DOI:
10.1039/C3DT00053B
(Perspective)
Dalton Trans., 2013,
42, 6676-6693
An alternative picture of alkali-metal-mediated metallation: cleave and capture chemistry
Received
7th January 2013
, Accepted 18th February 2013
First published on 18th February 2013
Abstract
This perspective article takes an alternative look at alkali-metal-mediated chemistry (exchange of a relatively inert C–H bond for a more reactive C–metal bond by a multicomponent reagent usually containing an alkali metal and a less electropositive metal such as magnesium or zinc). It pictures that the cleavage of selected C–H bonds can be accompanied by the capturing of the generated anion by the multi (Lewis acid)–(Lewis base) character of the residue of the bimetallic base. In this way small atoms or molecules (hydrides, oxygen-based anions) as well as sensitive organic anions (of substituted aromatic compounds, ethers or alkenes) can be captured. Cleave and capture reactions which occur in special positions on the organic substrate are also included.
 Robert E. Mulvey | Robert Emmet Mulvey was born in Glasgow, Scotland in 1959. He received his first degree (BSc, 1st class Honours) and his Ph.D. (in organolithium chemistry under the direction of Dr Ron Snaith) at the University of Strathclyde in 1981 and 1984 respectively. Following two years as a postdoctoral fellow at the University of Durham (under the direction of Professor Ken Wade), he returned to Strathclyde in 1986 and was promoted to a Professorship in 1995. A Fellow of the Royal Society of Edinburgh (FRSE), he has a long held fascination with polar organometallic chemistry, especially with the synergies at work when different metals are mixed within the same ligand environment. |
Introduction
Avant Garde metallation: mixing the palettes of Grignard and Schlenk
s-Block organometallic chemistry has attracted over one hundred years of continuous study since the pioneering works of the European masters, Grignard in organomagnesium chemistry (in recognition of his development of the halides “RMgX”, which now are popularly referred to as Grignard reagents)1 and Schlenk in organolithium chemistry2 (alkyl- and aryl-lithium compounds could similarly be called Schlenk reagents but the community has never adopted this terminology). The latter compounds have always been superior in metallation chemistry due to their higher reactivity, which transforms relatively inert, relatively non-polar carbon–hydrogen bonds into more reactive, more polar carbon–metal bonds that in turn can be used in subsequent carbon–carbon or carbon–heteroatom bond formation. For about 50 years, the art of metallation has been dominated by alkyl lithium compounds3 (most commonly, n-butyllithium) and sterically inflated lithium amides4 (inflated by branching near the amido nitrogen as in the three most popular examples, lithium diisopropylamide, LDA; lithium 2,2,6,6-tetramethylpiperidide, LiTMP; and lithium 1,1,1,3,3,3-hexamethyldisilazide, LiHMDS). However, contemporary metallation developing continually over the past decade has begun to challenge this dominance by taking these single-component basic compounds in combination with other components to create new metallating collages (or cocomplexes) that can often outperform the single-component compounds. Arguably, the Lochmann–Schlosser superbase, “BunLi·KOBut”,5 a combination of n-butyllithium and potassium t-butoxide was the prototype for the design of these multicomponent bases though with the odd exception (notably the excellent unimetallic superbase work of Caubere6), this idea of mixing components to create improved metallating reagents never caught on to any significant extent for a long time. However, it is now well recognised (though perhaps not well understood) that co-operative effects between two distinct metals, or more accurately between the multiple components (metals, anions, solvent ligands) that can co-exist on mixing distinct compounds together, can induce novel reactivities.7 Several research groups in exploiting this idea have made important advances in metallation chemistry. Among them, Knochel has developed turbo-Grignard reagents with empirical formulas such as “(TMP)MgCl·LiCl”,8 obviously a mixture of the amidomagnesium halide (TMP)MgCl (sometimes referred to as a Hauser base9) and the turbo-charging ingredient the ionic salt LiCl. Uchiyama and Kondo mixed the lithium amide LiTMP with the alkyl zinc compound But2Zn (prepared via ButLi and ZnCl2) to generate the zincate metallator “LiZn(But)2TMP”.10 The same metathesis approach has been used by Mongin in preparing all-amido modifications such as the cadmate “LiCd(TMP)3”11 by reacting the lithium amide with cadmium chloride in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. All of these multicomponent mixtures have in common a lithium chloride component and THF as solvent so the full picture of the co-operativity at work may involve both of these compounds though details of the active metallating species remain sketchy. Our own group have investigated different multicomponent systems though the bulk of research has focused on the sodium TMP-zincate [(TMEDA)Na(TMP)(But)Zn(But)]12 which, uniquely in comparison to those systems mentioned previously, has been generally utilised in hexane solvent. Several recent reviews13 covering both our and these related systems have surveyed their applications in deprotonative metallation of aromatic and heteroaromatic substrates. The excellent latest review14 by Knochel gives an outline of the chronological development of these multicomponent metallators and details in full the chemistry of his own extensively studied salt-supported organometallic reagents. There is no need to retrace old ground here. Instead, inspired by a paper in Nature Chemistry from 2010,15 this article brings a brand new perspective to the area of alkali-metal-mediated metallation (AMMM) chemistry by considering (some of) these processes as “cleave and capture” reactions. As this is a new emerging idea, this opening portrait of cleave and capture reactions within mixed-metal chemistry is restricted mainly to relevant work in our laboratory or that of Hevia (both within the University of Strathclyde), that has appeared since about 2008. However, it may emerge that this concept could equally apply to a gallery of other AMMM reactions reported by different research groups.
:1 stoichiometry. All of these multicomponent mixtures have in common a lithium chloride component and THF as solvent so the full picture of the co-operativity at work may involve both of these compounds though details of the active metallating species remain sketchy. Our own group have investigated different multicomponent systems though the bulk of research has focused on the sodium TMP-zincate [(TMEDA)Na(TMP)(But)Zn(But)]12 which, uniquely in comparison to those systems mentioned previously, has been generally utilised in hexane solvent. Several recent reviews13 covering both our and these related systems have surveyed their applications in deprotonative metallation of aromatic and heteroaromatic substrates. The excellent latest review14 by Knochel gives an outline of the chronological development of these multicomponent metallators and details in full the chemistry of his own extensively studied salt-supported organometallic reagents. There is no need to retrace old ground here. Instead, inspired by a paper in Nature Chemistry from 2010,15 this article brings a brand new perspective to the area of alkali-metal-mediated metallation (AMMM) chemistry by considering (some of) these processes as “cleave and capture” reactions. As this is a new emerging idea, this opening portrait of cleave and capture reactions within mixed-metal chemistry is restricted mainly to relevant work in our laboratory or that of Hevia (both within the University of Strathclyde), that has appeared since about 2008. However, it may emerge that this concept could equally apply to a gallery of other AMMM reactions reported by different research groups.
Discussion
The basis for cleave and capture
The multicomponent compositions of the new genre of metallating agents offer several Lewis acidic and Lewis basic enticements for attracting substrate molecules. Taking the widely studied sodium TMP-zincate, [(TMEDA)·Na(TMP)(But)Zn(But)] (Fig. 1), as a typical example, it contains two Lewis acidic sites and five Lewis basic sites, and within these sites there are choices as they belong to two different metal cations, two different anions, and a neutral bidentate donor ligand. Aside from enticing the substrate molecule to take part in a reaction, the metallator with its multi Lewis character is well equipped to capture the resulting anion following the reaction.
![Molecular structure of [(TMEDA)·Na(TMP)(But)Zn(But)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f1.gif) |
| | Fig. 1 Molecular structure of [(TMEDA)·Na(TMP)(But)Zn(But)]. | |
Small atom/molecule capture
Capture of hydride.
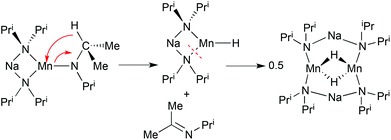
An interesting variation of this phenomenon is found with mixed-metal reagents based on the diisopropylamide (Pri2N−; DA) ligand. Lithium diisopropylamide [(LiNPri2)n; LDA] has dominated metallation chemistry for the past 30 or so years but arguably now has been overtaken by the new genre of complex TMP bases. Sterically more imposing TMP is a stronger Brønsted base than DA as evidenced by a comparison of pKa data for LiTMP (37.9)16 and LDA (34.4)16 though what these values are when the amides are part of a multicomponent assembly is more uncertain. One significant drawback of DA not applicable to the TMP system is the presence of β-hydrogen atoms. Delocalisation of the negative charge residing on the amido N can generate an imido C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond with concomitant elimination of a β-hydrogen as a hydride ion. Attempts to synthesise donor-free sodium tris(diisopropylamido) manganate [NaMn(NPri2)3] in a hexane/toluene solvent mixture promoted this process but following the scenario outlined above the eliminated hydride is captured by a fragment of the intended mixed metal amide that has lost one DA group.17 A balanced equation (Scheme 1) can be written for this novel hydride capture process where the byproduct is the known ketimine Me2CNPri (N-(propan-2-ylidene)propan-2-amine). The hydride capture product [Na2Mn2(μ-H)2(NPri2)4]·2toluene provides a beautiful example of molecular aesthetics.18 Viewing the structure (Fig. 2) the lay person may imagine a suit of armour or a robot from a science fiction movie. To the chemist it is a centrosymmetric structure of C2h symmetry with an eight atom metal–nitrogen ring in which sodium and manganese(II) atoms alternate. This chair-shaped (Na2Mn2N4) ring carries a 2+ charge which is balanced by two hydride ions that straddle the midpoint of the structure in a [Mn(μ-H)2Mn] double bridge. Unlike the manganese atoms, the sodium atoms do not bond with the hydride ions but complete their coordination requirements by forming contacts with the π-framework of toluene solvent molecules. The promise of this method as a hydride capture strategy is apparent by the fact that analogous reactions have been reported for sodium magnesiate and potassium magnesiate systems leading to the formation of [M2Mg2(μ-H)2(NPri2)4]·2toluene (where M = Na or K).19 Because of their topological relationship to conventional crown ether complexes, we refer to these isostructural hydride capture products as hydrido “inverse crowns”. To elaborate, the eight-atom (MNM′N)2 rings can be considered “hosts” to the hydride ion “guests” located in the hollow of the ring, akin to polyether rings hosting alkali metal cations in conventional crown ether complexes. The qualification “inverse” derives from the mutual interchange of Lewis acidic (alkali metal, divalent metal) and Lewis basic (hydrido) sites compared to those (alkali metal and oxygen atom, respectively) in conventional crown ether complexes. However it should be stressed that this is merely an aesthetic description which does not imply anything about the mechanism of formation of hydrido inverse crowns. It is highly likely that their structures are constructed via dimerization of short-lived monomeric species [e.g., Na(NPri2)2Mn(H)] as opposed to via preformed cationic rings [e.g., {Na2Mn2(NPri2)4}2+] capturing hydride ions. Hydride elimination is induced thermally in all of these examples by heating the reaction solution to reflux temperatures. At lower temperatures heterobimetallic tris-diisopropylamides are stable with (e.g., in [TMEDA)·NaMg(NPri2)3]) or without (e.g., in [{KMg(NPri2)3}∞]) auxiliary donor support.20 The novelty in this particular form of hydride capture lies in the lack of ionic hydride formation especially in the presence of highly electropositive s-block metals. Thermodynamically there is a strong compulsion to form [(M2+2H−)∞] or [(M+H−)∞] ionic salts due to their high lattice energies but their formation is suppressed here by the steric entrapment of the bulky [Na(NPri2)2] assemblies that shield opposite sides of the (Mn2H2) or (Mg2H2) ring (Fig. 3).
N bond with concomitant elimination of a β-hydrogen as a hydride ion. Attempts to synthesise donor-free sodium tris(diisopropylamido) manganate [NaMn(NPri2)3] in a hexane/toluene solvent mixture promoted this process but following the scenario outlined above the eliminated hydride is captured by a fragment of the intended mixed metal amide that has lost one DA group.17 A balanced equation (Scheme 1) can be written for this novel hydride capture process where the byproduct is the known ketimine Me2CNPri (N-(propan-2-ylidene)propan-2-amine). The hydride capture product [Na2Mn2(μ-H)2(NPri2)4]·2toluene provides a beautiful example of molecular aesthetics.18 Viewing the structure (Fig. 2) the lay person may imagine a suit of armour or a robot from a science fiction movie. To the chemist it is a centrosymmetric structure of C2h symmetry with an eight atom metal–nitrogen ring in which sodium and manganese(II) atoms alternate. This chair-shaped (Na2Mn2N4) ring carries a 2+ charge which is balanced by two hydride ions that straddle the midpoint of the structure in a [Mn(μ-H)2Mn] double bridge. Unlike the manganese atoms, the sodium atoms do not bond with the hydride ions but complete their coordination requirements by forming contacts with the π-framework of toluene solvent molecules. The promise of this method as a hydride capture strategy is apparent by the fact that analogous reactions have been reported for sodium magnesiate and potassium magnesiate systems leading to the formation of [M2Mg2(μ-H)2(NPri2)4]·2toluene (where M = Na or K).19 Because of their topological relationship to conventional crown ether complexes, we refer to these isostructural hydride capture products as hydrido “inverse crowns”. To elaborate, the eight-atom (MNM′N)2 rings can be considered “hosts” to the hydride ion “guests” located in the hollow of the ring, akin to polyether rings hosting alkali metal cations in conventional crown ether complexes. The qualification “inverse” derives from the mutual interchange of Lewis acidic (alkali metal, divalent metal) and Lewis basic (hydrido) sites compared to those (alkali metal and oxygen atom, respectively) in conventional crown ether complexes. However it should be stressed that this is merely an aesthetic description which does not imply anything about the mechanism of formation of hydrido inverse crowns. It is highly likely that their structures are constructed via dimerization of short-lived monomeric species [e.g., Na(NPri2)2Mn(H)] as opposed to via preformed cationic rings [e.g., {Na2Mn2(NPri2)4}2+] capturing hydride ions. Hydride elimination is induced thermally in all of these examples by heating the reaction solution to reflux temperatures. At lower temperatures heterobimetallic tris-diisopropylamides are stable with (e.g., in [TMEDA)·NaMg(NPri2)3]) or without (e.g., in [{KMg(NPri2)3}∞]) auxiliary donor support.20 The novelty in this particular form of hydride capture lies in the lack of ionic hydride formation especially in the presence of highly electropositive s-block metals. Thermodynamically there is a strong compulsion to form [(M2+2H−)∞] or [(M+H−)∞] ionic salts due to their high lattice energies but their formation is suppressed here by the steric entrapment of the bulky [Na(NPri2)2] assemblies that shield opposite sides of the (Mn2H2) or (Mg2H2) ring (Fig. 3).
 |
| | Scheme 1 Hydride capture via cleavage of a metal attached diisopropylamide ligand. Solvating toluene molecules are omitted. | |
![Molecular structure of the hydrido inverse crown [Na2Mn2(μ-H)2(NPri2)4]·2toluene.](/image/article/2013/DT/c3dt00053b/c3dt00053b-f2.gif) |
| | Fig. 2 Molecular structure of the hydrido inverse crown [Na2Mn2(μ-H)2(NPri2)4]·2toluene. | |
![Alternative view of [Na2Mn2(μ-H)2(NPri2)4]·2toluene showing the shielding of the (Mn2H2) ring.](/image/article/2013/DT/c3dt00053b/c3dt00053b-f3.gif) |
| | Fig. 3 Alternative view of [Na2Mn2(μ-H)2(NPri2)4]·2toluene showing the shielding of the (Mn2H2) ring. | |
Molecular carriers that can store, release and restore hydrogen are currently in high demand for technological innovations.21c Though these have synthetic possibilities (e.g., as novel reducing agents), interest is driven mainly by the potential of hydrogen to become the World's future generations energy resource, especially in mobile appliances such as motor cars. Lightweight molecules with high hydrogen content meet several requirements of an ideal hydrogen storage material. Therefore molecular s-block metal hydrides are particularly sought after if not as viable hydrogen storage materials themselves but as model systems towards developing such materials. While the hydrido inverse crown complexes described herein have too low hydrogen content (Harder has recently reported a β-diketiminate supported magnesium hydride cluster with ten hydride ions21a,b) for storage applications, the principle that co-operative mixed metal effects can be exploited for hydride capture is now established. The next challenge to the synthetic chemist is to refine this principle to build larger, more hydrogen rich materials.
Co-operative mixed metal effects have also been instrumental in transforming saturated diamines (R)NHCH2CH2NH(R) into unsaturated diazaethenes (R)NCHCHN(R) in a process thought to involve an intermediate hydride species.22 Subjecting a diamine (R = Pri) to a mixture of butyllithium and di-t-butylzinc leads to a dimetallated diamide which in turn releases H2 to generate the final diazaethene product (Scheme 2). DFT calculations show that lithium and zinc work co-operatively to develop a zinc-hydride intermediate: the former stabilises the breaking Zn–N bond, while the latter stabilises the making Zn–H bond which in turn proceeds to deprotonate the allylic CH2 on the diamine backbone to ultimately produce an NCHCHN unit and H2. Such multiple C–H/N–H bond activation is usually confined to redox active metals such as transition metals which can engage in oxidative addition and reductive elimination processes,23 but this work shows that a similar chemistry is possible with s-reactive metals through metal–ligand–metal′ co-operativity. Introducing an equivalent of n-butyllithium·TMEDA to the monolithiated diamine/di-t-butylzinc cocomplex in an attempt to lithiate the remaining N–H bond induces β-hydride elimination from a But group releasing a hydride ion and isobutene (Scheme 3).22 This hydride ion was captured in the molecular dilithium zincate species [{(TMEDA)·Li}2{(Pri)NCH2CH2N(Pri)}Zn(But)H] that could be isolated and crystallographically characterised (Fig. 4). The hydride capturing occurs via a Li2Zn triangle though the Zn⋯H bond is rather long, while the shorter Li–H bonds are asymmetrical in length. NMR spectroscopic experiments confirmed the retention of the Li–H bond (a 1JLi–H coupling of 13.3 Hz was detected). They also suggested the structure is dynamic with LiH dissociating from and reassociating with the monolithium zincate [(TMEDA)Li{(Pri)NCH2CH2NH(Pri)}Zn(But)2] thus prompting the description of the latter as a “molecular scaffold for the molecularisation of the usually insoluble polymeric LiH.” This follows as the dilithium hydridozincate exhibits good solubility in hexane.
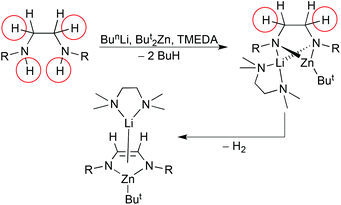 |
| | Scheme 2 Fourfold C–H/N–H bond activation of a diamine to a diazaethene via Li/Zn co-operativity. | |
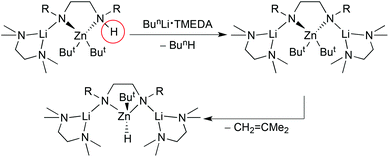 |
| | Scheme 3 Lithium zinc co-operative capture of a hydride ion generated by β-elimination from a t-butyl ligand. | |
![Molecular structure of the dilithium hydridozincate [{(TMEDA)·Li}2{(Pri)NCH2CH2N(Pri)}Zn(But)H].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f4.gif) |
| | Fig. 4 Molecular structure of the dilithium hydridozincate [{(TMEDA)·Li}2{(Pri)NCH2CH2N(Pri)}Zn(But)H]. | |
Capture of different oxygen-based anions
In mixed-metal chemistry the designation “inverse crown” was first coined for inverse crown “ethers” (ICEs) because captured oxygen-based anions filled their ring cavities. Early ICE examples combining alkali metals with either magnesium or zinc have been reviewed.24 Reported in 200825 were the first alkali metal–manganese(II) ICEs, [Li2Mn2(TMP)4O] and [Na2Mn2(HMDS)4O] (HMDS = 1,1,1,3,3,3-hexamethyldisilazide), the latter of which provided an important clue to a possible mechanism involved in inverse crown formation. Having a square-like architecture with metal atoms bisecting its N⋯N edges, the lithium–manganese ICE resembles the tetrameric structure of homometallic LiTMP26 but in addition the ICE complex has a captured oxide (O2−) ion at its core (Fig. 5). The molecular structure of [Na2Mn2(HMDS)4O] exhibits the same basic motif which appears to result from a redox reaction (Scheme 4). Evidence came from a combination of NMR and mass spectroscopic studies which identified the oxidatively coupled product Me3SiCH2CH2SiMe3, made from coupling two Me3SiCH2− anions, with the other half reaction reducing dry aerobic oxygen to oxide, which ends up captured in the ICE complex. Similar redox mechanisms may be operative in the ICE producing reactions of magnesium or zinc, through the possibility of competing mechanisms perhaps involving adventitious traces of water cannot be dismissed.
![Empty [(LiTMP)4] and filled (inverse crown, [Li2Mn2(TMP)4O]) cyclotetrameric metal amides.](/image/article/2013/DT/c3dt00053b/c3dt00053b-f5.gif) |
| | Fig. 5 Empty [(LiTMP)4] and filled (inverse crown, [Li2Mn2(TMP)4O]) cyclotetrameric metal amides. | |
![Proposed redox pathway for the formation of inverse crown ether [Na2Mn2(HMDS)4O].](/image/article/2013/DT/c3dt00053b/c3dt00053b-s4.gif) |
| | Scheme 4 Proposed redox pathway for the formation of inverse crown ether [Na2Mn2(HMDS)4O]. | |
Matching the general landscape of polar organometallic chemistry, the oxygen chemistry of these mixed-metal systems is extraordinarily complicated. Reactions can often be unpredictable and unplanned. Adventitious inclusion of oxygen or water can lead to serendipitous products that, in suitably crystalline cases, are often discovered in the first instance via X-ray crystallography. The extent of inclusion can also have a profound influence on the outcome. It is significant that the amide:oxide stoichiometry in the aforementioned ICE complexes is 4:1 even though in some cases excess oxygen was passed through the reaction solution. This hints that ICE complexes could be inert to further oxygen exposure preventing complete oxidation due to the substantial kinetic protection offered by the set of bulky TMP or HMDS ligands. A markedly contrasting 1:1, amide:oxide stoichiometric ratio is encountered in two isostructural “super” ICE complexes of common formula [{NaMg(NR2)(O)(THF)}6] (where NR2 = TMP or DA).27 Possessing S6 symmetry, their common structure could be regarded as a form of magnesium oxide [(MgO)∞: note this has a face-centred cubic structure of the sodium chloride type] which has been transformed into a discrete molecular hexagonal block by the attachment of bulky THF-solvated sodium amide monomers onto the edges of the top and bottom (MgO)3 faces, with those on the top alternating with those on the bottom (Fig. 6). The presence of THF was the most significant difference between the syntheses of these super ICEs and their 1:1 variants. Mass spectroscopic detection of ethene from the reaction mixture points suspicion at THF being the oxide source, consistent with other THF-cleavage reactions known to generate ethene and oxide amongst a host of different decomposition products. Hard organolithium bases generally deprotonate THF at the α-position, inducing a [π4s + π2s] cycloreversion to release ethene and an enolate “CH2CH–O−”. Whereas a redox mechanism is implicated in the formation of 1:1 ICEs, the participation of THF suggests that a metallation mechanism is more likely in the case of super ICEs. The loss of two Brønsted basic components from the original mixed metal mixture could be indicative of a twofold metallation of THF (there is precedence for a mixed metal multicomponent reagent acting as a di-base to effect a twofold metallation of the same substrate28), but given the low super ICE yields obtained (less than 20%) it is conceivable that several competitive processes are in operation leading to other as yet unidentified products.
![Molecular structure of the super ICE complex [{NaMg(DA)(O)(THF)}6].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f6.gif) |
| | Fig. 6 Molecular structure of the super ICE complex [{NaMg(DA)(O)(THF)}6]. | |
Spectacular related reactions emphasise further the complexity inherent in THF cleavage, especially when the composition of the mixed metal reagent is altered even in a seemingly small way. Oxide is captured in the ICE complex, [Na2Mg2(TMP)4O] – previously made via an oxygen exposure route in the absence of THF – when THF is cleaved by [(TMEDA)Na(TMP)(CH2SiMe3)Mg(TMP)] (Scheme 5).15 Only the presence of TMEDA and most significantly the exchange of an n-butyl ligand for the silylalkyl Me3SiCH2 distinguishes this mixed metal reagent from that which yielded the super ICE complex and ethene. This time, however, the carbon product of the THF cleavage could also be captured in a bimetallic trap which proved isolable. This surprising product was the 1,4-dimagnesiated butadiene [{(TMEDA)Na(μ-TMP)}2{1,4-[Mg(TMP)]2-C4H4}]. Metal entrapment of the s-trans-buta-1,3-diene fragment occurs via a combination of Mg–C σ bonds at the terminal sites and Na⋯CC π-type contacts. A new feature for a diene system, this synergic bonding is a recurring feature in metallated aromatic or alkene products where both the metallating metal (e.g., Mg) and alkali metal are retained in the structure. Since the mixed metal reagent residues located at the butadiene chain ends are each missing a Me3SiCH2− ligand this could be indicative of two monometallation reactions of alkyl basicity (at least ultimately) with concomitant evolution of Me4Si. Remarkably, six of the thirteen bonds in THF (four C–H and two C–O bonds) are cleaved in the reaction generating these ICE and dimagnesiated butadiene co-products, so if such metallations are occurring they must be accompanied by other events in what is clearly a complicated cascade process. The mechanism is yet to be elucidated. Significantly, exactly the same process is seen on substituting magnesium by the d-block metal manganese (in its +2 oxidation state) to form isostructural sodium-manganese ICE and 1,4-dimanganated butadiene complexes.15 Interestingly, the same 1,4-dimagnesiated butadiene product is observed when [(TMEDA)Na(TMP)(CH2SiMe3)Mg(TMP)] reacts with the sulfur homologue tetrahydrothiophene (THT; SC4H8), though the identity of the sulfur-containing byproduct (cf., the ICE complex in the THF cleavage reaction) could not be established.29 The cleave and capture reaction is noticeably slower (days instead of hours) with the larger heteroatom cycle, allowing detection of a possible intermediate on the reaction coordinate, [(TMEDA)Na(TMP)(C4H7S)Mg(TMP)], in which a mono-α-deprotonated THT molecule has been captured by the base residue (again the starting base minus the alkyl ligand). Transforming a 5-membered heterocycle to a 1,3-butadiene derivative is therefore a favourable reaction not dependent on the particular heteroatom or a particular combination of metals. We will return to the eccentricities of THF cleavage later in the article.
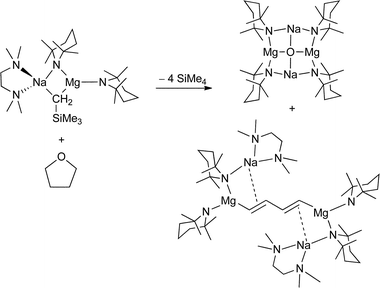 |
| | Scheme 5 Catastrophic cleavage of THF via a sodium TMP-magnesiate reagent. | |
Providing an interesting contrast with the hexameric magnesium oxide super ICEs, the zincate [(TMEDA)·Na(TMP)(But)Zn(But)] reacts with N,N-dimethylbenzylamine in the presence of an oxygen source to afford the zinc-rich dimer [{(TMEDA)·Na(TMP)Zn(But)Zn(O)But}2] (Scheme 6).30 Formally one molecule of zinc oxide (ZnO) has inserted into one Zn–C(But) bond of the zincate reagent and the structure then dimerises through a planar (ZnO)2 ring (Fig. 7). The source of the captured oxide could not be traced and no evidence was found to suggest the diamine was involved in this reaction. Other literature examples of bimetallic alkali metal–zinc compounds do not exhibit the same coordination framework nor is the 2:4, alkali metal:zinc stoichiometry usually observed (the most common such stoichiometries are 1:1 or 2:1, sometimes referred to as lower order and higher order zincates respectively). While this synthesis has not yet been reproduced, the existence of [{(TMEDA)·Na(TMP)Zn(But)Zn(O)But}2] opens up the possibility of capturing molecular fragments of zinc salts through multicomponent organometallic devices, thus setting another challenge to synthetic chemists.
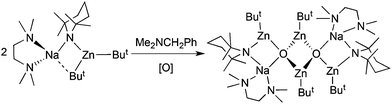 |
| | Scheme 6 Adventitious capture of oxide ions by sodium TMP-zincate. | |
![Molecular structure of the zinc-rich oxide containing complex [{(TMEDA)·Na(TMP)Zn(But)Zn(O)But}2].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f7.gif) |
| | Fig. 7 Molecular structure of the zinc-rich oxide containing complex [{(TMEDA)·Na(TMP)Zn(But)Zn(O)But}2]. | |
It is not uncommon for oxygen inclusion into organometallic compounds to be manifested in alkoxide (RO−) ligands. A nice example producing a rare polymeric ICE complex was obtained from deliberately exposing the trialkylmagnesiate [NaMg(CH2SiMe3)3] to oxygen in a hexane/benzene solvent mixture.31 Formulated as [{NaMg(OCH2SiMe3)(CH2SiMe3)2}∞], the polymer could be regarded as a co-complex between the homometallic alkoxide (NaOCH2SiMe3) and alkyl [Mg(CH2SiMe3)2] and thus could be viewed as an alkaline earth metal relation of the classic Lochmann–Schlosser, alkoxide–alkyl superbase (BunLi·KOBut), itself a co-complex between n-butyllithium and potassium t-butoxide. Mystery still surrounds the structure of (BunLi·KOBut) though it has served synthetic chemists for nearly fifty years.32 The structures of related compositions could hold clues towards solving this mystery. Possessing an eight-atom (NaCMgC)2 host ring that traps two alkoxide ligands O-bound to both metal centres, the ICE structure propagates through Na⋯Me(SiMe2) interactions to form a 2-dimensional infinite sheet arrangement (Fig. 8).
![Two-dimensional infinite sheet structure of [{NaMg(OCH2SiMe3)(CH2SiMe3)2}∞].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f8.gif) |
| | Fig. 8 Two-dimensional infinite sheet structure of [{NaMg(OCH2SiMe3)(CH2SiMe3)2}∞]. | |
Sensitive anion capture: synergic sedation
So far the article has dealt mainly with the capture of small atoms or molecules that have either been cleaved off an organic substrate or sourced from elemental oxygen or possibly moisture. As alluded to earlier, following a C–H deprotonation by the mixed-metal reagent, the residue of the base with its multi Lewis acidic metal grips is capable of capturing the resulting anion fully intact. This can be particularly advantageous when dealing with a sensitive anion such as that derived from an ether, of potential use in synthesis. Through the co-operative combination of different metal–ligand bonding, anions of this type can be stabilised as the following example illustrates.
THF, the best known most widely utilised cyclic ether, provides an excellent polar solvent for running reactions of polar organometallic compounds. Its usage applies to the chemistry of both the older established organolithium and Grignard reagents and the newer complex metallators of for example Knochel (e.g., TMPMgCl·LiCl; TMP2Mg·2LiCl; TMPZnCl·LiCl; TMP2Zn·2LiCl),14 Uchiyama/Kondo (But2ZnTMPLi, Bui3AlTMPLi),10,33 and Mongin (“LiZnTMP3”; “LiCdTMP3”).11,34 Organolithium reagents love affair with THF is based on more than good solubility properties. Often such reagents are activated by engaging with this Lewis base because its making of Li–O dative bonds is concomitant with the breaking of Li–C (or e.g., Li–N) aggregation bonds, leading to smaller, more reactive entities containing an RLi or R2NLi fragment as opposed to the full (RLi)n or (R2NLi)n aggregate. However, as in all relationships compromises must be made. This enhanced reactivity can quickly turn betrothal into betrayal with the hard organolithium base attacking THF by metallation (usually at the α-C atom) leading to a build up of negative charge next to the electron rich O atom with the inevitability of the relationship breaking down through ring opening of the heterocycle. The final decomposition products are condition dependent, but as mentioned before a [3 + 2] cycloreversion process generating ethene and the enolate of acetaldehyde is common.35 This process can usually only be slowed down by resorting to subambient temperatures (typically −78 °C). In striking contrast, using a mild AMMZn (alkali-metal-mediated zincation) reagent, a remarkable controlled ambient temperature metallation is possible. Remember that with the magnesiate or manganate reagent [(TMEDA)Na(TMP)(CH2SiMe3)M(TMP)] (M = Mg or Mn), THF is catastrophically cleaved into ICE and dimetallated butadiene products. However, the closely related zincate complex [(TMEDA)Na(TMP(CH2SiMe3)Zn(CH2SiMe3)] selectively deprotonates THF at the α-position and the reaction stops at that stage to produce the heterotrianionic complex [(TMEDA)Na(TMP)(C4H7O)Zn(CH2SiMe3)] (Scheme 7).36 Seemingly despite having gained a formal negative charge at its α-C atom, the hydrogen deficient C4H7O ring is sedated by its two metal grips through Zn–C and Na–O bonding (Fig. 9). Since the sodium–magnesium and sodium–manganese bases also have the capacity to capture the THF α-anion in a bimetallic grip, the identity of the divalent metal and specifically the stability of the metal–αC bond must be one kinetic key to the retention or opening of the heterocyclic ring. Zinc–carbon bonds have a higher degree of covalency than those of magnesium or manganese which would impart a greater stability on the sodium–zinc captured C4H7O ring, whereas the higher degree of ionicity of the two more electropositive divalent metals appears to incite ring opening of THF and subsequent bond breaking processes.
![Cleavage and capture of the THF α-anion via the sodium TMP-zincate, [(TMEDA)Na(TMP)Zn(CH2SiMe3)2].](/image/article/2013/DT/c3dt00053b/c3dt00053b-s7.gif) |
| | Scheme 7 Cleavage and capture of the THF α-anion via the sodium TMP-zincate, [(TMEDA)Na(TMP)Zn(CH2SiMe3)2]. | |
![Molecular structure of the sedated THF α-anion complex [(TMEDA)Na(TMP)(C4H7O)Zn(CH2SiMe3)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f9.gif) |
| | Fig. 9 Molecular structure of the sedated THF α-anion complex [(TMEDA)Na(TMP)(C4H7O)Zn(CH2SiMe3)]. | |
Further observations underscore the importance of the mixed-metal partnership in this chemistry. Though the sodium–zinc partnership gives access to a fully intact cycloanion of THF normally inaccessible to conventional metallating agents (e.g., alkyllithiums) under comparable conditions, the reaction producing it is inefficient requiring neat THF and a two-week stir to obtain a best yield of only about 52%.36 What a difference it can make switching from a sodium–zinc partnership to a lithium–aluminium partnership. To explain, exposing an equimolar amount of THF to the aluminate base “[(THF)Li(TMP)2Al(Bui)2]”, made in situ in hexane solution from a 2:2:1:1 mixture of BunLi, TMP(H), Bui2AlCl and THF, also cleaves and captures THF as a cycloanion, this time within [(THF)Li(TMP)(C4H7O)Al(Bui)2] (Scheme 8).37 This reaction is much faster (hours not days), stoichiometric in THF, and all the components of the base come in commercial bottle form unlike the BunNa and Zn(CH2SiMe3)2 precursors to the zincate base [(TMEDA)Na(TMP)(CH2SiMe3)Zn(CH2SiMe3)] which require to be pre-made. Trivalent aluminium is generally considered to form bonds to carbon of a similar polar covalency to magnesium–carbon bonds, making them more reactive than covalent zinc–carbon bonds, so one could have expected the aluminated THF to follow more the chemistry of magnesiated THF than zincated THF. However, other factors must contribute, notably the greater steric protection given by the four-coordinate Al (versus three-coordinate Mg) and the stronger grip on the O atom by Li (versus Na) reflected by a shorter Li–O bond (than Na–O bond). Furthermore, in general, the initial products of aluminate cleave and capture are more stable than either of their magnesiate or zincate counterparts. In explanation, whereas all three ate bases execute deprotonation via TMP basicity, generating a deprotonated substrate and TMP(H), in the magnesiate and zincate cases the reaction does not stop there but in a second step TMP(H) is deprotonated by the initial bimetallic product to reform an alkali metal–TMP–Mg (or Zn) bridge and eliminating irreversibly an alkane or silane (Scheme 9).38 Coordinative saturation (for Al, c.n. = 4) versus unsaturation (for Mg and Zn, c.n. = 3) is thought to be a decisive factor in this distinction. At the time of writing there is uncertainty as to the precise nature of the TMP-enriched base “[(THF)Li(TMP)2Al(Bui)2]” (a spin-off from Uchiyama/Kondo's mono-TMP base “Bui3AlTMPLi”), whether it exists as a single or mixed species.39 However its modus operandi in cleave and capture reactions seems clear. Deprotonation of its prey is accomplished by one TMP ligand with the resulting anion captured by the cationic [(THF)Li(TMP)Al(Bui)2]+ residue. Tetrahydrothiophene can be similarly aluminated and its resulting anionic form synergically sedated by the same base and captured in [(THF)Li(TMP)(C4H7S)Al(Bui)2] with the base residue chelating the heterocycle through Li–S and Al–C bonds (Fig. 10).37
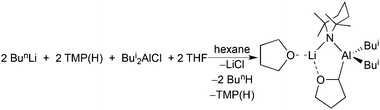 |
| | Scheme 8 Generation and capture of the α-cycloanion of THF via alkali-metal-mediated alumination. | |
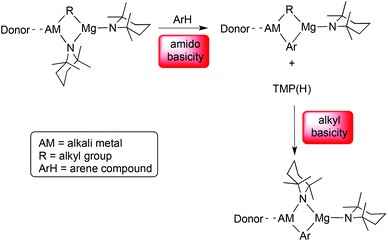 |
| | Scheme 9 Two-step deprotonation process in alkali-metal-mediated magnesiation via TMP-magnesiates. | |
![Molecular structure of the aluminated tetrahydrothiophene complex [(THF)Li(TMP)(C4H7S)Al(Bui)2].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f10.gif) |
| | Fig. 10 Molecular structure of the aluminated tetrahydrothiophene complex [(THF)Li(TMP)(C4H7S)Al(Bui)2]. | |
Another cyclic ether that has participated in cleave and capture chemistry is 1,4-dioxane, (O2C4H8). It was transformed to the alkoxy vinyl ether, CH2C(H)OCH2CH2O−, by reaction with the benzamide-ligated lithium aluminate [{PhC(O)NPri2}Li(TMP)(Bui)Al(Bui)2].40 This acyclic dioxane fragment as well as another molecule of intact dioxane was captured in the isolated product [{[PhC(O)NPri2]Li[O(CH2)2OC(H)CH2]Al(Bui)3}2(1,4-dioxane)] (Fig. 11). The mechanism proposed for this reaction was a stepwise process in which one dioxane molecule has a C–H bond cleaved by TMP to generate a C–Al bond while maintaining an O–Li dative bond, followed by a ring opening of the heterocyclic ring, and finally the new alkoxy nucleophile attacks the Al electrophile to construct a Li–O–Al bridge (Scheme 10). While appearing passive in this stepwise process, the benzamide which acts throughout as a carbonyl–O Lewis base towards Li proved essential for the dioxane ring opening. In the absence of the benzamide no ring opening was detected, instead a ligand distribution reaction appears to take place (Scheme 11) as suggested by isolation of the solvent (dioxane)-separated ion pair product [{(dioxane)4Li}+{Al(Bui)4}−], which could be regarded as a segregATE.
![Molecular structure of the aluminated dioxane complex [[PhC(O)NPri2]Li[O(CH2)2OC(H)CH2]Al(Bui)Al(Bui)2].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f11.gif) |
| | Fig. 11 Molecular structure of the aluminated dioxane complex [[PhC(O)NPri2]Li[O(CH2)2OC(H)CH2]Al(Bui)Al(Bui)2]. | |
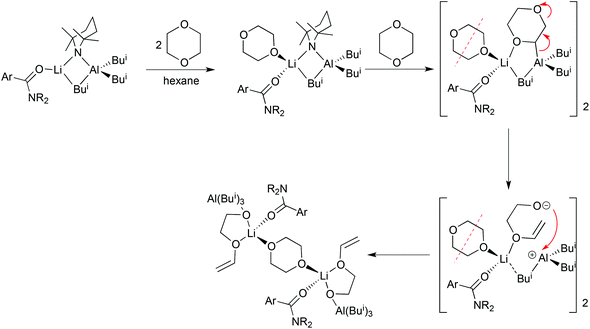 |
| | Scheme 10 Proposed pathway for formation of an aluminated ring opened dioxane complex. | |
![Dioxane-induced ligand redistribution of the monoamido-trialkylaluminate [Li(TMP)(Bui)Al(Bui)2].](/image/article/2013/DT/c3dt00053b/c3dt00053b-s11.gif) |
| | Scheme 11 Dioxane-induced ligand redistribution of the monoamido-trialkylaluminate [Li(TMP)(Bui)Al(Bui)2]. | |
Potassium lags well behind the other common Group 1 metals lithium and sodium in its utilisation in AMMM chemistry. However a glimpse of its vast potential has been caught in a reaction with ethene,36 which also underlines the differences between conventional organoalkali metal reagents and the new mixed-metal genre. Carbometallation, nucleophilic addition across the CC double bond, is common when this smallest alkene is treated with an alkyllithium reagent such as MeLi or BunLi (Scheme 12).41 Lack of steric hindrance and the relative non-acidity of the unactivated sp2C–H bonds favour this reaction over deprotonation. Proving that the new genre can be stronger bases less susceptible to nucleophilic complications, the potassium zincate [(PMDETA)K(TMP)(Me3SiCH2)Zn(Me3SiCH2)] smoothly metallates ethene at 50 °C in hexane solution, capturing the resulting vinyl anion in the potassium heterotrianionic zincate [(PMDETA)K(TMP)(CHCH2)Zn(Me3SiCH2)] (Scheme 12). Stabilised by three metal grips, the vinyl anion forms a σ-bond to zinc through the deprotonated C atom and an asymmetrical π-type interaction with potassium through its C2 unit (Fig. 12). In comparison with vinyllithium which is often utilised as a source of CH2CH−, this bimetallic vinyl complex has the significant advantages of good hydrocarbon solubility and non-pyrophoricity.
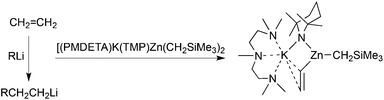 |
| | Scheme 12 Contrasting reactions of ethene with organolithium and potassium TMP-zincate reagents. | |
![Molecular structure of the vinyl captured bimetallic complex [(PMDETA)K(TMP)(CHCH2)Zn(Me3SiCH2)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f12.gif) |
| | Fig. 12 Molecular structure of the vinyl captured bimetallic complex [(PMDETA)K(TMP)(CHCH2)Zn(Me3SiCH2)]. | |
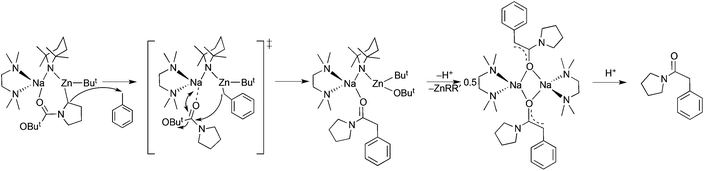
Other examples of synergic sedation using [(TMEDA)Na(TMP)(But)Zn(But)] as the cleave and capture agent are shown in Scheme 13.42 What the substrates all have in common are electron rich substituents containing highly basic heteroatoms which can entice metal atoms into dative unions while an electron-withdrawing acidifying effect is exerted upon the C–H bond nearest the substituent. These two related effects, coordination and inductive acidification, are the fundamental premises behind directed ortho-metallation (DoM), in which ortho C–H bonds are selectively converted to C–metal (nearly always C–Li) bonds.43 However the trade-off is that the electrophilicity of the substituent leaves it vulnerable to hostile nucleophilic attack from the metallating reagent, leading to unwanted side reactions and compromising synthetic efficiency. As a consequence DoM reactions executed by hard, highly polar alkyllithium or lithium amide reagents often require calming cryogenic conditions to reduce such decomposition. No such limitations apply to the DoM (or strictly here, AMMZn) reactions shown in Scheme 13 due to the softer, more sympathetic metallation provided by the TMP-zincate reagent which allows their execution at ambient temperature. In these reactions the ortho-C–Zn bonds are formed directly in one step thus circumventing the two-step low temperature lithiation/transmetallation (e.g., via ZnCl2) protocol generally essential for access to organozinc derivatives of relatively non-acidic organic substrates. Surviving zincation at ambient temperature in hexane solution, N-Boc pyrrolidine is deprotonated at an α-position with the anion captured within [(TMEDA)Na(TMP)(α-C4H7NBoc)Zn(But)].42a Synergic sedation in this case enhances the stability of this complex enabling it to be crystallised and structurally elaborated (Fig. 13), the first metallated N-Boc pyrrolidine intermediate to be so characterised. Following the trend observed for the other three structures in Scheme 13, the deprotonated pyrrolidine is held in place by a combination of zinc-anion (C) and sodium-neutral heteroatom (CO) bonding. Often used to protect the nucleophilic N atom during the preparation of C-substituted pyrrolidines, the Boc (t-butoxy carbonyl) group reveals its vulnerability when the reaction is repeated in toluene solution with the ButO− leaving group substituted by a benzyl anion generated from the solvent. In this multistep process (Scheme 14), N-Boc pyrrolidine acts initially as a Brønsted acid, then its resulting anion acts as a Brønsted base towards toluene, which, following the nucleophilic exchange, finally leads to a sodium pyrrolidine-substituted enolate.
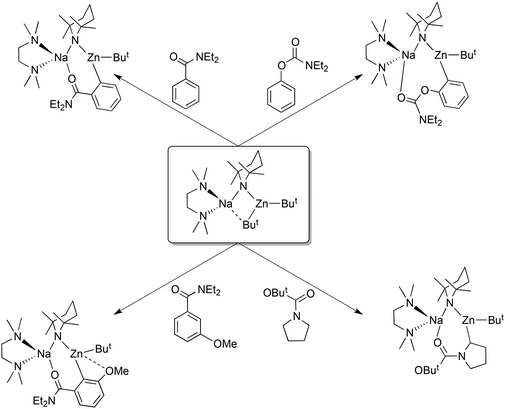 |
| | Scheme 13 Examples of alkali-metal-mediated ortho zincation. | |
![Molecular structure of the sodium zincate complex [(TMEDA)Na(TMP)(α-C4H7NBoc)Zn(But)] with a captured α-anion of N-Boc pyrrolidine.](/image/article/2013/DT/c3dt00053b/c3dt00053b-f13.gif) |
| | Fig. 13 Molecular structure of the sodium zincate complex [(TMEDA)Na(TMP)(α-C4H7NBoc)Zn(But)] with a captured α-anion of N-Boc pyrrolidine. | |
The four reaction examples in Scheme 13 also highlight a common limitation of AMMZn. Since only one deprotonated organic molecule ends up attached to it, the zinc centre coordinates to a mixture of three different anions, a situation that could complicate any tandem bond forming reactions (e.g., Negishi cross-couplings). Homoleptic zinc compositions containing lower order (e.g., Ar3Zn−) or higher order (e.g., Ar4Zn2−) anions (note that Organ has recently pinpointed higher order zincates as being especially significant as transmetallators in alkyl–alkyl Negishi coupling44) would be better suited for this purpose, so adapting AMMZn to produce homoleptic zincates remains an important challenge.
Inspiration for meeting this challenge can be found in a number of reactions. Ligand redistribution processes offer one possible solution. Changing the solvent can induce such processes as for example with the lithium heteroleptic zincate [(THF)2Li{MeO(o-C6H4)Zn(R)2}] (R = Me or But) which is stable in benzene or THF solution but rearranges to the higher order homoleptic composition [(THF)2Li2Zn{MeO(o-C6H4)}4] [the other products are thought to be {(THF)xLi2Zn(Me)4)} and Me2Zn(THF)x] when hexane is added (Scheme 15).38b Reagents with an appropriate ligand set can also produce homoleptic ates directly. The tris-(α-magnesiated) thiophene [(TMEDA)Na(C4H3S)3Mg(TMEDA)] can be prepared directly by reacting an all alkyl metal mixture with the sulfur heterocycle (Scheme 16), though it is also accessible via the TMP-magnesiate [(TMEDA)Na(TMP)(CH2SiMe3)Mg(TMP)] following a redistribution process.29 Noteworthy features of the structure of [(TMEDA)Na(C4H3S)3Mg(TMEDA)] (Fig. 14) include the asymmetrical bonding of the thiophene bridges (with the Mg in plane and the Na out of plane to the C4S ring reflecting their respective σ and π bonding predominance), the absence of any significant metal–sulfur bonding, and Mg as well as Na carrying a terminal TMEDA ligand.
![Synthesis of the asymmetrical structure of the homoleptic tetraanisolylzincate [(THF)2Li2Zn{MeO(o-C6H4)}4].](/image/article/2013/DT/c3dt00053b/c3dt00053b-s15.gif) |
| | Scheme 15 Synthesis of the asymmetrical structure of the homoleptic tetraanisolylzincate [(THF)2Li2Zn{MeO(o-C6H4)}4]. | |
![Synthesis of the tris(α-thiophenyl)magnesiate complex [(TMEDA)Na(C4H3S)3Mg(TMEDA)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-s16.gif) |
| | Scheme 16 Synthesis of the tris(α-thiophenyl)magnesiate complex [(TMEDA)Na(C4H3S)3Mg(TMEDA)]. | |
![Molecular structure of the sodium tris(α-thiophenyl)magnesiate complex [(TMEDA)Na(C4H3S)3Mg(TMEDA)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f14.gif) |
| | Fig. 14 Molecular structure of the sodium tris(α-thiophenyl)magnesiate complex [(TMEDA)Na(C4H3S)3Mg(TMEDA)]. | |
Alkali-metal-mediated alumination (AMMAl) is a particularly effective technique for deprotonating sensitive aromatic substrates in possession of halogen substituents. Halogen tolerant aluminations have been documented by Uchiyama and Knochel using the ate [LiAl(TMP)(Bui)3]33 and salt-activated [Al(TMP)3·3LiCl]45 respectively. Our aforementioned TMP-enriched aluminate “[Li(TMP)2Al(Bui)2]” also displays halogen tolerance when aluminating 4-halo-anisoles ortho to the methoxy substituent, which in turn have been quenched with halide electrophiles to form multi-halogenated anisoles (Scheme 17).46 Substituted aromatic compounds containing a mixture of chloro-, bromo-, and iodo-substituents are particularly appealing synthetically since the different relative strengths of the C–X (X = Cl, Br or I) bonds offer different opportunities for their selective functionalization. Sedation of the 4-halo-anisolyl ortho carbanions is accomplished by lithium and aluminium co-operating together through Li–O(Me) and Al–C interactions while the halo substituent dangles metal free (Fig. 15). However, such sedation is provisional on the position of the halide substituent relative to that of the methoxy group. Aluminating in between the two substituents of 3-iodoanisole places the electropositive metal and electronegative halogen next to each other.47 Their mutual attraction cannot be suppressed so elopement as a metal halide is inevitable, leaving the abandoned aromatic residue distressed as a benzyne. Conforming to the cleave and capture theme, both cleaved products can be captured, the benzyne with the external stimulus of 1,3-diphenylisobenzofuran through a Diels–Alder cyclisation, and lithium iodide without external stimulus sterically entrapped within a TMP(H)-solvated complex that restricts aggregation to the tetrameric state.
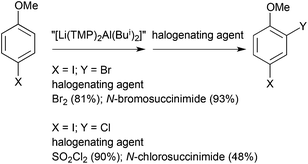 |
| | Scheme 17 Alkali-metal-mediated alumination of 4-halo-anisoles to give dihaloanisoles (yields in parentheses) following electrophilic halogenation. | |
![Molecular structure of the ortho-aluminated 4-iodo-anisole complex [(THF)Li(μ-TMP)(μ-{1-OMe-2-Al(Bui)2-4-I-C6H3})].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f15.gif) |
| | Fig. 15 Molecular structure of the ortho-aluminated 4-iodo-anisole complex [(THF)Li(μ-TMP)(μ-{1-OMe-2-Al(Bui)2-4-I-C6H3})]. | |
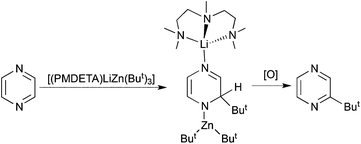
Diazines represent another important category of synthetically useful heterocycle. The sensitivity of their anionic forms generated upon metallation is reflected by the conditions, cryogenic temperatures, in situ performed electrophilic interceptions, and short reaction times, needed to avoid decomposition pathways. Changing the metallating agent from the lithium amide TMP to the more elaborate bimetallic amides “Li(TMP)·Zn(TMP)2” and (TMP)2Mg·2LiCl (in the presence of ZnCl2) enables the metallation/functionalisation of unsubstituted diazines and pyrazine/quinoxaline respectively under milder conditions as reported by Mongin48 and Knochel49 respectively. Hevia goes one step further in her metallation studies of pyrazine by synergically sedating its 2,5-dideprotonated form.50 Generated by reaction with stoichiometric [(THF)LiZn(TMP)(But)2] in THF solution, the sensitive dianion is captured and stabilised through a combination of covalent Zn–C and dative Li–N bonding within the crystalline solid [2,5-{(THF)2LiZn(TMP)But}2(C4H2N2)] (Fig. 16). Iodination of this intermediate via iodine is straightforward (Scheme 18). Interestingly changing the ate reagent from mixed alkyl-amido [(THF)LiZn(TMP)(But)2] to all alkyl [(PMDETA)LiZn(But)3] promotes t-butyl alkylation as opposed to deprotonation of pyrazine producing a dihydro intermediate which converts to 2-t-butyl pyrazine under hydrolysis/aerobic oxidation (Scheme 19). Such alkylation reactions are relatively common for diazines as the presence of the two electronegative N atoms generates a low lying LUMO.
![Molecular structure of the captured pyrazinyl dianion complex [2,5-{(THF)2LiZn(TMP)But}2(C4H2N2)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f16.gif) |
| | Fig. 16 Molecular structure of the captured pyrazinyl dianion complex [2,5-{(THF)2LiZn(TMP)But}2(C4H2N2)]. | |
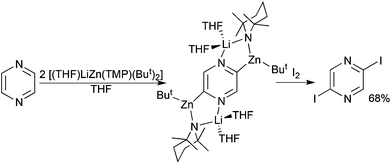 |
| | Scheme 18 Synthesis of 2,5-diiodopyrazine via a dizincated intermediate. | |
Cleave and capture in special positions
As nearly all useful organic compounds contain multiple C–H bonds, selectivity in C–H to C–metal transformations is of the utmost importance. Cleavage of a single C–H bond of choice enables subsequent functionalization at that specific position; otherwise metallation at multiple sites will lead to a wasteful distribution of products which may not be easy to separate. High reactivity normally goes hand in hand with low selectivity and vice versa so finding a metallating agent reactive enough to cleave one specific C–H bond and to do it selectively at the expense of all other C–H bonds in the molecule can be a colossal challenge. In the most widely applicable metallation tool developed to date, directed ortho-metallation (DoM),43 the aromatic substrate coerces the metallating agent to attack the C–H position adjacent to a directing group by offering it coordinative (Lewis basic heteroatoms for the Lewis acidic metal) and electronic (inductively-weakened Cδ−–Hδ+ polarised bonds for cleavage by the base anion) enticements. DoM can be accomplished by a broad range of directing groups though the degree of coercement (and thus the degree of difficulty in deprotonating ortho-C–H bonds) varies from group to group. ortho-Metallation (usually lithiation) of heteroatom-activated aromatic substrates is therefore commonplace. Covered in this section of the article are cleave and capture reactions executed by mixed metal reagents which produce novel regioselectivities that do not conform to this or similar patterns of reactivity.
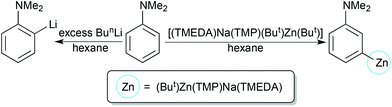
A striking example is provided by metallation of N,N-dimethylaniline (Scheme 20). Possessing a weakly directing Me2N group, this aromatic substrate only succumbs to BunLi under forcing conditions (refluxing a hexane solution overnight) in a standard ortho metallation.51 Surprisingly, changing the base to [(TMEDA)Na(TMP)(But)Zn(But)] re-directs the metallation to the meta position as evidenced initially by isolation of the crystalline metallated aniline intermediate [(TMEDA)Na(TMP)(m-C6H4-NMe2)Zn(But)]52 (yield 39%) (Fig. 17), which produced the 3-iodoaniline derivative quantitatively on quenching with iodine. A subsequent more complete study of the whole reaction solution with iodine identified a mixture of regioisomers of N,N-dimethyliodoaniline, with the meta isomer still the major product (total conversion, 72%; ortho:meta:para ratio 6:73:21), as quantified by NMR evidence.53 The most significant finding of this study established that it is the co-operativity between the different components of the mixed sodium–zinc base which accounts for this novel regioselectivity. Neither di-t-butylzinc nor sodium TMP, even with the addition of TMEDA, can cleave a hydrogen atom from N,N-dimethylaniline. Furthermore, trying to reproduce the zincation indirectly by first sodiating the aniline with the stronger alkyl base BunNa in the presence of TMEDA and cocomplexing with di-t-butylzinc, then adding TMP(H), produced a zincated anilide but of the ortho-isomer [(TMEDA)Na(TMP)(o-C6H4-NMe2)Zn(But)]. Analysis of this indirect zincation solution following iodine interception revealed iodinated anilides in a 16.6:1.6:1.0, ortho:meta:para ratio, that is, the mixture is now predominately ortho in character with only an insignificant quantity of meta product. Moreover it is likely that this meta product has its origin from a background retro reaction in which [(TMEDA)Na(o-C6H4-NMe2)Zn(But)2] reacts with TMP(H) to regenerate a small proportion of free aniline and the direct zincating reagent [(TMEDA)Na(TMP)(But)Zn(But)], the two starting materials behind the direct reaction that produces the meta isomer predominately (Scheme 21). A final general point to note about this sodiation/dialkylzinc cocomplexation versus direct sodium-mediated zincation comparison is that the former is an unclean reaction due to the aggressive reactivity of the alkyl sodium reagent; whereas the latter reaction is much cleaner as zinc's stronger electrophilicity attenuates sodium's reactivity and following the deprotonation a more covalent, more stable zinc–carbon bond forms as opposed to a more polarised, less stable sodium–carbon bond (Scheme 22).
![Molecular structure of the meta-metallated aniline complex [(TMEDA)Na(TMP)(m-C6H4-NMe2)Zn(But)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f17.gif) |
| | Fig. 17 Molecular structure of the meta-metallated aniline complex [(TMEDA)Na(TMP)(m-C6H4-NMe2)Zn(But)]. | |
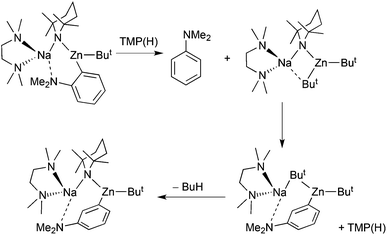 |
| | Scheme 21 Postulated conversion of ortho-zincated N,N-dimethylaniline to a meta analogue. | |
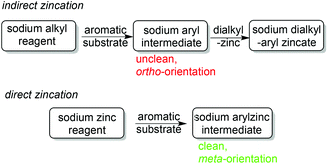 |
| | Scheme 22 General distinction between indirect and direct zincation of aromatic substrates. | |
This direct zincation (alkali metal and zinc in co-operation) versus indirect zincation (alkali metal and zinc not in co-operation) was also studied with toluene as the aromatic substrate. Interestingly, the former approach using [(TMEDA)Na(TMP)(But)Zn(But)] leads to a statistical mixture of meta and para regioisomers of deprotonated toluene captured in [(TMEDA)Na(C6H4Me)(TMP)Zn(But)].54 A combination of σ- and π-grips from zinc and sodium respectively hold the arene anions in place, which remarkably still retain their methyl (and ortho) hydrogen atoms (Fig. 18). Predictably this special regioselectivity is not repeated by the latter indirect approach of sequential sodiation (via butylsodium) and cocomplexation (via di-t-butylzinc), the isolated product being the benzylzincate [(TMEDA)2Na(CH2Ph)Zn(But)2] consistent with lateral (Me) deprotonation. The importance of the co-operation between the distinct metals was ratified by DFT studies of the direct reaction which revealed that of the four possible regioisomers of deprotonated toluene, the experimentally observed meta and para molecules are the most thermodynamically favoured.
![Molecular structure of the para-metallated toluene isomer [(TMEDA)Na(p-C6H4Me)(TMP)Zn(But)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f18.gif) |
| | Fig. 18 Molecular structure of the para-metallated toluene isomer [(TMEDA)Na(p-C6H4Me)(TMP)Zn(But)]. | |
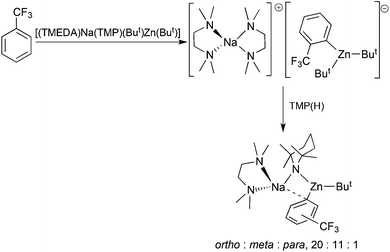
Changing the electron donating methyl substituent on the benzene ring to the strongly electron withdrawing trifluoromethyl (CF3) substituent complicates the regioselectivity of direct zincation by [(TMEDA)Na(TMP)(But)Zn(But)] (Scheme 23).55 The inductive power of CF3 is persuasive in directing deprotonation to the ortho position at 0 °C, manifested in the segregATE [{(TMEDA)2Na}+{Zn(o-C6H4-CF3)(But)2}−]. This compound is illuminating in that it portrays the kinetic intermediate of the two-step kinetic-thermodynamic mechanism behind alkali-metal-mediated zincation with TMP reacting first to generate TMP(H) and an aryl-dialkylzincate. Made either by raising the temperature to 25 °C or adding TMP(H) to the segregATE (mimicking the second step of the mechanism), the thermodynamic intermediate is the contacted ion pair molecule [(TMEDA)Na(C6H4-CF3)(TMP)Zn(But)] where the aryl component contains a mixture of ortho, meta and para isomers. The emergence of the last two isomers can be rationalised by a weakening of the ortho C–Zn bond due to the strong electron pulling power of the CF3 substituent.
Benzylic ethers are known to be poor ortho-directing molecules owing to the high acidity of the α-CH(R) unit that is sandwiched between the electronegative O atom and electron withdrawing aryl ring. The simplest example, benzyl methyl ether is metallated exclusively at the α-position (benzylic) on treatment with excess n-butyllithium in THF solution, but interception with D2O is temperature dependent.56 At −40 °C, deuteration occurs at the α-position; whereas at 25 °C, a Wittig rearrangement takes place to give 1-phenylethanol (Scheme 24). In comparison [(TMEDA)Na(TMP)(But)Zn(But)] again behaves anomalously as it deprotonates benzyl methyl ether exclusively at the ortho position in hexane solution, with the resultant anion captured in heterotrianionic [(TMEDA)Na(TMP)(o-C6H4-CH2OMe)Zn(But)] (Scheme 24).57 This anion acts as a bidentate ligand toward the metals through a Zn–C σ bond and a Na–O(Me) dative bond.
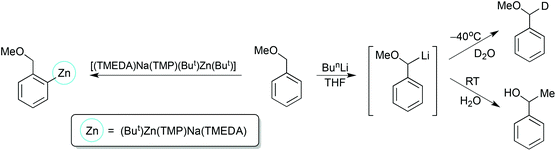 |
| | Scheme 24 Comparison of conventional lithiation and AMMZn methods towards deprotonation of benzyl methyl ether. | |
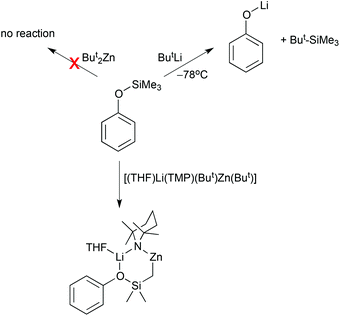
Another striking example where component co-operation in a mixed-metal system can access reactions essentially inaccessible via conventional organolithium reagents (and by implication via conventional two step metallation–transmetallation procedures) is provided by trimethylphenoxysilane, PhOSiMe3. Reaction of this alkoxysilane with t-butyllithium at −78 °C in THF solution promotes cleavage of the Si–O bond to generate the tetraalkylsilane ButSiMe3 and lithium phenolate, PhOLi (Scheme 25).58 The organozinc reagent But2Zn does not react at all with the alkoxysilane. In marked contrast, when the two metals are organised together in the lithium zincate [(THF)Li(TMP)(But)Zn(But)], a lithium mediated zincation takes place that promotes deprotonation at a lateral position converting the Si(Me)3 substituent to a Si(CH2)(Me)2 unit.59 This represents a new direct (single step) route to α-metallated silanes, which have many important synthetic applications. The product of this lateral zincation [(THF)Li(TMP)(PhSiOMe2CH2)Zn(But)] is structurally intriguing presenting a six-element (LiOSiCZnN) ring (Fig. 19). Note that in the absence of THF, the parent alkoxysilane takes it place bringing the two separate metal components together in a “pre-metallation” complex [(PhOSiMe3)Li(TMP)(But)Zn(But)] that metallates another equivalent of PhOSiMe3.
![Molecular structure of the laterally zincated alkylsilane [(THF)Li(TMP)(PhSiOMe2CH2)Zn(But)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f19.gif) |
| | Fig. 19 Molecular structure of the laterally zincated alkylsilane [(THF)Li(TMP)(PhSiOMe2CH2)Zn(But)]. | |
One of the most spectacular regioselective deprotonation reactions reported to date transformed the TMP anion from one of the strongest known Brønsted bases, as evidenced by the many examples covered in this article, to a Brønsted acid (that is, the anion loses a proton to become a dianion). This was the outcome of combining the potassium amide KTMP with the dialkylaluminium amide Bui2Al(TMP) and TMEDA in hexane solution, which, in effect, initiates a self-deprotonation with one TMP anion deprotonating another to generate a TMP2−(TMP*) dianion and TMP(H) (Scheme 26).60 Captured in the potassium aluminate [(TMEDA)K(TMP*)(Bui)Al(Bui)], the dianion is [with respect to TMP(H)] N,C-dideprotonated with one α-Me sidearm converted to a methylenic CH2 unit (Fig. 20). Mechanistically it is thought the TMP* dianion results from an intramolecular alumination within a nascent cocomplex involving the two distinct TMP-containing complexes. This reactivity is unprecedented and it remains to be established whether it is due to special structural features within this monovalent metal–trivalent metal partnership. It cannot be attributed to a general mixed-metal effect alone as like the homometallic alkali metal TMP complexes, recently reported hetero-alkali-metal TMP complexes61 also do not exhibit this N–H, C–H double deprotonation.
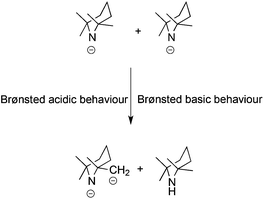 |
| | Scheme 26 Self deprotonation of the TMP anion. | |
![Molecular structure of the dianionic TMP aluminate complex [(TMEDA)K(TMP*)(Bui)Al(Bui)].](/image/article/2013/DT/c3dt00053b/c3dt00053b-f20.gif) |
| | Fig. 20 Molecular structure of the dianionic TMP aluminate complex [(TMEDA)K(TMP*)(Bui)Al(Bui)]. | |
Outlook
We are in the middle of a renaissance in metallation chemistry that could eventually see conventional organolithium reagents phased out in favour of new multicomponent bases. The design and optimisation of these multicomponent bases is still being perfected and much work remains to understand fully the co-operative effects at play between the different components which shape their special chemical profiles. Improvements in reducing the number of synthetic steps required for molecular functionalisation, in functional group tolerance, in kinetic stability, in selectivity and in various other aspects have already been documented, but economic factors may retard the progress of this revolution as far as large scale and industrial utilisation is concerned. Finding alternatives to expensive TMP, the active ingredient of most multicomponent bases known to date is a challenge that must be overcome in the future. Tangential to the main theme of alkali-metal-mediated metallation, this article describes selected recent examples from the alternative perspective of cleave and capture chemistry. Basically, from this viewpoint the multicomponent base functions as both a C–H bond cleave tool and a capture agent of the anions generated from this cleaving. Various hydride, oxygen-based, cyclic ether, and substituted aromatic anions have been captured through this approach. It is a new and thus immature idea, though in time it may grow to have important implications in the formation, stability and utilisation of sensitive anions in synthesis, in small molecule activation as well as in anion complexation chemistry.
Acknowledgements
I am greatly indebted to all of my co-workers and colleagues whose names appear in the publications cited for their intellectual insights, hard work and dedication, and most of all friendship during our development of this chemistry. It has been very much a team effort though four players Dr Eva Hevia, Dr Charlie O'Hara, Dr Stuart Robertson and Dr Jan Klett deserve special praise for their VIP performances. I owe Charlie and Stuart particular thanks for their help in preparing the figures and schemes used throughout the article. Extra thanks must also go to Jan for his marvellous cover artwork. Our work has been generously supported by the Royal Society, the EPSRC, the Nuffield Foundation and AstraZeneca for which we are extremely grateful.
References
-
(a) V. Grignard, Ann. Chim., 1901, 24, 433 CAS;
(b)
Grignard Reagents, ed. H. G. Richey Jr., John Wiley & Sons Ltd., Chichester, 2000 Search PubMed.
-
(a) W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1917, 50, 262 CrossRef CAS;
(b) T. T. Tidwell, Angew. Chem., Int. Ed., 2001, 40, 331 CrossRef CAS.
-
(a)
J. Clayden, Directed metallation of aromatic compounds, in The Chemistry of Organolithium Compounds, ed. Z. Rappoport and I. Marek, John Wiley & Sons Ltd., Chichester, 2004, ch. 10, p. 495 Search PubMed;
(b)
M. Schlosser, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, John Wiley & Sons Ltd., Chichester, 2nd edn, 2002, ch. 1, pp. 1–352 Search PubMed.
-
(a) K. Ziegler and H. Ohlinger, Justus Liebigs Ann. Chem., 1932, 495, 84 CrossRef CAS;
(b) M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854 CrossRef CAS;
(c) M. Hamell and R. Levine, J. Org. Chem., 1950, 15, 162 CrossRef CAS;
(d) U. Wannagat and H. Niederprüm, Chem. Ber., 1961, 94, 1540 CrossRef CAS;
(e) B. L. Lucht and D. Collum, J. Am. Chem. Soc., 1994, 116, 6009 CrossRef CAS.
-
(a)
F. Leroux, M. Schlosser, E. Zohar and I. Marek, in The Chemistry of Organolithium Compounds, ed. Z. Rappoport and I. Marek, John Wiley & Sons Ltd., Chichester, 2004, ch. 9, p. 457 Search PubMed.
- P. Caubere, Chem. Rev., 1993, 93, 2317 CrossRef CAS.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS.
- C. R. Hauser and H. G. Walker, J. Am. Chem. Soc., 1947, 69, 295 CrossRef CAS.
- Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539 CrossRef CAS.
-
(a) J.-M. L'Helgoual'ch, G. Bentabed-Ababsa, F. Chevallier, M. Yonehara, M. Uchiyama, A. Derdour and F. Mongin, Chem. Commun., 2008, 5375 RSC;
(b) F. Mongin and M. Uchiyama, Curr. Org. Chem., 2011, 15, 2340 CrossRef CAS.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184 CrossRef CAS.
-
(a) R. E. Mulvey, Organometallics, 2006, 25, 1060 CrossRef CAS;
(b) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802 CrossRef CAS.
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS.
- R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588 CrossRef CAS.
-
(a) H. Ahlbrecht and G. Schneider, Tetrahedron, 1986, 42, 4729 CrossRef CAS;
(b) A. Streitwieser, A. Facchetti, L. Xie, X. Zhang and E. C. Wu, J. Org. Chem., 2012, 77, 985 CrossRef CAS.
-
(a) V. L. Blair, L. M. Carella, W. Clegg, J. Klett, R. E. Mulvey, E. Rentschler and L. Russo, Chem.–Eur. J., 2009, 15, 856 CrossRef CAS;
(b) For an excellent review of hydride encapsulation in molecular alkali-metal clusters see: J. Haywood and A. E. H. Wheatley, Dalton Trans., 2008, 3378 RSC.
- Note that molecules of this type were included in a symposium on “Molecular Aesthetics”, held at ZKM (centre for art and media), Karlsruhe, Germany in July 2011. A book of this conference is due for release in 2013.
-
(a) D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O'Hara, R. E. Mulvey and R. B. Rowlings, Chem. Commun., 2002, 376 RSC;
(b) P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3354 CrossRef CAS.
- E. Hevia, F. R. Kenley, A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3347 CrossRef CAS.
-
(a) S. Harder, J. Spielmann, J. Intemann and H. Bandmann, Angew. Chem., Int. Ed., 2011, 50, 4156 CrossRef CAS;
(b) E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 9242 CrossRef CAS;
(c) For a review of early main group hydride compounds see: S. Harder, Chem. Commun., 2012, 48, 11165 RSC.
-
(a) R. Campbell, P. Garcia-Álvarez, A. R. Kennedy and R. E. Mulvey, Chem.–Eur. J., 2010, 16, 9964 CrossRef CAS;
(b) R. Campbell, D. Cannon, P. Garcia-Álvarez, A. R. Kennedy, R. E. Mulvey, S. D. Robertson, J. Sassmannshausen and T. Tuttle, J. Am. Chem. Soc., 2011, 133, 13706 CrossRef CAS.
- A. D. Bolig and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 14544 CrossRef CAS.
- R. E. Mulvey, Chem. Commun., 2001, 1049 RSC.
- A. R. Kennedy, J. Klett, R. E. Mulvey, S. Newton and D. S. Wright, Chem. Commun., 2008, 308 RSC.
- M. F. Lappert, M. J. Slade, A. Singh, J. L. Atwood, R. D. Rogers and R. Shakir, J. Am. Chem. Soc., 1983, 105, 302 CrossRef CAS.
- A. M. Drummond, L. T. Gibson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and T. Weightman, Angew. Chem., Int. Ed., 2002, 41, 2382 CrossRef CAS.
- L. Balloch, J. A. Garden, A. R. Kennedy, R. E. Mulvey, T. Rantanen, S. D. Robertson and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 6934 CrossRef CAS.
- V. L. Blair, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Chem.–Eur. J., 2010, 16, 8600 CrossRef CAS.
- L. Balloch, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, m252 Search PubMed.
- S. E. Baillie, V. L. Blair, E. Hevia and A. R. Kennedy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, m249 Search PubMed.
- L. Lochmann, Eur. J. Inorg. Chem., 2000, 1115 CAS.
- M. Uchiyama, H. Naka, Y. Matsumoto and T. Ohwada, J. Am. Chem. Soc., 2004, 126, 10526 CrossRef CAS.
-
(a) F. Chevallier, Y. S. Halauko, C. Pecceu, I. F. Nassar, T. U. Dam, T. Roisnel, V. E. Matulis, O. A. Ivashkevich and F. Mongin, Org. Biomol. Chem., 2011, 9, 4671 RSC;
(b) P. Garcia-Álvarez, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2011, 50, 9668 CrossRef.
- A. Maercker, Angew. Chem., Int. Ed., 1987, 26, 972 CrossRef.
- A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706 CrossRef CAS.
- E. Crosbie, P. Garcia-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2010, 49, 9388 CrossRef CAS.
-
(a) D. Nobuto and M. Uchiyama, J. Org. Chem., 2008, 73, 1117 CrossRef CAS;
(b) W. Clegg, B. Conway, E. Hevia, M. D. McCall, L. Russo and R. E. Mulvey, J. Am. Chem. Soc., 2009, 131, 2375 CrossRef CAS.
- R. E. Mulvey, D. R. Armstrong, B. Conway, E. Crosbie, A. R. Kennedy and S. D. Robertson, Inorg. Chem., 2011, 50, 12241 CrossRef CAS.
- J. Garcia-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2007, 2402 RSC.
-
J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, Elsevier, Oxford, 2002, ch. 7, pp. 273–280 Search PubMed.
-
(a) J. A. Garden, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. Commun., 2012, 48, 5265 RSC;
(b) L. Balloch, A. R. Kennedy, R. E. Mulvey, T. Rantanen, S. D. Robertson and V. Snieckus, Organometallics, 2011, 30, 145 CrossRef CAS.
-
(a) H. W. Gschwend and H. R. Rodriguez, Org. React., 1979, 26, 1 CAS;
(b)
G. C. Hartung and V. Snieckus, in The Directed ortho Metallation Reaction – A Point of Departure for New Synthetic Aromatic Chemistry in Modern Arene Chemistry, ed. D. Astruc, Wiley-VCH, New York, 2002, pp. 330–367 Search PubMed.
- L. C. McCann, H. N. Hunter, J. A. C. Clyburne and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 7024 CrossRef CAS.
- S. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 1501 CrossRef CAS.
- B. Conway, E. Crosbie, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. Commun., 2012, 48, 4674 RSC.
- E. Crosbie, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2012, 41, 1832 RSC.
- A. Seggio, F. Chevallier, M. Vaultier and F. Mongin, J. Org. Chem., 2007, 72, 6602 CrossRef CAS.
- Z. Dong, G. C. Clososki, S. H. Wunderlich, A. Unsinn, J. Li and P. Knochel, Chem.–Eur. J., 2009, 15, 457 CrossRef CAS.
- S. E. Baillie, V. L. Blair, D. C. Blakemore, D. Hay, A. R. Kennedy, D. C. Pryde and E. Hevia, Chem. Commun., 2012, 48, 1985 RSC.
- A. R. Lepley, W. A. Khan, A. B. Giumanini and A. G. Giumanini, J. Org. Chem., 1966, 31, 2047 CrossRef CAS.
- D. R. Armstrong, W. Clegg, S. H. Dale, E. Hevia, L. M. Hogg, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 3775 CrossRef CAS.
- D. R. Armstrong, L. Balloch, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and S. D. Robertson, Beilstein J. Org. Chem., 2011, 7, 1234 CrossRef CAS.
- D. R. Armstrong, J. Garcia-Álvarez, D. V. Graham, G. W. Honeyman, E. Hevia, A. R. Kennedy and R. E. Mulvey, Chem.–Eur. J., 2009, 15, 3800 CrossRef CAS.
- D. R. Armstrong, V. L. Blair, W. Clegg, S. H. Dale, J. Garcia-Álvarez, G. W. Honeyman, E. Hevia, R. E. Mulvey and L. Russo, J. Am. Chem. Soc., 2010, 132, 9480 CrossRef CAS.
- U. Azzena, L. Pilo and A. Sechi, Tetrahedron, 1998, 54, 12389 CrossRef CAS.
- L. Balloch, A. R. Kennedy, J. Klett, R. E. Mulvey and C. T. O'Hara, Chem. Commun., 2010, 46, 2319 RSC.
- T. F. Bates, S. A. Dandekar, J. L. Longlet, K. A. Wood and R. D. Thomas, J. Organomet. Chem., 2000, 595, 87 CrossRef CAS.
- E. Hevia, A. R. Kennedy, J. Klett and M. McCall, Chem. Commun., 2009, 3240 RSC.
- B. Conway, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and J. Garcia-Álvarez, Angew. Chem., Int. Ed., 2010, 49, 3182 CrossRef CAS.
- D. R. Armstrong, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem.–Eur. J., 2011, 17, 8820 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a