 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective oxidation of benzyl alcohol using in situ generated H2O2 over hierarchical Au–Pd titanium silicalite catalysts†
Inés
Moreno
ab,
Nicholas F.
Dummer
a,
Jennifer K.
Edwards
a,
Mosaed
Alhumaimess
a,
Meenakshisundaram
Sankar
a,
Raul
Sanz
b,
Patricia
Pizarro
b,
David P.
Serrano
bc and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
bDepartment of Chemical and Energy Technology, ESCET, Rey Juan Carlos University, c/ Tulipán s/n, Móstoles, 28933, Madrid, Spain
cIMDEA Energy Institute, Avda. Ramón de la Sagra, 3, Móstoles, 28935, Madrid, Spain
First published on 16th July 2013
Abstract
Benzyl alcohol was oxidized by an “in situ generated” hydrogen peroxy species, formed from a dilute mixture of hydrogen and oxygen, under mild conditions at a high rate over gold, palladium and gold–palladium nanoparticles supported on hierarchical titanium silicate materials. Hierarchical TS-1 supports were obtained from the crystallization of silanized protozeolitic units, being characterized by having a secondary porous system within supermicro/mesopore range and an enhanced surface area over a standard reference TS-1 material. The presence of the secondary porosity not only improves the accessibility to the active sites of the relatively large reactant molecules but also enhances the metal dispersion, leading to an improved catalytic performance for alcohol oxidation. The catalytic activity of metal loaded hierarchical TS-1 materials was found to be higher in reactions conducted in the presence of diluted hydrogen and oxygen, resulting in a 5-fold increase in the yield of benzaldehyde at 30 °C with an AuPd catalyst with secondary porosity. The improvement in rate observed is due to the oxidizing efficacy of in situ generated hydroperoxy species as compared to molecular oxygen alone as the terminal oxidant.
Introduction
Titanium silicate (TS-1)1 materials have emerged as effective catalysts for the oxidation of various substrates using environmentally benign oxidants, for example hydrogen peroxide.2–4 TS-1 possesses an MFI structure which consists of three dimensional pore system of 10 ring channels arranged sinusoidally (5.1 × 5.5 Å) with an intersecting straight 10-ring channel (5.3 × 5.6 Å).5 As with other such zeotype materials, which possess microporosity, they are limited in their application with bulky reactant molecules. The synthesis of hierarchical zeolite materials facilitates increased reactant diffusion towards the active sites within the pore structure.6,7 Preparation of such materials covers a wide range of strategies detailed in a review by van Donk et al.8Hierarchical TS-1 has been obtained with hard-templates based on porous carbon9,10 and soft templates such as surfactants or organosilane molecules.11,12 Recently, it has been reported the preparation of hierarchical TS-1 materials from the crystallization of silanized protozeolitic units.13,14 In this strategy, an organosilane compound is added to a solution of protozeolitic units, formed previously by a precrystallization step. These organosilanes anchor onto the external surface and thereby partially block the aggregative growth during the final hydrothermal treatment. After this crystallization step, a secondary porosity, in addition to the typical zeolite micropores, is formed when the organosilane compound is removed by calcination. These materials have been evaluated in alkene epoxidation, with both H2O2 and tertiary-butyl hydroperoxide (TBHP). The use of alkyl peroxides, typically with bulky structures, is generally ineffective with TS-1 due to the restrictive microporous structure.15,16 Enhancements in product yield were achieved with those materials with secondary porosity, where the increased surface area was related to an improved turnover frequency.14
Selective oxidation with TS-1 is generally carried out with hydrogen peroxide as an oxidant and is successful in a large number of applications.15,17–21 Typically, however, hydrogen peroxide is used as a stoichiometric oxidant which is considered expensive. Heterogeneous gold catalysts represent a promising alternative to the use of stoichiometric radical initiators, where oxygen can be activated and incorporated in the final product.22 Furthermore, in situ generation of hydroperoxy species with subsequent reaction to an oxygenated target represents a desirable, lower cost route. The recent review by Della Pina et al.23 highlighted advances in selective oxidation over gold catalysts with emphasis on the emerging industrial applications. Haruta et al.24 have demonstrated low temperature epoxidation of propene at the gas–solid interface. This process requires the use of sacrificial hydrogen to form a surface peroxy species in situ, which is able to complete the formation of propene oxide with high specificity. The catalyst used for this process involved gold nanoparticles supported on TiO2 and improvements in activity have been obtained more recently with supports including TS-1, Ti-MCM-41 and Ti-MCM-48.25–30
Selective oxidation in three phase systems with in situ generated hydroperoxy species is an emerging topic.31–36 However, the activity of the catalyst used must be sufficiently high to operate at the temperatures associated with direct hydrogen peroxide production which is low, and typically sub-ambient.37,38 Thermal decomposition of hydrogen peroxide occurs readily above ambient temperatures and rapidly in the presence of transition metals. The aerobic oxidation of benzyl alcohol to benzaldehyde under mild conditions has been shown to occur with the same catalyst systems as used for direct peroxide synthesis.38,39 These include supported gold–palladium nano-particles which have a high activity and selectivity to benzaldehyde. Generally, equimolar Au–Pd mixtures are considered to be advantageous as monometallic gold catalysts typically have poor activity. However, increasing the Pd content increases the formation of one of the by-products, toluene, which is undesirable. Attempts with Au–Pd supported on zeolitic materials for the oxidation of cyclohexane with in situ peroxy species has shown promise.32 However, the yields of cyclohexanol were found to be low; ca. 2%, more success was obtained with crotyl alcohol oxidation to crotonaldehyde over 2.5% Au–2.5% Pd/TS-1 catalyst with a yield of ca. 25% over 4 h at 60 °C.32 It was concluded that the rate of oxidation should be comparable to the production rate of peroxy species to ensure that the hydrogenation of the substrate (if applicable) is not dominant with Pd containing catalysts.
The use of on-site, small-scale hydrogen peroxide production for a range of applications is desirable with respect to storage and transportation costs of concentrated peroxide (typically 70 wt%). In this paper we report the application of in situ generated hydroperoxy species at low concentrations with hierarchical TS-1, prepared from silanized protozeolitic units, as a support to gold and palladium nano-particles for benzyl alcohol oxidation as a model reaction. These TS-1 zeolites, which possess a significant mesoporosity can improve both the accessibility of the reactants to the active sites as well as enhancing the metal dispersion and controlling the metal particle size. For this reason, the production rate of benzaldehyde from the oxidation of benzyl alcohol will be used to evaluate these hierarchical materials as catalysts with dual functionality.
Experimental
Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.016 TiO2:0.0355 TPAOH. Once the SiO2–TiO2 xerogel was obtained, it was impregnated with a 1 M TPAOH aqueous solution employing a TPAOH/xerogel mass ratio of 2.75 and, then, it was crystallized at 170 °C using heating by microwave radiation for 8 h in PTFE vessels under autogenous pressure.
:0.016 TiO2:0.44 TPAOH:28.5 H2O was prepared following the original recipe developed by Taramasso et al.1 This solution was precrystallized in a reflux system under stirring conditions at 90 °C for 24 h. The silanization agent (phenyl amino-propyltrimethoxysilane, PHAPTMS, Aldrich) was added at the desired loading (5, 8 and 12 mol%) in regard to the total silica content in the gel, the functionalization reaction being carried out under stirring at 90 °C for 6 h. Finally, the crystallization of the TS-1 zeolite was performed by microwave heating at 170 °C for 8 h in PTFE vessels under autogenous pressure. In all cases, the solid products obtained after the crystallization treatment were separated by centrifugation, washed several times with distilled water, dried (16 h, 110 °C) and calcined (in static air conditions 550 °C, 5 h).
:1.85 molar ratio Au:Pd), were prepared using the same procedure with appropriate amounts of HAuCl4·3H2O and PdCl2. Prior to use, samples were also calcined at 400 °C in static air for 3 h.
:0.016 TiO2:0.0355 TPAOH. Once the SiO2–TiO2 xerogel was obtained, it was impregnated with a 1 M TPAOH aqueous solution employing a TPAOH/xerogel mass ratio of 2.75 and, then, it was crystallized at 170 °C using heating by microwave radiation for 8 h in PTFE vessels under autogenous pressure.
:0.016 TiO2:0.44 TPAOH:28.5 H2O was prepared following the original recipe developed by Taramasso et al.1 This solution was precrystallized in a reflux system under stirring conditions at 90 °C for 24 h. The silanization agent (phenyl amino-propyltrimethoxysilane, PHAPTMS, Aldrich) was added at the desired loading (5, 8 and 12 mol%) in regard to the total silica content in the gel, the functionalization reaction being carried out under stirring at 90 °C for 6 h. Finally, the crystallization of the TS-1 zeolite was performed by microwave heating at 170 °C for 8 h in PTFE vessels under autogenous pressure. In all cases, the solid products obtained after the crystallization treatment were separated by centrifugation, washed several times with distilled water, dried (16 h, 110 °C) and calcined (in static air conditions 550 °C, 5 h).
:1.85 molar ratio Au:Pd), were prepared using the same procedure with appropriate amounts of HAuCl4·3H2O and PdCl2. Prior to use, samples were also calcined at 400 °C in static air for 3 h.
The materials 2.5 wt% Au–Pd/support were also prepared using the following deposition precipitation method (denoted DP). For the catalyst, a solution of HAuCl4·3H2O (2.5 mL, 2 g in 100 ml distilled water) and PdCl2 was diluted with water (50 mL). An aqueous sodium hydroxide solution (0.1 M) was added with stirring until pH = 9 was attained. Then, support (1 g) was added over the previous solution, with continuous stirring. The mixture was heated at 70 °C for 1 h maintaining the pH at 9. The solid was recovered by centrifugation and washed several times with distilled water. Finally, the catalyst was dried (110 °C, 16 h) and calcined at 400 °C in static air for 3 hours.
Catalysts used in this study and their preparative routes together with their codes are summarised in Table 1.
| Catalyst | Metala (wt%) | Preparation method | Support |
|---|---|---|---|
| a Per metal, IM = impregnation, DP = deposition precipitation. | |||
| AX | Au (2.5) | IM | TS-1 (XG-REF) |
| A12 | Au (2.5) | IM | TS-1 (LG-12%) |
| PX | Pd (2.5) | IM | TS-1 (XG-REF) |
| P12 | Pd (2.5) | IM | TS-1 (LG-12%) |
| APX | Au–Pd (2.5) | IM | TS-1 (XG-REF) |
| AP5 | Au–Pd (2.5) | IM | TS-1 (LG-5%) |
| AP8 | Au–Pd (2.5) | IM | TS-1 (LG-8%) |
| AP12 | Au–Pd (2.5) | IM | TS-1 (LG-12%) |
| DAPX | Au–Pd (2.5) | DP | TS-1 (XG-REF) |
| DAP12 | Au–Pd (2.5) | DP | TS-1 (LG-12%) |
Catalyst characterisation
Powder X-Ray Diffraction (XRD) patterns were measured using a Philips X'PERT MPD diffractometer operating with Cu Kα radiation with a step size and time of 0.02° and 10 s, respectively. The coordination of the titanium atoms in the samples, as well as the possible presence of extra-framework TiO2 phases, was checked by Diffuse Reflectance UV-VIS spectroscopy (DR UV-VIS) under ambient conditions with a Varian Cary-500 spectrophotometer equipped with a diffuse reflectance accessory and using BaSO4 as reference. The spectra were monitored in the range of 200–500 nm. Titanium content of the synthesized samples was estimated by means of inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis, performed in a Varian Vista AX system. Likewise, the total surface area was estimated according to the BET method by means of argon adsorption–desorption isotherms at 87 K, acquired with a Micrometrics ASAP 2010 instrument. The samples were out-gassed prior to the analysis at 300 °C under vacuum for at least 16 h. The pore size distribution and cumulative volume curves were calculated by applying the NL-DFT model with cylindrical pore geometry. TEM images were obtained in a Philips Technai 20 electron microscope operating at 200 kV and a resolution of 2.7 Å. The samples for TEM were prepared by dispersing the sample powder in acetone and drying a droplet of the suspension onto a carbon-coated copper grid.Catalyst testing
Turn over frequency (TOF) is defined as the total moles of benzaldehyde produced per hour divided by the total moles of metal present in the reaction.
Results and discussion
In this work, hierarchical titanium silicates materials (TS-1), prepared by silanization of protozeolitic units,13,14 were used as a support for gold and palladium nanocrystals (Au, Pd). For comparison purposes, a microporous conventional TS-1 zeolite prepared from amorphous SiO2–TiO2 xerogel was also employed as support.Catalyst characterization
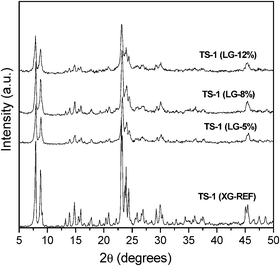 | ||
| Fig. 1 Powder XRD patterns of calcined TS-1 (XG-REF), TS-1 (LG-5%), TS-1 (LG-8%) and TS-1 (LG-12%) samples. | ||
The application of the NL-DFT model over the Ar adsorption–desorption isotherms (87 K) allows the pore size distribution in the micro- and mesopore region to be estimated within the complete micro/mesopore range. Fig. 2 illustrates the cumulative pore volume and pore size distribution (PSD) curves corresponding to the hierarchical TS-1 zeolites compared to the curves obtained for the reference zeolite. In all cases, a narrow peak with a maximum around 5.5 Å is detected in the PSD curve which agrees with the pore size of the MFI structure. However, in the pore size distribution curves corresponding to the hierarchical samples, an additional adsorption is detected between 10–60 Å, related to the presence of the secondary porosity in the supermicro/mesopore region, caused by the inhibitor growth effect of the silanization agent during the zeolite crystallization. The presence of this secondary porosity not only could increase the mass transport into the zeolite channels but could also improve the incorporation and dispersion of the metal particles onto the zeolitic surface, since they could be located into the secondary porosity while in the conventional microporous zeolite the metal particle are usually concentrated over the zeolitic crystal external surface.42
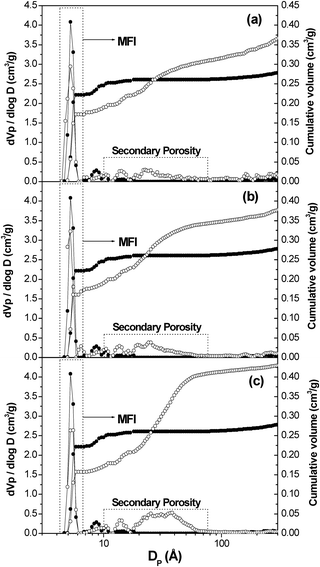 | ||
| Fig. 2 Cumulative pore curve and pore size distribution for TS-1 (XG-REF) (●) compared to (a) TS-1 (LG-5%) (○), (b) TS-1 (LG-8%) (○) and (c) TS-1 (LG-12%) (○) and estimated by applying NL-DFT method to Ar adsorption isotherms at 87 K. | ||
The textural properties of the TS-1 samples were obtained by applying the NL-DFT model from Ar adsorption–desorption isotherms (Table 2). The subscripts ZM and SP make reference to the zeolitic micropores and the secondary porosity, respectively. Hierarchical TS-1 samples prepared from silanized protozeolitic units were found to possess higher BET and secondary porosity surface areas than the reference zeolite. Thus, while for the reference TS-1 sample the latter values were ca. 470 m2 g−1 and ca. 53 m2 g−1, respectively, such values increased up to 656 m2 g−1 and 400 m2 g−1, respectively, for TS-1 (LG-12%). This increase of the BET surface area, which is exceptionally high when compared to the conventional MFI zeolites, corresponds to the surface area of the secondary porosity. A clear relationship is observed between the amount of silanization agent introduced into the synthesis gel and the modification degree of the hierarchical TS-1 textural properties.
| Material | % Ti | Si/Ti (mol) | S BET (m2 g−1) | S ZM (m2 g−1) | S SP (m2 g−1) | V ZM (cm3 g−1) | V SP (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| S BET = surface area by BET method, V = pore volume, ZM = zeolitic micropores, SP = secondary porosity. | |||||||
| TS-1 (XG-REF) | 1.32 | 59.0 | 469 | 416 | 53 | 0.222 | 0.075 |
| TS-1 (LG-5%) | 0.97 | 80.3 | 560 | 280 | 280 | 0.172 | 0.233 |
| TS-1 (LG-8%) | 1.39 | 56.0 | 633 | 263 | 371 | 0.161 | 0.239 |
| TS-1 (LG-12%) | 1.37 | 56.8 | 656 | 256 | 400 | 0.157 | 0.281 |
Finally, the morphology of the reference TS-1, TS-1 (XG-REF), and the hierarchical catalyst, TS-1 (LG-8%), were analyzed by TEM microscopy (Fig. 3). It can be observed that reference TS-1 (XG-REF) zeolite is formed by large particles with an average dimension of around 2 μm and regular and well defined edges. On the other hand, TS-1 (LG-8%) consists in slightly large aggregates with dimensions around 150–200 nm and sponge-like morphology. From images taken at higher magnifications it may be distinguished that these aggregates are formed by ultra-small crystalline domains (nano-units) with dimensions below or around 20–25 nm. The voids existing between these nano-units are related to the secondary porosity caused by the silanization agent. The presence of diffraction fringes in these units indicates their crystalline nature.
 | ||
| Fig. 3 TEM micrographs of TS-1 (XG-REF) and TS-1 (LG-8%) samples. | ||
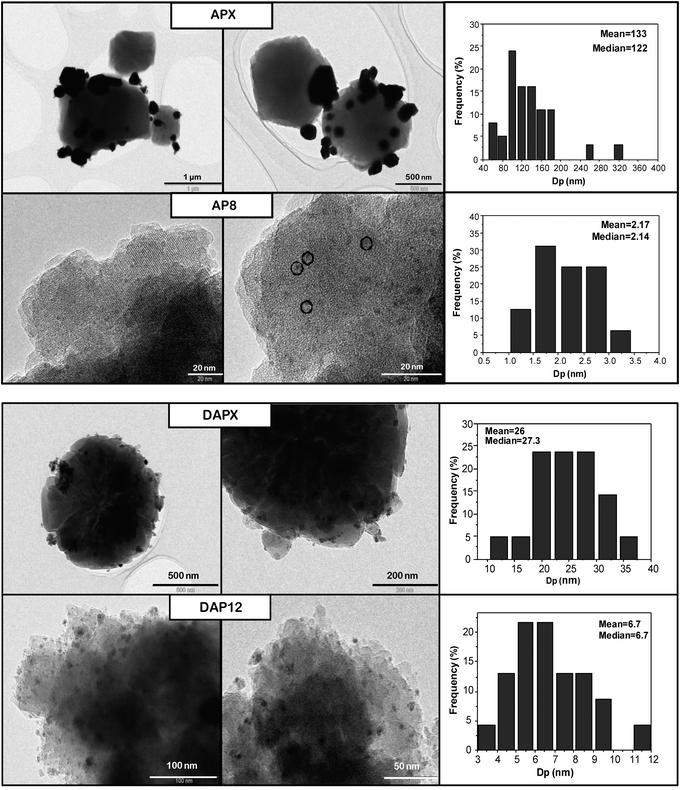 | ||
| Fig. 4 Representative TEM images and metal particle sizes of AuPd catalyst samples APX, AP8, DAPX and DAP12. | ||
Catalyst testing
The catalysts were investigated for solvent-free benzyl alcohol oxidation at 120 °C under moderate oxygen pressure and the results are illustrated in Table 3. They indicate that the secondary porosity of the hierarchical catalysts; AP5-12, DAP12 and P12 facilitated improved activity compared to standard TS-1 catalysts under these conditions. However, for catalysts AP5 and AP8 similar catalytic activity was observed. In general, the presence of an additional mesoporosity in zeolites improves the reactant and product diffusion leading to a higher conversion. This can be probed by pore size distributions and textural properties of the catalysts (Table 2) compared to the size of the benzyl alcohol molecule (length ca. 7.9 Å, width ca. 5.3 Å (calculated with Accelrys Materials Studio 6.0; Dmol3 geometry optimisation)). Moreover, it is possible that this secondary porosity enhances the degree of incorporation and dispersion of the metal particles. The respective benzaldehyde production rate and turn-over frequency (TOF) over APX was calculated to be 75 ± 0.5 mol h−1 kgcat−1 and 332 h−1 compared to catalyst AP12 with 135 ± 0.5 mol h−1 kgcat−1 and 615 h−1. However, the method of metal deposition and metal choice dominates both the level of conversion and benzaldehyde selectivity despite the obvious reduction in diffusion limitations. The combination of Au–Pd leads to increased catalyst activity, observed previously.45 When catalysts were prepared with deposition precipitation the selectivity to benzaldehyde is increased relative to those prepared via impregnation. Catalyst DAP12 achieved a benzaldehyde production rate of 171 ± 0.5 mol h−1 kgcat−1. Conversely, those prepared by impregnation were more active, however, with an increased amount of the by-product; toluene (Fig. 5). Toluene is formed by the disproportionation of benzyl alcohol to equimolar mixture of benzaldehyde and toluene.46 Disproportionation is observed in greater amount with impregnation catalysts, which we consider is related to the particle size of the metal nano-particle and potentially the Pd surface (Fig. 4). Such disproportionation of benzyl alcohol to benzaldehyde, toluene and water has been reported previously and occurs at elevated temperatures over Pd only or Pd containing catalysts.47 In addition, in some experiments a small amount of benzene is also observed which we consider may be formed by a hydrogenolysis pathway.| Catalyst | Conversion (%) | Selectivity (%) | TOF (h−1) | Benzaldehyde productiona (mol h−1 kgcat−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Benzene | Toluene | Benzaldehyde | Benzoic acid | Benzyl benzoate | ||||
| Conditions: 120 °C, 4 hours, 20 mg of catalyst, 0.0185 mol benzyl alcohol (2 g), PO2: 1.2 bar (17.4 psi).a ±0.5 mol h−1 kgcat−1. | ||||||||
| No catalyst | 0.4 | 5.5 | 0 | 93.7 | 0 | 0 | — | 1 |
| TS-1 (XG-REF) | 0.4 | 0 | 3.8 | 96.2 | 0 | 0 | — | 1 |
| TS-1 (LG-12%) | 0.2 | 0 | 0 | 100 | 0 | 0 | — | 1 |
| AX | 0.4 | 5.9 | 15.7 | 76.9 | 1.4 | 0 | 7 | 1 |
| A12 | 0.7 | 0 | 0 | 100 | 0 | 0 | 13 | 2 |
| APX | 52.0 | 0.3 | 28.2 | 61.4 | 9.8 | 0.3 | 332 | 75 |
| AP5 | 88.9 | 0.5 | 37.3 | 58.2 | 4.0 | 0.03 | 568 | 122 |
| AP8 | 88.2 | 0.6 | 37.2 | 60.7 | 1.3 | 0.2 | 564 | 126 |
| AP12 | 96.3 | 0.7 | 40.0 | 59.3 | 0.04 | 0 | 615 | 135 |
| DAPX | 50.9 | 1.7 | 8.6 | 85.7 | 4.1 | 0 | 325 | 103 |
| DAP12 | 78.4 | 2.1 | 2.4 | 92.3 | 3.2 | 0 | 501 | 171 |
| PX | 19.0 | 0.5 | 28.4 | 66.9 | 3.7 | 0.6 | 121 | 30 |
| P12 | 45.3 | 0.7 | 23.6 | 73.7 | 1.7 | 0.4 | 289 | 79 |
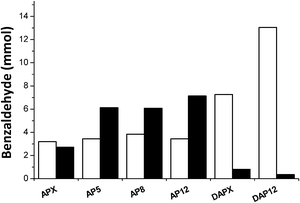 | ||
| Fig. 5 Moles of benzaldehyde formed through oxidation (□) and disproportionation (■) reactions at 120 °C over AuPd/TS-1 catalysts. | ||
Results from these initial studies indicate that these catalysts possess sufficient activity to perform oxidation reactions at much lower temperatures. Subsequent reactions were carried out under a partial pressure of oxygen in a water–methanol solvent mixture (Fig. 6 and Table S1, ESI†). These conditions were adopted as they are used for the direct synthesis of hydrogen peroxide by Edwards et al.,48 however, initially without hydrogen. The use of a water–methanol solvent mixture increases the solubility of the oxygen and hydrogen. In addition, methanol is unable to form peroxides under these conditions.49 At 2 °C, as expected, the catalytic activity was greatly reduced under these sub-ambient conditions. Comparison of catalysts APX and AP12 illustrates this as the benzaldehyde production rate was found to be 1 ± 0.5 mol h−1 kgcat−1 which is lower than the obtained using TS-1 supports used (ca. 5 ± 0.5 mol h−1 kgcat−1) (Table S1, ESI†). Increasing the reaction temperature to 30 °C (Table 4) the rate of disproportionation improved as well as benzaldehyde formation. However, comparison of catalysts APX and AP12 indicates that benzaldehyde selectivity can be improved with secondary porosity. Although, the benzaldehyde production rate was found to be 14 ± 0.5 mol h−1 kgcat−1 over the APX catalyst and this was comparable to the rate over AP8 and DAP12 of 11 ± 0.5 mol h−1 kgcat−1, respectively.
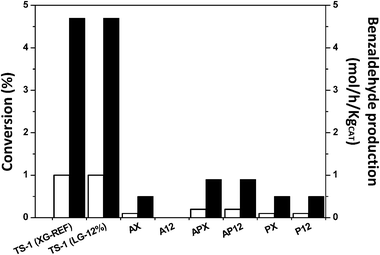 | ||
| Fig. 6 Benzyl alcohol conversion (□) and benzaldehyde production rate (■) at 2 °C over TS-1 and Au–Pd/TS-1 catalysts. Conditions: 0–2 °C, methanol (5.6 g), water (2.9 g), 2.3 mmol benzyl alcohol (0.25 g), 30 minutes, catalyst (10 mg), 25% O2/CO2 (10.4 bar, 150 psi). | ||
| Catalyst | Conversion (%) | Selectivity (%) | Benzaldehyde productiona (mol h−1 kgcat−1) | ||||
|---|---|---|---|---|---|---|---|
| Benzene | Toluene | Benzaldehyde | Benzoic acid | Benzyl benzoate | |||
| Conditions: 30 °C, methanol (5.6 g), water (2.9 g), 2.3 mmol benzyl alcohol (0.25 g), 30 minutes, catalyst (10 mg), 25% O2/CO2 (10.4 bar, 150 psi).a ±0.5 mol h−1 kgcat−1. | |||||||
| TS-1 (XG-REF) | 0.1 | 0 | 0 | 100 | 0 | 0 | 1 |
| TS-1 (LG-12%) | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 |
| AX | 0.1 | 0 | 0 | 100 | 0 | 0 | 1 |
| A12 | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 |
| APX | 4.7 | 0 | 31.9 | 61.2 | 5.1 | 1.8 | 14 |
| AP5 | 1.6 | 0 | 11.1 | 82.7 | 6.2 | 0 | 6 |
| AP8 | 2.4 | 0 | 3.6 | 96.4 | 0 | 0 | 11 |
| AP12 | 1.8 | 0 | 8.6 | 69.1 | 13.1 | 9.2 | 6 |
| DAPX | 2.8 | 0 | 14.6 | 80.7 | 3.1 | 1.6 | 11 |
| DAP12 | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 |
| PX | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 |
| P12 | 0.5 | 0 | 0 | 100 | 0 | 0 | 2 |
To gauge the potential of the catalysts to operate with hydrogen present and improve benzaldehyde yield with an in situ generated peroxy species as an oxidant, direct synthesis of H2O2 was performed. Fig. 7 and 8 illustrate the production rate of H2O2 synthesis at ca. 2 and 30 °C respectively and the resulting wt% of peroxide is shown in Tables S2 and S3 (ESI†). The un-modified TS-1 materials were found to be inactive at both temperatures used, as were the Au mono-metallic catalysts. Peroxide production over palladium mono-metallic catalysts PX and P12 indicates that secondary porosity is beneficial at both temperatures tested. The expected synergistic effect of Au–Pd48 on peroxide production is observable. At ca. 2 °C the production rate of H2O2 over catalysts APX and AP12 are 34 ± 0.5 and 25 ± 0.5 molH2O2 h−1 kgcat−1 respectively. These values are lower when compared to literature examples; for example over AuPd supported on acid washed carbon or TiO2 of 160 ± 0.5 and 110 ± 0.5 molH2O2 h−1 kgcat−1 respectively.50,51 However, at 30 °C (Fig. 8) the peroxide production rate is significantly increased. The rate of reaction is determined by the amount of peroxide remaining in the reaction media at 30 min. The catalyst may bring about an increase in the formation of H2O as a secondary product, via hydrogenation or combustion of the peroxide. We use the rate as a guide to indicate whether a catalyst formulation is likely to support peroxide production and facilitate benzyl alcohol oxidation at a given temperature.
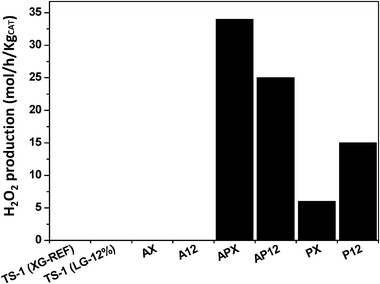 | ||
| Fig. 7 Production rate of hydrogen peroxide at 2 °C over TS-1 and Au–Pd/TS-1 catalysts. Conditions: 0–2 °C, methanol (5.6 g), water (2.9 g), 30 minutes, catalyst (10 mg), 5% H2/CO2 (28.96 bar, 420 psi), 25% O2/CO2 (10.4 bar, 150 psi). | ||
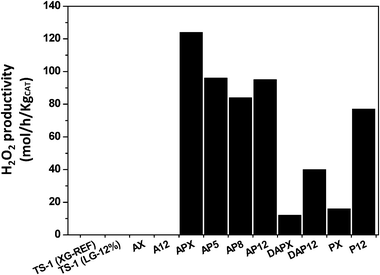 | ||
| Fig. 8 Production rate of hydrogen peroxide at 30 °C over TS-1 and Au–Pd/TS-1 catalysts. Conditions: 30 °C, methanol (5.6 g), water (2.9 g), 30 minutes, catalyst (10 mg), 5% H2/CO2 (28.96 bar, 420 psi), 25% O2/CO2 (10.4 bar, 150 psi). | ||
The addition of hydrogen to the benzyl alcohol reaction performed in the autoclave reactor at ca. 2 and 30 °C resulted in improvements in the benzaldehyde yield (Tables 5 and 6). Reaction at the lower temperature over mono-metallic gold catalysts (AX and A12) was comparable to reactions without hydrogen, i.e. a low rate comparable to data presented in Table 4, i.e. <5 molBA h−1 kgcat−1. This, we consider is simply due to the negligible formation of H2O2 or hydroperoxy species over these catalysts under these conditions. The presence of Pd in the catalyst as bi-metallic or mono-metallic nano-particles were able to facilitate hydroperoxy formation, as evidenced by the results in Fig. 7. However, the low reaction rate persists at 2 °C over these catalysts even with the in situ generation of small amounts of hydroperoxy species.
| Catalyst | Conversion (%) | Selectivity (%) | Benzaldehyde productiona (mol h−1 kgcat−1) | H2O2 residual concentration (wt%) | ||||
|---|---|---|---|---|---|---|---|---|
| Benzene | Toluene | Benzaldehyde | Benzoic acid | Benzyl benzoate | ||||
| Conditions: 0–2 °C, methanol (5.6 g), water (2.9 g), 2.3 mmol benzyl alcohol (0.25 g), 30 minutes, catalyst (10 mg), 5% H2/CO2 (28.96 bar, 420 psi), 25% O2/CO2 (10.4 bar, 150 psi).a ±0.5 mol h−1 kgcat−1. | ||||||||
| TS-1 (XG-REF) | 0.7 | 0 | 0 | 100 | 0 | 0 | 3 | 0 |
| TS-1 (LG-12%) | 1.1 | 0 | 0 | 100 | 0 | 0 | 5 | 0 |
| AX | 0.1 | 0 | 0 | 100 | 0 | 0 | 1 | 0 |
| A12 | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 | 0 |
| APX | 0.7 | 0 | 0 | 77.1 | 22.9 | 0 | 3 | 0.07 |
| AP12 | 0.7 | 0 | 0 | 63.0 | 27.0 | 0 | 2 | 0.03 |
| PX | 0.5 | 0 | 0 | 100 | 0 | 0 | 2 | 0.02 |
| P12 | 1.4 | 0 | 0 | 100 | 0 | 0 | 7 | 0.05 |
| Catalyst | Conversion (%) | Selectivity (%) | Benzaldehyde productiona (mol h−1 kgcat−1) | H2O2 residual concentration (wt%) | ||||
|---|---|---|---|---|---|---|---|---|
| Benzene | Toluene | Benzaldehyde | Benzoic acid | Benzyl benzoate | ||||
| Conditions: 30 °C, methanol (5.6 g), water (2.9 g), 2.3 mmol benzyl alcohol (0.25 g), 30 minutes, catalyst (10 mg), 5% H2/CO2 (28.96 bar, 420 psi), 25% O2/CO2 (10.4 bar, 150 psi).a ±0.5 mol h−1 kgcat−1. | ||||||||
| TS-1 (XG-REF) | 0.1 | 0 | 0 | 100 | 0 | 0 | 1 | 0 |
| TS-1 (LG-12%) | 0.2 | 0 | 0 | 100 | 0 | 0 | 1 | 0 |
| APX | 6.6 | 2.5 | 4.7 | 92.8 | 0 | 0 | 29 | 0.10 |
| AP5 | 2.0 | 0 | 6.2 | 93.8 | 0 | 0 | 9 | 0.10 |
| AP8 | 1.5 | 0 | 6.0 | 94.6 | 0 | 0 | 7 | 0.11 |
| AP12 | 7.0 | 1.4 | 4.6 | 92.6 | 1.5 | 0 | 31 | 0.13 |
| DAPX | 2.6 | 0 | 14.6 | 83.6 | 1.8 | 0 | 10 | 0.02 |
| DAP12 | 3.8 | 0 | 4.4 | 93.1 | 0 | 2.5 | 17 | 0.09 |
| PX | 2.4 | 0 | 3.7 | 96.3 | 0 | 0 | 11 | 0.06 |
| P12 | 3.4 | 0 | 13.1 | 82.8 | 1.9 | 2.3 | 13 | 0.08 |
Increasing the reaction temperature to 30 °C resulted in improvements in the benzaldehyde yield (Table 6). Previously, van der Pol et al.52 have shown that 2-octanol could be oxidised by addition of H2O2 at 30 °C over TS-1, although they reported a low total conversion of ca. 1.5% after 30 minutes. Therefore, the implication is that Ti centers are active to the formation of Ti–OOH species at such temperatures. However, below this temperature (20 °C), attempts to oxidize glycerol with H2O2 resulted in no reaction,53 which we also observed for reactions carried out at 2 °C. Diffusion limitations with respect to access of these Ti centers would appear to be a crucial aspect of any potential oxidation reaction. Over the catalysts APX and AP12 the production of benzaldehyde was found to be 29 ± 0.5 and 31 ± 0.5 mol h−1 kgcat−1 respectively. However, the rate was found to be low with catalyst AP5 and AP8, such that no conclusion can be drawn on the potential benefits of secondary porosity under these conditions. However, we consider that under these conditions the reduced diffusion limitations play a minor role when compared to the presence of metal nano-particles (Au–Pd, Pd). The stability of the peroxide over the larger metal particles of the reference based catalysts is high and potentially this facilitates the high oxidation rate observed (Table 6). The complexity of the reaction pathway is difficult to define with respect to adsorbed metal nano-particles, availability of Ti centers and the stability of the produced peroxy species. However, the high selectivity to benzaldehyde is encouraging with respect to the potential excess of hydrogen peroxide generated during the reaction. Selectivity to the secondary oxidation reaction product benzoic acid was low and the formation of benzyl benzoate low or negligible. Furthermore, no methyl benzoate, a potential condensation product from the reaction of methanol and benzoic acid, was found.
Comparing the AuPd catalysts prepared by DP (DAPX and DAP12), it can be seen that a combination of secondary porosity and smaller metal particle size facilitates both improved benzyl alcohol conversion and benzaldehyde selectivity. The formation of toluene is retarded in the case of using the hierarchical zeolite; potentially due to metal particle size differences compared to catalysts prepared by impregnation, further enhancing the benzaldehyde selectivity. Compared to the APX-12 series the smaller metal particles may play a role in the combustion of H2O2 hence the alcohol oxidation rate may be higher in the case of the smaller crystallites but the smaller metal particles being more active decrease the stability of H2O2. Thus in the overall objective of oxidising benzyl alcohol a balance must be found to introduce greater access to reactants and retain some form of stability of the peroxide species produced in situ in order to affect the greatest oxidation potential.
Conclusions
Hierarchical TS-1 materials were synthesised and used as supports for Au, Pd and AuPd nano-particles. These were tested with the oxidation of benzyl alcohol as a model reaction. The use of in situ generated H2O2 as an oxidant in this reaction would be attractive and the catalysts were evaluated for peroxide production at 2 and 30 °C. At sub-ambient conditions the rate of oxidation was found to be very low, due to a low production rate of H2O2. Increasing the reaction temperature to 30 °C improved the peroxide production rate and hence the benzaldehyde production rate. Comparison of reactions at 30 °C in the presence and absence of hydrogen indicated that the formation of peroxide species is highly advantageous with respect to oxidation rates. Benzaldehyde production rates of ca. 30 mol h−1 kgcat−1 could be achieved when the reaction was carried out with H2 and ca. 5 mol h−1 kgcat−1 with molecular oxygen. The presence of secondary porosity was found to improve the selectivity of benzaldehyde, particularly with the catalysts prepared by deposition precipitation, since the AuPd particle size was lower for catalysts with secondary porosity and led to better metal dispersion. Therefore, the use of highly active zeolitic material with secondary porosity enables this approach to be potentially broadened to a greater number of oxidisable reactants than previously considered.Acknowledgements
We thank the EPSRC and Al Jouf University (M. A.) for financial support. I. M. thanks Rey Juan Carlos University for fellowship support during the postdoctoral visit to Cardiff University.References
- M. Taramasso, G. Perego and B. Notari, US Pat., 4410501, 1983 Search PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS.
- M. G. Clerici, Met. Oxide Catal., 2009, 2, 705–754 CAS.
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Nature, 1978, 272, 437–438 CrossRef CAS.
- C. C. Freyhardt, M. Tsapatsis, R. F. Lobo, K. J. Balkus, Jr. and M. E. Davis, Nature, 1996, 381, 295–298 CrossRef CAS.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef CAS.
- R. Chal, C. Gerardin, M. Bulut and S. van Donk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- Y. Fang and H. Hu, Catal. Commun., 2007, 8, 817–820 CrossRef CAS.
- I. Schmidt, A. Krogh, K. Wienberg, A. Carlsson, M. Brorson and C. J. H. Jacobsen, Chem. Commun., 2000, 2157–2158 RSC.
- Y. Liu, W. Zhang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2001, 40, 1255–1258 CrossRef CAS.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef CAS.
- D. P. Serrano, R. Sanz, P. Pizarro and I. Moreno, Top. Catal., 2010, 53, 1319–1329 CrossRef CAS.
- D. Serrano, R. Sanz, P. Pizarro and I. Moreno, Chem. Commun., 2009, 1407–1409 RSC.
- U. Romano, A. Esposito, F. Maspero, C. Neri and M. G. Clerici, Stud. Surf. Sci. Catal., 1990, 55, 33–41 CrossRef CAS.
- A. Corma, P. Esteve, A. Martinez and S. Valencia, J. Catal., 1995, 152, 18–24 CrossRef CAS.
- M. S. Holm, E. Taarning, K. Egeblad and C. H. Christensen, Catal. Today, 2011, 168, 3–16 CrossRef CAS.
- F. Maspero and U. Romano, J. Catal., 1994, 146, 476–482 CrossRef CAS.
- R. A. Sheldon and J. Dakka, Erdoel, Erdgas, Kohle, 1993, 109, 520–522 CAS.
- C. B. Khouw, C. B. Dartt, J. A. Labinger and M. E. Davis, J. Catal., 1994, 149, 195–205 CrossRef CAS.
- H. Mimoun, in Peroxides (1983), John Wiley & Sons, Ltd., 2010, pp. 463–482 Search PubMed.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- Y. A. Kalvachev, T. Hayashi, S. Tsubota and M. Haruta, J. Catal., 1999, 186, 228–233 CrossRef CAS.
- B. S. Uphade, T. Akita, T. Nakamura and M. Haruta, J. Catal., 2002, 209, 331–340 CrossRef CAS.
- C. Qi, T. Akita, M. Okumura, K. Kuraoka and M. Haruta, Appl. Catal., A, 2003, 253, 75–89 CrossRef CAS.
- N. Yap, R. P. Andres and W. N. Delgass, J. Catal., 2004, 226, 156–170 CrossRef CAS.
- B. Taylor, J. Lauterbach and W. N. Delgass, Appl. Catal., A, 2005, 291, 188–198 CrossRef CAS.
- A. K. Sinha, S. Seelan, M. Okumura, T. Akita, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2005, 109, 3956–3965 CrossRef CAS.
- A. Itoh, Y. Kuroda, T. Kitano, Z. Guo, A. Kunai and K. Sasaki, J. Mol. Catal., 1991, 69, 215–222 CrossRef CAS.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Commun., 2007, 8, 247–250 CrossRef CAS.
- S. Ma, G. Li and X. Wang, Chem. Lett., 2006, 35, 428–429 CrossRef CAS.
- K. Mori, Y. Miura, S. Shironita and H. Yamashita, Langmuir, 2009, 25, 11180–11187 CrossRef CAS.
- M. S. Yalfani, S. Contreras, J. Llorca, M. Dominguez, J. E. Sueiras and F. Medina, Phys. Chem. Chem. Phys., 2010, 12, 14673–14676 RSC.
- S. Okada, S. Ikurumi, T. Kamegawa, K. Mori and H. Yamashita, J. Phys. Chem. C, 2012, 116, 14360–14367 CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917–1923 RSC.
- S. Meenakshisundaram, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. W. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341–356 RSC.
- M. A. Uguina, D. P. Serrano, G. Ovejero, R. Van Grieken and M. Camacho, Appl. Catal., A, 1995, 124, 391–408 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and Á. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS.
- B. Taylor, J. Lauterbach and W. N. Delgass, Catal. Today, 2007, 123, 50–58 CrossRef CAS.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 361–364 CrossRef CAS.
- P. Paredes Olivera, E. M. Patrito and H. Sellers, Surf. Sci., 1994, 313, 25–40 CrossRef CAS.
- D. I. Enache, D. Barker, J. K. Edwards, S. H. Taylor, D. W. Knight, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 407–411 CrossRef CAS.
- S. S. Hladyi, M. K. Starchevsky, Y. A. Pazdersky, M. N. Vargaftik and I. I. Moiseev, Mendeleev Commun., 2002, 45–46 CrossRef CAS.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem.–Eur. J., 2011, 17, 6524–6532 CrossRef CAS.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- M. Piccinini, E. Ntainjua N, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488–2492 RSC.
- J. K. Edwards, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512–8515 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS.
- A. J. H. P. van der Pol and J. H. C. van Hooff, Appl. Catal., A, 1993, 106, 97–113 CrossRef CAS.
- P. McMorn, G. Roberts and G. Hutchings, Catal. Lett., 1999, 63, 193–197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00493g |
| This journal is © The Royal Society of Chemistry 2013 |