 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the fate of the tris(pentafluorophenyl)borane radical anion in weakly coordinating solvents†
Elliot J.
Lawrence
a,
Vasily S.
Oganesyan
a,
Gregory G.
Wildgoose
*a and
Andrew E.
Ashley
b
aEnergy and Materials Laboratory, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, United Kingdom. E-mail: G.Wildgoose@uea.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, United Kingdom
First published on 22nd November 2012
Abstract
We report a kinetic and mechanistic study into the one-electron reduction of the archetypal Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, in dichloromethane and 1,2-difluorobenzene. Electrochemical experiments, combined with digital simulations, DFT computational studies and multinuclear NMR analysis allow us to obtain thermodynamic, kinetic and mechanistic information relating to the redox activity of B(C6F5)3. We show that tris(pentafluorophenyl)borane undergoes a quasi-reversible one-electron reduction followed by rapid chemical decomposition of the B(C6F5)3˙− radical anion intermediate via a solvolytic radical pathway. The reaction products form various four-coordinate borates of which [B(C6F5)4]− is a very minor product. The rate of the follow-up chemical step has a pseudo-first order rate constant of the order of 6 s−1. This value is three orders of magnitude larger than that found in previous studies performed in the donor solvent, tetrahydrofuran. The standard reduction potential of B(C6F5)3 is reported for the first time as −1.79 ± 0.1 V and −1.65 ± 0.1 V vs. ferrocene/ferrocenium in dichloromethane and 1,2-difluorobenzene respectively.
Introduction
The preparation of tris(pentafluorophenyl)borane, B(C6F5)3, was first reported by Massey and co-workers in 1963.1–3 It was noted that the compound formed strong adducts with a number of different Lewis bases. The Lewis acidity of B(C6F5)3 was later measured and determined to be intermediate between BF3 and BCl3.3–5 Unlike the boron trihalides, however, B(C6F5)3 is a relatively thermally stable solid that exhibits a good resistance to hydrolysis.6,7 B(C6F5)3 therefore offers an unprecedented ease of handling, combined with strong Lewis acidity and adequate steric bulk. It is for this reason that Piers and Chivers described B(C6F5)3 as “the ideal boron-based Lewis acid”.7B(C6F5)3 has been employed as a key component in a number of important applications relating to synthetic organic transformations,8–12 the preparation of weakly coordinating anions,13–15 and the activation of olefin polymerization catalysts.16–20 Since the pioneering work of Stephan et al.21 in 2006, B(C6F5)3 has become the archetypal Lewis acid in Frustrated Lewis Pair (FLP) chemistry22–25 – currently a highly active area of research with applications in hydrogenation reactions26,27 and small molecule activation.28–38
In addition to its interesting Lewis acidic properties, the ability of B(C6F5)3 to act as a one-electron oxidant was accidentally discovered by Norton's group in 1999.39 Erker and co-workers had previously demonstrated that B(C6F5)3 could be used to open zirconocycles to generate effective olefin polymerization catalysts.40 When Norton and co-workers attempted to extend this concept to heteroatom-substituted zirconocycles, they noted the partial oxidation of their catalyst. Soon afterwards, Green et al. also observed the one-electron oxidation of a η2-vinyl molybdenum complex in the presence of B(C6F5)3.41 Norton's group then went on to investigate the redox properties of B(C6F5)3 by reducing it using decamethylcobaltocene (Cp*2Co) and studying the resulting B(C6F5)3˙− intermediate via EPR and UV-vis spectroscopic methods.42 The rate of decomposition of the B(C6F5)3˙− species was determined to be ca. 5.7 × 10−3 s−1 at 23 °C using UV-vis spectrophotometry (λmax = 603 nm).42 However, this value should be treated with some caution given that the experiments were performed in the donor solvent THF and the formation of the (THF)·B(C6F5)3 adduct is well known.43
Despite there being an interest in the redox properties of B(C6F5)3, its direct electrochemical reduction initially proved to be difficult for two reasons. Early attempts to record the cyclic voltammetry of B(C6F5)3 were performed using either coordinating solvents, e.g. THF, and/or common supporting electrolyte salts of ClO4−, PF6− or BF4− that can react with B(C6F5)3. These experimental conditions resulted in ill-defined cyclic voltammograms at best, and only enabled predictions of the reduction potential of B(C6F5)3.39,44 The first direct voltammetric reduction of B(C6F5)3 was reported by this group and collaborators in 2011.45 This was achieved by virtue of a carefully selected system, comprising CH2Cl2 solvent and a non-coordinating electrolyte based on Kobayashi's anion, [nBu4N][B(3,5-(CF3)2C6H3)4].46 However, no further mechanistic or kinetic studies were undertaken at that time. In this report we address this by extracting mechanistic and kinetic parameters for the reduction of B(C6F5)3 in solvents of low donor strength, whilst also determining the reaction products. This will allow for a better understanding of the one-electron redox chemistry of B(C6F5)3.
Experimental section
Materials and general methods
All synthetic reactions and manipulations were performed under a dry N2 atmosphere (BOC Gases) using either a Saffron glovebox or standard Schlenk-line techniques on a dual manifold vacuum/inert gas line. All glassware was dried under vacuum at 170 °C before use. Diethyl ether and light petroleum either were dried via distillation over Na/benzophenone diketyl; toluene was dried via distillation over molten Na; dichloromethane was dried via distillation over CaH2; 1,2-difluorobenzene (DFB) was dried by stirring over P4O10 and triply distilled prior to use. All solvents were sparged with nitrogen gas to remove any trace of dissolved oxygen and stored in ampules over activated 3 Å molecular sieves. Bromopentafluorobenzene was purchased from Fluorochem and used without further purification. Mg turnings were purchased from Alfa Aesar and used as supplied. All other reagents were purchased from SigmaAldrich and were of the highest grade available and used without further purification. Deuterated NMR solvents (CDCl3, 99.8%; CD3CN, 99.9%) were purchased from Cambridge Isotope Laboratories Inc. and were dried over P4O10, degassed using a triple freeze–pump–thaw cycle and stored over activated 3 Å molecular sieves. B(C6F5)3 and [Li(OEt2)3][B(C6F5)4] were prepared according to literature methods.47,48 NMR spectra were recorded using a Bruker Advance DPX-300 MHz spectrometer. Chemical shifts are reported in ppm and are referenced relative to appropriate standards; 19F is relative to CFCl3, 11B is relative to Et2O·BF3. For NMR experiments performed in non-deuterated solvents a C6D6 insert was used. ESI-MS spectra were recorded using a Shimadzu LCMS 2010EV spectrometer in negative ESI mode. EPR spectra were recorded using a Bruker ER200D spectrometer fitted with a dual-mode (ER4116M) X-band cavity and interfaced to an EMX control system. A flow-through cryostat used in conjunction with a Eurotherm (B-VT-2000) variable temperature controller provided temperatures ranging from 80–180 K.Preparation of [nBu4N][B(C6F5)4] electrolyte
A solution of [Li(OEt2)3][B(C6F5)4] (17.16 g, 18.9 mmol) in dry, degassed CH2Cl2 (50 mL) was added to a solution of tetrabutylammonium chloride (5.25 g, 18.9 mmol) in dry, degassed CH2Cl2 (50 mL). Rapid formation of a fine off-white precipitate resulted. The reaction mixture was left to stir overnight before filtration and removal of the solvent in vacuo to yield the crude product (16.97 g, 97%) as an off-white solid. [nBu4N][B(C6F5)4] was recrystallized three times according to the method of LeSuer et. al. prior to use as a supporting electrolyte.491H NMR (400 MHz, CDCl3): δ 3.11 (m, 8H), 1.58 (m, 8H), 1.36 (m, 8H), 0.96 (t, J = 8 Hz) ppm; 11B NMR (96 MHz, CDCl3): δ −16.7 ppm; 19F NMR (282 MHz, CDCl3): δ −132.5 (m, 8F, o-F), −162.8 (m, 4F, p-F), −166.7 (m, 8F, m-F) ppm.Electrochemistry
All electrochemical experiments were performed under an inert atmosphere using an Autolab PGSTAT 30 computer-controlled potentiostat. Cyclic voltammetry (CV) was performed using a three-electrode configuration consisting of either a Pt macrodisk working electrode (GoodFellow, Cambridge, UK; 99.99%; area 1.4 ± 0.5 × 10−3 cm2) or a Pt microdisk working electrode (GoodFellow, Cambridge, UK; 99.99%; radius 30.5 ± 0.5 μm), combined with a Pt wire counter electrode and a Ag wire pseudoreference electrode. The Pt working electrodes were polished between experiments using successive grades of alumina slurries (from 1.0 to 0.3 μm), rinsed in distilled water and subjected to brief ultrasonication to remove any adhered alumina microparticles. The electrodes were then dried in an oven at 120 °C to remove any residual traces of water. The Pt working electrode areas were calibrated for each experiment using a 5.0 mM ferrocene solution in either CH3CN or CH2Cl2 solvent containing 0.1 M [nBu4N][PF6] as the supporting electrolyte. The Pt macrodisk working electrode area was accurately determined by construction of a Randles–Sevcik plot from cyclic voltammograms recorded at varying scan rates (50–750 mV s−1).50 The Pt microdisk working electrode area was accurately determined from the steady state current, measured using linear sweep voltammetry (scan rate = 5 mV s−1).50 The Ag wire pseudoreference electrodes were calibrated to the ferrocene/ferrocenium couple in CH2Cl2 at the end of each run to allow for any drift in potential, following IUPAC recommendations.51 All electrochemical measurements were performed at ambient temperatures under an inert N2 atmosphere in either CH2Cl2 or 1,2-difluorobenzene (DFB) containing 0.05 M [nBu4N][B(C6F5)4] as the supporting electrolyte, and iR-compensated using positive-feedback to within 85 ± 5% of the solution uncompensated resistance. CV simulations were performed using DigiElch – Professional (v 7.030) software.Computational modelling
All calculations were performed using the Gaussian 09 computational package.52 Geometry optimisation, vibration frequencies and spin distribution calculations have been carried out using the three-parameter exchange functional of Becke53 (B3) and the correlation functional of Lee, Yang, and Parr (LYP), B3LYP.54 In each case an all electron 6-311+G(d,p) basis set has been implemented. Structures were geometry optimised in the gas phase with the default convergence criteria and confirmed as minima through frequency calculations. Zero-point energies and thermodynamic properties were calculated at 298.15 K/1 atm. All calculations have been performed at spin-unrestricted level of theory.Results and discussion
Electrochemical experiments
The direct voltammetric reduction of B(C6F5)3 was explored at a macrodisk electrode using cyclic voltammetry (Fig. 1 and ESI 1†) with weakly coordinating electrolyte systems comprising of a solution of [nBu4N][B(C6F5)4] in either CH2Cl2 or DFB low-donor solvents. Similar voltammetric behaviour was observed in either solvent. Upon first scanning towards more negative potentials a reduction wave was observed at −1.82 and −1.67 V vs. Cp2Fe0/+ (at 100 mV s−1) for CH2Cl2 and DFB respectively. At slow scan rates the reduction wave appears to be irreversible, with no corresponding oxidation peak observed upon reversing the scan direction. However, at faster scan rates (up to 5000 mV s−1) a small oxidation wave was observable as the scan rate was increased.![Overlaid cyclic voltammograms of B(C6F5)3 in DFB (5.1 mM, 0.05 M [nBu4N][B(C6F5)4]) recorded at scan rates of 100–5000 mV s−1 at a Pt macrodisk working electrode.](/image/article/2013/DT/c2dt31622f/c2dt31622f-f1.gif) | ||
| Fig. 1 Overlaid cyclic voltammograms of B(C6F5)3 in DFB (5.1 mM, 0.05 M [nBu4N][B(C6F5)4]) recorded at scan rates of 100–5000 mV s−1 at a Pt macrodisk working electrode. | ||
The observed voltammetric behaviour is indicative of an EC process, using Testa–Reinmuth notation.55 B(C6F5)3 undergoes a heterogeneous, electrochemically quasi-reversible reduction (E-step) at the electrode. This is rapidly followed by an irreversible, homogeneous chemical step in the solution (C-step) to form an electroinactive product. As the scan rate is increased, the kinetics of the chemical follow-up step begin to be outrun on the voltammetric timescale, and the re-oxidation of the B(C6F5)3˙− intermediate back to the neutral B(C6F5)3 parent compound is observed (Scheme 1).
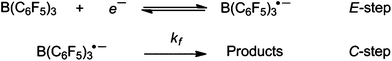 | ||
| Scheme 1 Postulated EC mechanism of B(C6F5)3 reduction. | ||
Upon closer inspection, additional small reduction and corresponding oxidation waves are also observed at more cathodic potentials than the main B(C6F5)3 reduction peak. Their broad, symmetric wave shape appears to be characteristic of surface-adsorbed species. In light of the NMR analysis of the reaction products (discussed below) we tentatively attribute this to the formation of radical species on the electrode surface during the decomposition process of B(C6F5)3˙−.
In order to quantitatively investigate the mechanism of B(C6F5)3 reduction, we first need to determine the number of electrons (n) involved in the reduction process. The diffusion coefficient (D) of the neutral B(C6F5)3 is also required. Values of n and D were determined simultaneously by performing potential-step chronoamperometry at a microdisk electrode and numerically fitting the experimental data using the Shoup–Szabo approximation.56 This accurately predicts the current–time response over the entire time domain to a maximum error of less than 0.6% provided that both the concentration of the redox active species and the radius of the microelectrode are known.56 Chronoamperomograms were recorded for the reduction of B(C6F5)3 in both CH2Cl2 and DFB, and are shown in Fig. 2 and ESI 2† along with the Shoup–Szabo best fits calculated in Origin™. The Shoup–Szabo best fits confirm that B(C6F5)3 undergoes a one-electron (n = 1) reduction in both solvent systems, with diffusion coefficients of 8.5 ± 0.1 × 10−6 and 3.9 ± 0.1 × 10−6 cm2 s−1 for CH2Cl2 and DFB respectively. The difference in the value of the diffusion coefficient between CH2Cl2 and DFB likely reflects the greater viscosity of DFB.
![Experimental chronoamperogram (crosses) and Shoup–Szabo best fit (solid line) for the reduction of B(C6F5)3 in CH2Cl2 (4.8 mM, 0.05 M [nBu4N][B(C6F5)4]) at a 31 μm radius Pt microdisk.](/image/article/2013/DT/c2dt31622f/c2dt31622f-f2.gif) | ||
| Fig. 2 Experimental chronoamperogram (crosses) and Shoup–Szabo best fit (solid line) for the reduction of B(C6F5)3 in CH2Cl2 (4.8 mM, 0.05 M [nBu4N][B(C6F5)4]) at a 31 μm radius Pt microdisk. | ||
To confirm the diffusion coefficients of B(C6F5)3, steady-state (scan rate = 5 mV s−1) linear sweep voltammetry was performed at a microdisk electrode in both solvent systems (ESI 3†). Assuming a one-electron reduction process, the diffusion coefficient of B(C6F5)3 can be determined from the measured steady-state limiting current,50 and was found to be 8.4 ± 0.1 × 10−6 and 4.7 ± 0.1 × 10−6 cm2 s−1 for CH2Cl2 and DFB respectively. Considering the experimental error encountered in accurately measuring a steady state current, these D values are in excellent agreement with those obtained using chronoamperometry.
Digital simulation of mechanistic and kinetic parameters
Having ascertained both the number of electrons transferred during the reduction of B(C6F5)3 and its diffusion coefficient in the solvents studied, we next performed digital simulations of the experimentally observed cyclic voltammetric data in order to extract kinetic and thermodynamic parameters. A variety of plausible mechanisms for the decay of B(C6F5)3˙− were simulated. These included unimolecular chemical decomposition to form further redox active products (ECE), disproportionation (DISP), and bimolecular radical recombination (EC2 or EC2E) mechanisms. None of these mechanisms were found to fit the observed voltammetry. However, the voltammetric reduction of B(C6F5)3 produced very good fits between simulation and experiment, as shown in Fig. 3a and 3b, when modelled as an EC process (Scheme 1). The globally optimized parameters for the electrochemical reduction (standard potential, E0; charge transfer coefficient, α; and standard electron transfer rate constant, k0) and the pseudo-first order rate constant, kf, for the homogeneous chemical decay step are given in Table 1.![Experimental (solid line) and simulated (open circles) overlaid cyclic voltammograms of B(C6F5)3 (4.9 mM, 0.05 M [nBu4N][B(C6F5)4]) at scan rates of ν = 100, 500, 1000, 2000 and 5000 mV s−1 for dichloromethane (top) and 1,2-difluorobenzene (bottom) electrolyte solvents. Inset: plots comparing simulated (open circles) and experimental (closed circles) peak potentials (Ep) and peak currents (Ip) vs. the logarithm of scan rate (ν).](/image/article/2013/DT/c2dt31622f/c2dt31622f-f3.gif) | ||
| Fig. 3 Experimental (solid line) and simulated (open circles) overlaid cyclic voltammograms of B(C6F5)3 (4.9 mM, 0.05 M [nBu4N][B(C6F5)4]) at scan rates of ν = 100, 500, 1000, 2000 and 5000 mV s−1 for dichloromethane (top) and 1,2-difluorobenzene (bottom) electrolyte solvents. Inset: plots comparing simulated (open circles) and experimental (closed circles) peak potentials (Ep) and peak currents (Ip) vs. the logarithm of scan rate (ν). | ||
| Parameter | CH2Cl2 | DFB |
|---|---|---|
| E 0/V vs. Cp2Fe0/+ | −1.79 ± 0.01 | −1.65 ± 0.01 |
| α | 0.49 ± 0.02 | 0.52 ± 0.02 |
| k 0/10−3 cm2 s−1 | 13 ± 0.3 | 11 ± 0.2 |
| DB(C6F5)3/10−6 cm2 s−1 | 8.5 ± 0.1 | 3.9 ± 0.1 |
| DB(C6F5)3˙−/10−6 cm2 s−1 | 8.4 ± 0.1 | 3.9 ± 0.1 |
| k f/s−1 | 6.1 ± 0.1 | 7.7 ± 0.2 |
The values for the standard reduction potential of B(C6F5)3 are within the error range of the value predicted by Cummings et al.44 and the value reported in our earlier work for the direct measurement of the B(C6F5)3 reduction potential.45 The ca. 200 mV difference between the value we previously reported and those herein reflects the subtle but important difference between the mid-peak potential, Emid, that we reported previously, and the thermodynamic standard potential, E0, obtained via simulation. The modest value of the standard electron transfer rate constant (k°) suggests that the reduction of B(C6F5)3 is an electrochemically quasi-reversible process (vide infra). However, the chemical reactivity of B(C6F5)3˙− limits the observation of the corresponding (oxidative) back peak, except at relatively fast scan rates. B(C6F5)3˙− undergoes a rapid follow-up chemical reaction with pseudo-first order rate constants (kf) of 6.1 ± 0.1 and 7.7 ± 0.1 s−1 in CH2Cl2 and DFB respectively. These values obtained in weakly coordinating solvents are ca. three orders of magnitude larger than the decomposition rate constant reported by Norton et al. using EPR measurements in the donor solvent, THF.42 Indeed the follow-up reaction in CH2Cl2 or DFB occurs so rapidly as to preclude any kinetic measurements using EPR techniques.57
Computational modelling of B(C6F5)3˙−
Given the Lewis acidity of B(C6F5)3 it is somewhat curious that the rate of electron transfer, k0, is only of the order of 10−2 cm s−1 – i.e. it exhibits quasi-reversible electron transfer kinetics. We therefore performed DFT computational modelling of the B(C6F5)3 and B(C6F5)3˙− species to ascertain the optimised (gas phase) geometry, SOMO, and comparative charge and spin density distributions. For spin-unrestricted type of calculations the unpaired molecular orbital of the B(C6F5)3˙− complex is best represented by a spin down (beta) SOMO shown in ESI 4a.†Fig. 4 shows the resulting spin density distribution in B(C6F5)3˙− and ESI 4b and ESI 4c† show the charge distribution based on Mulliken electron population analysis of B(C6F5)3 and B(C6F5)3˙− respectively.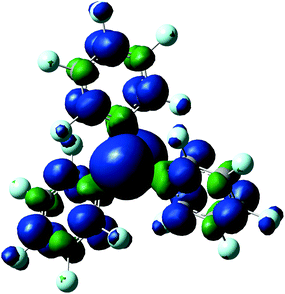 | ||
| Fig. 4 The spin density distribution calculated using the spin-unrestricted B3LYP 6-311+G(d,p) basis set for the corresponding SOMO of the B(C6F5)3˙− radical anion. | ||
The optimised geometries of B(C6F5)3 and B(C6F5)3˙− reveal little deviation from planarity around the trigonal planar boron centre, although the torsional angle between the aryl rings and the central plane containing the boron atom is reduced from 37°, in the case of B(C6F5)3, to 34°, in the B(C6F5)3˙− species. This is due to delocalisation of some spin density onto the perfluoroaryl rings, within the SOMO.
Marcus theory describes the rate of adiabatic electron transfer in terms of the reorganisation energy (λ). This is comprised of contributions from inner (λi) and outer (λo) sphere electron transfer. λi describes changes in bond strength and bond angles during electron transfer, and λo depends on the reorientation of solvent dipoles and electronic polarization within the solvent molecules.50 Given that DFT calculations indicate there is no significant change between the structures of B(C6F5)3 and B(C6F5)3˙−, we infer that the solvent reorganisation energy (λo) is the rate-limiting factor during electron transfer. The relationship between Marcus theory and Butler–Volmer kinetics applied in our voltammetric simulation are described via the charge transfer coefficient, α:
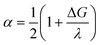
Given that we obtain values of α that are close to 0.5 in either solvent system (Table 1), it is confirmed that the reorganisation energy is very much larger than the Gibbs energy for this reaction.
DFT modelling shows that, in its reduced form, both spin and charge density are predominantly located on the central boron atom of the B(C6F5)3˙− radical anion. Together with the indication that solvent reorganisation is strongly coupled to the electron transfer, these findings may indicate that decomposition of the B(C6F5)3˙− radical anion predominantly proceeds via solvolysis at the boron centre to form four-coordinate borate species (vide infra).
NMR and MS characterisation of B(C6F5)3˙− decomposition products
Finally, we attempted to elucidate the reaction products resulting from the decay of B(C6F5)3˙−via11B and 19F NMR spectroscopy. A colourless solution of B(C6F5)3 (49 mg, 0.1 mmol) in dry, degassed CH2Cl2 or DFB (4 mL) was added to a saturated brown/yellow solution of Cp*2Co (33 mg, 0.1 mmol) in either CH2Cl2 or DFB (4 mL) under an inert N2 atmosphere. Immediately upon mixing, the solution initially turned a deep blue colour, indicative of the B(C6F5)3˙− radical anion, which then very rapidly formed a dark yellow/brown solution upon standing. An aliquot was taken and NMR analysis performed directly on the reaction mixture using a C6D6 insert. Then, the solvent was removed and rigorously dried in vacuo to yield a brown residue, which was taken up in either CDCl3 or CD3CN (0.8 mL) for further NMR analysis.The 11B NMR (96.3 MHz, CH2Cl2 with C6D6 insert) spectrum obtained after the chemical reduction of B(C6F5)3 in CH2Cl2 reveal a mixture of five radical decomposition products formed via reaction with the solvent. These are listed in Table 2. The identity of each product has also been tentatively assigned, where possible, by comparison with known literature values.58–60 The doublet observed at δ −0.52 ppm has a coupling constant of 77 Hz, hence we assign this to an as yet unidentified four-coordinate borate species containing one B–H bond (vide infra) representing ca. 18% of the products formed.
The corresponding 19F NMR (282 MHz, CH2Cl2 with C6D6 insert) spectrum of this same sample is complex. Five signals were observed as doublets of multiplets (arising from second-order spin–spin coupling) between δ −132.0 and −135.9 ppm, corresponding to ortho-F nuclei on the aryl rings. A further series of broad overlapping multiplets were observed from δ −162.0 to −165.5 ppm and from δ −165.7 to −168.4 ppm, corresponding to aryl fluorine nuclei in the para- and meta-positions respectively. Whilst these latter overlapping signals could not be assigned, the ortho-F signals are listed in Table 3 together with their relative product distribution determined by integration of the peaks. A tentative assignment has been made by comparison to literature values.58–60
| δ/ppm | Multiplicity | Relative % product distribution | Assignment |
|---|---|---|---|
| a Ref. 59. b Ref. 60. c Ref. 58. d See Experimental section. e Speculative – see text. | |||
| −132.4 | dm | 39 ± 1 | [ClB(C6F5)3]−a |
| −133.7 | dm | 14 ± 2 | [HB(C6F5)3]−b |
| −134.4 | dm | 21 ± 2 | [Cl2B(C6F5)2]−c |
| −135.4 | m | 6 ± 1 | [B(C6F5)4]−d |
| −135.8 | dm | 20 ± 3 | [HClB(C6F5)2]−e |
The 19F peak at δ −135.8 ppm is as yet unassigned, but it is likely to correspond to the unidentified product giving rise to the doublet at δ −0.52 ppm in the 11B NMR spectrum. Based on chemical intuition, if we speculate that this is in fact a product of the form [HClB(C6F5)2]− then it forms ca. 20% of the product distribution (based on integration of the 19F NMR signals).
When the reduction of B(C6F5)3 is performed in DFB three signals are observed in the 11B NMR spectrum (96.3 MHz, DFB with C6D6 insert) at: δ −3.88 (sharp s, unassigned), −13.28 (s, [B(C6F5)4]−), and −0.28 to 1.16 (br m, unassigned) ppm. The relative product distribution of these peaks is ca. 55, 5, and 40% respectively. The broad multiplet between −0.28 and 1.16 ppm comprising ca. 40% of products most likely corresponds to several structurally related products giving rise to overlapping signals. This is further borne out upon examination of the 19F NMR spectrum (282 MHz, DFB with C6D6 insert) whereby a complex series of overlapping muliplets is observed between δ −131.7 to −135.4 (dm, ortho-aryl F), −161.4 to −164.7 (tm, para-aryl F), and −166.0 to −166.6 (m, meta-aryl F) ppm, indicative of multiple products containing fluorinated aryl rings. No significant change in any of the NMR spectra was observed upon exchanging the solvent to CDCl3 or CD3CN.
The lack of NMR data for borate and borane species in DFB hindered full assignment of the products. We can only assign the tetrakis(pentafluorophenyl)borate species with any certainty. However, given that the rate of decomposition of B(C6F5)3˙− is pseudo-first order and similar in either solvent system, it is likely that solvolysis occurs in DFB as it does in dichloromethane – the solvent being in vast excess in both cases. Comparison of the 11B and 19F NMR spectra of authentic samples of [tBu4N][BF4] and [tBu4N][FB(C6F5)3] revealed no evidence for the formation of any reaction products containing B–F bonds in either the dichloromethane or DFB solvent systems. Hence, we speculate that the unidentified (major) products of the decomposition of B(C6F5)3˙− in DFB are likely to be borate species of the form [(C6F5)3−xB(C6H4F)x]−. Furthermore, 1H NMR (CDCl3) analysis of the products from either solvent reaction system revealed no evidence of H-abstraction from the Cp*2Co.
Whilst the decomposition of the B(C6F5)3˙− radical anion via a solvolytic pathway may not be surprising, the key point to note is that, in contradiction to Norton's earlier suggestion,42 decamethylcobaltocenium tetrakis(pentafluorophenyl)borate is the very minor (ca. 5%) product of this reaction. Further, whilst CH2Cl2 is known to be prone to radical attack, DFB is usually considered to be less susceptible.61 Yet, the B(C6F5)3˙− radical anion intermediate must be a sufficiently reactive species as to decompose via solvolytic pathways at a similar rate in either weakly coordinating solvent.
Interestingly mass spectrometric characterisation (ESI-MS) of the reaction products from either DFB or dichloromethane could only detect one product with molecular ion peaks at m/z values of 678.90 (100%, M−), 677.80 (24.98%) and 679.90 (25.75%) Da. This is indicative of the tetrakis(pentafluorophenyl)borate anion; however, given the likelihood of fragmentation and recombination reactions in the mass spectrometer this observation must be interpreted with some caution.
Conclusions
We have studied the direct voltammetric reduction of tris(pentafluorophenyl)borane, B(C6F5)3, in two weakly coordinating solvents, dichloromethane and 1,2-difluorobenzene. In either case cyclic voltammetric combined with digital simulations indicate that the reaction follows an EC mechanism whereby the electro-generated B(C6F5)3˙− radical anion undergoes a rapid chemical step in solution to form redox inactive products. Multinuclear NMR analysis of these products generated by the chemical reduction of B(C6F5)3 with decamethylcobaltocene are indicative of the formation of several four-coordinate borate species arising from solvolytic radical reaction pathways. Solvolysis resulting in reactivity predominantly at the boron centre is further supported by spin density and charge distribution DFT calculations.Chronoamperometry at a microdisk electrode has allowed us to ascertain that the reduction of B(C6F5)3 is indeed a one-electron process and report diffusion coefficients in each solvent.
Values of the pseudo-first order rate constants for the chemical decomposition of the B(C6F5)3˙− radical anion were determined to be 6.1 ± 0.1 s−1 and 7.7 ± 0.2 s−1 in dichloromethane and 1,2-difluorobenzene respectively. The thermodynamic standard potential, E°, of B(C6F5)3 was also extracted for the first time with values of −1.79 ± 0.1 V and −1.65 0.1 V vs. ferrocene/ferrocenium in dichloromethane and 1,2-difluorobenzene respectively. These values are in close agreement with previous estimates based on either the reduction peak potentials44 or the measured mid-peak potentials,45 which do not strictly correspond to the thermodynamic potential, E°.
The rate of decomposition of the radical anion is sufficiently fast in solvents of low donor strength that we were unable to measure a signal from B(C6F5)3˙− using EPR experiments, even at low temperatures – in stark contrast to previous reports using strong donor solvents.42
Thus, almost fifty years after tris(pentafluorophenyl)borane was first discovered, we are able to report thermodynamic and kinetic parameters relating to its redox properties in selected weakly donor solvents. Once again, we emphasise the importance of carefully considering the choice of solvent when attempting to study the chemistry of this and its related electrophilic and Lewis acidic boranes.
Acknowledgements
EJL thanks the EPSRC for financial support via a Doctoral Training Account studentship. GGW thanks the Royal Society for support via a University Research Fellowship. AEA thanks Imperial College, London for support via a Junior Research Fellowship and the Royal Commission for the Exhibition of 1851 for a Research Fellowship. The authors thank Dr Myles R. Cheesman and the Henry Welcome Unit for Biological EPR, University of East Anglia for assistance with EPR studies.Notes and references
- A. G. Massey, A. J. Park and F. G. A. Stone, Proc. Chem. Soc., London, 1963, 212 CAS.
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS.
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1966, 5, 218–225 CrossRef CAS.
- M. A. Beckett, D. S. Brassington, S. J. Coles and M. B. Hursthouse, Inorg. Chem. Commun., 2000, 3, 530–533 CrossRef CAS.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- G. Erker, Dalton Trans., 2005, 1883 RSC.
- W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS.
- J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS.
- J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS.
- K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527–538 CrossRef CAS.
- R. E. LaPointe, G. R. Roof, K. A. Abboud and J. Klosin, J. Am. Chem. Soc., 2000, 122, 9560–9561 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS.
- A. Bernsdorf, H. Brand, R. Hellmann, M. Köckerling, A. Schulz, A. Villinger and K. Voss, J. Am. Chem. Soc., 2009, 131, 8958–8970 CrossRef CAS.
- X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10015–10031 CrossRef CAS.
- E. Y. X. Chen and T. J. Marks, Chem. Rev.-Columbus, 2000, 100, 1391–1434 CAS.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. H. Hughes and M. Bochmann, Organometallics, 2002, 21, 451–453 CrossRef CAS.
- M. Bochmann, S. J. Lancaster, M. D. Hannant, A. Rodriguez, M. Schormann, D. A. Walker and T. J. Woodman, Pure Appl. Chem., 2003, 75, 1183–1196 CrossRef CAS.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS.
- D. W. Stephan, Dalton Trans., 2009, 3129 RSC.
- A. L. Kenward and W. E. Piers, Angew. Chem., Int. Ed., 2008, 47, 38–41 CrossRef CAS.
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367–2378 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701 RSC.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., 2007, 119, 5056–5059 CrossRef.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS.
- M. Ullrich, K. S.-H. Seto, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 2335 RSC.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS.
- G. C. Welch, J. D. Masuda and D. W. Stephan, Inorg. Chem., 2005, 45, 478–480 CrossRef.
- S. D. Tran, T. A. Tronic, W. Kaminsky, D. Michael Heinekey and J. M. Mayer, Inorg. Chim. Acta, 2011, 369, 126–132 CrossRef CAS.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef.
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS.
- R. C. Neu, E. Otten, A. Lough and D. W. Stephan, Chem. Sci., 2010, 2, 170–176 RSC.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS.
- C. B. Caputo and D. W. Stephan, Organometallics, 2011, 31, 27–30 CrossRef.
- C. J. Harlan, T. Hascall, E. Fujita and J. R. Norton, J. Am. Chem. Soc., 1999, 121, 7274–7275 CrossRef CAS.
- J. Karl, G. Erker and R. Fröhlich, J. Am. Chem. Soc., 1997, 119, 11165–11173 CrossRef CAS and references therein.
- C. J. Beddows, A. D. Burrows, N. G. Connelly, M. Green, J. M. Lynam and T. J. Paget, Organometallics, 2000, 20, 231–233 CrossRef.
- R. J. Kwaan, C. J. Harlan and J. R. Norton, Organometallics, 2001, 20, 3818–3820 CrossRef CAS.
- L. H. Doerrer, A. J. Graham, D. Haussinger and M. L. H. Green, J. Chem. Soc., Dalton Trans., 2000, 813–820 RSC.
- S. A. Cummings, M. Iimura, C. J. Harlan, R. J. Kwaan, I. V. Trieu, J. R. Norton, B. M. Bridgewater, F. Jäkle, A. Sundararaman and M. Tilset, Organometallics, 2006, 25, 1565–1568 CrossRef CAS.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS.
- W. E. Geiger and F. Barrière, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS.
- S. J. Lancaster, ChemSpider SyntheticPages, 2003, http://cssp.chemspider.com/215.
- E. Martin, D. L. Hughes and S. J. Lancaster, Inorg. Chim. Acta, 2010, 363, 275–278 CrossRef CAS.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS.
- R. G. Compton and C. E. Banks, Understanding Voltammetry, Imperial College Press, 2nd revised edn, 2011 Search PubMed.
- G. Gritzner and J. Kůta, Electrochim. Acta, 1984, 29, 869–873 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant and others, Gaussian, Inc., Wallingford, CT, 2004.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. C. Testa and W. H. Reinmuth, Anal. Chem., 1961, 33, 1320–1324 CrossRef CAS.
- D. Shoup and A. Szabo, J. Electroanal. Chem., 1982, 140, 237–245 CrossRef CAS.
- The reaction between Cp*2Co and B(C6F5)3 and subsequent decomposition of the radical anion occurred too rapidly (in less than 5 s) to be able to measure the EPR signal of the B(C6F5)3˙− radical anion. This was despite our best efforts, including freezing one or both solutions to liquid nitrogen temperatures prior to mixing and then allowing to thaw to −50 °C in the spectrometer. Visually this corresponded to a rapid (almost immediate) colour change, on mixing, from a blue solution to a yellow solution. This indicated formation of the decamethylcobaltocenium cation, even at −78 °C.
- C. Jiang, O. Blacque and H. Berke, Organometallics, 2009, 28, 5233–5239 CrossRef CAS.
- C. Bibal, C. C. Santini, Y. Chauvin, C. Vallée and H. Olivier-Bourbigou, Dalton Trans., 2008, 2866 RSC.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS.
- T. R. O'Toole, J. N. Younathan, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1989, 28, 3923–3926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammogram, chronoamperometry, linear sweep voltammetry, DFT calculations (PDF). See DOI: 10.1039/c2dt31622f |
| This journal is © The Royal Society of Chemistry 2013 |