 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous hydrolysis and hydrogenation of cellobiose to sorbitol in molten salt hydrate media
Jianrong
Li
,
Helena S. M. P.
Soares
,
Jacob A.
Moulijn
and
Michiel
Makkee
*
Catalysis Engineering, ChemE, Delft University of Technology, Julianalaan 136, 2628 BL, Delft, The Netherlands. E-mail: m.makkee@tudelft.nl; Fax: +31 15 278 5006; Tel: +31 15 278 1391
First published on 12th March 2013
Abstract
The hydrolysis and hydrogenation of cellobiose (4-O-β-D-glucopyranosyl-D-glucose) in ZnCl2·4H2O solvent was studied to optimize the conditions for conversion of lignocellulose (the most abundant renewable resource) into sorbitol (D-glucitol). Water at neutral pH does not allow hydrolysis of cellobiose under the conditions of the experiments (up to 125 °C, 4 h), but relatively fast hydrogenation of cellobiose into 3-β-D-glucopyranosyl-D-glucitol over a Ru/C catalyst in the presence of H2 takes place. In ZnCl2·4H2O hydrolysis does take place in the range of temperatures studied (75–125 °C) and simultaneously hydrogenation (H2, Ru/C), though at a lower rate than that in neutral water. Thus, a one-pot conversion of cellobiose into sorbitol is possible. The hydrolysis is the rate limiting reaction, but the selectivity to sorbitol is determined by the rate of hydrogenation. Under optimal conditions the yield of sorbitol is ≥95%. The kinetic pathways are discussed.
Introduction
The sustainable production of fuels and chemicals from biomass has attracted broad interest from the catalysis and chemical engineering community.1,2 At present much attention is given to the so-called 2nd generation of biomass resources, viz., lignocellulosics, mainly because of their abundant presence without direct competition with the food chain. Cellulose as the most abundant fraction of lignocellulosics especially deserves attention (i) because of its crystalline structure and since the glucosidic linkages are in the β configuration, it is much more stable in most processes, for instance under fermentation conditions; and (ii) because of its simple, regular structure and well-defined composition, it is well suited for the production of platform molecules for the chemical industry.3–9Conversion of cellulose into different platform chemicals is a hot topic.10,11 These platform chemicals include glucose, hexitols (sorbitol), HMF,12 gluconic acid, alkyl glycosides, ethylene glycol,13 and levulinic acid.14 Conversion of cellulose into sorbitol through glucose is possible by a combination of hydrolysis and hydrogenation.
Hydrolysis of glycosidic bonds in cellulose to glucose can be achieved with hot water;3,4 with concentrated or diluted mineral acids such as H2SO4,15–17 HCl; solid acid catalysts such as, Nafion, Amberlyst, H-mordenite (H-zeolite), carbon based solid acids,18,19 and sulfonated silica–carbon nanocomposites which were found to be more active and selective than the pure carbon based solid acid catalysts;20 phosphate salts;21 enzymes;22 ionic liquids such as 1-butyl-3-methylimidazolium chloride;23,24 basic solvents such as NaOH in water;25 inorganic molten salt hydrates such as ZnCl2·4H2O and LiCl·4H2O.6,26
Inorganic molten salt hydrates as solvents for cellulose have been studied earlier by Fischer et al.27,28 Zhang and coworkers used ionic liquids in the hydrolysis of cellulose to glucose and further dehydration of D-glucose viaD-fructose into 5-hydroxymethylfurfural (HMF).29,30
Hydrogenation catalysts commonly used in the literature are supported metal catalysts,31 such as ruthenium on carbon,32–38 ruthenium on carbon nanotubes,39,40 Ru-loaded zeolites,41 nickel on carbon nanofibers,42 tungsten carbide on carbon nanotubes;43 ruthenium nano-particles and promoted nickel catalysts were also reported as catalysts for conversion of cellulose into polyols.44,45 In this study Ru/C catalysts have been selected, because in contrast to RANEY®-type Ni catalysts in the hydrogenation of glucose no leaching was observed.37
In previous work, we have selected molten salt hydrate ZnCl2·4H2O as the solvent and 5 wt% ruthenium on carbon as a hydrogenation catalyst for the study of cellulose conversion. We reported the conversion of cellulose into sorbitol (D-glucitol) and subsequent dehydration into isosorbide in molten salt hydrate ZnCl2·4H2O, as illustrated in Fig. 1.6
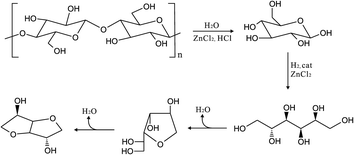 | ||
| Fig. 1 Conversion of cellulose into isosorbide. The reaction pathway consists of hydrolysis of cellulose to glucose, hydrogenation of glucose to sorbitol, dehydration of sorbitol to sorbitan (only 1,4-anhydrosorbitol is shown in the scheme), and sequential dehydration of sorbitan to isosorbide.6 | ||
The yield of glucose from cellulose was over 90%. Compared to reported processes converting biomass, this number is high. Nevertheless the development of a practical process calls for an intensive programme because (i) the intermediate product glucose is prone to degradation, calling for a precise engineering of the process; and (ii) the separation of this type of products from the solvent, ZnCl2·4H2O, has not been reported in the literature.
In this paper, we focus on the production of sorbitol and the conditions for obtaining high sorbitol yield. Primary study of this work is a part of a PCT international application.46
In the hydrolysis step the glucose produced is a thermally rather unstable molecule. At higher temperature isomerization to fructose occurs and large amounts of degradation products are formed.4 An advantage of the reaction scheme, as illustrated in Fig. 1, is that glucose is converted into the much more stable sorbitol. It is self-evident that the engineering of coupling of the two reactions, glucose formation and conversion, is crucial.
An exploratory study aimed at optimizing the conversion of the solid cellulose into glucose is expected to be very time-consuming. When a simple model compound could be used the research would be greatly facilitated. An obvious choice is cellobiose, (4-O-β-D-glucopyranosyl-D-glucose), which is the repeat unit of cellulose ((1,4-O-β-glucopyranosyl)n−1-D-glucose).
Several articles on a one-pot conversion of cellobiose or cellulose into sorbitol have been reported. Deng et al. used 68% concentrated HNO3 pretreated carbon nano-tube supported ruthenium as catalyst, and successfully converted cellobiose into sorbitol, a sorbitol yield of 87% was achieved at 185 °C after 3 hours reaction.3 Luo et al. showed that in water at elevated temperature in the presence of Ru/C catalysts up to 20% hexitols were produced from cellulose.4 Yan et al. reported the use of ruthenium nano-clusters as a hydrogenation catalyst under acidic conditions (pH = 2, HCl is generated during the formation of Ru nanoparticles) and 100% sorbitol yield was obtained at 120 °C after 12 hours reaction with a catalyst to cellobiose ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 (on weight basis).5 Fukuoka and Dhepe studied one-pot conversion of cellulose into sugar alcohols by supported metal catalysts and reported 30% sugar alcohols yield after 24 hours at 50 bar hydrogen and 190 °C with a catalyst to cellulose ratio of 1:2.47
:500 (on weight basis).5 Fukuoka and Dhepe studied one-pot conversion of cellulose into sugar alcohols by supported metal catalysts and reported 30% sugar alcohols yield after 24 hours at 50 bar hydrogen and 190 °C with a catalyst to cellulose ratio of 1:2.47
For a practical process the yield of sorbitol has to be high (typically >90%) and the catalyst to substrate ratio should be economically acceptable, the rates of the conversion steps involved (dissolution cellulose, hydrolysis, hydrogenation steps) should be satisfactory. The results of the model compound study should be representative for cellulose conversion. In this paper, we report an exploratory optimization study on the one-pot conversion of cellobiose into sorbitol in ZnCl2·4H2O solvent. The reaction pathway and the kinetics network will be discussed.
Experimental
Materials
Zinc chloride (≥98.0%), D-(+)-glucose (>99.5%), ruthenium 5 wt% on carbon and D-(−) fructose (≥99%) were obtained from Sigma-Aldrich. D-Sorbitol (D-glucitol) (≥99.5%) and D-(+)-cellobiose (≥99.0%) were obtained from Fluka.Reactions
The reactions were carried out in a mini-multi-batch reactors set-up (ProSense); the reactors (autoclaves) have a volume of 16 ml, are made of Hastelloy C, equipped with a magnetic stirrer (stirring rate up to 2000 rpm) and a K-type thermocouple to measure the temperature of the liquid (Tliquid). A separate heater is available allowing a temperature of up to 300 °C, and the maximum operation pressure is 100 bar.Analysis and characterization methods
:25), respectively.
Results
Cellobiose characterization
SEM and XRD results showed that cellobiose has a high crystallinity; using Scherrer's equation, we calculated that the crystals had a lateral dimension of 140 nm. TGA data showed that the cellobiose crystals contained very little free moisture, about 0.6% weight. In the region of 235–360 °C, cellobiose lost main part of its weight, corresponding with the thermal decomposition/degradation of cellobiose.Hydrolysis
In demi-water cellobiose did not react under the conditions applied (up to 125 °C, 4 h). In ZnCl2 hydrate hydrolysis of cellobiose occurred. The results of a typical example at 95 °C at varying reaction times are given in Fig. 2. Besides glucose, mannose and fructose are observed, showing the mild isomerization/epimerization activity of the system. Furthermore, 5-hydroxymethylfurfural (HMF) is observed, probably formed by dehydration of fructose. Some “unknown” products are observed. The structures of the unknowns have not yet been identified. The amount of byproducts (HMF and unknowns) increases with the reaction time.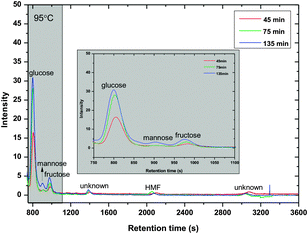 | ||
| Fig. 2 Hydrolysis of cellobiose at 95 °C at varying reaction times (0.5 g cellobiose, 6 g ZnCl2·4H2O). | ||
The conversion at various temperatures is shown in Fig. 3. At around 80 °C the conversion was limited, but the selectivity to glucose was high. No byproducts were formed, only glucose was observed. At 95 °C the rate was 2–3 times higher, but byproducts were observed (Fig. 2), resulting in loss of selectivity.
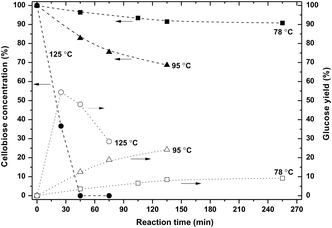 | ||
| Fig. 3 Product distribution of the hydrolysis of cellobiose at varying times and temperatures (0.5 g cellobiose, 6 g ZnCl2·4H2O). The dashed lines are guidance for the eyes. | ||
Full conversion was only achieved at 125 °C but at the expense of a low selectivity to glucose. At this temperature the glucose yield went through a maximum, illustrating the serial kinetic pathway with glucose as the main primary product. The maximum yield of glucose was only 55%.
The loss of selectivity as a function of temperature and conversion can be best seen in Fig. 4. Clearly, at high temperature the conversion is high but the selectivity is disappointing. The relatively high glucose selectivity point at 125 °C corresponds to, compared to the low temperature data, a short reaction time; at prolonged reaction time the glucose selectivity drops enormously, as is shown in Fig. 4.
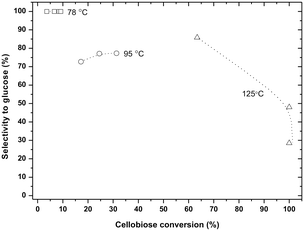 | ||
| Fig. 4 Plot of selectivity to glucose versus conversion for the hydrolysis of cellobiose (0.5 g cellobiose, 6 g ZnCl2·4H2O). The dashed lines are guidance for the eyes. The data correspond to those in Fig. 3. | ||
Simultaneous hydrolysis and hydrogenation
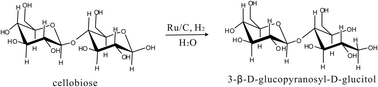 | ||
| Fig. 5 Cellobiose conversion into 3-β-D-glucopyranosyl-D-glucitol. | ||
Typical results are shown in Fig. 6. A clean reaction was observed with only one product, being 3-β-D-glucopyranosyl-D-glucitol. After 135 min at 95 °C, 90% conversion was reached with 100% selectivity to 3-β-D-glucopyranosyl-D-glucitol.
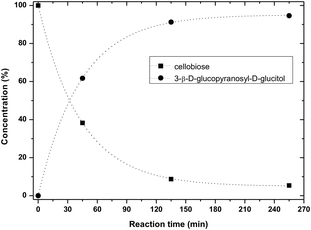 | ||
| Fig. 6 Cellobiose hydrogenation in water at 95 °C (0.5 g cellobiose, 6 g water, 0.025 g Ru/C, PH2 40 bar). The dashed lines are guidance for the eyes. | ||
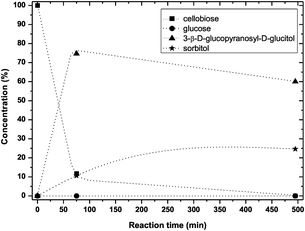 | ||
| Fig. 7 Cellobiose conversion at 95 °C (0.5 g cellobiose, 6 g ZnCl2·4H2O, 0.125 g Ru/C, PH2 40 bar). The dashed lines are guidance for the eyes. | ||
Apparently, 15% of the cellobiose or the intermediate product glucose were converted into byproducts. Clearly, compared to hydrogenation rate the rate of hydrolysis is low, in agreement with the earlier observation that in hydrolysis full conversion is only achieved at 125 °C. Fig. 8 shows the results of simultaneous hydrolysis and hydrogenation at this temperature. Already after 65 minutes 90% yield of sorbitol was realized. At longer reaction time full conversion was observed. Again 3-β-D-glucopyranosyl-D-glucitol as one of the intermediates was formed and the profile shows a maximum. At this temperature the concentration of 3-β-D-glucopyranosyl-D-glucitol did not exceed 10%. The reaction is proceeding in a remarkably clean way. A yield of 95% of sorbitol was obtained with only 5% of byproducts (mannitol and un-assigned products).
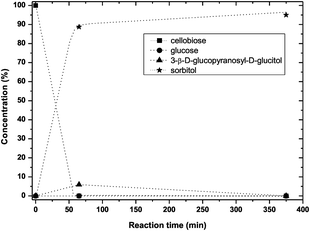 | ||
| Fig. 8 Product distribution for simultaneous hydrolysis and hydrogenation at a high catalyst loading at 125 °C (0.5 g cellobiose, 6 g ZnCl2·4H2O, 0.125 g Ru/C, PH2 40 bar). The dashed lines are guidance for the eyes. | ||
The interplay between hydrolysis and hydrogenation can be illustrated by tuning the rate of hydrogenation. Hydrogenation can be switched on by adding a catalyst.
The effect of different amounts of catalyst on the reaction results at 95 °C is shown in Fig. 9. The higher the loading of Ru/C, the larger is the rate of conversion of cellobiose. At 95 °C with 25 wt% catalyst, the conversion reached 88% after 75 min; with 50 wt% catalyst, full conversion of cellobiose was observed after 75 min. Although high conversions can be obtained, the selectivity to sorbitol was not high. Only 13% of sorbitol and 85% of 3-β-D-glucopyranosyl-D-glucitol were obtained when the catalyst amount was 25 wt% with respect to the cellobiose weight with a reaction time of 75 min.
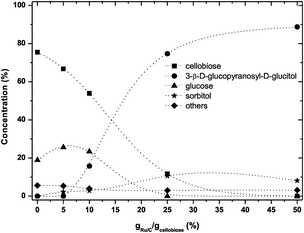 | ||
| Fig. 9 Effect of catalyst loading on the product distribution at 95 °C (0.5 g cellobiose, 6 g ZnCl2·4H2O, varying amounts of Ru/C, PH2 40 bar, 75 min). The dashed lines are guidance for the eyes. | ||
At 125 °C, when the amount of catalyst was varied the product distribution dramatically changes as is illustrated in Fig. 10. At low catalyst loading significant amounts of glucose and excessive amounts (>60%) of degradation products (“others”) were observed, whereas at highest catalyst loading essentially a clean production of sorbitol (95% of yield) was observed.
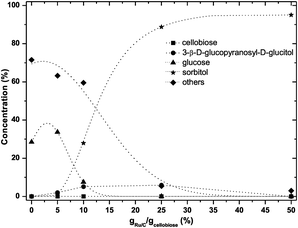 | ||
| Fig. 10 Effect of catalyst loading on the product distribution at 125 °C (0.5 g cellobiose, 6 g ZnCl2·4H2O, varying amounts of Ru/C, PH2 40 bar, 75 min). The dashed lines are guidance for the eyes. | ||
Discussion
The simultaneous hydrolysis and hydrogenation of cellobiose has a spectacular impact on the selectivity of the reacting system. In Fig. 11 we summarize at 125 °C the beneficial influence of coupling hydrolysis with hydrogenation. In the absence of favorable hydrogenation conditions (H2, Ru/C) extensive side reactions are observed (epimerization to mannose and isomerization to fructose, followed by degradation of fructose to HMF and char, unknowns). When the reaction is carried out under favorable hydrogenation conditions the reaction product is very clean. Obviously, ZnCl2·4H2O is a good solvent for dissolving cellulose, but it causes degradation reactions of glucose. Fortunately, hydrogenation in this solvent is possible, producing the more stable sorbitol and, as a consequence, a clean reaction is feasible if we push sufficiently the hydrogenation reaction rate. It should be noted that with the simultaneous hydrolysis and hydrogenation, the carbon-balance is closed and the char formation is avoided.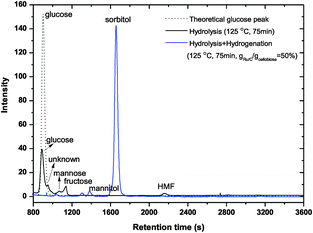 | ||
| Fig. 11 HPLC patterns showing the influence of hydrogenation on the occurrence of glucose degradation reactions at 125 °C (hydrolysis and hydrogenation conditions, see Experimental). | ||
The data for water as solvent show that hydrolysis of cellobiose is not occurring in a neutral environment. This observation is not surprising and agrees with the conclusion that cellobiose will be dissolved, but cannot be hydrolysed in water under the conditions of this study. Hydrogenation does occur and 3-β-D-glucopyranosyl-D-glucitol is formed,3 where the C–O bond in the pyranose ring structure of cellobiose is adsorbed at the catalyst surface, followed by bond breaking and reaction with two activated H atoms.51 An alternative mechanism would be the hydrogenation of the aldehyde bond in the open chain form.
The rate of hydrogenation is high and this route might be an attractive route to the β-isomer of the well-known sugar-free sweetener, isomalt.48,49
In ZnCl2·4H2O under hydrogenation conditions 3-β-D-glucopyranosyl-D-glucitol is also observed, but in addition sorbitol is produced. At lower temperature (95 °C) 3-β-D-glucopyranosyl-D-glucitol is the main product, whereas at higher temperature (125 °C) sorbitol is more predominant, suggesting that under our reaction conditions hydrolysis is the limiting reaction.
The reaction pathways from cellobiose to sorbitol have been studied previously. Yan and coworkers5 proposed that under acidic conditions, cellobiose is firstly hydrolyzed to glucose, subsequently glucose is hydrogenated to sorbitol with a hydrogenation catalyst. Deng and coworkers3 proposed for their system (nitric acid, Ru/C, H2) a different pathway, first hydrogenation takes place of cellobiose to 3-β-D-glucopyranosyl-D-glucitol, followed by consecutive hydrolysis resulting in sorbitol formation.
We conclude that in the ZnCl2 hydrate solvent hydrolysis of cellobiose to glucose and hydrogenolysis of cellobiose to 3-β-D-glucopyranosyl-D-glucitol proceed simultaneously. By tuning the conditions, the acidity, the temperature, the reaction time and the hydrogenation catalyst activity the selectivity can be chosen. We found that the hydrolysis requires higher temperature than the hydrogenation reaction.
We propose the pathway as presented in Fig. 12.
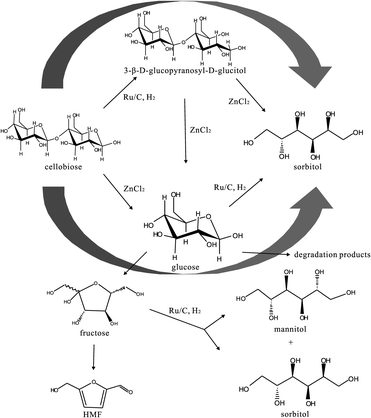 | ||
| Fig. 12 Scheme of the main hydrolysis and the hydrogenation conversion pathway of cellobiose in ZnCl2·4H2O. | ||
According to this scheme, the first step could be either the hydrogenation reaction to form 3-β-D-glucopyranosyl-D-glucitol, followed by the hydrolysis to sorbitol and glucose, or the hydrolysis to two glucose molecules, followed by hydrogenation to sorbitol.
The question arises which of the two pathways to sorbitol is the most important. In order to get information on the relative reactivity we studied the reactivity of the methyl ether, 1-β-O-methylcellobiose (for structure, see Fig. 13):
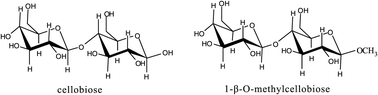 | ||
| Fig. 13 Structures of cellobiose and 1-β-O-methylcellobiose. | ||
Hydrogenation reactivity of glucose and methyl glucose have been compared.49 The reactivity of methyl glucose appeared to be relatively low. It is well-known that the methyl group hinders the ring opening of the pyranose structure.50,51 From our observation that the hydrogenation of methylcellobiose hardly occurred with respect to the hydrogenation of cellobiose, we conclude that the hydrogenation takes place via a ring opening hydrogenolysis mechanism.51 This nicely agrees with the conclusion that the hydrogenation step involves ring structure opening, though a mechanism involving a sequential keto-group hydrogenation is not ruled out. Thus the pathway involves either hydrolysis, followed by hydrogenation or hydrogenation followed by hydrolysis. From the observation that at mild temperature the rate of hydrogenation of cellobiose far exceeds the rate of hydrolysis we tentatively conclude that in our solvent the hydrolysis is the limiting reaction. Referring to the kinetic scheme in Fig. 12, we conclude that both pathways to sorbitol, via 3-β-D-glucopyranosyl-D-glucitol followed by hydrolysis or via direct hydrolysis of cellobiose to glucose followed by hydrogenation, are operative, but the first one is kinetically most important.
It should be noted that the selectivity of the process is critically dependent on the conditions. If the hydrogenation activity is not high enough, glucose will undergo isomerization and extensive degradation reactions. Several products will be formed such as HMF (dehydration product of fructose),29,30 mannitol (hydrogenation product of fructose) and to some extent mannose and fructose. Similarly, if the reaction conditions are changed, the reaction depth might be changed accordingly, and sorbitol might undergo extensive sequential reactions, as for instance reported in the work of Huber et al.52,53 The strategy and importance of tuning parameters for hydrolysis of cellulose has also been reported recently by Van de Vyver et al.54 To control the reaction depth of this work, tuning of the ZnCl2 hydrate medium concentration, the hydrogenation catalyst amount, reaction temperature and reaction time can be done. In addition, adding extra mineral acid such as HCl has been reported, which accelerates the rate determining step, i.e. the hydrolysis step.6,41 However, addition of HCl leads to an increase of the rate of degradation reactions and it might interfere with the hydrogenation catalysis. For a better understanding and evaluation of the ZnCl2 hydrate medium effects, it was decided to use no additional HCl.
In this study sorbitol was the desired product. The kinetic scheme shows that by tuning the conditions the severity of hydrolysis and of hydrogenation can be tuned independently of each other. Thus, the technology presented is flexible and in principle allows the production of different platform molecules, such as glucose, 3-β-D-glucopyranosyl-D-glucitol, sorbitol, and HMF.
For the hydrogenation step the heterogeneous catalyst Ru/C has been selected. The catalyst stability is critical for the process economics. Recently it was reported by Op de Beeck et al.,55 that the stability of the Ru/C hydrogenation catalyst is an issue, because of deposition of polymeric byproducts. Investigations regarding the catalyst stability should have high priority.
Cellobiose has been studied as a model for cellulose. The results suggest that when cellulose is reacted under combined hydrolysis and hydrogenation conditions the oligomers formed are quickly transformed by hydrogenations into the 3-β-D-(poly)-glucopyranosyl-D-glucitol analogues and, as a consequence, the selectivity to sorbitol will be comparable with that presented here for cellobiose. In an exploratory study this was shown to be the case.46
Conclusions
Simultaneous hydrolysis and hydrogenation of cellobiose reduces following reactions as degradation and isomerization, allowing high conversion of cellobiose with extremely high selectivity to sorbitol (95% sorbitol yield). Optimizing the reaction conditions (amount of catalyst, hydrogen partial pressure, the reaction temperature, composition of the molten salt hydrate) allows producing sorbitol at high yield. Reaction pathways have been assigned.The technology presented here allows a flexible production of platform molecules from cellulose. The potential sweetener 3-β-D-glucopyranosyl-D-glucitol can be easily produced from cellulose by hydrogenation of cellobiose.
Acknowledgements
The authors would like to thank BIOeCON for the financial support; Prof. Dr. Ir. Herman van Bekkum is acknowledged for his stimulating discussions.Notes and references
- N. Li, G. A. Tompsett, T. Zhang, J. Shi, C. E. Wyman and G. W. Huber, Green Chem., 2011, 13, 91–101 RSC.
- P. Gallezot, Top. Catal., 2010, 53, 1209–1213 CrossRef CAS.
- W. Deng, M. Liu, X. Tan, Q. Zhang and Y. Wang, J. Catal., 2010, 271, 22–32 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS.
- R. M. de Almeida, J. Li, C. Nederlof, P. O'Connor, M. Makkee and J. A. Moulijn, ChemSusChem, 2010, 3, 325–328 CrossRef.
- P. Yang, H. Kobayashi and A. Fukuoka, Cuihua Xuebao, 2011, 32, 716–722 CAS.
- V. Jollet, F. Chambon, F. Rataboul, A. Cabiac, C. Pinel, E. Guillon and N. Essayem, Green Chem., 2009, 11, 2052–2060 RSC.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 38–50 CAS.
- F. Ilgen, D. Ott, D. Kralisch, C. Reil, A. Palmberger and B. Koenig, Green Chem., 2009, 11, 1948–1954 RSC.
- M.-Y. Zheng, A.-Q. Wang, N. Ji, J.-F. Pang, X.-D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, M. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- S. Deguchi, K. Tsujii and K. Horikoshi, Green Chem., 2008, 10, 623–626 RSC.
- Q. Xiang, Y. Y. Lee, P. O. Pettersson and R. W. Torget, Appl. Biochem. Biotechnol., 2003, 105–108, 505–514 CrossRef CAS.
- H. Zhao, J. H. Kwak, Z. C. Zhang, H. M. Brown, B. W. Arey and J. E. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS.
- M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS.
- S. Van de Vyver, L. Peng, J. Geboers, H. Schepers, F. de Clippel, C. J. Gommes, B. Goderis, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1560–1563 RSC.
- A. Charmot and A. Katz, J. Catal., 2010, 276, 1–5 CrossRef CAS.
- M. A. Torlopov, D. V. Tarabukin, S. V. Frolova, T. P. Shcherbakova and V. V. Volodin, Khim. Rastit. Syr'ya, 2007, 69–76 CAS.
- I. A. Ignatyev, C. Van Doorslaer, P. G. N. Mertens, K. Binnemans and D. E. De Vos, ChemSusChem, 2010, 3, 91–96 CrossRef CAS.
- R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schueth, ChemSusChem, 2010, 3, 266–276 CrossRef CAS.
- G. Bonn, H. Binder, H. Leonhard and O. Bobleter, Monatsh. Chem., 1985, 116, 961–971 CrossRef CAS.
- N. J. Cao, Q. Xu and L. F. Chen, Appl. Biochem. Biotechnol., 1995, 51–52, 21–28 CrossRef.
- H. Leipner, S. Fischer, E. Brendler and W. Voigt, Macromol. Chem. Phys., 2000, 201, 2041–2049 CrossRef CAS.
- S. Fischer, H. Leipner, K. Thuemmler, E. Brendler and J. Peters, Cellulose, 2003, 10, 227–236 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X.-d. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- J. M. Robinson, C. E. Burgess, M. A. Bently, C. D. Brasher, B. O. Horne, D. M. Lillard, J. M. Macias, H. D. Mandal, S. C. Mills, K. D. O'Hara, J. T. Pon, A. F. Raigoza, E. H. Sanchez and J. S. Villarreal, Biomass Bioenergy, 2004, 26, 473–483 CrossRef CAS.
- G. Liang, C. Wu, L. He, J. Ming, H. Cheng, L. Zhuo and F. Zhao, Green Chem., 2011, 13, 839–842 RSC.
- E. Crezee, B. W. Hoffer, R. J. Berger, M. Makkee, F. Kapteijn and J. A. Moulijn, Appl. Catal., A, 2003, 251, 1–17 CrossRef CAS.
- B. W. Hoffer, E. Crezee, P. R. M. Mooijman, A. D. van Langeveld, F. Kapteijn and J. A. Moulijn, Catal. Today, 2003, 79–80, 35–41 CrossRef CAS.
- P. Gallezot, N. Nicolaus, G. Fleche, P. Fuertes and A. Perrard, J. Catal., 1998, 180, 51–55 CrossRef CAS.
- H. Wang, L. Zhu, S. Peng, F. Peng, H. Yu and J. Yang, Renewable Energy, 2012, 37, 192–196 CrossRef CAS.
- W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS.
- C. Du, L. Zhou and S. Zhao, Adv. Mater. Res., 2011, 236–238, 432–436 CrossRef CAS.
- Y. Zhu, Z. N. Kong, L. P. Stubbs, H. Lin, S. Shen, E. V. Anslyn and J. A. Maguire, ChemSusChem, 2010, 3, 67–70 CrossRef CAS.
- A. Shrotri, A. Tanksale, J. N. Beltramini, H. Gurav and S. V. Chilukuri, Catal. Sci. Technol., 2012, 2, 1852–1858 CAS.
- M. Makkee, J. A. Moulijn, J. Li, P. O'Connor, J. C. Rasser, A. E. Rosheuvel and R. M. de Almeida, PCT Int. Appl., WO 2012035160 A1, 2012 Search PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- http://www.polyol.org/fap/fap_isomalt.html .
- J. Kuszmann, G. Medgyes and S. Boros, Carbohydr. Res., 2004, 339, 2407–2414 CrossRef CAS.
- J. F. Ruddlesden, A. Stewart, D. J. Thompson and R. Whelan, Faraday Discuss. Chem. Soc., 1981, 72, 397–411 RSC.
- M. Makkee, A. P. G. Kieboom and H. Van Bekkum, Carbohydr. Res., 1985, 138, 225–236 CrossRef CAS.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS.
- H. Zhang, Y. Cheng, T. P. Vipute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 CAS.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |