 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the selectivity of a supported gold catalyst in solvent- and radical initiator-free aerobic oxidation of cyclohexene†
Daniil S.
Ovoshchnikov‡
a,
Baira G.
Donoeva‡
ab,
Bryce E.
Williamson
a and
Vladimir B.
Golovko
*ab
aDepartment of Chemistry, University of Canterbury, Christchurch 8140, New Zealand. E-mail: vladimir.golovko@canterbury.ac.nz; Fax: +64 3 364 2110; Tel: +64 3 364 2442
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 17th December 2013
Abstract
The selectivity of supported gold catalysts in aerobic oxidation of cyclohexene under solvent-free conditions without addition of a radical initiator was tuned by either WO3 or the metal–organic framework MIL-101, used as a support/co-catalyst. WO3 was found to promote the formation of cyclohexene oxide via reaction of cyclohexenyl hydroperoxide with cyclohexene, while MIL-101 catalysed conversion of cyclohexenyl hydroperoxide to 2-cyclohexen-1-one.
Introduction
The epoxidation of alkenes is an important industrial process for production of fine chemicals.1 Traditionally, it involves the use of stoichiometric oxygen donors,2 peroxycarboxylic acids3 or hydrogen peroxide,4 which either produces environmentally undesirable wastes or, in the case of H2O2, increases the cost of the process. Utilization of molecular oxygen is highly desirable to make epoxidation “green” and cost-effective. Another important way to reduce the environmental impact of a reaction is to conduct it under solvent-free conditions.5Since the discovery of the exceptional catalytic activity of gold in low-temperature oxidation of CO by Haruta6 and in hydrochlorination of ethylene by Hutchings,7 gold catalysts have proven to be efficient in a number of different reactions: cyclizations, rearrangements, selective hydrogenation, C–C coupling reactions, and selective oxidation of alcohols and olefins.8–10 Among other reactions, aerobic oxidation of cyclohexene was shown to be catalysed by supported nanosized gold.11–14 However, the reaction required the use of t-butyl hydroperoxide as a radical initiator,13 and the high yield of cyclohexene oxide was achieved only by conducting the reaction in a specific solvent.11 Under solvent- and radical initiator-free conditions, mainly products of allylic oxidation are formed.14,15
One of the approaches to improve the selectivity of the catalysed reaction towards the desired product is to utilize a bifunctional catalyst.16 An elegant example of such an approach is the phenol hydrogenation process catalysed by palladium deposited on Al2O3: while the palladium nanoparticles activate the hydrogen, the support, being a Lewis acid, activates the substrate and stabilizes cyclohexanone, preventing its over-hydrogenation to cyclohexanol.17
Recently, we showed that metallic gold nanoparticles formed from triphenylphosphine stabilized Au clusters catalyse formation of cyclohexenyl hydroperoxide in the aerobic oxidation of cyclohexene.15 In the present study, we investigated the effects of the supports and co-catalysts on the activity and selectivity of the Au-based catalysts for aerobic oxidation of cyclohexene under solvent-free conditions, without the addition of a radical initiator. We showed how the selectivity of this reaction can be tuned towards formation of either cyclohexene oxide or 2-cyclohexen-1-one as major products.
Experimental
Materials
Gold (99.99%), sodium borohydride (>97%), hydrochloric acid (37%), nitric acid (65%), dichloromethane (99.8%), n-decane (99%), n-hexane (98%), triphenylphosphine (98%), cyclohexene (99%, inhibitor-free), 2-cyclohexen-1-ol (95%), 2-cyclohexen-1-one (98%) and cyclohexene oxide (98%) were purchased from Sigma-Aldrich and Merck. Oxygen (99.7%) was obtained from BOC gases. The titanium(IV) oxide (P25, TiO2), silicon dioxide (Aerosil OX 50, SiO2) and tungsten(VI) oxide (nanopowder, <100 nm particle size, WO3) were purchased from Evonik and Sigma-Aldrich. All materials were used without further purification.Catalyst preparation
Triphenylphosphine-stabilized (PPh3-stabilized) gold clusters, [Au9(PPh3)8](NO3)3 (Au9) and Au101(PPh3)21Cl5 (Au101, approximate composition), were synthesized following previously described methods.18,19 Successful preparation and purity of clusters were confirmed by 1H and 31P NMR, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA).As-made gold clusters were deposited onto the oxide supports from CH2Cl2 solution. Typically, a calculated amount of gold cluster dissolved in CH2Cl2 (10 mL, 1–2 mg mL−1) was added dropwise to a vigorously stirred slurry of SiO2, TiO2 or WO3 (500 mg) in CH2Cl2 (15 mL). The mixture was stirred for 30 min and the solid was collected by centrifugation. A colourless supernatant solution confirmed complete cluster deposition. The catalysts were washed with CH2Cl2 (20 mL) and dried under vacuum at room temperature.
In the case of deposition of Au9 on WO3, for loadings greater than 0.1 wt%, n-hexane (50 mL) was slowly added to the slurry to ensure cluster deposition. Solids were collected by centrifugation, washed with n-hexane and dried under vacuum at room temperature.
The metal–organic framework MIL-101 was synthesized following a previously described method20 and characterized using PXRD, TGA and surface area measurements (see the ESI†).
Catalyst characterization
NMR spectra of gold clusters were recorded using an Oxford/Varian AS500 500 MHz NMR spectrometer. TGA was performed using Alphatech SDT Q600. TEM images were taken using a Philips CM200 instrument at 200 kV. Samples were deposited on holey carbon coated copper grids (300 mesh) from n-hexane. Typically, at least 100 particles were counted to calculate the average particle diameter. The gold content of the catalysts was determined quantitatively by atomic absorption spectroscopy (AAS) using a Varian SpectrAA 220 FS instrument. Diffuse reflectance UV-vis (DR UV-vis) spectra were recorded on a GBC Cintra 404 spectrophotometer.Catalytic testing
Cyclohexene oxidation was performed in a glass reactor equipped with a reflux condenser. Except where specified below, the system was flushed with oxygen three times and remained connected to an O2-filled rubber balloon throughout the reaction. Typically, a mixture of cyclohexene (5 mL), catalyst (50 mg) and n-decane (0.2 M, as an internal standard) was magnetically stirred (500 rpm) at 65 °C. After 16 hours, the reactor was cooled down to room temperature, the condenser was rinsed with acetone (5 mL) and the reaction mixture was separated from the solid catalyst by centrifugation. The catalyst was washed with acetone and dried under vacuum before recycling.The liquid samples were analysed via gas chromatography (GC) using a Shimadzu GC-2010 equipped with an Rxi-5SilMS capillary column (30 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID). Products were identified by gas chromatography-mass spectrometry (GC-MS) using a Shimadzu GCMS-QP2010. Quantitative analysis of reaction mixtures was performed via GC-FID using calibration solutions of commercially available products. The concentration of cyclohexenyl hydroperoxide was determined using iodometric titration in 80% acetic acid.21 Pre-treatment of the reaction mixture with PPh3 (ref. 22) did not change the concentration of 2-cyclohexen-1-one compared to an untreated mixture, indicating that cyclohexenyl hydroperoxide was not decomposing during GC analysis and therefore allowing us to plot a calibration curve for cyclohexenyl hydroperoxide on the basis of comparison of GC data and iodometric titration results.
Results and discussion
Preparation of Au/WO3, Au/SiO2 and Au/TiO2
The NMR spectra of synthesized clusters closely match previously published spectra: the 31P spectrum of Au9 clusters in CD2Cl2 has a singlet at δ = 56.9 ppm; the 1H spectrum of Au101 in CD2Cl2 shows a broad phenyl resonance, centred at 7.1 ppm, with minimal signal (7.52 ppm) due to unbound AuClPPh3.17,18 TGA showed 52% (Au9) or 26% (Au101) weight loss of the starting mass in comparison to calculated values of 52% and 21%, respectively, based on the loss of triphenylphosphine ligands (see the ESI† for details). The diameters of gold cores for Au101 and Au9 were 1.5 nm and 0.9 nm, respectively, as determined by TEM, which is in accordance with previously published data.17,18 We have chosen non-porous nanopowders of TiO2, SiO2 and WO3 as supports with similar morphologies – particle sizes below 100 nm and surface areas ≤50 m2 g−1.Two series of catalysts with target gold loadings ranging from 0.1 wt% to 0.5 wt% were prepared. The actual loadings, established using AAS, are shown in Table 1, where the two-digit prefixes in the sample designations are indicative of the approximate loadings in wt%. It was found that clusters readily adhere from CH2Cl2 solution to SiO2 and TiO2 at all target loadings.
| Catalyst | Gold loading, AAS (wt%) | Nanoparticle diameter, TEM (nm) | |
|---|---|---|---|
| Mean ± s.e.f | s.d.f | ||
| a Recovered after the 1st catalytic cycle. b Recovered after the 2nd catalytic cycle. c Recovered after the 3rd catalytic cycle. d No gold particles were detected by bright-field TEM. e Ref. 15. f s.e. – standard error of the mean, s.d. – standard deviation of the distribution. | |||
| 0.5Au101/WO3 | 0.52 | 2.24 ± 0.08 | 0.7 |
| 0.5Au101/WO3a | 0.47 | 5.08 ± 0.27 | 2.1 |
| 0.5Au101/WO3b | 0.47 | 5.39 ± 0.17 | 1.8 |
| 0.1Au101/WO3 | 0.097 | 2.25 ± 0.05 | 0.5 |
| 0.1Au101/WO3a | 0.070 | 4.40 ± 0.17 | 1.6 |
| 0.3Au9/WO3 | 0.31 | <1d | — |
| 0.3Au9/WO3a | 0.28 | 5.13 ± 0.11 | 1.5 |
| 0.3Au9/WO3b | 0.28 | 5.79 ± 0.25 | 2.1 |
| 0.3Au9/WO3c | 0.28 | 6.13 ± 0.21 | 2.0 |
| 0.1Au9/WO3 | 0.093 | <1d | — |
| 0.1Au9/WO3a | 0.087 | 7.93 ± 0.27 | 2.2 |
| 0.5Au101/SiO2 | 0.44 | 1.97 ± 0.04 | 0.6 |
| 0.5Au101/SiO2a | 0.29 | 5.06 ± 0.18 | 2.0 |
| 0.1Au101/SiO2e | 0.12 | 1.59 ± 0.04 | 0.4 |
| 0.1Au101/SiO2a,e | 0.068 | 4.94 ± 0.15 | 1.5 |
| 0.3Au9/SiO2 | 0.27 | <1d | — |
| 0.3Au9/SiO2a | 0.20 | 6.27 ± 0.17 | 1.6 |
| 0.3Au9/SiO2b | 0.20 | 6.85 ± 0.23 | 2.3 |
| 0.1Au9/SiO2 | 0.098 | <1d | — |
| 0.1Au9/SiO2a,e | 0.073 | 9.6 ± 0.6 | 3.9 |
In the case of WO3, for target loadings greater than 0.1 wt%, successful deposition of Au9 required addition of n-hexane, and maximum actual loadings of only ca. 0.3 wt% could be achieved. Deposition of Au101 on WO3 was readily achieved without the use of n-hexane for all target loadings.
After deposition, the mean diameter of Au101 clusters, as determined by TEM, slightly increases (Table 1). We were unable to detect as-deposited Au9 clusters using bright-field TEM, which indicates that the size of metal core remains below 1 nm. However, during the course of catalytic cyclohexene oxidation, both types of clusters sinter to form particles with mean diameters ranging from ca. 4 to 10 nm (Table 1 and Fig. 1). The mean diameter of the nanoparticles formed from Au101 during the catalytic cycle is decreasing with lower gold loading. In contrast, Au9 clusters form bigger nanoparticles at lower loading. This could suggest different sintering mechanisms for two clusters – Au9 acts as feedstock for nanoparticle growth, while Au101 clusters collide and agglomerate through surface diffusion. We suggest that agglomeration of Au9 clusters occurs similarly to the process of crystal formation in which lower concentration of precursor leads to the creation of smaller amount of nucleation sites and thus to bigger crystals. Thus, at high gold loadings, the majority of Au9 clusters rapidly sinter into stable nanoparticles with mean diameters of 4–6 nm, while at lower surface concentrations, clusters initially form fewer nanoparticles that act as nucleation sites that keep growing, consuming the remaining Au9 clusters to eventually form 8–10 nm particles. As agglomeration of Au101 proceeds via cluster collision, the degree of sintering should decrease with the lower surface concentration of clusters.
 | ||
| Fig. 1 Representative TEM images and particle size distributions of the catalysts. (A) 0.5Au101/WO3 as deposited. Catalysts recovered after the 1st catalytic cycle: (B) 0.5Au101/WO3, (C) 0.3Au9/WO3, (D) 0.3Au9/SiO2. | ||
Aerobic oxidation of cyclohexene under solvent-free conditions
Table 2 summarizes the catalytic activity of Au9 and Au101 deposited on TiO2, SiO2 and WO3 at various gold loadings for the aerobic oxidation of cyclohexene, which was conducted under solvent-free conditions without the addition of a radical initiator and using molecular oxygen as the only oxidant.| Catalyst | Conversion, % | Selectivity, % | |||
|---|---|---|---|---|---|
| Cy-oxide | Cy-ol | Cy-one | CyOOH | ||
| a Reaction conditions: cyclohexene (5 mL), n-decane (0.2 M) as an internal standard, catalyst (0.05 g), O2 (~1 atm), 65 °C, 16 h, glass reactor. b Ref. 15. | |||||
| Blank | 2 | — | — | — | — |
| TiO2 | 2 | — | — | — | — |
| SiO2 | 2 | — | — | — | — |
| WO3 | 9 | 33 | 30 | 4 | 32 |
| 0.5Au9/TiO2 | 2 | — | — | — | — |
| 0.5Au101/TiO2 | 2 | — | — | — | — |
| 0.5Au101/WO3 | 50 | 26 | 18 | 17 | 19 |
| 0.1Au101/WO3 | 36 | 35 | 23 | 12 | 18 |
| 0.3Au9/WO3 | 50 | 27 | 20 | 16 | 17 |
| 0.1Au9/WO3 | 33 | 34 | 24 | 11 | 21 |
| 0.5Au101/SiO2b | 48 | 6 | 15 | 24 | 38 |
| 0.1Au101/SiO2b | 39 | 7 | 11 | 17 | 54 |
| 0.3Au9/SiO2 | 43 | 7 | 12 | 19 | 51 |
| 0.1Au9/SiO2b | 25 | 5 | 7 | 12 | 68 |
Cyclohexene conversion and product distribution depend strongly on the nature of the support. TiO2-based catalysts are inactive, showing conversions comparable to that of the reaction in the absence of a catalyst (blank). Clusters deposited on SiO2 show high conversions of cyclohexene, with cyclohexenyl hydroperoxide (CyOOH) being the main product. Other products were 2-cyclohexen-1-one (Cy-one), 2-cyclohexen-1-ol (Cy-ol) and cyclohexene oxide (Cy-oxide). WO3-supported clusters have comparable activity, but the main product is cyclohexene oxide.
Unsupported gold nanoparticles have previously been shown to have high catalytic activity in some liquid-phase reactions;23 thus, it is important to investigate whether the observed catalytic activity should be attributed to supported or leached gold species. As seen from the AAS studies (Table 1), gold does leach into solution from as-prepared samples during the first catalytic cycle.
However, no such leaching was detected during subsequent recyclability tests. We suggest that as clusters agglomerate into bigger particles, leaching of gold species stops. This suggestion correlates with the fact that the degree of leaching during the first cycle decreases with higher loading. A higher surface density of gold clusters accelerates agglomeration and hence fewer non-sintered clusters have time to leach into solution. Because the catalyst retains ca. 90% of its catalytic activity during catalytic cycles 2–5 (Fig. 2), while leaching becomes undetectable on these stages, we conclude that the activity is very predominantly attributable to the supported, agglomerated particles and that gold species leached during the first cycle are essentially catalytically inactive. The methodology of hot filtering is typically used to determine whether the catalyst is homogeneous or heterogeneous.24 In our case, the reaction did not slow down upon Au/WO3 removal using a 0.2 μm filter after 6 h, which, according to the method, should indicate the homogeneous nature of the catalysis. However, it is known that cyclohexenyl hydroperoxide can catalyse the autoxidation of cyclohexene.25 Therefore, the hot filtering test is not suitable for distinguishing between heterogeneous and homogeneous catalysis for cyclohexene oxidation when cyclohexenyl hydroperoxide is formed in a sufficient amount.
 | ||
| Fig. 2 Recyclability of 0.3Au9/WO3. Conversion of cyclohexene (black) and selectivity towards cyclohexene oxide (grey). | ||
To provide further evidence that leached gold species are inactive in cyclohexene oxidation, we subjected the catalysts to a pair of tests under reaction-like conditions in which gold leaching would occur but cyclohexenyl hydroperoxide formation would be suppressed. In the first test, an initial reaction was performed at 65 °C for 6 h using as-made catalyst, but the reactor was filled with argon instead of oxygen. The solid catalyst was then removed by hot filtration and the liquid reaction mixture was subjected to typical reaction conditions (65 °C or 16 h) with the reactor now filled with ca. 1 atm oxygen. The existence of gold leachate in the reaction mixture was confirmed using ICP-MS, but no detectable cyclohexene conversion was observed. In the second test, cyclohexenyl hydroperoxide formation was suppressed by addition of n-hexane. An experiment was conducted under oxygen atmosphere (ca. 1 atm) using the mixture of cyclohexene (50 μL) and n-hexane (2 mL). After 6 h, the liquid phase was separated by hot filtering and added to 5 mL of cyclohexene. When subjected to typical reaction conditions, the mixture showed no cyclohexene conversion over 16 h. In contrast, a solid catalyst, isolated on the first stage of the second test, catalysed the oxidation of cyclohexene in the mixture of n-hexane (2 mL) and cyclohexene (5 mL), giving 11% conversion.
Investigation of the catalysts' bifunctionality
The similarity in morphology and size of the gold nanoparticles formed on SiO2 and WO3 supports during the catalytic reaction is indicated by TEM and DR UV-vis spectroscopy. In the latter, the bands attributed to the localized surface plasmon resonance have similar shapes, positions and intensities for both types of catalysts (Fig. 3).26–28 Interestingly, SiO2- and WO3-based catalysts with similar gold loadings have comparable turnover frequencies (TOFs) calculated from the initial reaction rates (Fig. 4). | ||
| Fig. 3 DR UV-Vis spectra of the catalysts. (a) 0.3Au9/WO3 as deposited. Catalysts recovered after the 1st catalytic cycle: (b) 0.3Au9/SiO2, (c) 0.3Au9/WO3 and (d) 0.5Au101/WO3. | ||
 | ||
| Fig. 4 Reaction profile of cyclohexene oxidation catalysed by 0.3Au9/WO3 (A) and 0.3Au9/SiO2 (B). Cyclohexene conversion (■); left ordinate. Yield of cyclohexene hydroperoxide (◊), cyclohexene oxide (○), 2-cyclohexen-1-one (×), 2-cyclohexen-1-ol (△); right ordinate. | ||
The product evolution profiles for both types of catalysts have similar cyclohexenyl hydroperoxide accumulation stages. Based on these data, we suggest that gold particles catalyse cyclohexene conversion to cyclohexenyl hydroperoxide (reaction I, Scheme 1), while WO3 catalyses reaction of cyclohexenyl hydroperoxide with cyclohexene, producing cyclohexene oxide and 2-cyclohexen-1-ol (reaction II, Scheme 1).
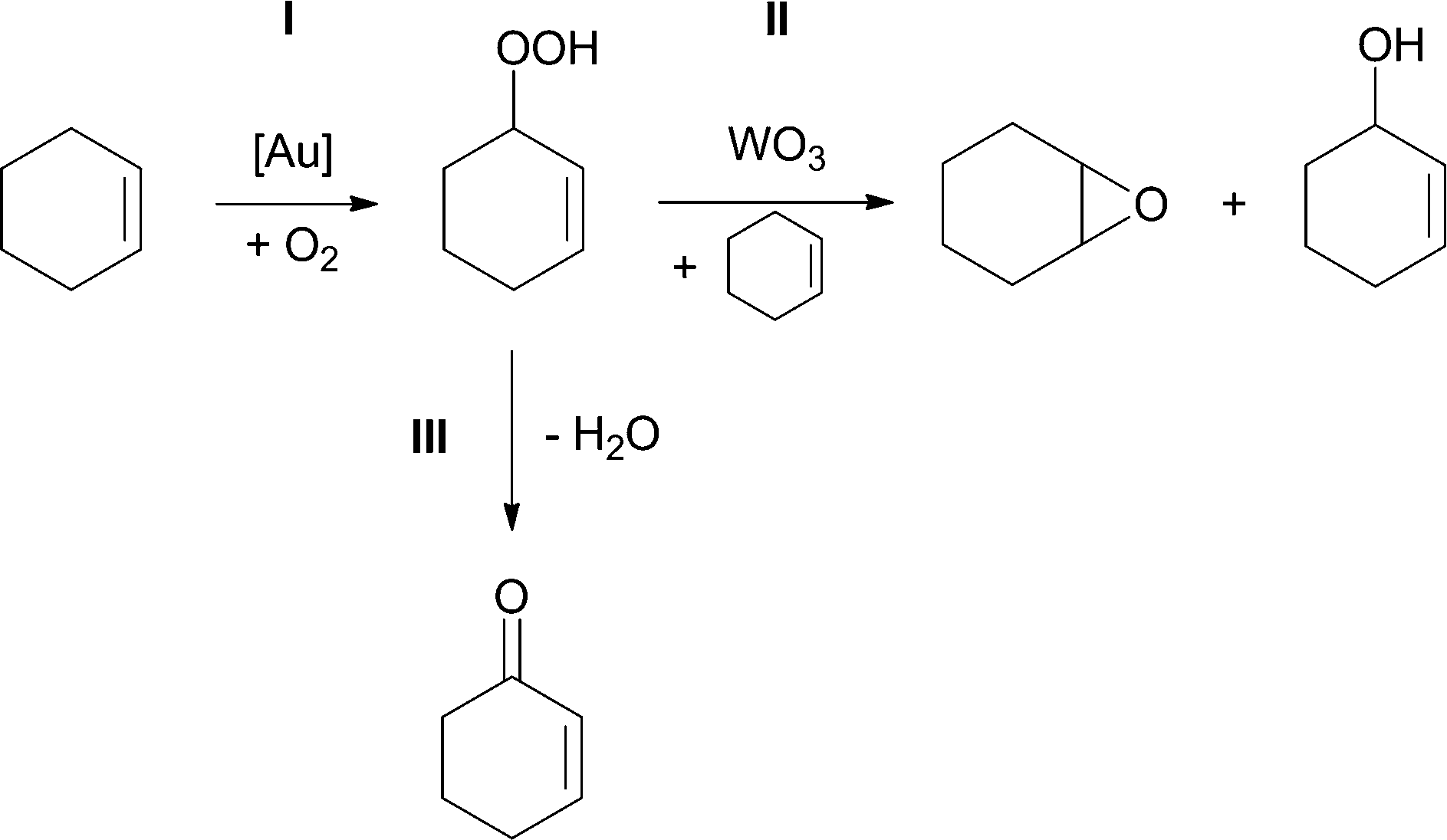 | ||
| Scheme 1 Proposed mechanism for cyclohexene oxidation. | ||
To support this hypothesis, we conducted a series of experiments in which pure WO3 powder was mixed with 03Au9/SiO2 at different ratios (Fig. 5A). Interestingly, the addition of just 2 wt% of WO3 (1 mg) changes the selectivity of the reaction, with cyclohexene oxide becoming the main product. With 10 wt% of WO3 (5 mg), the distribution of products is almost identical to that for 03Au9/WO3. It is possible, however, that in this series of experiments, some gold species leach from silica-based catalyst and adsorb on tungsten oxide, thus forming a catalytic system with high selectivity towards cyclohexene oxide.
 | ||
| Fig. 5 Effect of the co-catalyst on the selectivity in cyclohexene oxidation. Co-catalyst: WO3 (A) or MIL-101 (B). | ||
To exclude this possibility, we performed a reaction with the silica-based catalyst and hot filtered the reaction mixture into a vial containing tungsten oxide (5 mg), which was then collected by centrifugation. Inasmuch as potentially impregnated WO3 was inactive in the conversion of the fresh cyclohexene, we conclude that the observed change of selectivity upon addition of WO3 to the Au/SiO2 catalyst should be attributed to properties of pure WO3 acting as a co-catalyst rather than to a synergistic effect between support and Au nanoparticles. The discovered ability of WO3 to activate cyclohexenyl hydroperoxide, promoting its reaction with cyclohexene, is consistent with reports on activation of H2O229 and alkyl hydroperoxides30 by WO3 through the formation of peroxo complexes of tungsten which are highly active and selective in the epoxidation of olefins.31
We also found that the use of a different co-catalyst can shift selectivity of Au/SiO2 towards allylic oxidation products. For such a co-catalyst, we have chosen the metal–organic framework MIL-101, which was recently reported as a catalyst for allylic oxidation of cyclohexene with molecular oxygen, with 2-cyclohexen-1-one being the main product.32,33 Results of catalytic testing of the mixture consisting of pure MOF and 0.3Au9/SiO2 in different ratios show the effect of altering the selectivity of reaction similar to the one found for WO3, but with 2-cyclohexen-1-one becoming the main product (Fig. 5B). Reaction catalysed by the mixture of MIL-101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3Au9/SiO2 (5:45, mg:mg) gives the maximum 2-cyclohexen-1-one yield of 16%, which is twice the yield we have been able to achieve with the pure MOF.
:0.3Au9/SiO2 (5:45, mg:mg) gives the maximum 2-cyclohexen-1-one yield of 16%, which is twice the yield we have been able to achieve with the pure MOF.
Conclusions
We have prepared a series of SiO2-, TiO2- and WO3-supported gold catalysts by deposition of Au9 and Au101 clusters. Under the conditions of solvent-free aerobic oxidation of cyclohexene, clusters sinter into larger nanoparticles with mean diameters ranging from ca. 4 to 10 nm, depending on the gold loading. Leaching of gold species occurs during the first catalytic cycle but becomes undetectable on further stages. We have shown that leached gold species are inactive in cyclohexene oxidation.We found that cyclohexenyl hydroperoxide, the formation of which is catalysed by Au nanoparticles, can be converted to other products in the presence of different heterogeneous co-catalysts. Cyclohexene oxide is formed via reaction of cyclohexenyl hydroperoxide with cyclohexene catalysed by WO3, either present as a support or introduced to the reaction as a co-catalyst physically admixed with silica-supported gold catalyst. Selectivity towards formation of 2-cyclohexen-1-one is shifted by using MIL-101 as a co-catalyst for gold supported on SiO2.
We have shown that careful choice of support or co-catalyst for supported gold nanoparticles can tune the selectivity of cyclohexene oxidation towards cyclohexene oxide or 2-cyclohexen-1-one under solvent-free conditions without addition of a radical initiator and using oxygen as the only oxidant.
Acknowledgements
We would like to thank Campbell McNicoll and Tim Kemmitt from Callaghan Innovation NZ for their help with PXRD and surface area measurements. This work was supported by the University of Canterbury and the MacDiarmid Institute. B. D. thanks the MacDiarmid Institute for the research scholarship and D. O. thanks the University of Canterbury for UC Doctoral scholarship.Notes and references
- R. A. Sheldon and H. van Bekkum, in Fine Chem. Heterog. Catal., Wiley-VCH Verlag GmbH, 2007, pp. 473–551 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, 1981 Search PubMed.
- N. Prileschajew, Ber. Dtsch. Chem. Ges., 1909, 42, 4811–4815 CrossRef CAS.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- C. H. A. Tsang, Y. Liu, Z. Kang, D. D. D. Ma, N.-B. Wong and S.-T. Lee, Chem. Commun., 2009, 5829–5831 RSC.
- Z.-Y. Cai, M.-Q. Zhu, J. Chen, Y.-Y. Shen, J. Zhao, Y. Tang and X.-Z. Chen, Catal. Commun., 2010, 12, 197–201 CrossRef CAS PubMed.
- B. G. Donoeva, D. S. Ovoshchnikov and V. B. Golovko, ACS Catal., 2013, 2986–2991 CrossRef CAS.
- G. A. Somorjai and M. Yang, Top. Catal., 2003, 24, 61–72 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef CAS.
- F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem., 2008, 2008, 106–111 CrossRef.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS.
- H. A. Liebhafsky and W. H. Sharkey, J. Am. Chem. Soc., 1940, 62, 190–192 CrossRef CAS.
- G. B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 189, 39–66 CrossRef CAS.
- Y. Mikami, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2013, 3, 58–69 CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- S. M. Mahajani, M. M. Sharma and T. Sridhar, Chem. Eng. Sci., 1999, 54, 3967–3976 CrossRef CAS.
- A. Henglein, Langmuir, 1999, 15, 6738–6744 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2002, 107, 668–677 CrossRef.
- R. Bera and S. Koner, Inorg. Chim. Acta, 2012, 384, 233–238 CrossRef CAS PubMed.
- A. T. Bolsoni, J. S. dos Santos, M. D. Assis and H. P. Oliveira, J. Non-Cryst. Solids, 2011, 357, 3301–3306 CrossRef CAS PubMed.
- K. Kamata, K. Yonehara, Y. Sumida, K. Hirata, S. Nojima and N. Mizuno, Angew. Chem., Int. Ed., 2011, 50, 12062–12066 CrossRef CAS PubMed.
- N. V. Maksimchuk, O. V. Zalomaeva, I. Y. Skobelev, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Proc. R. Soc. A, 2012, 468, 2017–2034 CrossRef CAS.
- I. Y. Skobelev, A. B. Sorokin, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2013, 298, 61–69 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: TGA of gold clusters. Characterization of MIL-101, including PXRD patterns, TGA and nitrogen physisorption analysis data. See DOI: 10.1039/c3cy01011b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |