 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A well-defined model system for the chromium-catalyzed selective oligomerization of ethylene†
Wesley H.
Monillas
,
John F.
Young
,
Glenn P. A.
Yap
and
Klaus H.
Theopold
*
Department of Chemistry and Biochemistry, Center for Catalytic Science and Technology, University of Delaware, Newark, Delaware 19716, USA. E-mail: theopold@udel.edu; Fax: +1 3028316335; Tel: +1 3028311546
First published on 6th March 2013
Abstract
The chromium(I) dinitrogen complex [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-N2) catalyzes the selective trimerization of ethylene to 1-hexene at ambient pressure and temperature, and in the absence of any cocatalyst. After the conversion of the substrate, the catalyst cleanly converts to another chromium(I) species, namely [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-C2H4), which is not catalytically active. Binuclear metallacycles containing Cr(II) have been prepared as candidates for catalytically active intermediates; however they are not kinetically competent to explain the catalysis. Turning thus to mononuclear metallacycles featuring Cr(III), a chromacyclopentane, chromacyclopentene and chromacyclopentadiene have been prepared as models of catalytic intermediates. Of these, the latter also catalyzes the trimerization of ethylene. These results support the proposal that selective ethylene oligomerization catalysis involves an interplay between Cr(I) ethylene complexes and mononuclear Cr(III) metallacycles.
Introduction
α-Olefins are valuable precursors for a wide range of chemical processes.1 Owing to the variety of their applications, selective syntheses, e.g. of 1-hexene or 1-octene, are inherently preferable to the statistical (Schulz–Flory) mixtures resulting from the widely practiced ethylene oligomerization reactions based on the Cossee mechanism.2 Originally discovered in the '60s,3,4 there has been a recent flurry of interest in the selective trimerization of ethylene to 1-hexene, predominantly catalyzed by chromium complexes.5–40 While the differential reactivity of metallacycles of different ring sizes is widely held to be the cause of this unusual selectivity, many questions about the mechanistic details remain; among these are the formal oxidation states and charges of the intermediates, the role of mononuclear versus binuclear catalysts, and the exact nature of the product forming step(s). Most relevant catalysts require activation by cocatalysts (e.g. MAO), which complicate spectroscopic detection or isolation of intermediates.41 There exists thus a need for well-defined compounds that catalyze ethylene trimerization in isolation. This contribution describes such a system.Results and discussion
Catalysis
Some time ago we reported the synthesis and characterization of several compounds featuring chromium in the rare formal oxidation state +I;42,43 the most reactive of these is the side-on dinitrogen complex [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-N2) (1, (i-Pr2Ph)2nacnac = 2,4-pentane-N,N′-bis(2,6-diisopropylphenyl)diketiminate).441 reacts cleanly with ethylene to eventually yield [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-C2H4) (2) as the sole organometallic product;43 however, before the latter is formed all available ethylene is oligomerized to 1-hexene and smaller amounts of 1-butene. Thus, structurally characterized 1 is a catalyst – or a catalyst precursor – for the selective trimerization of ethylene, which does not require a cocatalyst (see Scheme 1).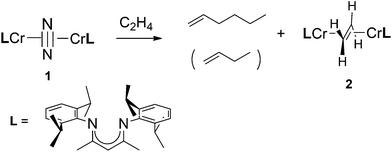 | ||
| Scheme 1 Chromium catalyzed trimerization of ethylene. | ||
Monitoring (by 1H NMR) the reaction of 1 with an excess of ethylene at ambient temperature in a sealed NMR tube revealed the gradual replacement of the broad and isotropically shifted resonances of 1 by those of a yet unidentified paramagnetic compound (the catalyst resting state), while ethylene was being converted to 1-hexene. Only during the final stages of the reaction, when the ethylene concentration became very small, did the characteristic resonances of 2 appear. Interestingly, isolated 2 did not react with ethylene; thus it is not an intermediate in the catalytic reaction. However, as long as the ethylene concentration was maintained constant and significant, catalysis continued unabated and the activity showed no appreciable diminution over the course of two days at ambient temperature.
Table 1 lists the results of catalytic runs under various conditions of temperature, pressure and catalyst concentration. The data indicate that the rate of hexene production is first order in catalyst concentration ([1], see runs 1 and 8–11) and second order in ethylene pressure (runs 1–5). While non-integer orders in ethylene are fairly common for this type of reaction,7 a log(TOF)/log(PC2H4) plot exhibited a slope of 2.05(9). We note that the resulting rate law does not per se require a termolecular turnover-limiting step as part of the catalytic cycle.45
| Runa | Catalyst (mmol) | Solvent (mL) | C2H4 pressure (psi) | Temperature (°C) | Turnover (mmol C6/mmol cat/h) |
|---|---|---|---|---|---|
| a Description of typical method can be found in the Experimental section. b Turnover for reactions at 40 °C calculated before catalyst poisoning. | |||||
| 1 | 0.031 | 10 | 15 | 23 | 0.76 |
| 2 | 0.031 | 10 | 40 | 23 | 5.7 |
| 3 | 0.031 | 10 | 50 | 23 | 11.4 |
| 4 | 0.031 | 10 | 60 | 23 | 26.9 |
| 5 | 0.093 | 30 | 215 | 23 | 172.5 |
| 6 | 0.031 | 10 | 15 | 40 | 1.2 |
| 7 | 0.031 | 10 | 40 | 40 | 8.4b |
| Catalyst (M) | Rate (M h−1) | ||||
| 8 | 0.0016 | 15 | 23 | 0.00064 | |
| 1 | 0.0031 | 15 | 23 | 0.00234 | |
| 9 | 0.0041 | 15 | 23 | 0.00322 | |
| 10 | 0.0062 | 15 | 23 | 0.00485 | |
| 11 | 0.0093 | 15 | 23 | 0.00810 | |
1-Hexene was the only C6 product detected under these conditions. Lower pressure and higher temperature favored the formation of 1-butene. For example, the C6/C4 selectivity at 23 °C increased from 4 at 15 psi to 15 at 50 psi. Only traces of octene were detected by GC at long reaction times (>48 h). Notably, there was no polyethylene formed in any of these reactions. Elevated temperatures resulted in slightly increased rates of hexene formation; however, they also led to catalyst deactivation via formation of 2. We realize, of course, that the activities reported here are minute, and pale in comparison with commercially used catalysts. However, our point is that this system is well-characterized – cleanly progressing from one structurally characterized compound to another – and does not require any co-catalyst or activator. As such it lends itself to mechanistic scrutiny and the results of such an examination may be of general relevance to selective catalytic oligomerizations.
Attempts to isolate the spectroscopically observed intermediate formed during the catalytic reaction – e.g. by cooling solutions under ethylene – were not successful, nor were we able to trap this species by the addition of potential ligands (e.g.pyridine) to catalytically active solutions. However, borrowing an experiment used by Mashima et al.,46 we found that addition of a methylene chloride solution of bromine (Br2) to a reaction in progress produced [(i-Pr2Ph)2nacnacCr(μ-Br)]2 and 1,4-dibromobutane (identified by retention time in the GC and by GCMS). The formation of the latter at a minimum suggests that the intermediate results from coupling of two ethylene units; it is most likely a metallacycle of some type.
These observations are consistent with the commonly suggested mechanism for the selective trimerization of ethylene by transition metal catalysts (see Scheme 2). According to this proposal,47 coordination of two molecules of ethylene to each chromium(I), followed by reductive coupling, forms a mononuclear metallacyclopentane containing Cr(III). The latter is a candidate for the yet unidentified species spectroscopically observed during turnover. Insertion of another ethylene into the relatively stable chromacyclopentane yields the homologous chromacycloheptane. Due to its greater conformational flexibility, β-hydrogen elimination and reductive elimination of the resulting alkenyl hydride are kinetically favored over further ring expansion, thus explaining the selectivity for 1-hexene.
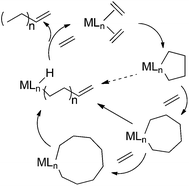 | ||
| Scheme 2 Mechanisms for selective ethylene oligomerization. | ||
The binuclear nature of both 1 and 2 raised the question whether a bimetallic mechanism might be operative. Among the various lines of evidence arguing against such a pathway we mention but two at this juncture. First, the lack of reactivity of 2 with added ethylene would seem to be an obstacle to such a mechanism. Furthermore, we have shown that binuclear chromium alkyl/aryl hydrides, such as might be generated by such a mechanism, are resistant to reductive elimination,42,48 militating against such a transformation as the product forming step of the catalytic cycle. However, a concerted 1–5 hydrogen shift has been discussed as an alternative chain transfer step.
Synthesis of potential bimetallic intermediates
Crucial intermediates in a bimetallic reaction mechanism would be dinuclear metallacycles in which carbon chains of various lengths (C4, C6, C8) bridge a dichromium fragment. Accordingly, we set out to prepare compounds of this type.49–51 Alkylation of [(i-Pr2Ph)2nacnacCr(μ-I)]2 with 1,ω-dilithioalkanes indeed yielded such compounds, after some optimization (see Scheme 3).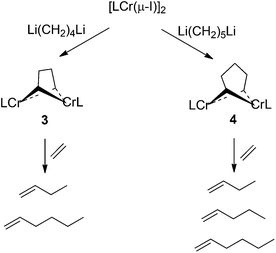 | ||
| Scheme 3 Synthesis and ethylene reactivity of binuclear metallacycles. | ||
The required dilithioalkyl reagents were synthesized using a procedure reported by Negishi.52 For example, Li(CH2)4Li was prepared by lithium/halide exchange between 1,4-diiodobutane and 4.2 equivalents of t-butyl lithium. Although the slight excess of t-butyl lithium ensured full conversion, upon quenching with diethyl ethersolvent at room temperature, lithium ethoxide (LiOEt) and ethylene are formed.53 The LiOEt side product yielded an undesirable chromium ethoxide complex that was observed crystallographically in our initial attempts to alkylate [(i-Pr2Ph)2nacnacCr(μ-I)]2 with Li(CH2)4Li. Therefore, the lithiation procedure was modified to eliminate the formation of lithium ethoxide. The lithium/iodide exchange reaction was performed at −78 °C and the diethyl ether solvent was removed under vacuum while the solution warmed from −78 °C to 0 °C. The crude product was extracted with pentane and filtered under nitrogen to remove any traces of LiOEt.54 Removal of the pentane solvent under vacuum yielded a gummy white precipitate which was dissolved and stored at −30 °C in Et2O as a clear yellow solution. The use of the dilithioalkyls generated by this procedure did not produce any chromium complexes containing ethoxide moieties. However, several attempts to synthesize 1,6-dilithiohexane by this route were unsuccessful, presumably due to formation of cyclohexane or polymerization;55 accordingly the synthesis of a bimetallacyclooctane complex was eventually abandoned.
Reaction of the [(i-Pr2Ph)2nacnacCr(μ-Cl)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56 with Li(CH2)4Li at a variety of temperatures (−78 °C to RT) was always incomplete, giving mixtures of the starting material and the desired bimetallacycle. It was then found that addition of Li(CH2)4Li to iodide [(i-Pr2Ph)2nacnacCr(μ-I)]2 at room temperature resulted in complete conversion to the desired product, namely [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)2] (3). The molecular structure of 3 was determined by X-ray diffraction and the result is shown in Fig. 1. The homologous bimetallacycloheptane [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)3] (4) was synthesized in analogous fashion viaalkylation of the iodide precursor with 1,5-dilithiopentane (Li(CH2)5Li) at room temperature. The structure of 4, as determined by X-ray diffraction, is depicted in Fig. 2.
56 with Li(CH2)4Li at a variety of temperatures (−78 °C to RT) was always incomplete, giving mixtures of the starting material and the desired bimetallacycle. It was then found that addition of Li(CH2)4Li to iodide [(i-Pr2Ph)2nacnacCr(μ-I)]2 at room temperature resulted in complete conversion to the desired product, namely [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)2] (3). The molecular structure of 3 was determined by X-ray diffraction and the result is shown in Fig. 1. The homologous bimetallacycloheptane [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)3] (4) was synthesized in analogous fashion viaalkylation of the iodide precursor with 1,5-dilithiopentane (Li(CH2)5Li) at room temperature. The structure of 4, as determined by X-ray diffraction, is depicted in Fig. 2.
![The molecular structure of [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)2] (3); hydrogen atoms and isopropyl groups have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–Cr2, 2.5450(15); Cr1–N1, 2.101(6); Cr1–N2, 2.067(5); Cr1–C1, 2.259(7), Cr1–C4, 2.164(8); Cr2–N3, 2.062(5); Cr2–N4, 2.011(6); Cr2–C1, 2.158(8); Cr2–C4, 2.288(7), C1–C2; 1.516(11), C2–C3, 1.367(11); C3–C4, 1.483(11); N1–Cr1–N2, 89.1(2); N3–Cr2–N4, 89.4(2); C1–Cr1–C4, 75.2(3), C1–Cr2–C4, 74.7(3); Cr1–C1–Cr2, 70.4(2); Cr1–C4–Cr2, 69.7(2).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f1.gif) | ||
| Fig. 1 The molecular structure of [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)2] (3); hydrogen atoms and isopropyl groups have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–Cr2, 2.5450(15); Cr1–N1, 2.101(6); Cr1–N2, 2.067(5); Cr1–C1, 2.259(7), Cr1–C4, 2.164(8); Cr2–N3, 2.062(5); Cr2–N4, 2.011(6); Cr2–C1, 2.158(8); Cr2–C4, 2.288(7), C1–C2; 1.516(11), C2–C3, 1.367(11); C3–C4, 1.483(11); N1–Cr1–N2, 89.1(2); N3–Cr2–N4, 89.4(2); C1–Cr1–C4, 75.2(3), C1–Cr2–C4, 74.7(3); Cr1–C1–Cr2, 70.4(2); Cr1–C4–Cr2, 69.7(2). | ||
![The molecular structure of [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)3] (4); hydrogen atoms and isopropyl groups have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–Cr2, 2.5567(8)(15); Cr1–N1, 2.122(2); Cr1–N2, 2.075(2); Cr1–C1, 2.266(3), Cr1–C5, 2.194(3); Cr2–N3, 2.091(3); Cr2–N4, 2.138(2); Cr2–C1, 2.172(3); Cr2–C5, 2.258(3), C1–C2; 1.500(4), C2–C3, 1.434(6); C3–C4, 1.436(6); C4–C5, 1.553(5); N1–Cr1–N2, 89.42(8); N3–Cr2–N4, 89.14(8); C1–Cr1–C5, 83.78(10), C1–Cr2–C5, 84.47(11); Cr1–C1–Cr2, 70.27(8); Cr1–C5–Cr2, 70.04(8).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f2.gif) | ||
| Fig. 2 The molecular structure of [(i-Pr2Ph)2nacnacCr]2(μ-CH2)2(CH2)3] (4); hydrogen atoms and isopropyl groups have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–Cr2, 2.5567(8)(15); Cr1–N1, 2.122(2); Cr1–N2, 2.075(2); Cr1–C1, 2.266(3), Cr1–C5, 2.194(3); Cr2–N3, 2.091(3); Cr2–N4, 2.138(2); Cr2–C1, 2.172(3); Cr2–C5, 2.258(3), C1–C2; 1.500(4), C2–C3, 1.434(6); C3–C4, 1.436(6); C4–C5, 1.553(5); N1–Cr1–N2, 89.42(8); N3–Cr2–N4, 89.14(8); C1–Cr1–C5, 83.78(10), C1–Cr2–C5, 84.47(11); Cr1–C1–Cr2, 70.27(8); Cr1–C5–Cr2, 70.04(8). | ||
The C4 bimetallacycle 3 crystallized in the monoclinic space group P21 with a co-crystallized molecule of hexamethyldisiloxane (solvent of recrystallization). C5 bimetallacycle 4 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a co-crystallized molecule of pentane. The molecular structures of 3 and 4 are very similar, except for the incrementally larger ring size of 4. Both feature terminal methylene groups that bridge the two chromium atoms; in that sense they may be described as a dichromabicyclo[1.1.2]hexane (3) and dichromabicyclo[1.1.3]heptane (4). Extreme electron deficiency – discounting metal–metal bonding, the individual chromium atoms feature 10-valence electron configurations – drive the formation of 3-center/2-electron M–C–M bonds. This structural feature has ample precedent in Cr(II) chemistry; for example, we had earlier prepared compounds of the type [Cp*Cr(μ-R)]2, which show strong similarities in gross structure, metal–metal distances, and magnetism.57 The local coordination environments around the Cr atoms in 3 and 4 deviate from the preferred square planar geometry for four coordinate Cr(II) complexes.58 The Cr2C2-cores of the molecules adopt a butterfly shape and the relevant Cr–C distances exhibit a short/long pattern, rendering this fragment somewhat unsymmetric. The non-coordinated carbon atoms (i.e., C(2), and C(3) of 3 and C(2), C(3), and C(4) of 4) exhibit positional disorder; thus their C–C single bonds appear unrealistically short.59 The Cr–Cr distances in 3 and 4 are 2.5451(15) and 2.5567(8) Å, respectively, and are therefore within the range of weak Cr–Cr bonding. Both compounds have identical effective magnetic moments of 2.6(1)μB/Cr at room temperature, which is considerably lower than expected of a typical high-spin Cr(II) ions (d4, S = 2). These values are reminiscent of similar bridging Cr(II) alkyls and consistent with either antiferromagnetic coupling,60 metal–metal bonding, or a superposition of both.61
with a co-crystallized molecule of pentane. The molecular structures of 3 and 4 are very similar, except for the incrementally larger ring size of 4. Both feature terminal methylene groups that bridge the two chromium atoms; in that sense they may be described as a dichromabicyclo[1.1.2]hexane (3) and dichromabicyclo[1.1.3]heptane (4). Extreme electron deficiency – discounting metal–metal bonding, the individual chromium atoms feature 10-valence electron configurations – drive the formation of 3-center/2-electron M–C–M bonds. This structural feature has ample precedent in Cr(II) chemistry; for example, we had earlier prepared compounds of the type [Cp*Cr(μ-R)]2, which show strong similarities in gross structure, metal–metal distances, and magnetism.57 The local coordination environments around the Cr atoms in 3 and 4 deviate from the preferred square planar geometry for four coordinate Cr(II) complexes.58 The Cr2C2-cores of the molecules adopt a butterfly shape and the relevant Cr–C distances exhibit a short/long pattern, rendering this fragment somewhat unsymmetric. The non-coordinated carbon atoms (i.e., C(2), and C(3) of 3 and C(2), C(3), and C(4) of 4) exhibit positional disorder; thus their C–C single bonds appear unrealistically short.59 The Cr–Cr distances in 3 and 4 are 2.5451(15) and 2.5567(8) Å, respectively, and are therefore within the range of weak Cr–Cr bonding. Both compounds have identical effective magnetic moments of 2.6(1)μB/Cr at room temperature, which is considerably lower than expected of a typical high-spin Cr(II) ions (d4, S = 2). These values are reminiscent of similar bridging Cr(II) alkyls and consistent with either antiferromagnetic coupling,60 metal–metal bonding, or a superposition of both.61
Reactivity with ethylene
It is difficult to imagine how any bimetallic mechanism for the oligomerization of ethylene catalysed by 1 could circumvent 3 as an intermediate, and 4 is surely representative of the kind of bimetallacycle involved in such a putative mechanism. Accordingly, their reactivity with ethylene and kinetic competency vis-à-vis the catalysis described above is of interest. Preliminary 1H-NMR spectroscopic studies of the reaction of 3 with 1 atm of ethylene in C6D6 indicated the presence of α-olefin(s) as well as the starting material 3 after 1 day at room temperature. GC/MS analysis confirmed the presence of 1-hexene and 1-butene. However, quantitation of the products from this reaction revealed that only 11% of 1-hexene and 4% of 1-butene had been generated (Table 2) based on the starting material 3. A similar reaction at 40 °C generated only ∼6% hexene and ∼14% butene. At higher temperatures more 1-butene is observed, presumably due to shift from ring expansion to β-hydrogen elimination. Even after two weeks at room temperature, the amount of 1-hexene formed did not reach a single turnover (100% yield). At an ethylene pressure of 250 psi (17 atm) larger amounts of 1-hexene were generated; however, higher molecular weight oligomers (C10–C20) and an insoluble white precipitate (presumed to be polyethylene) were also observed. To summarize, while the reaction of 3 with ethylene yielded 1-hexene and 1-butene, their rate of formation was much slower than during the catalysis with 1. The reaction also showed different selectivity and a more varied product profile. Based on these observations alone, it seems unlikely that 3 is an intermediate in the catalytic oligomerization. However, the formation of 1-hexene from 3 is intriguing and merits some further investigation.A catalytic reaction of dimetallacycloheptane 4 with ethylene would presumably first produce the odd numbered α-olefins 1-pentene and possibly 1-heptene, which would be followed by production of even numbered α-olefins. GC/MS analysis of the volatile products of a reaction of 4 with ethylene (1 atm) showed that the only odd-numbered α-olefin produced was pentene (<1.0% yield), while no heptene was detected. However, significant amounts of 1-butene and 1-hexene were formed (see Table 2). This suggested that the bimetallacycles decompose – inter alia – via β-hydrogen elimination, but that they do not undergo further ring expansion by insertion of ethylene. The formation of even-numbered olefins suggests that the decomposition of the bimetallacycles generates a catalyst for the oligomerization of ethylene. The nature of this species was addressed next.
Both 3 and 4 decompose slowly at ambient temperature in solution, and this process becomes faster at elevated temperatures. 1H-NMR decomposition studies in C6D6 at 65 °C showed that 3 and 4 completely decomposed within two hours and that several paramagnetic products were formed in this reaction. One of the organometallic products observed in the 1H-NMR during the thermolysis of 3 was the known hydride complex [(i-Pr2Ph)2nacnacCr(μ-H)]2 (5).42 We reasoned that one β-hydrogen elimination of 3 would form an intermediate of the type [(i-Pr2Ph)2nacnacCr]2(μ-H)(μ-CH2CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2); the latter might have a choice between suffering another β-hydrogen elimination to form 5 and eliminating butadiene, or undergoing reductive elimination of 1-butene and yielding two Cr(I) fragments. However, analysis by 1H-NMR and GC/MS did not show any butadiene; only butene was detected by GC/MS. Since no free butadiene was found, despite the formation of 5, it was hypothesized that another paramagnetic compound formed during the decomposition reaction might be a butadiene complex. In separate 1H-NMR experiments in C6D6 or d8-THF, 5 was exposed to 1 atm of butadiene and left to react overnight at room temperature. 5 completely disappeared and resonances for butene and a paramagnetic compound that exactly matched the unassigned 1H-NMR resonances noted during the decomposition of 3 were observed (Scheme 4).
CH2); the latter might have a choice between suffering another β-hydrogen elimination to form 5 and eliminating butadiene, or undergoing reductive elimination of 1-butene and yielding two Cr(I) fragments. However, analysis by 1H-NMR and GC/MS did not show any butadiene; only butene was detected by GC/MS. Since no free butadiene was found, despite the formation of 5, it was hypothesized that another paramagnetic compound formed during the decomposition reaction might be a butadiene complex. In separate 1H-NMR experiments in C6D6 or d8-THF, 5 was exposed to 1 atm of butadiene and left to react overnight at room temperature. 5 completely disappeared and resonances for butene and a paramagnetic compound that exactly matched the unassigned 1H-NMR resonances noted during the decomposition of 3 were observed (Scheme 4).
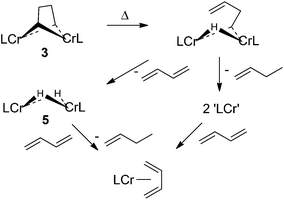 | ||
| Scheme 4 Decomposition of 3 and formation of butadiene complex 5. | ||
In hopes of determining if this new paramagnetic compound was indeed a butadiene complex, 1 was exposed to 1 atm of butadiene in THF; this reaction indeed yielded a monomeric Cr butadiene adduct, namely (i-Pr2Ph)2nacnacCr(η4-C4H6)(THF) (6). The molecular structure of 6 was determined by X-ray diffraction; its molecular structure is shown in Fig. 3. 6 exhibits square pyramidal geometry and contains a cis-η4-bonded butadiene molecule and a coordinated molecule of THF in the apical position (the basal plane is defined by the nacnac ligand and the butadiene). The bond distances of the butadiene's erstwhile double bonds (C1–C2 and C1A–C2A, 1.378(4) Å) are identical to the formal single bond distance (C2–C2A, 1.379(5)), indicating a modest transfer of electron density to the diene, which is consistent with other reported Cr-(η4-1,3-butadiene) complexes.62–65 However, the relative Cr–C distances still favour a description as a π-complex rather than as the valence tautomeric metallacyclopentene.66 The room temperature effective magnetic moment of 6, μeff(295) = 3.8(1)μB, is consistent with a quartet (S = 3/2) ground state. The chromium in 6 has a formal oxidation state of +I, giving it a d5 electronic configuration; 6 apparently adopts an intermediate spin state.
![The molecular structure of (i-Pr2Ph)2nacnacCr(η4-C4H6)(THF) (6); hydrogen atoms have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–N1, 2.0991(17); Cr1–C1, 2.202(3); Cr1–C2, 2.167(2); Cr1–O1, 2.222)(3); C1–C2, 1.378(4); C2–C2A, 1.379(5); N1–Cr1–N1A, 88.54(9); N1–Cr1–C1, 96.46(9); N1–Cr1–C2, 103.64(9); N1–Cr1–O1, 92.8(3).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f3.gif) | ||
| Fig. 3 The molecular structure of (i-Pr2Ph)2nacnacCr(η4-C4H6)(THF) (6); hydrogen atoms have been omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–N1, 2.0991(17); Cr1–C1, 2.202(3); Cr1–C2, 2.167(2); Cr1–O1, 2.222)(3); C1–C2, 1.378(4); C2–C2A, 1.379(5); N1–Cr1–N1A, 88.54(9); N1–Cr1–C1, 96.46(9); N1–Cr1–C2, 103.64(9); N1–Cr1–O1, 92.8(3). | ||
We note that 6 also provides a model for the binding of two molecules of ethylene to Cr(I); it may be considered an analogue of the proposed LnCr(C2H4)2 intermediate of ethylene trimerization proceeding by a mononuclear metallacycle mechanism (see Scheme 1). The notion that 3 decomposes to form a butadiene complex was confirmed, since the 1H-NMR spectrum of 6 matched the unassigned paramagnetic resonances observed in the decomposition of 3. To summarize, 3 decomposes to equal parts of 5 and 6. This result implies that there are multiple pathways to generate Cr(I) fragments, which may initiate oligomerization catalysis. The observation of butene during thermolysis of 3 is also consistent with the detection of pentene during the reaction of 4 with ethylene, and it suggests that decomposition of these bimetallacycles can lead to catalytically active Cr(I) fragments.
Finally, in a separate 1H-NMR experiment in THF-d8, 6 was exposed to 1 atm of ethylene, whereupon it produced 1-hexene and 1-butene (confirmed by GC/MS analysis). Mechanistically, dissociation of butadiene followed by the binding of ethylene would lead to the formation of 1-hexene and then regenerate a Cr(I) fragment that can either continue in trimerization or rebind butadiene. These results suggest that labile monomeric Cr(I) compounds can selectively oligomerize ethylene and it provides some support for a monomeric metallacycle mechanism for ethylene trimerization. This notion will be further explored in the next section.
Synthesis of mononuclear model compounds
As the binuclear hypothesis based on Cr(I) olefin complexes and Cr(II) bimetallacycles became discredited (see above), a premium was placed on preparing possible intermediates of a mononuclear mechanism, which would presumably involve Cr(III) metallacycles. Notably, we tried to devise synthetic routes to a mononuclear chromacyclobutane, which might be a credible candidate for the active intermediate observed during catalysis. A severe obstacle to the synthesis of e.g. (i-Pr2Ph)2nacnacCr(CH2)4 was presented by our finding that the (i-Pr2Ph)2nacnacCr-fragment does not readily support the preparation of trivalent chromium dialkyls of the type (i-Pr2Ph)2nacnacCrR2, whereas the corresponding methyl-substituted nacnac ligand affords ready access to compounds of the composition (Me2Ph)2nacnacCrR2 (R = Me, Bn, CH2SiMe3).67 Indeed, whereas treatment of (Me2Ph)2nacnacCrIIICl2(THF)2 with lithium alkyls produces Cr(III) dialkyls, similar treatment of (i-Pr2Ph)2nacnacCrIIICl2(THF)2 or [(i-Pr2Ph)2nacnacCrIIICl(μ-Cl)]2 with such reagents invariably leads to reduction and formation of Cr(II) alkyls of the type (i-Pr2Ph)2nacnacCrR (R = CH2SiMe3) or [(i-Pr2Ph)2nacnacCr(μ-R)]2 (R = Me).68 In view of this dichotomy, we chose to initially attempt the synthesis (Me2Ph)2nacnacCr(CH2)4. However, as detailed elsewhere, reaction of (Me2Ph)2nacnacCrIIICl2(THF)2 with Li(CH2)4Li actually yielded the bimetallacycle [(Me2Ph)2nacnacCr]2(μ-CH2)2(CH2)2] (an analogue of 3), presumably due to the greater reducing power of the dilithio reagent. To overcome this problem, we decided to make the chromium(III) ion in the halide precursor more electron rich by substituting it with stronger σ-donors. Gratifyingly, this was met with success. Treatment of (Me2Ph)2nacnacCrIIICl2(py)2 with Li(CH2)4Li afforded the mononuclear chromacyclobutane (Me2Ph)2nacnacCr(CH2)4(py) (7). The molecular structure of 7 was determined by X-ray diffraction, and the result is shown in Fig. 4.![The molecular structure of (Me2Ph)2nacnacCr(CH2)4(py) (7); hydrogen atoms, except those on the alkyl chain have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–C1 2.076(5), Cr–C4 2.120(5), Cr–N1 2.048(4), Cr–N2 2.126(4), Cr–N3, 2.138(4), C1–Cr–C4 83.2(2), N1–Cr–N2, 88.99(16).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f4.gif) | ||
| Fig. 4 The molecular structure of (Me2Ph)2nacnacCr(CH2)4(py) (7); hydrogen atoms, except those on the alkyl chain have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–C1 2.076(5), Cr–C4 2.120(5), Cr–N1 2.048(4), Cr–N2 2.126(4), Cr–N3, 2.138(4), C1–Cr–C4 83.2(2), N1–Cr–N2, 88.99(16). | ||
7 is a rare example of a mononuclear chromacyclopentane. It differs from the putative catalytic intermediate only by virtue of the coordinated pyridine. However, the latter apparently makes all the difference. 7 does not catalyze the trimerization of ethylene, nor does it decompose readily upon heating. We believe that the strongly coordinated pyridineligand prevents binding and insertion of ethylene as well as decomposition by β-elimination. Attempts to remove the pyridine by adding various Lewis acids (AlMe3, B(C6F5)3, BF3) did not yield tractable products, nor did these additives facilitate reactions of 7 with ethylene. In summary, 7 may be a suggestive structural model for a catalytic intermediate, but the strong coordination by pyridine blocks further reactivity (such as binding and insertion of ethylene or even β-hydrogen elimination). Its ancillary ligand also differs from the one which gives rise to catalysis. Accordingly, 7 does not by itself provide convincing evidence for the catalytic activity of mononuclear metallacycles.
We then turned our attention to the preparation of compounds that might serve as models for some of the species proposed to constitute the catalytic cycle (Scheme 1), while employing the same ancillary ligand as the actual catalytic system. First up, we wondered about an analogue of the mononuclear ethylene complex (i-Pr2Ph)2nacnacCr(C2H4), which may enjoy a fleeting existence after the elimination/substitution of the α-olefin product. In the presence of excess ethylene, it would be expected to form (i-Pr2Ph)2nacnacCr(C2H4)2 to begin another turnover, while lack of substrate presumably leads to irreversible formation of 2. We hypothesized that a sterically more demanding π-acceptor might stabilize such a 1:1 complex, and indeed the reaction of 1 with one equivalent of diphenylacetylene produced the mononuclear alkyne adduct (i-Pr2Ph)2nacnacCr(η2-C2Ph2) (8). The molecular structure of 8 as determined by X-ray diffraction is shown in Fig. 5. The diphenylacetylene fragment is bound nearly perpendicularly to the plane of the ligand. Both C1 and C2 were found to be located at similar distances from the chromium at distances of 1.957(4) Å and 1.963(4) Å, respectively, with a Cr-centroid distance of 1.846(6) Å. The C–C distance of the diphenylacetylene was found to have been elongated by 0.115 Å from free diphenylacetylene to a distance of 1.320(5) Å.69 The latter distance is the strikingly similar to the CC double bond distance in stilbene, which measures 1.321(5) Å.70 The diphenylacetylene fragment in 8 is no longer linear but features an average C–C–Cipso angle of 137.3(4)°. This value is within the range of other structurally characterized chromium η2-diphenylacetylene complexes, which possess a C–C–Cipso angles in the range of 136.3–155.9°.71 These structural changes in the diphenylacetylene fragment, and the fact that 8 has an effective magnetic moment of 3.9(1)μB, suggests that the chromium center of 8 is in the +III oxidation state, with corresponding twofold reduction of the diphenylacetylene ligand, resulting in a more appropriate description as a metallacyclopropene.
![The molecular structure of (i-Pr2Ph)2nacnacCr(η2-C2Ph2) (8); hydrogens have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–N1, 1.980(2); Cr1–C7, 1.944(4); Cr1–C8, 1.970(4); C7–C8, 1.307(5); N1–Cr1–N1A, 90.78(14); N1–Cr1–C7, 130.52(8); N1–Cr1–C8, 132.17(8).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f5.gif) | ||
| Fig. 5 The molecular structure of (i-Pr2Ph)2nacnacCr(η2-C2Ph2) (8); hydrogens have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr1–N1, 1.980(2); Cr1–C7, 1.944(4); Cr1–C8, 1.970(4); C7–C8, 1.307(5); N1–Cr1–N1A, 90.78(14); N1–Cr1–C7, 130.52(8); N1–Cr1–C8, 132.17(8). | ||
If 8 is to be a model of a 1:1 ethylene complex, it might be expected to rapidly react with additional ethylene. We already have a model of sorts for the ensuing bis(ethylene) complex, in the form of butadiene complex 6, but the covalent linkage of the two olefins in butadiene prevents it from modelling the subsequent reductive coupling step. On this score, 8 delivers. Monitoring the addition of ethylene to a THF-d8 solution of 8 by 1H NMR spectroscopy showed formation of a new paramagnetic chromium compound, but no formation of hexene or any other olefin. Isolation and determination of the structure of the new compound (see Fig. 6) showed it to be the five-membered metallacycle (i-Pr2Ph)2nacnacCr(CPh)2(CH2)2 (9), which can the thought of as resulting from the insertion of ethylene into one of the Cr–C bonds of the metallacyclopropene 8, or alternatively as the product of a reductive coupling of a coordinated alkyne and alkene. 9 adopts a square pyramidal geometry with a THF molecule occupying one of the basal sites. The C3–C4 distance of 1.354(4) Å is consistent with a CC double bond. The C1–C2 distance (1.526(4) Å) and C2–C3 distance (1.508(4) Å) on the other hand are characteristic of C–C-single bonds, consistent with the description of 9 as a metallacyclopentene. In addition, the hydrogen atoms on C1 and C2 can be located in the difference map and their positions refined.
![The molecular structure of (i-Pr2Ph)2nacnacCr(CPh)2(CH2)2 (9); hydrogen atoms, except for those on the alkyl chain, have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–N1, 2.137(2); Cr–N2, 2.052(2); Cr–O1, 2.1260(18); Cr–C1, 2.091(3); Cr–C4, 2.051(3); C1–C2, 1.526(4); C2–C3, 1.508(4); C3–C4, 1.354(4); N1–Cr–N2, 88.87(10); N1–Cr–O1, 86.15(8); N2–Cr–C1, 93.35(11); C1–Cr–O1, 85.56(10); C4–Cr–N1, 120.82(10); C4–Cr–N2, 107.19(10); C4–Cr–O1, 88.30(9); C4–Cr–C1, 80.11(11).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f6.gif) | ||
| Fig. 6 The molecular structure of (i-Pr2Ph)2nacnacCr(CPh)2(CH2)2 (9); hydrogen atoms, except for those on the alkyl chain, have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–N1, 2.137(2); Cr–N2, 2.052(2); Cr–O1, 2.1260(18); Cr–C1, 2.091(3); Cr–C4, 2.051(3); C1–C2, 1.526(4); C2–C3, 1.508(4); C3–C4, 1.354(4); N1–Cr–N2, 88.87(10); N1–Cr–O1, 86.15(8); N2–Cr–C1, 93.35(11); C1–Cr–O1, 85.56(10); C4–Cr–N1, 120.82(10); C4–Cr–N2, 107.19(10); C4–Cr–O1, 88.30(9); C4–Cr–C1, 80.11(11). | ||
Although the THF molecule coordinated to chromium in 9 might be expected to be substitutionally labile, the compound does not react further with additional ethylene. In particular, it does not catalyse the oligomerization of ethylene in THF (as 1 does). We note the possibility that the large steric demand of the phenyl substituents is responsible for this reluctance, effectively blocking the coordination of additional ethylene. On the other hand, 9 is already the second mononuclear chromacycle (besides 7) that does not produce any higher olefins under ethylene. Fortunately, the situation is about to improve.
Diphenylacetylene is a rather ‘large’ stand-in for ethylene by any reckoning, which in turn led us to exploring smaller alkynes. Accordingly, exposure of a THF solution of 1 to 2-butyne resulted in the rapid formation of a white precipitate. Permitting the reaction to continue yielded a large amount of the white precipitate and effected a slow color change of the solution from brown to green-grown. 13C NMR spectroscopy proved that the white precipitate was hexamethylbenzene, i.e. the product of a catalytic cyclotrimerization of the alkyne.72,73 Removal of the organic product by filtration and standard work-up of the filtrate afforded the five-membered metallacycle (i-Pr2Ph)2nacnacCr(η2-C4Me4)(THF) (10). The molecular structure of 10 has been determined by X-ray diffraction, and is shown in Fig. 7.
![The molecular structure of (i-Pr2Ph)2nacnacCr(η2-C4Me4)(THF) (10); hydrogen atoms have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–N1, 2.019(2); Cr–N2, 2.126(2); Cr–O1, 2.1102(19); Cr–C1–2.081(3); Cr–C4, 2.007(3); C1–C2, 1.359(4); C2–C3, 1.485(4); C3–C4, 1.347(4); N1–Cr–N2, N1–Cr–C1, 96.42(10); N2–Cr–O1, 87.05(8); C4–Cr–N1, 98.97(10); C4–Cr–N2, 105.98(10); C4–Cr–O1, 97.10(10); C4–Cr–C1, 82.56(12).](/image/article/2013/DT/c3dt00109a/c3dt00109a-f7.gif) | ||
| Fig. 7 The molecular structure of (i-Pr2Ph)2nacnacCr(η2-C4Me4)(THF) (10); hydrogen atoms have been removed for clarity. Selected interatomic distances [Å] and angles [°]: Cr–N1, 2.019(2); Cr–N2, 2.126(2); Cr–O1, 2.1102(19); Cr–C1–2.081(3); Cr–C4, 2.007(3); C1–C2, 1.359(4); C2–C3, 1.485(4); C3–C4, 1.347(4); N1–Cr–N2, N1–Cr–C1, 96.42(10); N2–Cr–O1, 87.05(8); C4–Cr–N1, 98.97(10); C4–Cr–N2, 105.98(10); C4–Cr–O1, 97.10(10); C4–Cr–C1, 82.56(12). | ||
The compound is a metallacyclopentadiene (or ‘metallole’), which presumably results from reductive coupling of two alkynes bound to chromium. The compound possesses square pyramidal geometry with one terminus of the butadienediyl ligand in the axial position. As expected, the coordinated C4Me4-fragment is planar and exhibits clear bond length alternations, as C1–C2 and C3–C4 were found to be 1.359(4) Å and 1.347(4) Å, respectively, while C2–C3 was found to be 1.485(4) Å. The effective magnetic moment of 10 is 3.8(1)μB, consistent with chromium in the +III oxidation state. Compound 10 is the resting state of the cyclotrimerization catalyst, as its exposure to 2-butyne reinitiates the catalysis and yields more hexamethylbenzene.
More to the point, 10 is another mononuclear chromacycle, which (unlike 9) is not encumbered by excessive steric hindrance, and the coordinated THF of which is apparently subject to displacement by additional substrate. Therefore it might be expected to react with ethylene as well. Gratifyingly then, 10 was indeed found to be a catalyst for the selective oligomerization of ethylene. Exposure of a THF-d8 solution of 10 to ethylene yielded 1-hexene, ethylene complex 2, and an unidentified organic compound (presumably 1,2,3,4-tetramethylhexatriene).
Conclusions
We have shown that a well-characterized chromium(I) dinitrogen complex (i.e., 1) catalyzes the selective trimerization of ethylene to 1-hexene in homogeneous THF solution, albeit slowly. To our knowledge, this is the only example of a catalyst for the trimerization of ethylene that unambiguously starts and finishes in the formal oxidation state Cr(I).74,75 Since the catalyst functions in the absence of any cocatalysts, which often obscure assignments of structure and oxidation state,76,77 this system presents an excellent chance to address fundamental questions regarding a commercially important and mechanistically intriguing catalytic reaction. For example, we have prepared relevant binuclear Cr(II) metallacycles and shown that they are not intermediates in the catalysis. By extrapolation, and absent any unambiguous evidence for a role of binuclear sites in this type of catalysis, we suggest that the alternative mononuclear Cr(III) metallacycles are most likely intermediates in selective oligomerization catalysis. While the independent synthesis of the most likely intermediate in our system – i.e., (i-Pr2Ph)2nacnacCr(CH2)4(THF) – proved elusive, we have prepared several closely related analogues of it. Of these, two proved inactive, but for cogent reasons. However, the mononuclear chromacyclopentadiene (i-Pr2Ph)2nacnacCr(η2-C4Me4)(THF) (10) reacted with ethylene to catalytically produce 1-hexene and enter the same reaction channel as 1. To the extent that these results may prove general, they strongly support the notion that chromium catalysts for selective ethylene oligomerization alternate between mononuclear Cr(I) ethylene complexes and Cr(III) metallacycles. Our results also show that neutral compounds are competent catalysts for the selective oligomerization.78 However, the activity of this catalyst is rather low by comparison, and we surmise that cationic, more electrophilic intermediates are the mainstays of more active ones.Experimental
General considerations
All manipulations of compounds were carried out using standard Schlenk, high vacuum line, and glovebox techniques under an atmosphere of N2. Solvents were purchased from Fisher Scientific, degassed, and dried by passing through activated alumina. THF-d8 and C6D6 were purchased from Cambridge Isotopes Laboratory and stored under vacuum over Na/K alloy. All other reagents were purchased from Aldrich or Acros and dried using standard procedures as needed. [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-N2) (1), [(i-Pr2Ph)2nacnacCr]2(μ–η2:η2-C2H4) (2), and [(i-Pr2Ph)2nacnacCr(μ-X)]2 (X = Cl, I) were prepared by previously published procedures.431H NMR spectra were taken on a Bruker DRX-400 spectrometer and were referenced to the residual protons of the solvent. FTIR spectra were taken on Mattson Alpha Centauri or Mattson Genesis Series spectrometers. UV/vis spectra were recorded using a HP 8453 spectrophotometer. Mass spectra were obtained by the University of Delaware Mass Spectrometry Facility. Elemental analyses were performed by Desert Analytics. Room-temperature molar magnetic susceptibilities (χm) in the solid state were determined using a Johnson Matthey magnetic susceptibility balance. They were corrected for diamagnetism using Pascal constants and converted into effective magnetic moments (μeff).
GC/MS measurements for oligomerizations were taken on a HP G1800A GCD. To determine the response factor for the GC 1 mmol of 1-pentene, 1-hexene, 1-heptene, 1-octene, and nonane were placed in a 50 mL volumetric flask and then filled to 50 mL with THF. The flask was shaken and 1 μL was injected into the GC, the average area for three runs was calculated and the ratio of the GC/MS response to the α-olefin to nonane was calculated and plotted versus carbon number. The slope of the linear trendline allowed for the calculation of the RF for the 1-hexene; the RF for nonane was arbitrarily set to 1.0. The raw integrals for the 1-hexene analyses were divided by the RF to obtain the corrected integrals.
Typical oligomerization procedure for reactions <90 psi
A thick walled reactor was heated to 250 °C overnight to remove all traces of moisture and fitted with a cap consisting of an inlet port, gauge, and connections for vacuum and ethylene. The reactor was evacuated and backfilled with ethylene three times. The reactor was charged with 1 atm of ethylene. In the nitrogen dry box the requisite amount of catalyst was dissolved in 10 mL THF; nonane was added as the standard. A syringe containing the aforementioned mixture was removed from the dry-box and the solution mixture was injected into the reactor. The reactor pressure was raised to the necessary pressure and the reaction mixture was stirred. At various times throughout the oligomerization an aliquot was removed from the reactor and injected in the GC.Typical oligomerization procedure for reaction at 215 psi
In the dry-box, 90 mg (93 mmol) of catalyst was added to a 300 mL Parr reactor followed by 90 mL THF and the nonane standard. Pentane was also added as an additional check. The reactor assembly was brought of the dry-box and the connection line was purged with ethylene for 2 minutes. The reaction was opened to ethylene and the pressure was regulated to 215 psi and stirred for certain time periods. When the set time was reached the ethylene pressure was released, the solution is exposed to air and an aliquot injected into the GC. This procedure was repeated for each time frame.Typical oligomerization procedure for the reactions of 3 and 4 with ethylene
In a nitrogen-filled dry box the bimetallacycle was dissolved in 10 mL THF and nonane was added via a micropipette to a glass ampoule. The ampule was removed from the nitrogen dry box and placed onto a high vacuum line. After three freeze–pump–thaw cycles 1 atm of ethylene was backfilled into the vessel. After 24 h the reaction mixture was exposed to air, filtered with celite, and then 1 μL was injected onto the GC/MS.Procedure for the reaction done at 250 psi
In a nitrogen-filled dry-box, 180 mg (0.154 mmol) of 3 was added to a 300 mL Parr reactor followed by 30 mL THF and the nonane standard. The reactor assembly was removed out of the dry-box and the connection line was purged with ethylene for 2 minutes. The reactor was opened to ethylene and the pressure was regulated to 250 psi (17 atm) and the solution stirred for 24 h. After 24 h the ethylene pressure was released and reaction mixture was exposed to air, filtered with celite, and then 1 μL was injected onto the GC/MS.Preparation of 1,4-dilithiobutane Li(CH2)4Li
10 mL of diethyl ether was added to a Schlenk flask in a nitrogen-filled glove box. The flask was then cooled to −78 °C using a dry ice/acetone bath. 4.2 equivalents of 1.7 M t-BuLi in pentane (11.95 mL, 20.3 mmol) were added to the cold ether solution and allowed to stir for 15 minutes. One equivalent of 1,4-diiodobutane (1.50 g, 4.84 mmol) was then added via syringe. The reaction was allowed to stir for 1 h at −78 °C. Afterwards, the diethyl ether was removed under vacuum at −78 °C. The reaction was warmed over 1 hour to 0 °C, while maintaining a dynamic vacuum. The resulting white/yellow product was kept under vacuum for an additional hour at 0 °C. The precipitate was then extracted, in an inert atmosphere, with 100 mL of pentane in 50 mL portions. The gummy white precipitate was then dissolved in dry ethyl ether giving a translucent yellow solution which was stored at −30 °C.Preparation of (μ-tetramethylene) [2,4-pentane-N,N′-bis(2,6-diisopropylphenyl)ketiminato] dichromium(II), [(i-Pr2Ph)2nacnacCr]2[(μ-CH2)2(CH2)2] (3)
To a solution of [(i-Pr2Ph)2nacnacCr(μ-I)]2 (0.200 g, 0.168 mmol) in 70 mL pentane, 0.480 M Et2O solution of Li(CH2)4Li (0.385 mL, 0.185 mmol) was added dropwise. The solution rapidly changed color from green to brown. The solution was stirred at room temperature for 3 hours. The solvent was removed under vacuum, leaving a crude brown powder. Pentane was then added to the crude reaction mixture and cooled to −30 °C for 1 hour. The mixture was filtered and the pentane solvent was removed under a vacuum leaving a crude brown powder. This was recrystallized from hexamethyldisiloxane by slow cooling to −30 °C, yielding X-ray quality crystals (0.095 g, 49% yield). 1H-NMR (400 MHz, C6D6): 11.25 (br), 10.09 (br), 7.46 (br), 4.43 (br), 3.14 (br), 1.79 (br), 1.49 (br), 1.34 (br), 1.14 (br), 0.22 ((CH3)3Si)2O) ppm. IR (KBr): 3057 (w), 2959 (s), 2927 (s), 2867 (m), 1525 (s), 1434 (s), 1310 (s), 1253 (s), 1170 (m), 1097 (w), 1055 (m), 930 (w), 845 (m), 793 (m), 758 (m) cm−1. UV/Vis (pentane): λmax (ε) = 455 (1326 M−1 cm−1) nm. μeff = 2.6(1)μB/Cr (294 K). Mp: 100 °C dec. Mass spectrumm/z (%): 469 (100%) (C29H41N2Cr+), 938 (28%) (C58H82N4Cr2+). Anal. Calcd for C64H108N4OSi2Cr2: C, 69.77; H, 9.14; N, 5.04. Found: C, 69.55; H, 8.68; N, 4.78. The compound decomposes slowly at room temperature.Preparation of μ-pentamethylene) [2,4-pentane-N,N′-bis(2,6-diisopropylphenyl)ketiminato] dichromium(II), [(i-Pr2Ph)2nacnacCr]2[(μ-CH2)2(CH2)3] (4)
To a solution of [(i-Pr2Ph)2nacnacCr(μ-I)]2 (0.200 g, 0.168 mmol) in 70 mL pentane, 0.480 M Et2O solution of Li(CH2)5Li (0.385 mL, 0.185 mmol) was added dropwise. The solution rapidly changed color from green to brown. The solution was stirred at room temperature for 3 hours. The solvent was removed under vacuum leaving a crude brown powder. Pentane was then added to the crude reaction mixture and cooled to −30 °C for 1 hour. The mixture was filtered and the pentane solvent was removed under a vacuum leaving a crude brown powder. This was recrystallized from hexamethyldisiloxane by slow cooling to −30 °C, yielding X-ray quality crystals (0.093 g, 47% yield). 1H-NMR (400 MHz, C6D6): 10.81 (br), 9.56 (br), 7.03 (br), 4.36 (br), 3.07 (br), 1.85, 1.52, 1.32, 0.22 ((CH3)3Si)2O) ppm. IR (KBr): 3057 (w), 2959 (s), 2925 (m), 2867 (m), 1525 (s), 1434 (s), 1311 (s), 1254 (s), 1171 (m), 1098 (m), 1056 (m), 1042 (m), 932 (w), 846 (m), 793 (s), 760 (m) cm−1. UV/Vis (toluene): λmax (ε) = 477 (125 M−1 cm−1) nm. μeff = 2.6(1)μB/Cr (294 K). Mp: 95 °C dec. Mass spectrumm/z (%): 469 (100%) (C29H41N2Cr+). Anal. Calcd for C65H110N4OSi2Cr2: C, 70.72; H, 9.46; N, 4.78. Found: C, 69.66; H 8.42; N, 4.75. The compound decomposes slowly at room temperature.Preparation of [2,4-pentane-N,N′-bis(2,6-diisopropylphenyl) ketiminato](η4-cis-butadiene)(tetrahydrofuran) chromium(I), (i-Pr2Ph)2nacnacCr(η4-C4H6)(THF) (6)
1 (0.400 g 0.336 mmol) (0.400 g 0.336 mmol) was placed in ampoule with a stir bar and 50 mL THF and attached to a vacuum manifold. Three freeze–pump–thaw cycles were performed, followed by back filling with 1 atm of 1,3-butadiene. The solution was stirred for 6 hours at room temperature resulting in a color change from brown to dark red. Butadiene and solvent were removed by vacuum transfer, and the ampoule was then sealed and brought back into the dry-box. The crude solid was recrystallized from pentane. X-ray quality crystals were grown by slow cooling of a solution in pentane to −30 °C (100% conversion by 1H-NMR). 1H-NMR (400 MHz, C6D6): 11.35 (br), 9.0 (br), 7.82 (br), 3.44 (br), 1.63 (s), 1.42 (m), 1.35 (s), 0.87 (s), −0.75 (br), −4.29 (br) ppm. IR 3054 (w), 2959 (s), 2867 (s), 1518 (s), 1405 (s), 1359 (s), 1316 (s), 1260 (m), 1227 (m), 1174 (m), 1100 (m), 1020 (m), 932 (m), 867 (m), 790 (s), 758 (s), 437 (w) cm−1. UV/Vis (pentane): λmax (ε) = 475 (716 M−1 cm−1) nm. μeff = 3.8(1)μB (294 K). Mp: 108 °C dec. Mass spectrumm/z (%): 596.1 (12%) (C37H55N2OCr+), 524.2 (100%) (C33H47N2Cr+).Preparation of (2,6-Me2Ph)2nacnacCr(CH2)4(py) (7)
(2,6-Me2Ph)2nacnacCrCl2(py) (0.200 g, 0.394 mmol) was partially dissolved in 150 mL pentane giving a dark brown solution. The solution was chilled to −30 °C and 1,4-dilithiobutane (0.39 M, 0.394 mmol) was added. The color deepened as the reaction stirred for 3 hours while warming up to room temperature. The solvent was removed under a reduced pressure. The crude solid was extracted with pentane and the extract filtered, concentrated, and cooled to −30 °C to yield 3 (0.082 g, 42%). 1H NMR (400 MHz, C6D6): 12.2 (s), 9.80 (b), 6.99 (s), 4.80 (s), 3.59 (b), 2.11 (s), 1.52 (s), 1.40 (s), 1.19 (s), 0.91 (s), 0.28 (s) ppm. IR (KBr): 2961 (m), 2923 (m), 2855 (w), 1624 (2), 1547 (m), 1524 (m), 1540 (m), 1392 (s), 1261 (s), 1183 (m), 1095 (m), 1022 (m), 800 (s), 765 (m), 555 (w) cm−1. UV/Vis (pentane): λmax (ε) = 422 (1665 M−1 cm−1) nm, 482 (416 M−1 cm−1) nm, 668 (142 M−1 cm−1) nm. μeff = 3.9(1)μB (294 K). Mp = 92–95 °C (dec).Preparation of (i-Pr2Ph)2nacnacCr(η2-C2Ph2) (8)
(LiPrCr)2(μ2–η2:η2-N2) (1) (0.200 g, 0.206 mmol) was dissolved in 20 mL THF and 0.074 mg (0.416 mol) of diphenylacetylene was added and the solution was stirred for 4 hours. The THF was removed and the product was extracted with pentane. Concentration and cooling to −30 °C yielded brown crystals of 8 (0.197 g, 74% yield). 1H NMR (400 MHZ, THF-d8): 143.6 (br), 39.9 (br), 20.4 (br), 17.1 (br), 10.0 (br), 6.64 (br), 2.80 (br), −4.55 (br) ppm. IR (KBr): 3056 (w), 2961 (s), 2926 (m), 2867 (m), 1589 (w), 1526 (s), 1489 (m), 1462 (m), 1436 (m), 1383 (s), 1373 (s), 1315 (s), 1257 (m), 1174 (m), 1103 (m), 1056 (m), 1024 (m), 933 (w), 856 (w), 796 (m), 759 (m), 697 (m) cm−1. UV/vis (pentane): λmax (ε) = 390 (4657 M−1 cm−1) nm. μeff (293 K) = 3.9(1)μB. Mp: 211 °C (dec). Mass spectrumm/z (%): 469.3 (70) [M+ − C2Ph2], 178.1 (100) [C2Ph2]. Anal. Calcd for C43H51N2Cr: C, 79.71; H, 7.94; N, 4.33. Found: C, 79.41; H, 7.65; N, 4.37.Preparation of (i-Pr2Ph)2nacnacCr(CPh)2(CH2)2 (9)
(i-Pr2Ph)2nacnacCr(η2-C2Ph2) (8) (0.100 g, 0.154 mmol) was dissolved in 20 mL, excess ethylene was added and the solution was stirred for up to 24 hours. The THF was removed and the product was extracted with pentane. Concentration and cooling to −30 °C yielded light brown crystals of 9 (0.140 g, 60% yield). 1H NMR (400 MHZ, THF-d8): 29.78 (br), 22.84 (br), 14.86 (br), 11.01 (br), 7.70 (br), 4.73 (br), −6.66 (br), −13.01 (br) ppm. IR (KBr): 3057 (w), 3016 (w), 2960 (s), 2926 (s), 2866 (m), 1522 (s), 1490 (m), 1472 (m), 1465 (m), 1457 (s), 1437 (s), 1419 (m), 1384 (s), 1374 (s), 1339 (w), 1313 (m), 1256 (m), 1172 (w), 1101 (w), 1025 (w), 933 (w), 795 (m), 756 (m), 698 (m), 668 (w) cm−1. UV/vis (pentane): λmax (ε) = 370 (4200 M−1 cm−1), 454 (1195 M−1 cm−1) nm. μeff (293 K) = 3.4(1)μB. Mp: 192 °C (dec). Mass spectrumm/z (%): 469.2 (51) [M+ − C16H14 − C4H8O]. Anal. Calc. for C59H75N2CrO: C, 80.50; H, 8.59; N, 3.18. Found: C, 78.81; H, 7.86; N, 3.18.Preparation of (i-Pr2Ph)2nacnacCr(η2-C4Me4)(THF) (10)
(LiPrCr)2(μ2–η2:η2-N2) (1) (0.200 g, 0.206 mmol) was dissolved in 20 mL THF and 2-butyne (0.07 mL, 0.850 mmol) was added and the solution was stirred 2 hours. The THF was removed and the product was extracted with pentane. Concentration and cooling to −30 °C yielded green crystals of 5, which always contained some residual hexamethylbenzene (0.080 g, ∼30% yield). 1H NMR (400 MHZ, THF-d8): 11.00 (br), 9.13 (br), 8.44 (br), 7.46 (br), 6.92 (br), 5.46 (br), 5.11 (br), 4.48 (br), 3.16 (br) ppm. IR (KBr): 2962 (s), 2925 (s), 2866 (m), 1653 (w), 1539 (m), 1524 (s), 1464 (s), 1436 (s), 1385 (s), 1368 (m), 1317 (m), 1260 (m), 1172 (m), 1099 (s), 1057 (m), 1024 (s), 935 (w), 797 (s), 761 (w) cm−1. UV/vis (pentane): λmax (ε) = 380 (4077 M−1 cm−1), 748 (317 M−1 cm−1) nm. μeff (293 K) = 3.8(1)μB. Mp: 73 °C (dec). Mass spectrumm/z (%): 577.3 (69) [M+ − C4H8O]. Anal. Calc. for C41H61CrN2O: C, 75.77; H, 9.46; N, 4.31. Found: C, 73.58; H, 9.36; N, 4.28.Crystallographic structure determinations
A summary of crystal data collection and refinement parameters for compounds 3, 4, 6, 7, 8, 9 and 10 can be found in Table 3. Suitable crystals were selected, mounted with viscous oil and cooled to the data collection temperature. Data were collected on a Bruker APEX CCD diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073). Unit cell parameters were obtained from 60 data frames, 0.3° ω, from three different sections of the Ewald sphere. No symmetry higher than triclinic was observed for 4 and 10. The systematic absences in the data and the unit cell parameters were consistent to P21 and P21/m for 3, and Pnma and Pn21a (Pna21) for 6 and 8, and, uniquely, P21/n for 7, and P21/c for 9. The centrosymmetric space group options for 4 and 10 (P), and 6 and 8 (Pnma), and the noncentrosymmetric space group for 3 (P21) yielded chemically reasonable and computationally stable results of refinement. The data-sets were treated with SADABS absorption corrections based on redundant multiscan data.79 The structures were solved using direct methods and refined with full-matrix, least-squares procedures on F2. A close inspection of the packing diagram did not reveal any overlooked symmetry for 3 and the Flack parameter80 refined to 0.03(3) indicating that the true hand of the data was determined. Compound 4 consistently deposits as small, weakly-diffracting crystals resulting in lower-than-ideal 2θ range. Two symmetry unique but chemically identical compound molecules were found in the asymmetric unit of 8. The metallacyclic carbon atoms and the isopropyl moieties displayed unresolvable disorder causing a larger than usual Ueq range for carbon atoms and associated hydrogen atoms. Compound 6 was located disordered at a mirror plane. Solvent molecules were found cocrystallized in the asymmetric units of 3 (hexamethyldisiloxane), 4 (pentane), 7 (pentane) and 9 (pentane). Disordered pentane molecules of crystallization in 7 with net occupancy of 1.5 per unit-cell, in two void spaces located away from the compound molecule, and two molecules of co-crystallized pentane per compound molecule in 9 were treated as diffused contributions.81 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms on metallacycles were located from the difference map, treated with chemically sensible restraints, and refined with a riding model. All other hydrogen atoms were treated as idealized contributions with 1.2–1.5Ueq of the associated carbon atom. Atomic scattering factors are contained in the SHELXTL 6.12 program library.
| 3 | 4 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|
| a Quantity minimized: Rw(F2) = Σ[w(Fo2 − Fc2)2]/Σ[(wFo2)2]1/2. | |||||||
| Formula | C68H108Cr2N4OSi2 | C68H104Cr2N4 | C37H55CrN2O | C31.88H42.50CrN3 | C43H51CrN2 | C59H75CrN2O | C41H61CrN2O |
| Fw | 1157.76 | 1081.55 | 595.83 | 519.69 | 647.86 | 880.21 | 649.92 |
| Space group | P21 |
P |
Pnma | P21/n | Pnma | P21/c |
P |
| Color | Brown | Brown | Red | Green | Orange | Brown | Green |
| a (Å) | 13.404(5) | 13.187(4) | 16.702(3) | 11.960(6) | 21.020(4) | 13.834(4) | 8.924(3) |
| b (Å) | 14.089(5) | 13.567(4) | 20.310(4) | 14.191(7) | 18.707(3) | 17.440(5) | 11.555(4) |
| c (Å) | 18.924(7) | 18.405(6) | 9.768(2) | 16.977(8) | 19.091(4) | 20.034(5) | 19.287(7) |
| α (°) | 90 | 88.750(5) | 90 | 90 | 90 | 90 | 106.796(6) |
| β (°) | 109.268(7) | 74.798(4) | 90 | 97.191(5) | 90 | 104.087(5) | 96.988(6) |
| γ (°) | 90 | 88.291(4) | 90 | 90 | 90 | 90 | 100.378(7) |
| V (Å3) | 3374(2) | 3175.8(17) | 3313.5(11) | 2859(2) | 7507(2) | 4688(2) | 1840.8(12) |
| Z, Z′ | 2, 1 | 2, 1 | 4, 0.5 | 4, 1 | 8, 2 | 4, 1 | 2, 1 |
| D (calcd) (g cm−3) | 1.140 | 1.131 | 1.194 | 1.207 | 1.146 | 1.247 | 1.173 |
| μ (MoKα) (mm−1) | 0.400 | 0.383 | 0.376 | 0.424 | 0.335 | 0.288 | 0.343 |
| Temp, K | 120(2) | 120(2) | 120(2) | 120(2) | 180(2) | 120(2) | 120(2) |
| Data/params | 11746/732 |
13753/679 |
4153/322 | 4952/337 | 6835/474 | 8258/500 | 6485/420 |
| GOF on F2 | 1.025 | 1.059 | 1.049 | 1.081 | 1.036 | 1.054 | 1.049 |
| R(F) (%) | 7.42 | 5.84 | 5.36 | 8.41 | 5.56 | 6.09 | 5.27 |
| R w(F2) (%) | 12.77 | 16.20 | 13.33 | 18.15 | 15.24 | 16.82 | 14.18 |
| Flack param | 0.03(3) | — | — | — | — | — | — |
Acknowledgements
This work was supported by the U.S. National Science Foundation, through grants CHE-0616375 and CHE-0911081 to KHT.Notes and references
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 2003 Search PubMed.
- D. Vogt, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996, pp. 245–258 Search PubMed.
- R. M. Manyik, W. E. Walker and T. P. Wilson, Union Carbide Corporation, US, Continuous processes for the production of ethylene polymers and catalysts suitable therefore, US Patent, 3300458, 1967 Search PubMed.
- R. M. Manyik, W. E. Walker and T. P. Wilson, J. Catal., 1977, 47, 197–209 CrossRef CAS.
- J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641–3668 CrossRef CAS.
- D. F. Wass, Dalton Trans., 2007, 816–819 RSC.
- T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS.
- B. R. Aluri, N. Peulecke, S. Peitz, A. Spannenberg, B. H. Muller, S. Schulz, H. J. Drexler, D. Heller, M. H. Al-Hazmi, F. M. Mosa, A. Wohl, W. Muller and U. Rosenthal, Dalton Trans., 2010, 39, 7911–7920 RSC.
- B. H. Muller, N. Peulecke, S. Peitz, B. R. Aluri, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wohl and W. Muller, Chem.–Eur. J., 2011, 17, 6935–6938 Search PubMed.
- B. H. Muller, N. Peulecke, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, R. Schmidt, A. Wohl and W. Muller, Organometallics, 2012, 31, 3695–3699 Search PubMed.
- W. Muller, A. Wohl, S. Peitz, N. Peulecke, B. R. Aluri, B. H. Mueller, D. Heller, U. Rosenthal, M. H. Al-Hazmi and F. M. Mosa, ChemCatChem, 2010, 2, 1130–1142 Search PubMed.
- S. Peitz, B. R. Aluri, N. Peulecke, B. H. Muller, A. Wohl, W. Muller, M. H. Al-Hazmi, F. M. Mosa and U. Rosenthal, Chem.–Eur. J., 2010, 16, 7670–7676 Search PubMed.
- S. Peitz, N. Peulecke, B. R. Aluri, S. Hansen, B. H. Muller, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wohl and W. Muller, Eur. J. Inorg. Chem., 2010, 1167–1171 CrossRef CAS.
- S. Peitz, N. Peulecke, B. R. Aluri, B. H. Muller, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wohl and W. Muller, Organometallics, 2010, 29, 5263–5268 Search PubMed.
- S. Peitz, N. Peulecke, B. R. Aluri, B. H. Muller, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wohl and W. Muller, Chem.–Eur. J., 2010, 16, 12127–12132 CrossRef CAS.
- S. Peitz, N. Peulecke, B. H. Muller, A. Spannenberg, H. J. Drexler, U. Rosenthal, M. H. Al-Hazmi, K. E. Al-Eidan, A. Wohl and W. Muueller, Organometallics, 2011, 30, 2364–2370 Search PubMed.
- A. Wohl, W. Muller, S. Peitz, N. Peulecke, B. R. Aluri, B. H. Muller, D. Heller, U. Rosenthal, M. H. Al-Hazmi and F. M. Mosa, Chem.–Eur. J., 2010, 16, 7833–7842 Search PubMed.
- S. Licciulli, I. Thapa, K. Albahily, I. Korobkov, S. Gambarotta, R. Duchateau, R. Chevalier and K. Schuhen, Angew. Chem., Int. Ed., 2010, 49, 9225–9228 CrossRef CAS.
- I. Thapa, S. Gambarotta, I. Korobkov, R. Duchateau, S. V. Kulangara and R. Chevalier, Organometallics, 2010, 29, 4080–4089 CrossRef CAS.
- R. Duchateau, S. Gambarotta, S. Licciulli, K. Albahily, S. V. Kulangara and I. Thapa, Abstr. Pap. Am. Chem. Soc., 2011, 241, 1155-INOR Search PubMed.
- S. Licciulli, K. Albahily, V. Fomitcheva, I. Korobkov, S. Gambarotta and R. Duchateau, Angew. Chem., Int. Ed., 2011, 50, 2346–2349 Search PubMed.
- K. Albahily, S. Gambarotta and R. Duchateau, Organometallics, 2011, 30, 4655–4664 CrossRef CAS.
- K. Albahily, V. Fomitcheva, Y. Shaikh, E. Sebastiao, S. I. Gorelsky, S. Gambarotta, I. Korobkov and R. Duchateae, Organometallics, 2011, 30, 4201–4210 Search PubMed.
- K. Albahily, S. Licciulli, S. Gambarotta, I. Korobkov, R. Chevalier, K. Schuhen and R. Duchateau, Organometallics, 2011, 30, 3346–3352 Search PubMed.
- I. Thapa, S. Gambarotta, I. Korobkov, M. Murugesu and P. Budzelaar, Organometallics, 2012, 31, 486–494 Search PubMed.
- Y. Shaikh, K. Albahily, M. Sutcliffe, V. Fomitcheva, S. Gambarotta, I. Korobkov and R. Duchateau, Angew. Chem., Int. Ed., 2012, 51, 1366–1369 Search PubMed.
- K. Albahily, Z. Ahmed, S. Gambarotta, E. Koc, R. Duchateau and I. Korobkov, Organometallics, 2011, 30, 6022–6027 Search PubMed.
- Y. Shaikh, J. Gurnham, K. Albahily, S. Gambarotta and I. Korobkov, Organometallics, 2012, 31, 7427–7433 Search PubMed.
- L. E. McDyre, T. Hamilton, D. M. Murphy, K. J. Cavell, W. F. Gabrielli, M. J. Hanton and D. M. Smith, Dalton Trans., 2010, 39, 7792–7799 RSC.
- A. F. R. Kilpatrick, S. V. Kulangara, M. G. Cushion, R. Duchateau and P. Mountford, Dalton Trans., 2010, 39, 3653–3664 RSC.
- Y. Qi, Q. Dong, L. Zhong, Z. Liu, P. Y. Qiu, R. H. Cheng, X. L. He, J. Vanderbilt and B. P. Liu, Organometallics, 2010, 29, 1588–1602 Search PubMed.
- I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 Search PubMed.
- T. W. Hey and D. F. Wass, Organometallics, 2010, 29, 3676–3678 CrossRef CAS.
- S. K. Kim, T. J. Kim, J. H. Chung, T. K. Hahn, S. S. Chae, H. S. Lee, M. Cheong and S. O. Kang, Organometallics, 2010, 29, 5805–5811 Search PubMed.
- Z. Mohamadnia, E. Ahmadi, M. N. Haghighi and H. Salehi-Mobarakeh, Catal. Lett., 2011, 141, 474–480 Search PubMed.
- P. Y. Qiu, H. Cheng, B. P. Liu, B. Tumanskii, R. J. Batrice, M. Botoshansky and M. S. Eisen, Organometallics, 2011, 30, 2144–2148 CrossRef CAS.
- P. W. N. M. van Leeuwen, N. D. Clement and M. J. L. Tschan, Coord. Chem. Rev., 2011, 255, 1499–1517 CrossRef CAS.
- E. Ahmadi, Z. Mohamadnia and M. N. Haghighi, Catal. Lett., 2011, 141, 1191–1198 Search PubMed.
- S. F. Liu, R. Pattacini and P. Braunstein, Organometallics, 2011, 30, 3549–3558 CrossRef CAS.
- L. H. Do, J. A. Labinger and J. E. Bercaw, Organometallics, 2012, 31, 5143–5149 Search PubMed.
- T. E. Stennett, M. F. Haddow and D. F. Wass, Organometallics, 2012, 31, 6960–6965 Search PubMed.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Angew. Chem., Int. Ed., 2007, 46, 6692–6694 CrossRef CAS.
- W. H. Monillas, G. P. A. Yap, L. A. MacAdams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090–8091 CrossRef CAS.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Inorg. Chim. Acta, 2011, 369, 103–119 Search PubMed.
- A. A. Frost and R. G. Pearson, Kinetics and Mechanism, John Wiley & Sons, Inc., New York, 1953 Search PubMed.
- R. Arteaga-Muller, H. Tsurugi, T. Saito, M. Yanagawa, S. Oda and K. Mashima, J. Am. Chem. Soc., 2009, 131, 5370–5371 CrossRef CAS.
- J. R. Briggs, J. Chem. Soc., Chem. Commun., 1989, 674–675 RSC.
- L. A. MacAdams, G. P. Buffone, C. D. Incarvito, J. A. Golen, A. L. Rheingold and K. H. Theopold, Chem. Commun., 2003, 1164–1165 RSC.
- K. H. Theopold and R. G. Bergman, Organometallics, 1982, 1, 1571–1579 CrossRef CAS.
- J. R. Moss and L. G. Scott, Coord. Chem. Rev., 1984, 60, 171–190 CrossRef CAS.
- C. P. Casey and J. D. Audett, Chem. Rev., 1986, 86, 339–352 CrossRef CAS.
- E. Negishi, D. R. Swanson and C. J. Rousset, J. Org. Chem., 1990, 55, 5406–5409 CrossRef CAS.
- W. F. Bailey and E. R. Punzalan, J. Org. Chem., 1990, 55, 5404–5406 CrossRef CAS.
- C. W. Kamienski and D. H. Lewis, J. Org. Chem., 1965, 30, 3498–3504 Search PubMed.
- W. F. Bailey, R. P. Gagnier and J. J. Patricia, J. Org. Chem., 1984, 49, 2098–2107 CrossRef CAS.
- V. C. Gibson, C. Newton, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Eur. J. Inorg. Chem., 2001, 1895–1903 CrossRef CAS.
- R. A. Heintz, R. L. Ostrander, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1994, 116, 11387–11396 CrossRef CAS.
- A. R. Hermes, R. J. Morris and G. S. Girolami, Organometallics, 1988, 7, 2372–2379 CrossRef CAS.
- P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement – A Crystallographers's Guide to SHELXL, Oxford University Press, Oxford, 2006 Search PubMed.
- R. L. Carlin, Magnetochemistry, Springer, Berlin, 1986 Search PubMed.
- P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884–4899 CrossRef CAS.
- K. Angermund, P. Betz, A. Dohring, P. W. Jolly, C. Kruger and K. U. Schonfelder, Polyhedron, 1993, 12, 2663–2669 CrossRef CAS.
- A. Dohring, J. Gohre, P. W. Jolly, B. Kryger, J. Rust and G. P. J. Verhovnik, Organometallics, 2000, 19, 388–402 CrossRef.
- N. F. Wang, D. J. Wink and J. C. Dewan, Organometallics, 1990, 9, 335–340 CrossRef CAS.
- D. W. Norman, M. J. Ferguson, R. McDonald and J. M. Stryker, Organometallics, 2006, 25, 2705–2708 Search PubMed.
- C. Elschenbroich and A. Salzer, Organometallics, A Concise Introduction, VCH, Weinheim, 1992 Search PubMed.
- L. A. MacAdams, G. P. Buffone, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 2005, 127, 1082–1083 CrossRef CAS.
- K. H. Theopold, L. A. MacAdams, C. Puttnual, G. P. Buffone and A. L. Rheingold, Polym. Mater. Sci. Eng., 2002, 86, 310 Search PubMed.
- The CC distance of free diphenylacetylene is 1.205(4) Å taken from the following publications retrieved from the CSD: (a) V. D. Samarskaya, R. M. Myasnikova and A. I. Kitaigorodskij, Kristallografiya, 1968, 13, 616 Search PubMed; (b) A. A. Epriritu and J. G. White, Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem., 1978, 147, 177 Search PubMed; (c) I. E. Zanin, M. Y. Antipin and T. Y. Struchlov, Kristallografiya, 1991, 36, 411 CAS; (d) R. Thomas, S. Lakshmi, S. K. Pati and G. U. Kulkami, J. Phys. Chem. B, 2006, 110, 24674 CrossRef CAS.
- A search of the CSD showed 14 structures of stilbene with an average CC distance of 1.321(5) Å (range: 1.288–1.341 Å).
- A search of the CSD showed 10 structures with a η2-diphenylacetylene fragment with a C–C–Cipso angle of 145.3(19)° (range: 136.3–155.9°). Selected examples: (a) D. J. Wink, J. R. Fox and N. J. Cooper, J. Am. Chem. Soc., 1985, 107, 5012 CrossRef CAS; (b) I. M. Barlett, N. G. Connelly, A. G. Orpen and M. J. Quayle, Chem. Commun., 1996, 2583 RSC; (c) A. A. Danopoulos, G. Wilkinson, T. K. N. Sweet and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1996, 271 RSC; (d) C. J. Adams, I. M. Bartlett, N. G. Connelly, D. J. Harding, O. D. Hayward, A. J. Martin, M. J. Quayle and P. H. Reiger, J. Chem. Soc., Dalton Trans., 2002, 4281 RSC.
- Y. Yamamoto, Curr. Org. Chem., 2005, 9, 503–519 CrossRef CAS.
- L. S. Zhou, S. Li, K. Kanno and T. Takahashi, Heterocycles, 2010, 80, 725–738 Search PubMed.
- L. E. Bowen, M. F. Haddow, A. G. Orpen and D. F. Wass, Dalton Trans., 2007, 1160–1168 RSC.
- A. J. Rucklidge, D. S. McGuinness, R. P. Tooze, A. M. Z. Slawin, J. D. A. Pelletier, M. J. Hanton and P. B. Webb, Organometallics, 2007, 26, 2782–2787 CrossRef CAS.
- A. Jabri, P. Crewdson, S. Gambarotta, I. Korobkov and R. Duchateau, Organometallics, 2006, 25, 715–718 CrossRef CAS.
- C. Temple, A. Jabri, P. Crewdson, S. Gambarotta, I. Korobkov and R. Duchateau, Angew. Chem., Int. Ed., 2006, 45, 7050–7053 CrossRef CAS.
- T. Agapie, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2007, 129, 14281–14295 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122.
- H. D. Flack, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1983, 39, 876–881 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † CCDC 698322 and 918023–918028. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt00109a |
| This journal is © The Royal Society of Chemistry 2013 |