DOI:
10.1039/C3DT32430C
(Paper)
Dalton Trans., 2013,
42, 6645-6654
Hydrogen bonded anion ribbons, networks and clusters and sulfur–anion interactions in novel radical cation salts of BEDT-TTF with sulfamate, pentaborate and bromide†
Received
11th October 2012
, Accepted 26th February 2013
First published on 28th February 2013
Abstract
The novel radical cation salt (BEDT-TTF)3(sulfamate)2·2H2O (BEDT-TTF = bis(ethylenedithio)tetrathiafulvalene) is semiconducting with donor stacks comprised of pairs of partially oxidized molecules and a single more highly oxidized molecule which is twisted out of the stack by ca. 30°. Hydrogen bonded pairs of sulfamate ions are linked into parallel ribbons by further hydrogen bonding between sulfamates and bridging water molecules. In contrast, the BEDT-TTF salt with pentaborate contains infinite layers formed of a network of hydrogen bonded pentaborate anions. Two new bromide salts of BEDT-TTF are reported, one is a semiconducting 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt in which the bromide is integrated among the BEDT-TTF donors, while the other contain a square of four bromide ions linked together by hydrogen bonding to a centrally located H5O2+ cation for every five BEDT-TTF molecules.
:1 salt in which the bromide is integrated among the BEDT-TTF donors, while the other contain a square of four bromide ions linked together by hydrogen bonding to a centrally located H5O2+ cation for every five BEDT-TTF molecules.
Introduction
Radical cation salts of planar conjugated organosulfur systems are being intensively investigated on account of the wide range of electrical properties found in these systems, with a view to incorporating these into useful materials.1 The salts are commonly prepared by electrocrystallisations of donors such as TTF 1,2 EDT 23 and BEDT-TTF 34–6 and their substituted derivatives7 as well as from related systems such as 4.8 BEDT-TTF has been used extensively as a substrate, and has led to salts with a range of stoichiometries and crystal packing modes with conducting, semiconducting and superconducting properties. Anions incorporated into salts have varied from single atom species such as chloride, to linear, tetrahedral and octahedral species, such as I3−, AuCl2−, ClO4−, ReO4−, PF6− and metal[tris(oxalate)] anions,9 as well others which form two-dimensional networks, e.g. with mixed metal oxalate networks,10 and more complex species such as the Keggin type anions and anions with Kagome topology.11 For the BEDT-TTF salts a number of distinct packing modes for the donors have been observed and analysed.5,6 Since several superconducting salts have anionic layers e.g. formed from [Cu(NCS)2],12 we turned our attention to anions that had the potential to form layers through inter-anion hydrogen bonds. Here we report the first BEDT-TTF salts containing the sulfamate anion and the pentaborate anion, which form hydrogen bonded ribbons and layers respectively. The sulfamate anion has not been exploited as a counterion with BEDT-TTF, TTF or related materials. The related hydrogen sulfate and hydrogen selenate anions form 3:2 salts with BEDT-TTF which contains hydrogen bonded anion pairs,13,14 but the sulfamate has an extra hydrogen atom which could conceivably lead to the formation of two dimensional structures. A 1:1 salt of BEDT-TTF with hydrogen sulfate has also been reported.15 As far as we are aware there are no borate related salts of BEDT-TTF reported. Furthermore, we also report two new phases containing bromide ions including a 1:1 salt where the bromide ion is in close contact with the sulfur atoms of BEDT-TTF cations, and another salt containing the [Br4(H5O2)]3− complex anion.
Results and discussion
(BEDT-TTF)3(SO3NH2)2(H2O)2
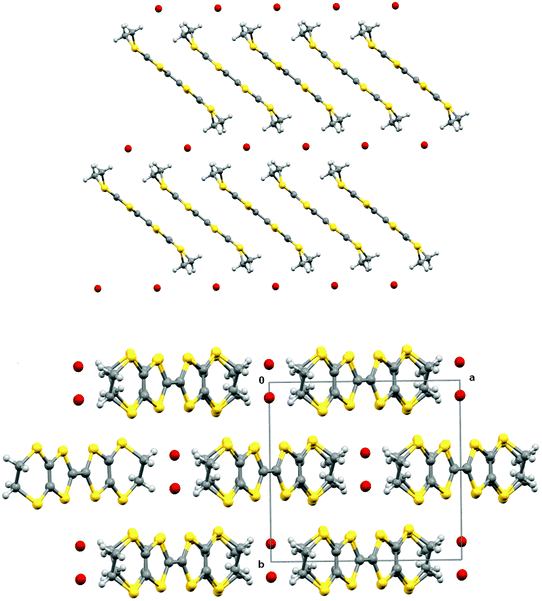
Electrocrystallisation of BEDT-TTF with tetrabutylammonium sulfamate in dichloromethane or THF over three weeks gave black shiny needles whose solid state structure was determined by X-ray crystallography at 120 K. The crystal system is triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and the unit cell contains three nearly planar BEDT-TTF species, one of which lies across a crystallographic centre of symmetry, two sulfamate anions and two water molecules. All hydrogen atom positions were included in the crystal structure refinement. Stacks of BEDT-TTF molecules extend along the crystallographic b axis with a repeating pattern involving three donor molecules (Fig. 1). The stacks are separated from each other in the c direction by a set of parallel ribbons composed of hydrogen bonded sulfamate anions and water molecules aligned in the a direction, and repeated in the b direction, thus filling the ab plane (Fig. 2). The sulfamate nitrogen has pyramidal bonding geometry with the sum of bond angles at nitrogen equal to 333°. Within a ribbon, sulfamate anions are hydrogen bonded in pairs by two centrosymmetrically related N–H⋯O bonds (2.24(3) Å), and each pair is linked to the next pair by a pair of centrosymmetrically related N–H⋯O hydrogen bonds (N–H⋯O: 2.25(3) Å) and by hydrogen bonding to two bridging water molecules (O⋯H–OH: 2.02(5) and 2.06(4) Å). Thus each sulfamate oxygen and hydrogen atom is involved in hydrogen bonding. However, there is no hydrogen bonding between the ribbons, because they are isolated from each other by the ends of donor molecules which narrow the channel between stacks at these points (Fig. 3). The edges of the ribbons are formed by the oxygen atoms of the water molecules, which are almost 4 Å apart. The donor and anion/water layers are tightly packed with three short contacts between sulfamate oxygen atoms and hydrogen atoms of the donors’ ethylene bridges (2.33(3)–2.47(3) Å) which have O⋯H–C angles greater than 150°.
, and the unit cell contains three nearly planar BEDT-TTF species, one of which lies across a crystallographic centre of symmetry, two sulfamate anions and two water molecules. All hydrogen atom positions were included in the crystal structure refinement. Stacks of BEDT-TTF molecules extend along the crystallographic b axis with a repeating pattern involving three donor molecules (Fig. 1). The stacks are separated from each other in the c direction by a set of parallel ribbons composed of hydrogen bonded sulfamate anions and water molecules aligned in the a direction, and repeated in the b direction, thus filling the ab plane (Fig. 2). The sulfamate nitrogen has pyramidal bonding geometry with the sum of bond angles at nitrogen equal to 333°. Within a ribbon, sulfamate anions are hydrogen bonded in pairs by two centrosymmetrically related N–H⋯O bonds (2.24(3) Å), and each pair is linked to the next pair by a pair of centrosymmetrically related N–H⋯O hydrogen bonds (N–H⋯O: 2.25(3) Å) and by hydrogen bonding to two bridging water molecules (O⋯H–OH: 2.02(5) and 2.06(4) Å). Thus each sulfamate oxygen and hydrogen atom is involved in hydrogen bonding. However, there is no hydrogen bonding between the ribbons, because they are isolated from each other by the ends of donor molecules which narrow the channel between stacks at these points (Fig. 3). The edges of the ribbons are formed by the oxygen atoms of the water molecules, which are almost 4 Å apart. The donor and anion/water layers are tightly packed with three short contacts between sulfamate oxygen atoms and hydrogen atoms of the donors’ ethylene bridges (2.33(3)–2.47(3) Å) which have O⋯H–C angles greater than 150°.
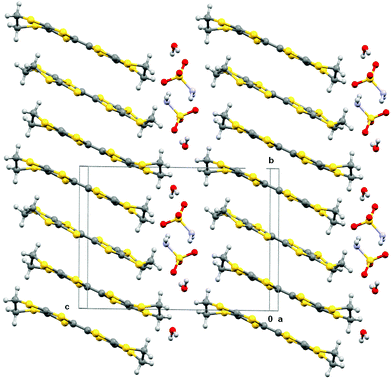 |
| | Fig. 1 Crystal packing of (BEDT-TTF)3(SO3NH2)2(H2O)2 viewed down the a axis. | |
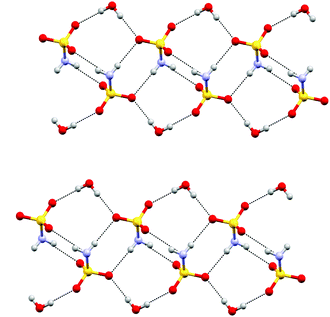 |
| | Fig. 2 Layer structure formed by sulfamate anions and water molecules in (BEDT-TTF)3(SO3NH2)2·(H2O)2, the a axis is horizontal and the b axis is vertical. | |
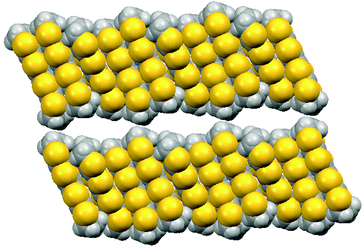 |
| | Fig. 3 Space-filling view of the donor packing viewed along the a axis, with the b axis horizontal and the c axis vertical, showing the distinct channels which are filled by the ribbons of sulfamate ions and water molecules (which are omitted for clarity). | |
The planar BEDT-TTF molecules are stacked along the b axis with every third molecule twisted out from the other two, which is a very unusual arrangement for a BEDT-TTF salt. Thus, a pair of centrosymmetrically related donors (Type I) alternates with a donor molecule (Type II) which lies on a centre of symmetry and whose main axis lies at ca. 31° to that of the Type I pair (Fig. 4(a)). The components of the Type I pair do not lie directly above one another but offset to the side (Fig. 4(b)). There are many short S⋯S contacts in the interstack direction. For sheets comprising Type I molecules 75% of the sulfur atoms are involved in short contacts <3.6 Å, two as short as 3.2850(8) and 3.3157(8) Å, and two more at 3.4040(8) and 3.4105(8) Å. For the sheets comprising Type II molecules also 75% of the sulfur atoms are involved in short contacts, with values of 3.4055(9) and 3.4504(8) Å (Fig. 5). The ethylene bridges are well ordered in all cases, assisted by the weak hydrogen bonding to sulfamate, with the Type I donor having two different dithiin ring conformations, one near to a half chair and the other close to an envelope, while for the centrosymmetric Type II molecule this conformation is closer to a half chair. The offset between the two Type I molecules leads to a pair of H⋯S contacts between the ethylene bridge of one molecule and the dithiin sulfur of the other of 2.89 Å, and there are no S⋯S contacts between the pair less than 3.84 Å. Between Type I and Type II molecules in a stack, the shortest contacts are from a bridge H atom to two S atoms (2.90 and 2.97 Å) and the intervening C atom (2.78 Å), and there is a S⋯C contact at the other side of the stack (3.402 Å) (Fig. 5). The shortest three S⋯S contacts between these molecules lie in the range (3.627–3.687 Å). Guionneau et al. have established a relation between the bond lengths of the TTF section of BEDT-TTF and the charge it carries.16 Using their formula the charges on the Type I and Type II donor molecules are estimated as +0.54 and +0.82, and thus for the whole unit cell these total +1.9, close to the expected value of +2.0. The error on the calculated molecular charges is ca. ±0.1. Raman spectroscopy has also been used to estimate the oxidation state of BEDT-TTF donors. Thus, measurements on multiple crystals identified symmetrical stretching modes of the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at ν = 1421, 1460, 1476 and 1490 cm−1, which correspond to the oxidation states of the donor molecules to be in the region of +0.5 to +0.9, according to the method of Wang et al.17 These two independent methods suggests that the centrosymmetrically arranged pair of Type I donor molecules each carry a charge of ca. +0.5, while the Type II donor molecule which lies on a centre of symmetry has a charge of close to +1.
C bond at ν = 1421, 1460, 1476 and 1490 cm−1, which correspond to the oxidation states of the donor molecules to be in the region of +0.5 to +0.9, according to the method of Wang et al.17 These two independent methods suggests that the centrosymmetrically arranged pair of Type I donor molecules each carry a charge of ca. +0.5, while the Type II donor molecule which lies on a centre of symmetry has a charge of close to +1.
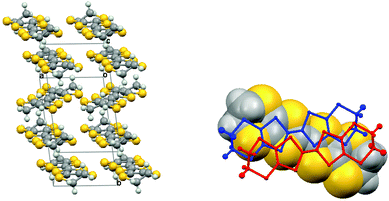 |
| | Fig. 4 (a) View of the donor stacking arrangement in (BEDT-TTF)3(SO3NH2)2 (H2O)2 with centrosymmetric pairs (Type I) and single molecules (Type II) alternating along the b stacking axis; (b) Overlap of the two centrosymmetrically related Type I donor molecules (wireframe) with the Type II donor (space-filling) behind viewed down the b axis. | |
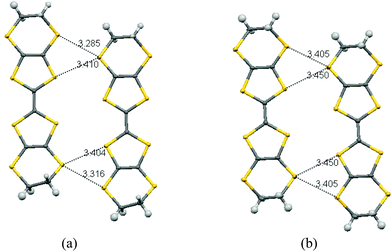 |
| | Fig. 5 Short contacts within sheets of donor molecules (a) Type I donors, (b) Type II donors. The view is along the b axis with the a axis horizontal and the c axis near to vertical. | |
Crystals of the (BEDT-TTF)3(SO3NH2)2(H2O)2 are semi-conducting, with a room temperature conductivity of ca. 14 S cm−1. The temperature dependence of the resistivity is shown in Fig. 6. It is notable that the activation energy is not constant. At room temperature it is rather small, 20 meV, but gradually increases upon cooling in the range 220–240 K, and below 220 K the activation energy is 109 meV. Our structural studies above refer to the low temperature range. Comparing the crystal structures at 120 K and 300 K shows no major structural phase transition. However, at 300 K the very large anisotropic displacement parameters of the three oxygen atoms of the sulfamate suggest the onset of some rotation of the SO3 group. This may be related to flexing of the ethylene bridges of the neighbouring BEDT-TTF molecules which at 120 K are well ordered. This order–disorder transition may be the origin of the electrical phase transition observed. The observed semiconducting behaviour is in agreement with the non-uniform charge distribution among the BEDT-TTF molecules. Bond lengths from X-ray data and peaks in the Raman spectra are consistent with charge ordering in this material.16–18
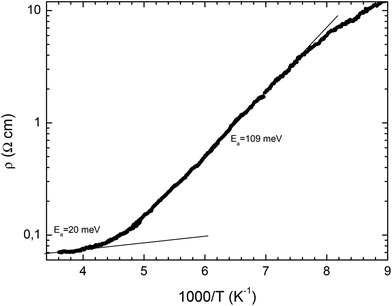 |
| | Fig. 6 Plot of resistivity against 1000/T for (BEDT-TTF)3(SO3NH2)2(H2O)2. | |
(BEDT-TTF)2(B5O6(OH)4)
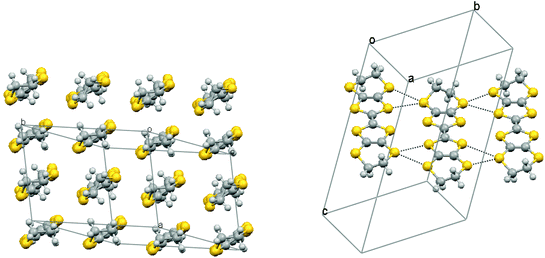
A few small crystals of a 2:1 radical cation salt with the pentaborate anion (B5O6(OH)4)− were obtained serendipitously from an electrocrystallisation of BEDT-TTF with α-methylbenzylammonium phenylpropionate, and were characterised by X-ray crystallography at 120 K. This anion has probably formed from borax leaking into the cell from solder in the electrode housing or from the glass. The pentaborate anion has been reported as salts with both inorganic and organic cations, and crystals of some of the former have non-linear optical properties.19 To our knowledge this is the first BEDT-TTF salt with a simple borate-derived anion, though salts with boron containing anions such as BF4−, a cobalt bis(carbollide) derivative and the Keggin-type anion [BW12O40]5− are known,20–22 and boron has been incorporated into an organosulfur donor as a β-diketonatoboron difluoride group.23 The structure was refined in the triclinic space group P, and contains two BEDT-TTF molecules and one (B5O6(OH)4)− anion in the asymmetric unit. The crystal structure consists of stacks of BEDT-TTF molecules along the a axis, with further stacks lying side by side in the −b and +b directions (Fig. 7). These banks of stacks are separated from the next set of stacks by a continuous layer formed by hydrogen bonded (B5O6(OH)4)− anions in the ab plane (Fig. 7). Two pairs of centrosymmetrically related hydrogen bonds (O⋯H: 1.90 and 1.91 Å) connect the anions together to form rows along b. Further hydrogen bonds (O⋯H: 1.99 Å) link these rows together to form the infinite sheet. A range of hydrogen bonded networks of pentaborate ions have been reported in salts with non-metal cations,24,25 but the network observed here appears to be unique.
![Crystal packing in (BEDT-TTF)2(B5O6(OH)4) viewed down the [1 1 0] direction with the c axis horizontal (left), and a hydrogen bonded layer of (B5O6(OH)4)− anions in the ab plane (right).](/image/article/2013/DT/c3dt32430c/c3dt32430c-f7.gif) |
| | Fig. 7 Crystal packing in (BEDT-TTF)2(B5O6(OH)4) viewed down the [1 1 0] direction with the c axis horizontal (left), and a hydrogen bonded layer of (B5O6(OH)4)− anions in the ab plane (right). | |
There is some disorder in the conformations of three out of four ethylene bridges in the BEDT-TTF units, which is not unusual. This structure belongs to the β′′ family of BEDT-TTF salts, and more specifically designated as β210×2 due to periodicity and position of the dislocations in the stacking arrangement.5 There are rows of donors aligned side by side in the [1 1 0] direction which are composed alternatively of the two unique donor molecules (Fig. 8). There are short S⋯S contacts between these molecules (3.325(3), 3.325(3), 3.503(3) and 3.538(3) Å) to one side and (3.286(3), 3.374(3) 3.406(3) and 3.560(3) Å) to the other side. In contrast, the closest S⋯S contact between rows is >3.7 Å. Estimation of the charge on the two independent donor molecules from their bond length analysis16 gives values of +0.64 and +0.52, which would be consistent with a roughly equal division of the +1.0 charge between the two donors. Crystals were too small to obtain conductivity data.
 |
| | Fig. 8 (a) The β′′ packing of donors in (BEDT-TTF)2(B5O6(OH)4), and (b) the side to side short S⋯S contacts (3.325–3.560 Å) between donors. | |
(BEDT-TTF)·Br and (BEDT-TTF)5[Br4(H5O2)]
A number of phases of BEDT-TTF with the bromide anion have been reported, for example the solvates of a 2:1 phase with ethylene glycol and water: (BEDT-TTF)2Br·(ethylene glycol)26 and δ-(BEDT-TTF)2Br·3H2O.27,28 Further phases reported are the 3:2 phase (BEDT-TTF)3Br229 and two crystalline forms of its hydrate β′′-(BEDT-TTF)3Br2·2H2O,30 and Kozlov has prepared a 1:1 phase (BEDT-TTF)Br by chemical oxidation.31 Here we report two further phases which have been isolated by electrocrystallisation, one is a 1:1 bromide salt while the other is more complex containing five BEDT-TTFs and a complex anion composed of four bromides grouped around a H5O2+ unit.
Black rod-like crystals of the BEDT-TTF·Br phase were obtained during electrocrystallisation experiments of BEDT-TTF in a mixed solvent system of 1,2,4-trichlorobenzene–ethanol (3:2) with tetrabutyl 1R-(+)-camphorsulfonate. The latter was prepared from the parent acid and tetrabutylammonium hydroxide which is the likely source of the bromide impurity. The presence of bromide in the crystal was also confirmed by energy dispersive X-ray analysis. Crystal structure determination at 173 K showed the crystal to be monoclinic, space group C2/c, with four donors and four bromides in the unit cell. The donor molecule sits on a centre of symmetry and the bromide ion on a two-fold axis. The donor cation is planar apart from the terminal rings which adopt the commonly observed near envelope conformation. The donor molecules are arranged in stacks along the c axis, with their molecular planes lying at ca. 40° to this axis. The ac plane is formed of such parallel stacks with bromide ions lying between the ethylene bridges of the donor cations (Fig. 9). However, the relative disposition of molecules in adjacent layers is controlled by the C centering, so that the central sulfur atoms of donor cations in an adjacent layer neighbour the bromides (Fig. 10). Thus, in contrast to most BEDT-TTF salts, the anions are not segregated from the donors but are incorporated among them. The local environment of a bromide ion is shown in Fig. 11. Each bromide makes contacts with four hydrogen atoms belonging to four different donor molecules (Br⋯H: 2.84 and 2.87 Å), and also with two sulfur atoms (Br⋯S: 3.507 Å) belonging to dithiole rings of adjacent donor cations in the neighbouring stack. The S⋯Br bridges between adjacent donor cations are ca. 0.2 Å shorter than any S⋯S contacts within the stack.
 |
| | Fig. 9 Crystal packing of BEDT-TTF·Br showing a single layer viewed down the b axis with c axis horizontal (above), and viewed down the c axis and showing the offset between donor cations in successive layers (below). | |
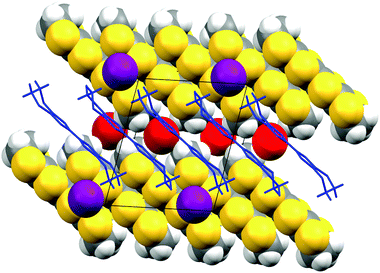 |
| | Fig. 10 View of the relative organisation of the ions in adjacent layers of BEDT-TTF·Br, viewed down the b axis with the c axis horizontal, the front layer (donor drawn as blue capped sticks and bromides in purple) against the rear layer (drawn in space filling mode with bromides in red.). | |
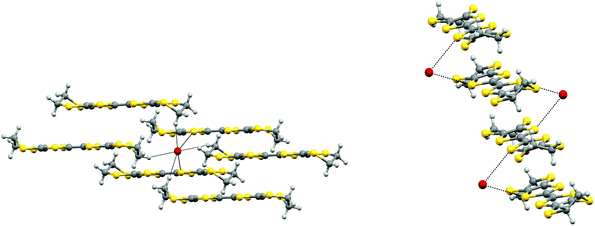 |
| | Fig. 11 Environment of the bromide ion in BEDT-TTF·Br showing its contacts to two S atoms and four H atoms (left) and the short S⋯Br contacts (3.507 Å) from one stack of donor cations to bromide anions (right). | |
Within a stack, adjacent molecules are significantly offset on their longest axis so that the central CC double bond of one donor lies over the terminal dithiin ring of the next donor cation. The S⋯S and C⋯C contacts within a stack are not particularly short: 3.687–3.794 Å and 3.578–3.596 Å respectively. However, there are short S⋯S contacts between stacks (3.293, 3.410, 3.452 and 3.544 Å). Assignment of the donor as a monocation is supported by analysis16 of the bond lengths in the TTF portion of the donor which predict a charge of +1.0. Furthermore, Raman spectroscopy shows symmetrical CC bond stretching frequencies at 1420 and 1450 cm−1 consistent with a monocation structure.17 Mori has also referred to this phase.32 Crystals of the (BEDT-TTF)·Br are semiconducting, with a room temperature conductivity of ca. 2.7 × 10−4 S cm−1. The temperature dependence of the resistivity is shown in Fig. 12. It is notable that the activation energy is not constant; for the region from room temperature to 200 K the activation energy is 260 meV, whilst below 192 K the activation energy is 130 meV. Our structural studies for this compound were performed at 173 K; the sudden change in resistance at T = 192 K could indicate a phase change, however a room temperature crystal structure determination shows a very similar unit cell and molecular packing arrangement.
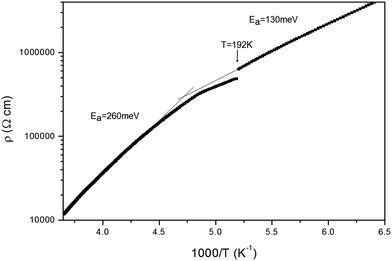 |
| | Fig. 12 Plot of resistivity against 1000/T for (BEDT-TTF)Br. | |
Very small crystals of a quite different salt of BEDT-TTF with bromide arose from further electrocrystallisation experiments involving tetrabutylammonium bromide and 1S-(−)-camphorsulfonic acid. This new hydrated BEDT-TTF bromide phase contains a [Br4(H5O2)]3− cluster for every five BEDT-TTF molecules. The crystal structure is triclinic in space group P and is shown in Fig. 13 and 14. The cluster contains four bromide ions organised in an approximate square which are bridged by hydrogen bonding to a [H2O⋯H+⋯OH2] unit (Fig. 14). The central hydrogen is likely to be disordered between two positions which correspond to a hydrogen bonded water and hydroxonium cation. Furthermore, the H5O2+ unit is disordered 50:50 between two orientations, related by a mirror plane through the four bromide ions, and maintaining the hydrogen bonding to each bromide ion.
![Crystal packing arrangement of (BEDT-TTF)5(Br4(H5O2)) viewed down the a axis with the donor stacking along the c axis (left) and arrangement of [Br4(H5O2)]3 anions in the ac plane (right). Only one of the two orientations of the (H5O2)− anion is shown.](/image/article/2013/DT/c3dt32430c/c3dt32430c-f13.gif) |
| | Fig. 13 Crystal packing arrangement of (BEDT-TTF)5(Br4(H5O2)) viewed down the a axis with the donor stacking along the c axis (left) and arrangement of [Br4(H5O2)]3 anions in the ac plane (right). Only one of the two orientations of the (H5O2)− anion is shown. | |
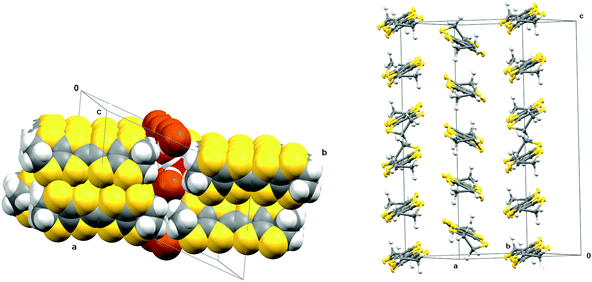 |
| | Fig. 14 Donor stacking arrangement in (BEDT-TTF)5(Br4(H5O2)) viewed down the stacking axis (left), and showing the tilting pattern of the donor molecules in successive stacks along the c axis (right). | |
There are four crystallographically unique donor molecules and two further ones which each lie on a centre of symmetry, thus giving five donor molecules per anion cluster. There are two crystallographically unique stacks of donors lying side by side, each of which shows a ABCCBABCCBA type of stacking where A is a donor lying on a centre of symmetry. Further stacks lie in the +a and −a directions, forming an array of donor molecules in the ac plane. Molecules in adjacent stacks are tilted in opposite directions as in the α- and θ-phases of BEDT-TTF salts, but in this case the periodicity is much higher with five donors in the repeat unit. Arrays are separated by layers composed of the anion clusters (Fig. 13). The inter-donor S⋯S contacts are shorter between stacks than within them. Thus, within a stack S⋯S contacts are >3.9 Å, while between stacks the shortest contacts are ca. 3.5 Å. The cluster of four bromides around H5O2+ appears not to have been observed before, but there are several examples for the corresponding motif with chlorides.33 Mori et al. have reported a structure involving a 2D network of chloride anions and H5O2+ cations in (BEDT-TTF)3Cl2.5(H5O2).34 The Raman spectrum of the (BEDT-TTF)5(Br4(H5O2)) salt, in which the five donor molecules bear a total charge of +3, showed a broad peak centred around 1440 cm−1 suggesting that there may be a spread of charges between the five donors. In contrast, the salt (BEDT-TTF)5Cl3(H2O)5 which also possesses this rather rare +0.6 average charge per donor,35 showed Raman bands at 1462 and 1467 cm−1 though these do not fit so well with the values predicted for such a charge (ca. 1458 and 1485 cm−1).17
Conclusion
The crystal packing arrangements of the donors in the four salts described are very different, and are dictated by the anion structure and whether they form hydrogen bonded ribbons, networks or simple layers. In these cases the anion layers lie outside the donor stacks but in the case of BEDT-TTF·Br the bromide makes contacts to some sulfur atoms. Future work will involve a deeper study of the physical properties of these materials.
Experimental
X-Ray crystallography
Diffraction data were measured at low temperature using MoKα radiation on a Bruker–Nonius FR591 rotating anode diffractometer equipped with 10 cm confocal mirrors, except for ET·Br which was measured on a Bruker SMART system equipped with a fine focused X-ray source. Structures were solved and refined with the SHELX36 suite of programs using the XSEED interface,37 and illustrations made with the Mercury38 and POV-Ray programs.39
(BEDT-TTF)3(SO2NH2)2(H2O)2.
Electrocrystallisation of BEDT-TTF (10 mg) in a solution of tetrabutylammonium sulfamate (40 mg, 0.12 mmol) in dichloromethane (25 ml) at room temperature with a current of 0.1 mA for two weeks, followed by increasing the current for a further week to 1.0 mA gave shiny black needles of the radical cation salt on the anode, m.p. 218–220 °C. Raman/cm−1: 1421, 1460, 1476, 1490.40 Use of THF as solvent also produced the same crystalline material.
Crystal data for (BEDT-TTF)3(SO3NH2)2(H2O)2: 3C10H8S8·2SO3NH2·2H2O, Mr = 1382.14, triclinic, a = 6.57440(10), b = 11.5052(3), c = 16.3531(4) Å, α = 90.0380(10), β = 93.6690(10), γ = 96.9530(10)°, V = 1225.29(5) Å3, Z = 1, P, Dc = 1.873 g cm−3, μ(MoKα) = 1.183 mm−1, T = 120(2) K, 5602 unique reflections, 4944 with F > 4σ(F), R = 0.0335, wR = 0.0676. Cell parameters at 300 K: a = 6.761(8), b = 11.861(14), c = 16.726(16) Å, α = 90.86(4), β = 91.18(5), γ = 100.95(5)°, V = 1316(3) Å3.
(BEDT-TTF)2(B5O6(OH)4).
Electrocrystallisation of BEDT-TTF (10 mg) in a solution of (S)-alpha-methylbenzylammonium rac-2-phenylpropionate (40 mg, 0.12 mmol) in dichloromethane (25 ml) at room temperature with a current of 0.1 mA for 5 days gave dark needles on the anode.
Crystal data for (BEDT-TTF)2(B5O6(OH)4): (C10H8S8)2(B5O6(OH)4), Mr = 987.53, triclinic, a = 9.1142(3), b = 11.5777(4), c = 18.1484(7) Å, α = 97.136(2), β = 101.350(2), γ = 104.651(2)°, V = 1785.72(11) Å3, Z = 2, P, Dc = 1.84 g cm−3, μ(MoKα) = 1.02 mm−1, T = 120(2) K, 8141 unique reflections, 5418 with F > 4σ(F), R = 0.093, wR = 0.187.
(BEDT-TTF)Br.
Electrocrystallisation of BEDT-TTF (10 mg) in a solution of tetrabutylammonium 1R-(+)-camphorsulfonate (40 mg, 0.08 mmol) in 1,2,4-trichlorobenzene–ethanol (60:40 v/v, 25 ml) at room temperature with a current of 0.1 mA for two weeks gave dark rods on the anode. These were carefully collected, washed with acetone and dried in air, m.p. 206 °C; Raman/cm−1ν4 = 1420, ν3 = 1450.40
Crystal data for (BEDT-TTF)·Br: C10H8S8·Br, Mr = 464.55, monoclinic, a = 12.867(3), b = 11.036(2), c = 11.270(2) Å, β = 103.66(3)°, V = 1555.0(5) Å3, Z = 4, C2/c, Dc = 1.98 g cm−3, μ(MoKα) = 3.70 mm−1, T = 173(2) K, 1131 unique reflections, 1099 with F > 4σ(F), R = 0.028, wR = 0.070. Cell parameters at 294 K: a = 12.8238(16), b = 11.0175(10), c = 11.2402(11) Å, β = 103.712(11)°, V = 1542.8(3) Å3.
(BEDT-TTF)5[(Br)4(H5O2)].
Electrocrystallisation of BEDT-TTF (10 mg) in a solution of tetrabutylammonium bromide (26 mg, 0.08 mmol) and 1R-(+)-camphorsulfonic acid (19 mg, 0.08 mmol) in 1,2,4-trichlorobenzene–ethanol (60:40 v/v, 25 ml) at room temperature with a current of 0.1 mA for two weeks gave small dark plates on the anode. These were carefully collected, washed with acetone and dried in air, m.p. 209 °C. The Raman spectrum shows a broad signal at ca. 1440 cm−1.40
Crystal data for (BEDT-TTF)5(Br4(H5O2)): 5C10H8S8·(Br4(H5O2)), Mr = 2280.11, triclinic, a = 11.1312(2), b = 18.0651(4), c = 20.7862(4) Å, α = 75.8770(10), β = 89.9950(10), γ = 72.0850(10)°, V = 3844.19(13) Å3, Z = 2, P, Dc = 1.97 g cm−3, μ(MoKα) = 3.22 mm−1, T = 120(2) K, 17619 unique reflections, 15225 with F > 4σ(F), R = 0.050, wR = 0.114.
Electrical transport measurements
(BEDT-TTF)3(SO3NH2)2(H2O)2.
Electrical conductivity were performed in the range 100–320 K, using a measurement cell attached to the cold stage of a closed cycle helium refrigerator. Four Au wires 25 μm diameter were attached on the crystal with carbon paste to contacts in a four-in-line configuration along the long crystal axis. The sample was checked for unnested to nested voltage ratio, as defined by Schafer et al.41 that was below 5%. Measurements were done imposing through the sample a current of 1 μA at low frequency (77 Hz) and measuring the voltage drop with a lock-in amplifier.
(BEDT-TTF)Br.
Two-probe DC transport measurements were made along the long axis of a crystal using an Oxford Instruments Maglab System 2000. Gold wires 25 μm diameter were attached to the crystal, and the attached wires were connected to an eight-pin integrated circuit plug with carbon conductive cement (RS Components). The resistance of the contacts was assumed to be negligible since the crystal had a high intrinsic resistance.
Acknowledgements
We thank the EPSRC for grant EP/C510488/1 and for a studentship (ACB), the EPSRC National Crystallography Service for datasets, the EPSRC Mass Spectrometry Service for measurements. We wish to acknowledge the use of the EPSRC funded Chemical Database Service at Daresbury.42 The work in Portugal was supported by FCT through grant PTDC/QUI-QUI/101788/2008. JW thanks Profs E.C. Constable and C. Housecroft for a study period at the University of Basel, Switzerland. The work has benefited from support from ESF COST action D35. We thank Mr Brian O'Neill and Mr Peter Moore for construction of constant current sources.
References
- C. Simao, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artes, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef CAS; M. Mas-Torrent, P. Hadley, S. T. Bromley, J. Veciana and C. Rovira, Transistors, 2010, 175 Search PubMed.
- M. Herranz, L. Sanchez and N. Martin, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1133 CrossRef CAS; M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef; M. R. Bryce, W. Davenport, L. M. Goldenberg and C. Wang, Chem. Commun., 1998, 945 RSC.
- W. Biberacher, H. Mueller, C. P. Heidmann, C. Probst, A. Lerf, K. Andres, V. Kakoussis, G. C. Papavassiliou, J. Riede and H. U. Hummel, Synth. Met., 1991, 42, 2377 CrossRef CAS; K. Kikuchi, I. Mochiduki, A. Yamada, H. Saitoh, K. Saito, I. Ikemoto and K. Murata, Synth. Met., 1991, 42, 1921 CrossRef.
- J. M. Williams, M. A. Beno, H. H. Wang, P. C. W. Leung, T. J. Emge, U. Geiser and K. D. Carlson, Acc. Chem. Res., 1985, 18, 261 CrossRef CAS; P. Day, C. R. Chim., 2003, 6, 301 CrossRef.
- T. Mori, Bull. Chem. Soc. Jpn., 1998, 71, 2509 CrossRef CAS.
- T. Mori, H. Mori and S. Tanaka, Bull. Chem. Soc. Jpn., 1999, 72, 179 CrossRef CAS; T. Mori, Bull. Chem. Soc. Jpn., 1999, 72, 2011 CrossRef; T. Mori, Chem. Rev., 2004, 104, 4947 CrossRef.
- J. D. Wallis and J.-P. Griffiths, J. Mater. Chem., 2005, 15, 347 RSC; S. J. Krivickas, A. Ichikawa, K. Takahashi, H. Tajima, J. D. Wallis and H. Mori, Synth. Met., 2011, 161, 1563 CrossRef CAS; F. Riobe and N. Avarvari, Chem. Commun., 2009, 3753 RSC; C. Rethore, N. Avarvari, E. Canadell, P. Auban-Senzier and M. Fourmigue, J. Am. Chem. Soc., 2005, 127, 5748 CrossRef; A. Saad, O. Jeannin and M. Fourmigue, New J. Chem., 2011, 35, 1004 RSC; S. A. Baudron, N. Avarvari, P. Batail, C. Coulon, R. Clérac, E. Canadell and P. Auban-Senzier, J. Am. Chem. Soc., 2003, 125, 11583 CrossRef; A. M. Madalan, C. Réthoré, M. Fourmigué, E. Canadell, E. B. Lopes, M. Almeida, P. Auban-Senzier and N. Avarvari, Chem.–Eur. J., 2010, 16, 528 CrossRef.
- J. I. Yamada, M. Watanabe, T. Toita, H. Akutsu, S. I. Nakatsuji, H. Nishikawa, I. Ikemoto and K. Kikuchi, Mol. Cryst. Liq. Cryst., 2003, 390, 121 Search PubMed; E. I. Zhilyaeva, A. M. Flakina, R. N. Lyubovskaya, I. V. Fedyanin, K. A. Lyssenko, M. Y. Antipin, R. B. Lyubovskii, E. I. Yudanova and J. Yamada, Synth. Met., 2006, 156, 991 CrossRef CAS; J. I. Yamada and H. Akutsu, Crystals, 2012, 2, 812 CrossRef.
- M. Kurmoo, A. W. Graham, P. Day, S. J. Coles, M. B. Hursthouse, J. L. Caulfield, J. Singleton, F. L. Pratt and W. Hayes, J. Am. Chem. Soc., 1995, 117, 12209 CrossRef CAS; L. Martin, P. Day, S. Nakatsuji, J. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369 RSC.
- E. Coronado, J. R. Galán-Mascarós, A. I. Coldea, P. Goddard, J. Singleton, J. D. Wallis, S. J. Coles and A. Alberola, J. Am. Chem. Soc., 2010, 132, 9271 CrossRef CAS; E. Coronado, J. R. Galán-Mascarós, C. J. Gomez-Garcia and V. Laukhin, Nature, 2000, 408, 447 CrossRef.
- J. R. Galán-Mascarós, C. Giménez-Saiz, S. Triki, C. J. Gómez-Garcia, E. Coronado and L. Ouahab, Angew. Chem., Int. Ed. Engl., 1995, 34, 1460 CrossRef CAS; S. A. Baudron, P. Batail, C. Coulon, R. Clerac, E. Canadell, V. Laukhin, R. Melzi, P. Wzietek, D. Jerome, P. Auban-Senzier and S. Ravy, J. Am. Chem. Soc., 2005, 127, 11785 CrossRef.
- A. M. Kini, U. Geiser, H. H. Wang, K. D. Carlson, J. M. Williams, W. K. Kwok, K. G. Vandervoort, J. E. Thompson and D. L. Stupka, Inorg. Chem., 1990, 29, 2555 CrossRef CAS.
- L. C. Porter, H. H. Wang, M. M. Miller and J. M. Williams, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2201 CrossRef CAS; A. Miyazaki, T. Enoki and G. Saito, Synth. Met., 1995, 70, 793 CrossRef; N. D. Kushch, E. B. Yagubskii, V. E. Korotkov, R. P. Shibaeva, L. I. Buravov, A. Y. Zvarykina, V. N. Laukhin and A. G. Khomenko, Synth. Met., 1991, 42, 2131 CrossRef.
- A. I. Kotov, L. I. Buravov, E. B. Yagubskii, S. S. Khasanov, L. V. Zorina, R. P. Shibaeva and E. Canadell, Synth. Met., 2001, 124, 357 CrossRef CAS.
- H. Y. Chen, Q. Fang, P. Li, W. T. Yu, C. Y. Xu, J. B. Zhang and D. Q. Zhang, Synth. Met., 2011, 161, 365 CrossRef CAS.
- P. Guionneau, C. J. Kepert, G. Bravic, D. Chasseau, M. R. Truter, M. Kurmoo and P. Day, Synth. Met., 1997, 86, 1973 CrossRef CAS.
- H. H. Wang, J. R. Ferraro, J. M. Williams, U. Geiser and J. A. Schlueter, J. Chem. Soc., Chem. Commun., 1994, 1893 RSC.
- K. Yakushi, Crystals, 2012, 2, 1291 CrossRef CAS.
- D. M. Schubert and C. B. Knobler, Eur. J. Glass Sci. Technol. B, 2009, 50, 71 Search PubMed; X. Chen, C. Yang, X. Chang, H. Zang and W. Xiao, J. Alloys Compd., 2009, 492, 543 CrossRef CAS; M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse and S. Varma, J. Organomet. Chem., 2007, 692, 2832 CrossRef; P. Becker, P. Held and L. Bohaty, Cryst. Res. Technol., 2000, 35, 1251 CrossRef; V. Domenech, J. Solans and X. Solans, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 643 CrossRef; G. Heller and B. Bichowski, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1978, 33B, 20 Search PubMed.
- K. Armbruster, P. Bele, H. Brunner, C. Benming, S. Dahm, R. Geiger, I. Heinen, H. J. Keller and D. Schweitzer, Synth. Met., 1991, 42, 2025 CrossRef CAS; A. Miyazaki, I. Ichikawa, T. Enoki and G. Saito, Bull. Chem. Soc. Jpn., 1997, 70, 2647 CrossRef.
- O. Kazheva, G. Alexandrov, A. Kravchenko, V. Starodub, I. Lobanova, I. Sivaev, V. Bregadze, L. Buravov and O. Dyachenko, Solid State Sci., 2008, 10, 1734 CrossRef CAS.
- C. J. Gomez-Garcia, C. Gimenez-Saiz, S. Triki, E. Coronado, P. Le Magueres, L. Ouahab, L. Ducasse, C. Sourisseau and P. Delhaes, Inorg. Chem., 1995, 34, 4139 CrossRef CAS; C. J. Gomez-Garcia, C. Gimenez-Saiz, S. Triki, E. Coronado, L. Ducasse, P. Le Magueres, L. Ouahab and P. Delhaes, Synth. Met., 1995, 70, 783 CrossRef.
- M. Guerro, T. Roisnel and D. Lorcy, Tetrahedron, 2009, 65, 6123 CrossRef CAS.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse, D. A. Knox and J. L. Timmis, Dalton Trans., 2010, 39, 3944 RSC.
- M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan and D. M. Schubert, Cryst. Growth Des., 2006, 6, 538 CAS.
- N. P. Karpova, S. V. Konovalikhin, O. A. Dyachenko, R. N. Lyubovskaya and E. I. Zhilyaeva, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 62 CAS; E. I. Zhilyaeva, R. N. Lyubovskaya, N. P. Onishchuk, S. V. Konovalikhin and O. A. Dyachenko, Izv. Akad. Nauk. SSSR, Ser. Khim., 1990, 1438 Search PubMed.
- M. Luo, T. Ishida, A. Kobayashi and T. Nogami, Synth. Met., 1998, 96, 97 CrossRef CAS.
- X. H. Bu, L. Y. Wu and P. Coppens, Z. Kristallogr.– New Cryst. Struct., 1997, 212, 105 Search PubMed; Q. C. Zhang, P. J. Wu, Y. Li and D. Zhu, Synth. Met., 1998, 98, 129 CrossRef CAS; Q. C. Zhang, P. J. Wu, K. J. Jiang, R. S. Ding, Y. Q. Wang and D. Zhu, Chin. Chem. Lett., 1999, 10, 179 Search PubMed.
- S. S. P. Parkin, E. M. Engler, V. Y. Lee and R. R. Schumaker, Mol. Cryst. Liq. Cryst., 1985, 119, 375 CrossRef CAS.
- H. Urayama, G. Saito, A. Kawamoto and J. Tanaka, Chem. Lett., 1987, 1753 CrossRef CAS; S. S. Khasanov, L. V. Zorina, R. B. Shibaeva, S. I. Pesotskii, M. V. Kartsovnik, L. F. Veiros and E. Canadell, Synth. Met., 2002, 131, 41 CrossRef.
- M. E. Kozlov, K. I. Pokhodnia and A. A. Yurchenko, Spectrochim. Acta, Part A, 1989, 45, 437 CrossRef.
- T. Mori, Chem. Rev., 2004, 104, 4947 CrossRef CAS ; note the reference to BEDT·TTF·Br, the same phase as reported above, should be to a Private Communication from T. Mori.
- L. Dobrzycki, M. Chruszcz, W. Minor and K. Wozniak, CrystEngComm, 2007, 9, 152 RSC; A. A. Hoser, L. Dobrzycki, M. J. Gutmann and K. Wozniak, Cryst. Growth Des., 2010, 10, 5092 Search PubMed; J. L. Atwood, S. G. Bott, K. D. Robinson, E. J. Bishop and M. T. May, J. Crystallogr. Spectrosc. Res., 1991, 21, 459 CrossRef CAS.
- H. Mori, I. Hirabayashi, S. Tanaka and Y. Maruyama, Bull. Soc. Chim. Jpn., 1993, 66, 2156 CrossRef CAS.
- B. Zhang, Y. X. Yao and D. B. Zhu, Synth. Met., 2001, 120, 671 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef.
- L. J. Barbour, X-Seed – A software tool for supramolecular crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
-
Persistence of Vision Raytracer (POV-Ray), Persistence of Vision Pty. Ltd., Williamstown, Victoria, Australia, 2004, http://www.povray.com/.
- See ESI.†.
- D. E. Schafer, F. Wudl, G. A. Thomas, J. P. Ferraris and D. O. Cowan, Solid State Commun., 1974, 14, 347 CrossRef CAS.
- “The United Kingdom Chemical Database Service”, D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence