experiments with even higher initial amounts of sulfide were not successful due to increased precipitation of CdS and observation of large fractions of protein aggregates during SEC. The rather high standard deviation for the values listed in Table 1 (columns 3 and 4) are in a large part due to the incomplete separation of monomer and multimer peaks, which still persisted after intensive optimization of the separation procedure.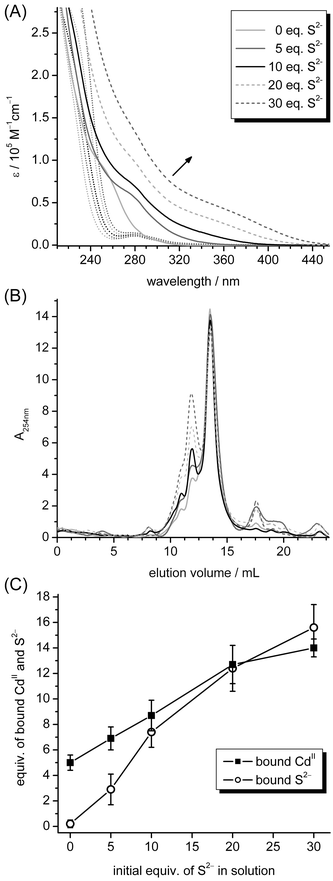 | ||
| Fig. 3 Titration of 10 μM Zn5cicMT2 in a buffer containing 10 mM Tris-HCl, 10 mM NaCl, pH 7.3 with increasing amounts of CdII ions in the presence of 0, 5, 10, 20, or 30 equiv. of S2−. (A) UV spectra of respective starting points, i.e. Zn5cicMT2 in the presence of 0–30 equiv. of S2− ions (dotted lines), and the final spectra after the addition of CdII ions (solid and dashed lines). (B) Elution profiles of the final titration mixtures from (A) when applied to a size exclusion chromatography column. The peak at 13.5 mL belongs to the monomeric protein species. The legend is the same as in (A). (C) Plot of final amounts of bound CdII and S2− ions quantified in the peak at 13.5 mL against the equiv. of S2− ions initially added to the solution before titration with CdII ions. | ||
| Initial S2− | CdII/MTa | CdII/MTb | S2−/MTc | CdIIadd/S2−d | CdII/Stote |
|---|---|---|---|---|---|
| a Equiv. of CdII ions added until no further changes of ε275 nm are observed in the UV spectra. b [CdII] from F-AAS after the purification of the titration mixture with size exclusion chromatography. c [S2−] from 2-PDS and methylene blue assays after the purification of the titration mixture with size exclusion chromatography. d Values for CdIIadd are calculated from (column 3 − 5 CdII), and values for S2− are taken from column 4. e Values for CdII are taken from column 3, and values for Stot are calculated from (column 4 + 14 SCys). | |||||
| 0 | 5.0 ± 0.1 | 5.0 ± 0.6 | 0.2 ± 0.3 | [0.15 ± 0.22] | 0.35 ± 0.04 |
| 5 | 7.8 ± 0.8 | 6.9 ± 0.9 | 2.9 ± 1.2 | 0.64 ± 0.28 | 0.41 ± 0.17 |
| 10 | 9.1 ± 0.3 | 8.7 ± 1.2 | 7.4 ± 1.2 | 0.50 ± 0.11 | 0.41 ± 0.09 |
| 20 | 11.8 ± 1.1 | 12.7 ± 1.5 | 12.4 ± 1.8 | 0.62 ± 0.12 | 0.48 ± 0.09 |
| 30 | 15.4 ± 2.2 | 14.0 ± 0.7 | 15.6 ± 1.8 | 0.58 ± 0.07 | 0.47 ± 0.06 |
Calculating the ratio between additionally incorporated CdII ions (CdIIadd) and bound S2− ions (Table 1, column 5) it is noticeable that the values range between 0.50 and 0.66; hence for each additional CdII ion roughly 2 S2− ions are incorporated. The overall charge of Cd5cicMT2 is already −6 and in this way will increase further by two more negative charges for each additionally incorporated sulfide ion. This is against expectations, as such an increase in negative charges should destabilize the cluster assembly because of charge repulsion between the ligands and hence impede the formation of larger clusters. Nevertheless, similar ratios can be calculated based on the CdII and S2− ion contents given in ref. 20 and hence seem to reflect a general trend. The structural implication of this, however, remains unclear. Another possibility is that some sort of charge compensation occurs in solution in the form of counter ions such as Na+ or by protonation of sites. Although never observed for “inorganic” CdS clusters with additional organic thiolate ligands, protonation of the sulfide ions to result in hydrogen sulfide, HS−, might be another possibility to reduce the overall charge of the cluster.30 Examining the ratios between bound CdII ions and the combined content of sulfide ions and Cys thiolate groups it is apparent that recruitment of sulfide ions as additional ligands increases not only the total CdII binding capacity but also the efficiency of binding as fewer ligands in the form of sulfide ions or thiolate groups are needed per bound CdII ion in these larger aggregates (Table 1, column 6). The ratio CdII:Stot in the absence of sulfide ions amounts to 0.35(4) and hence agrees well, e.g., with the value of 0.35 obtained for mammalian Cd7MT. In the presence of 20 or 30 equiv. of sulfide this ratio rises to 0.48(9) and 0.47(6), which is closer to the value of the sulfide containing higher molecular mass PC complex of yeast, i.e. 0.42, mentioned in the Introduction. The ratios calculated from the data in ref. 20 show a less clear picture with values ranging between 0.20 and 0.41. However, it has to be emphasized that these values are derived from recombinantly expressed forms, which had not been experimentally “forced” to adopt the form with maximum binding capacity and hence a comparison of values has to be carried out with caution.
Successful sulfide incorporation follows a distinctive pathway
So far all experiments were performed adhering to a specific sequence of component additions, i.e. apo- or Zn5cicMT2 was first incubated with the required amount of sulfide ions and only then CdII ions were added. To evaluate the importance of this procedure for the efficiency of sulfide incorporation and accordingly the formation of larger CdII/S-thiolate clusters the order of addition was varied. In one experiment, Zn5cicMT2 was first incubated with the required amount of CdII, i.e. 0–18 equiv., and only then 10 equiv. of S2− ions were added. In the second experiment, the required amount of CdII, i.e. 0–15 equiv., was first mixed with 10 equiv. of S2− ions followed by the addition of Zn5cicMT2 as the last component. The progress of all titrations was followed with UV, CD, and MCD spectroscopy (Fig. 4). The UV spectra of the titration endpoints, i.e. the points at which additional equiv. of CdII do not lead to further changes of the spectra, show as previously observed that the formation of sulfide containing CdII–thiolate clusters leads to the evolution of absorption bands around 275 nm and higher wavelengths. This is clearly seen when the spectra of the Cd5cicMT2 (“MT2 + xCdII”, Fig. 4A black line) and the Cd9S7cicMT2 forms (“MT2 + 10 S2− + xCdII”, Fig. 4A gray line) are compared. However, when the sequence of additions was altered, both resulting spectra show molar absorptivities in the spectral range above 275 nm that lie between the spectra of the sulfide-free and sulfide-containing species. Both the latter species are shown in Fig. 2B and E, respectively. The CD spectrum of Cd9S7cicMT2 is distinctively different from that of the sulfide-free Cd5cicMT2 species and can be used as a reliable marker for the presence of a sulfide containing cluster. Thus, while the final CD spectrum of the titration “xCdII + 10 S2− + MT2” is closely similar to that of Cd5cicMT2, the spectrum for the experiment “MT2 + xCdII + 10 S2−” shows CD bands with ellipticity values between the sulfide-free and the sulfide-containing form (Fig. 4B). Inspection of the corresponding MCD spectra reveals that, similar to the CD spectra, the “xCdII + 10 S2− + MT2” and Cd5cicMT2 spectra are virtually identical. However, the MCD spectrum of “MT2 + xCdII + 10 S2−” has the same profile as that of Cd5cicMT2 but shows significantly lower magnetic ellipticity values of its extrema (Fig. 4C). Consequently, although the UV and CD spectra of the titration “MT2 + xCdII + 10 S2−” seem to indicate the formation of a certain amount of sulfide containing clusters, the magnetic ellipticity envelope is more similar to sulfide-free Cd5cicMT2. There is only a minor shoulder at about (−)277 nm observed next to the most intense MCD band at (−)262 nm. Its presence may indicate a low fraction of a protein form hosting a cluster with a low amount of sulfide ions. Therefore, to characterize the final products formed in more detail the titration mixtures were purified with size exclusion chromatography to remove unbound CdII and S2− ions as well as protein aggregates. Analysis of the fractions at 13.5 mL, i.e. of the monomeric protein forms, for their CdII and S2− contents revealed for the titration experiment “MT2 + xCdII + 10 S2−” a final average stoichiometry of Cd5.0(1.2)S0.3(6)cicMT2 and for “xCdII + 10 S2− + MT2” the average composition Cd5.4(5)S0.4(1)cicMT2. Hence, it appears that both variations of the sequence of component additions do not enable sulfide incorporation into the cluster. The results are even more unambiguous when the same experiments are performed with apo-cicMT2 instead of Zn5cicMT2 (ESI†). Only when apo-cicMT2 is first incubated with sulfide ions before CdII ions are added sulfide incorporation is observed. All other sequences of CdII and sulfide additions result in UV spectra that are nearly identical to that obtained for Cd5cicMT2.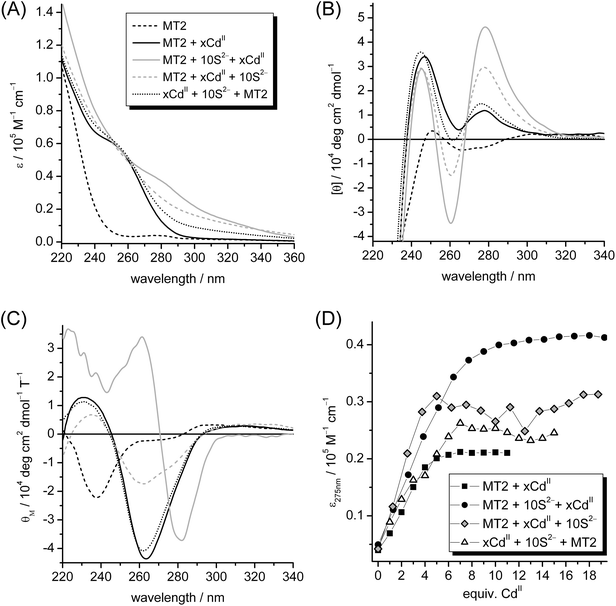 | ||
| Fig. 4 UV (A), CD (B), and MCD (C) spectra of the endpoints of titration of 10 μM Zn5cicMT2 in a buffer containing 10 mM Tris-HCl, 10 mM NaCl, pH 7.3 with CdII in the presence of 10 equiv. of S2− varying the order of component additions (see Materials and methods). For comparison, the spectra of Zn5cicMT2 (black dashed line) and Cd5cicMT2 (black line), i.e. Zn5cicMT2 titrated with 5 equiv. of CdII, are additionally shown. (D) Plot of the molar absorptivity at 275 nm against the equiv. of CdII ions added for different titration points leading to the final UV spectra shown in (A). | ||
To analyse the maximum binding capacity for CdII ions, for each titration the molar absorptivity at 275 nm was plotted against the number of CdII ions added (Fig. 4D). As expected, for the titration in the absence of sulfide ions the maximum absorptivity is reached at around five equiv. CdII and for the titration, in which incubation of the protein with sulfide ions occurred prior to CdII addition, around nine CdII ions are required to reach the maximum binding capacity. In the experiment, in which the components were mixed in the order “MT2 + xCdII + 10 S2−”, saturation is achieved after the addition of five CdII as in the experiments without sulfide ions. For the sequence “xCdII + 10 S2− + MT2” seven equiv. of CdII are required (Fig. 4D). The larger variations observed in the plots of the latter titrations compared to those in the “MT2 + xCdII” and “MT2 + 10 S2− + xCdII” experiments reflect the difference in sample preparations (Materials and methods).
Overall, it is intriguing that when the first CdII is added to apo- or Zn5cicMT2 and then sulfide ions are provided, no sulfide is incorporated into the cluster. Hence it appears that the sulfide must be already coordinated to the CdII ions for successful incorporation. On the other hand if the first CdII and sulfide ions are incubated for 10 min also no incorporation of sulfide ions is observed. It has been reported in the literature that the formation of the mineral ZnS from aqueous solution proceeds first via neutral complexes with 1:1 stoichiometry and additional aqua ligands that subsequently aggregate to larger negatively charged complexes, e.g. Zn4S64−.49 Hence it might be mechanistically feasible that initially formed analogous neutral 1:1 CdS complexes can be bound by the protein, while the aggregated negatively charged higher molar mass complexes are not incorporated. Consequently, it would be the kinetically controlled formation of Cd/S aggregates that determines if sulfide incorporation into the MT takes place. However and as mentioned above, as CdII ions are incorporated into the protein even after pre-incubation with sulfide ions cicMT2 must be able to compete with the preformed Cd/S complexes for CdII ions. A feasible mechanism would be the replacement of aqua or other less thiophilic ligands by Cys-S ligands of the protein.
Sulfide incorporation as a general feature of MTs?
In previous reports, the occurrence of sulfide ions in MTs was only analysed in recombinantly expressed proteins as isolated from E. coli or in MTs from yeast, both grown in metal supplemented growth media.20,22 To probe the sulfide incorporation ability of MTs further, CdII-MT forms were titrated with additional amounts of sulfide ions leading indeed to the detection of sulfide ions in MTs; however, the CdII content remained constant as no additional CdII ions were provided.21 In the present study, we attempted to analyse the sulfide incorporation abilities of mammalian MTs in more detail. For this, we titrated 10 μM rabbit liver Zn7MT2A with CdII ions in the absence and presence of 10 equiv. of S2− as performed analogously for Zn5cicMTs, and monitored the progress of CdII incorporation with UV spectroscopy (Fig. 5). Clearly, the titration of Zn7MT2A with CdII ions in the presence of sulfide leads to the evolution of additional transitions red-shifted from the intense LMCT bands around 250 nm indicative of CdII–thiolate cluster formation, although the intensity of these additional transitions is considerably weaker compared to the titration of Zn5cicMT2. As expected, the absorptivity at 250 nm reaches a maximum value after the addition of seven equiv. of CdII for the titration of rabbit liver Zn7MT2A in the absence of sulfide ions. In the presence of sulfide, the CdII binding capacity seems to be increased, resulting in saturation with metal ions after approximately 10 equiv. of CdII ions. To analyse the formed complexes further, both samples were again purified with size exclusion chromatography and the content of CdII and S2− ions was determined. This procedure gives a final average composition of Cd6.7(4)S0.2(1)MT2A for the titration performed in the absence of sulfide ions and Cd8.4(4)S3.0(3)MT2A for the form obtained after titration with CdII ions in the presence of sulfide. It is hence obvious that sulfide ions can only marginally increase the binding capacity of rabbit liver MT2A for CdII compared to cicMT2. In addition, less efficient CdII binding is reflected in the ratio between additionally coordinated CdII ions and incorporated sulfide ions, which amounts to 0.47(5) and is thus slightly lower than the values of 0.50–0.66 obtained for cicMT2, indicating that less CdII ions are coordinated per additional sulfide ion. Also the ratio between the total amount of bound CdII ions and the combined number of Cys-thiolate and sulfide ligands remains constant with 0.37(4) compared to 0.35 in the sulfide-free Cd7MT2A form.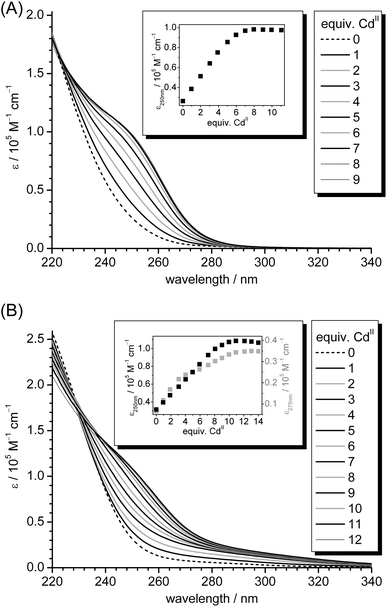 | ||
| Fig. 5 (A) UV spectra of the titration of 10 μM reconstituted rabbit liver Zn7MT2A in a buffer containing 10 mM Tris-HCl, 10 mM NaCl, pH 7.5 with increasing amounts of CdII ions. The inset shows the increase of molar absorptivity of the LMCT band at 250 nm plotted against the number of CdII ions added to the solution. (B) UV spectra of the analogous titration as shown in (A), but in the presence of 10 equiv. of S2− ions. In the inset the molar absorptivities at 250 and 275 nm are plotted against the number of CdII ions. | ||
To investigate the presence of sulfide ions in mammalian MTs in vivo the native form of rabbit liver MT2A, isolated and purified from CdII-supplemented animals by the group of Prof. Milan Vašák (Institute of Inorganic Chemistry, University of Zurich) as described,32,33 was analysed with a combination of F-AAS, 2-PDS, and the methylene blue assay. As a result the native content of sulfide ions was below 0.2 per MT2A, and thus within the error range of the method, and the general formula Zn2.4Cd4.6MT2A was determined, being in line with previous investigations that never revealed the occurrence of any sulfide ions in mammalian MTs.50 It has to be considered however that although the native protein was never exposed to acidic conditions during the purification procedure, which self-evidently would release any sulfide ions present in the form of H2S, it was exposed to ion exchange chromatography with DEAE cellulose, which has been proposed to remove sulfide ions from MT preparations.22 Accordingly, also our experiments could not finally prove the absence of sulfide ions from native preparations of mammalian MTs.
Conclusions
Building up on our previous results that the size of the CdII–thiolate cluster from the plant MT cicMT2 can be increased in the presence of sulfide ions resulting in a monomeric protein form with the average composition Cd9S7cicMT2,30 we investigated the increase in cluster size for the ZnII-form, the ability of cicMT2 to form and host even larger cluster structures when more sulfide ions are supplied, and tried to shed light on a possible mechanism of CdII/S-thiolate cluster formation.Our studies revealed that upon exposure of the fully metal loaded ZnII- and CdII-containing MII5cicMT2 forms to sulfide ions, no sulfide was incorporated into the protein structure (see above and the ESI†). In contrast, sulfide ions together with additional metal ions were incorporated into the protein structure, forming the MII8/9S7cicMT2 species, during the metal-induced protein folding process starting from apo-cicMT2. The observed replacement of bound ZnII ions in Zn5cicMT2 by more strongly bound CdII in the absence of sulfide has already been reported for MTs from various species.44,51–53 Here we show that while the addition of CdII to Zn5cicMT2 followed by sulfide results in Cd5cicMT2, the reverse order leads to the formation of Cd9S7cicMT2 (Fig. 6A). As metal exchange in MTs is a fast process, the former result reflects the formation of the Cd5cicMT2 species and its subsequent exposure to sulfides (see above). We picture that the addition of CdII to a mixture of Zn5cicMT2 and sulfide ions results in a transient CdS complex, where besides S also other ligands, e.g. aqua ligands, are present. This complex then competes for the zinc sites in the protein leading to the formation of the Cd9S7cicMT2 species. Regardless of the operating mechanism, in all instances CdS or ZnS complexes must be formed prior to sulfide and metal incorporation into the MT structure. However, the question remains whether sulfide incorporation into MTs has any physiological relevance. The underlying concept of recruiting sulfide ions as additional ligands has been already documented for the PC complexes of yeast and plants and in general it might be a rather cost-efficient way for an organism to increase the metal ion binding capacity of a protein. Nevertheless, clearly more biological studies are needed to shed light on the presence of sulfide ions in MTs.
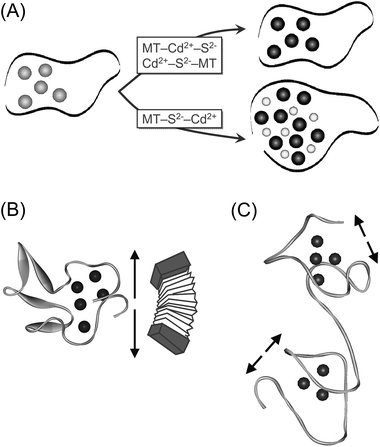 | ||
| Fig. 6 (A) Proposed pathway of CdII/S-thiolate cluster formation in dependence of the sequence of MT, CdII, and S2− additions. The protein backbone is depicted as a black ribbon, ZnII ions as gray, CdII ions as black, and S2− ions as smaller light gray spheres. (B) Schematic presentation of the structural arrangement of the metal–thiolate cluster proposed for Cd5cicMT2. The arrows indicate the experimentally observed possibility of cluster expansion in the presence of sulfide and additional CdII ions, which might be facilitated by the long Cys-free linker region similar to the movement of an accordion. (C) The absence of Cys-free linker regions within the two domains in mammalian MTs might, on a purely hypothetical basis, inhibit significant expansion of the clusters as illustrated by the shorter arrows. The structure was prepared using the coordinates of rat Zn2Cd5MT2 (PDB code 4MT2). | ||
In addition, we compared the results obtained with cicMT2 to the ability of a mammalian MT, i.e. rabbit liver MT2A, to use sulfide to increase its CdII binding capacity. Interestingly, cicMT2 is able to form a much larger CdII/S-thiolate cluster structure relative to its CdII–thiolate cluster in the absence of sulfide ions than observed for rabbit liver MT2A under the same in vitro conditions. On a purely hypothetical basis, it is intriguing to search for a structural explanation of this increased metal ion binding capacity by consulting the most obvious difference in the amino acid structure of cicMT2 and plant MTs from the MT1, MT2, and MT3 subgroups in general, compared to the sequences of mammalian MTs, i.e. the long Cys-free amino acid stretch between the N- and C-terminal Cys-rich regions, which seem to contain even β-sheets as secondary structural elements.54 As Cd5cicMT2 as well as Cd9S7cicMT2 were shown to presumably form a hairpin-like structure with a single cluster (Fig. 6B),29,30 larger cluster structures can be easily envisioned to be hosted in such an arrangement with the linker region functioning as an adjustable hinge not unlike the ends of an accordion. The amino acid sequences of mammalian MTs however enclose two separate metal–thiolate clusters and do not contain longer linker regions within the amino acid stretches forming a cluster. Accordingly, steric reasons can be envisioned that prevent expansion of these clusters, a view that would nicely explain the finding that incorporation of sulfide ions only marginally increases the cluster size of rabbit liver MT2A (Fig. 6C). The hypothetical idea of expandable metal–thiolate clusters in response to physiological requirements would also shed light on a possible function of the enigmatic long Cys-free linker regions observed in most plant MT forms. Nevertheless, the final answer awaits further analysis of native MT forms, which need to be carefully isolated and purified.
Acknowledgements
We thank Prof. Milan Vašák (Institute of Inorganic Chemistry, University of Zurich) for supplying us with a sample of purified rabbit liver MT2A and for numerous helpful discussions. Financial support from the Swiss National Science Foundation is gratefully acknowledged (SNSF-Professorship to EF).References
- P.-A. Binz and J. H. R. Kägi, in Metallothionein IV, ed. C. Klaassen, Birkhäuser Verlag, Basel, 1999, pp. 7–13 Search PubMed.
- M. Margoshes and B. L. Vallee, J. Am. Chem. Soc., 1957, 79, 4813–4814 CrossRef CAS.
- B. L. Vallee, in Metallothionein, ed. J. H. R. Kägi and M. Nordberg, Birkhäuser Verlag, Basel, 1979, pp. 19–40 Search PubMed.
- C. Jacob, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3489–3494 CrossRef CAS.
- A. H. Robbins, D. E. McRee, M. Williamson, S. A. Collett, N. H. Xuong, W. F. Furey, B. C. Wang and C. D. Stout, J. Mol. Biol., 1991, 221, 1269–1293 CAS.
- S. G. Bell and B. L. Vallee, ChemBioChem, 2009, 10, 55–62 CrossRef CAS.
- C. Cobbett and P. B. Goldsbrough, Annu. Rev. Plant Biol., 2002, 53, 159–182 CrossRef CAS.
- W.-J. Guo, M. Meetam and P. B. Goldsbrough, Plant Physiol., 2008, 146, 1697–1706 CrossRef CAS.
- W. Maret, J. Biol. Inorg. Chem., 2011, 16, 1079–1086 CrossRef CAS.
- G. Mir, J. Domènech, G. Huguet, W.-J. Guo, P. B. Goldsbrough, S. Atrian and M. Molinas, J. Exp. Bot., 2004, 55, 2483–2493 CrossRef CAS.
- R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8428–8430 CrossRef CAS.
- M. Vašák and D. W. Hasler, Curr. Opin. Chem. Biol., 2000, 4, 177–183 CrossRef.
- E. Freisinger, Dalton Trans., 2008, 6663–6675 RSC.
- E. Grill, E.-L. Winnacker and M. H. Zenk, Science, 1985, 230, 674–676 CAS.
- J. C. Steffens, D. F. Hunt and B. G. Williams, J. Biol. Chem., 1986, 261, 13879–13882 CAS.
- A. Murasugi, C. Wada and Y. Hayashi, J. Biochem., 1981, 90, 1561–1564 CAS.
- A. Murasugi, C. Wada and Y. Hayashi, J. Biochem., 1983, 93, 661–664 CAS.
- R. N. Reese, R. K. Mehra, E. B. Tarbet and D. R. Winge, J. Biol. Chem., 1988, 263, 4186–4192 CAS.
- J. Domènech, R. Orihuela, G. Mir, M. Molinas, S. Atrian and M. Capdevila, J. Biol. Inorg. Chem., 2007, 12, 867–882 CrossRef.
- M. Capdevila, J. Domènech, A. Pagani, L. Tío, L. Villarreal and S. Atrian, Angew. Chem., Int. Ed., 2005, 44, 4618–4622 CrossRef CAS.
- L. Tío, L. Villarreal, S. Atrian and M. Capdevila, Exp. Biol. Med., 2006, 231, 1522–1527 Search PubMed.
- R. Orihuela, F. Monteiro, A. Pagani, M. Capdevila and S. Atrian, Chem.–Eur. J., 2010, 16, 12363–12372 CrossRef CAS.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS.
- D. E. Barañano, C. D. Ferris and S. H. Snyder, Trends Neurosci., 2001, 24, 99–106 CrossRef.
- T. C. Bartholomew, G. M. Powell, K. S. Dodgson and C. G. Curtis, Biochem. Pharmacol., 1980, 29, 2431–2437 CrossRef CAS.
- H. Beinert, Eur. J. Biochem., 2000, 267, 5657–5664 CrossRef CAS.
- W. Maret, Biochemistry, 2004, 43, 3301–3309 CrossRef CAS.
- F. J. Munoz, R. V. Ullán, E. Labrador, B. Dopico and F. J. Muñoz, Physiol. Plant., 1998, 104, 273–279 CAS.
- X. Wan and E. Freisinger, Metallomics, 2009, 1, 489–500 RSC.
- X. Wan and E. Freisinger, Inorg. Chem., 2013, 52, 785–792 CrossRef CAS.
- J. E. Tropea, S. Cherry and D. S. Waugh, Methods Mol. Biol., 2009, 498, 297–307 CAS.
- M. Kimura, N. Otaki and M. Imano, in Metallothionein, eds. J. H. R. Kägi and M. Nordberg, Birkhäuser Verlag, Basel, 1979, pp. 163–168 Search PubMed.
- J. H. R. Kägi, S. R. Himmelhoch, P. D. Whanger, J. L. Bethune and B. L. Vallee, J. Biol. Chem., 1974, 249, 3537–3542 Search PubMed.
- A. O. Pedersen and J. Jacobsen, Eur. J. Biochem., 1980, 106, 291–295 CrossRef CAS.
- S. Damodaran, Anal. Biochem., 1985, 145, 200–204 CrossRef CAS.
- T. W. Thannhauser, Y. Konishi and H. A. Scheraga, Anal. Biochem., 1984, 138, 181–188 CrossRef CAS.
- W. Bae and R. K. Mehra, J. Inorg. Biochem., 1998, 70, 125–135 CrossRef CAS.
- R. N. Reese and D. R. Winge, J. Biol. Chem., 1988, 263, 12832–12835 CAS.
- I. G. Dance, A. Choy and M. L. Scudder, J. Am. Chem. Soc., 1984, 106, 6285–6295 CrossRef CAS.
- V. S. Gurin, J. Phys. Chem., 1996, 100, 869–872 CrossRef CAS.
- V. S. Gurin, Solid State Commun., 1998, 108, 389–392 CrossRef CAS.
- T. Türk, U. Resch, M. A. Fox and A. Vogler, J. Phys. Chem., 1992, 96, 3818–3822 CrossRef.
- J. Domènech, A. Tinti, M. Capdevila, S. Atrian and A. Torreggiani, Biopolymers, 2007, 86, 240–248 CrossRef.
- E. Freisinger and M. Vašák, Met. Ions Life Sci., 2013, 11, 339–371 CrossRef.
- S. Pérez-Rafael, A. Kurz, M. Guirola, M. Capdevila, O. Palacios and S. Atrian, Metallomics, 2012, 4, 342–349 RSC.
- D. J. Plocke and J. H. R. Kägi, Eur. J. Biochem., 1992, 207, 201–205 CrossRef CAS.
- A. Murasugi, C. Wada and Y. Hayashi, Biochem. Biophys. Res. Commun., 1981, 103, 1021–1028 CrossRef CAS.
- A. Murasugi, C. Wada Nakagawa and Y. Hayashi, J. Biochem., 1984, 96, 1375–1379 CAS.
- G. W. Luther, S. M. Theberge and D. T. Rickard, Geochim. Cosmochim. Acta, 1999, 63, 3159–3169 CrossRef CAS.
- M. Vašák, personal communication.
- M. Beltramini, K. Lerch and M. Vasak, Biochemistry, 1984, 23, 3422–3427 CrossRef CAS.
- H. Willner, M. Vašák and J. H. R. Kägi, Biochemistry, 1987, 26, 6287–6292 CrossRef CAS.
- E. A. Peroza and E. Freisinger, J. Biol. Inorg. Chem., 2007, 12, 377–391 CrossRef CAS.
- E. Freisinger, J. Biol. Inorg. Chem., 2011, 16, 1035–1045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3dt32438a |
| This journal is © The Royal Society of Chemistry 2013 |