 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic water oxidation with cobalt-containing tungstobismutates: tuning the metal core†
Fabio
Evangelisti
,
Pierre-Emmanuel
Car
,
Olivier
Blacque
and
Greta R.
Patzke
*
Institute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: greta.patzke@aci.uzh.ch; Fax: +41 44 635 6802; Tel: +41 44 635 4691
First published on 14th October 2013
Abstract
A new type of tungstobismutate water oxidation catalyst (WOC) with a disordered Co|W core, [{Co(H2O)3}2{CoBi2W19O66(OH)4}]10− (1) was tested for visible-light-driven performance and compared to a series of isostructural Co- and Mn-containing polyoxometalates with variable transition metal contents, ([Co2.5(H2O)6{Bi2W19.5O66(OH)4}]8− (2) and [Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]6− (3)). All compounds were structurally characterized, and no indications for significant decomposition under catalytic conditions for visible-light-driven water oxidation ([Ru(bpy)3]2+ as photosensitizer (PS) and S2O82− as electron acceptor in different buffer systems) were found. For the first time, subtle differences in the core disorder patterns of isostructural POM-WOCs were revealed to be decisive for the catalytic activity of (1) (maximum TON of 21 with 97% oxygen yield for 115 μM (1)). Performance comparison of the POM series sheds new light on the structure–activity relationships for targeted POM-WOC construction. Indeed, the Co disorder differences between (1) and (2) exclusively affect the sterically more accessible external site of the two crystallographically independent Co core positions which has a 25% higher Co occupancy in (1). This points to a stereoselective reaction pattern for the tetranuclear POM core of WOC (1) which might open up novel construction strategies for the economic redesign of sandwich-type POM-WOCs. In parallel, we demonstrate for the POM series (1)–(3) that electrochemical measurements under catalytic conditions are a promising and convenient pre-screening strategy for WOC activity. Furthermore, POM/PS complex formation of (1) with [Ru(bpy)3]2+ is investigated in detail, and the different roles of Mn- and Co-centers in POM-WOC synthesis are compared.
Introduction
Artificial photosynthesis is a promising strategy to solve our worldwide energy and climate problems in a sustainable manner.1 Water oxidation is the most challenging step in the process, and nature's cubane-related {Mn3CaO4} core of Photosystem II (PS II) is by far the most inspirational motif for the design of water oxidation catalysts (WOCs).2 This places polyoxometalate (POM) clusters, which are preferably formed by W, Mo and V in their highest oxidation states,3 as structurally versatile hosts for a wide variety of tailored metal–oxo cores in the focus of current WOC research.4 POM-WOC design is a complex multi-parameter task with structure–activity relationships (SAR) and catalyst stability as representative crucial issues.5 We introduce a new efficient Co/Bi–POM-WOC, [{Co(H2O)3}2{CoBi2W19O66(OH)4}]10−, emerging from an isostructural series of sandwich-type Co- and Mn-POMs. Comparison of their WOC performance demonstrates that subtle variations of the metal-oxo core exert a tremendous influence on the observed catalytic activity. Special emphasis was placed on three essential criteria for efficient POM-WOC design: (1) SAR for POM core/shell construction, (2) POM/photosensitizer interactions and (3) WOC stability, i.e. drawing the line between homogeneous or heterogeneous catalysis.6(1) First, the crucial importance of SAR for POM catalyst construction is evident from the wide variety of mainly empirically discovered Co-, Ni-,7 and Ru-containing POM-WOCs with [Co4(H2O)2(PW9O34)2]10− and [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− among the most prominent examples.8 The growing class of Co-POMs illustrates the present challenges particularly well. Although manifold new members of this POM family are being discovered,9 only a small fraction of them are visible-light-driven WOCs with a preference for sandwich-type Co-POMs.4h,10–13 Recently, however, a new Keggin-type K7[CoIIICoII(H2O)W11O39] WOC was reported, which amplified the family of POM-WOCs towards smaller mixed-valent clusters.11 Comparison of this novel POM-WOC with Keggin-type analogues points to an essential interplay of the central {CoIIIO4} unit with the catalytically active peripheral {CoIIO6} octahedron which is orientationally disordered.11 Small molybdate clusters, such as [CoMo6O24H6]3− and [Co2Mo10O38H4]6−, have also been identified as promising WOCs, thus demonstrating that neither a critical POM size nor multi-site metal cores are mandatory for catalytic activity.4k,12 Not only the key requirements for POM-WOC shell construction still remain unclear to a large extent, but the type and oxidation state of the POM belt atoms require further systematic SAR explorations as well. Ni-POMs serve as an example: whereas the [Ni5(OH)6(OH2)3(Si2W18O66)]12− polyanion with a clamshell-shaped geometry exhibits WOC activity,7 the Ni-analogue of the catalytically active sandwich-type [Co4(H2O)2(SiW9O34)2]12− does not show any WOC properties under analogous test conditions.13 Mn-POMs are even more elusive targets for WOC construction: although PSII-inspired molecular Mn-WOCs attract a lot of attention, no efficient Mn-POM-WOCs have been reported so far to the best of our knowledge.5b This renders POM-WOC discovery a multi-parameter problem which has mostly been solved with empirical approaches to date.
(2) Next, the high negative charge of POMs poses another challenge for their combination with often positively charged photosensitizer (PS) molecules in homogeneous water oxidation systems. Crystalline POM hybrids of [PW11O39]7− and [PW12O40]3− with tris(bipyridine)ruthenium(III), [Ru(bpy)3]2+, one of the most widely used PS types, were first obtained through slow diffusion techniques.14 Soon after, the model system [β-Mo8O26]4−/[Ru(tbbpy)(biH2)]2+ confirmed the presence of supramolecular aggregates in solution.15 Next, our studies into the [Co4(H2O)2(SiW9O34)2]12− and [{Ru3O3(H2O)Cl2}(SiW9O34)]7− POM-WOCs under catalytic conditions provided clear spectroscopic evidence for their formation of POM-PS complexes which could be recycled several times for new WOC runs.13 Detailed flash photolysis investigations of primary interactions of [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− with [Ru(bpy)3]2+/3+ pointed to static quenching in POM/PS ion paired species (ratios ca. 1/4) as an adverse process in the photocatalytic cycle.16 In contrast, POM/PS hybrid complex formation with [Ru(bpy)3]2+ has recently been applied as a synthetic concept to construct new WOCs from the polyoxomolybdates [Mo6O19]2−, [Mo5S2O23]4− and α-[Mo8O26]4−.17 The complexity of these observations demonstrates that POM/PS interactions require detailed studies for each newly found POM-WOC.
(3) Finally, the high structural flexibility of POMs often comes at the expense of their stability. Possible metal leaching from the core region might give rise to subsequent formation of an active nanoparticular catalyst, as recently postulated for the formation of CoOx nanocatalysts from [Co4(H2O)2(PW9O34)2]10− as a precatalyst under electrochemical conditions.18 In parallel, laser flash photolysis studies on [Co4(H2O)2(PW9O34)2]10− under visible-light-driven water oxidation conditions indicated that the Co-POM serves as a precatalyst for an active molecular species.19 Most recent electrochemical studies on this model Co-POM-WOC finally revealed that subtle variations of the reaction conditions for micro- and nanomolar amounts of catalyst species render the true WOC extremely difficult to assign, i.e. the distinction between homo- and heterogeneous catalysis reaches the limits of standard analytical methods.20 Targeted applications of POMs as heterogeneous electrocatalysts, such as [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− on carbon paste electrodes, are an elegant alternative.21 However, the search for genuinely homogeneous POM-WOCs and related molecular catalysts remains essential for understanding their reaction pathways through in situ spectroscopy22 and theoretical calculations23 on the way to clear-cut SAR for informed design.
In the following, we introduce [{Co(H2O)3}2{CoBi2W19O66(OH)4}]10− (Co/Bi-POM (1)) as a POM-WOC with high stability under visible-light-driven water oxidation conditions. Unexpectedly, systematic comparison of Co/Bi-POM (1) with isostructural POMs showed that neither the closely related Co-POM [Co2.5(H2O)6{Bi2W19.5O66(OH)4}]8− nor its Mn-POM analogue, [Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]6−, are active WOCs. The outstanding catalytic properties of Co/Bi-POM (1) among the series were investigated with a variety of analytical methods and reference experiments in search of the active catalytic species. The results shed new light on guidelines for systematic POM-WOC construction.
Results and discussion
Synthetic parameters
Three isostructural sandwich POMs with disordered M2+|W cores were obtained from solution methods through systematic variations of literature protocols in order to compare the influence of cores with different Co2+|W ratios to Mn2+|W cores on the catalytic activity.24,25 Replacement of pure aqueous media by acetate solutions (pH 4.5, 40 mM) was applied to access two isostructural sandwich-type Co-POMs, namely Na10[{Co(H2O)3}2{CoBi2W19O66(OH)4}]·37H2O (Co/Bi-POM (1)) and Na8[Co2.5(H2O)6{Bi2W19.5O66(OH)4}]·32H2O (Co/Bi-POM (2)). Their isostructural Mn-analogue Na6[Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]·36H2O (Mn/Bi-POM (3)) was obtained from the same protocol24 by replacing the Co-based precursor with MnSO4·2H2O. In the following, crystal structures of compounds (1)–(3) are discussed in detail and compared to the present state of investigations on this POM type.Structure of Na10[{Co(H2O)3}2{CoBi2W19O66(OH)4}]·37H2O (Co/Bi-POM (1))
The sandwich-type Co/Bi-POM (1) crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). The two identical lacunary β-{BiW9O33} units of the [{Co(H2O)3}2{CoBi2W19O66(OH)4}]10− polyanion are connected via two disordered {WO2|CoO2} groups W(10)|Co(2) and two {Co(H2O)3} (Co(1)) moieties which form the tetranuclear metal core. The two crystallographically equivalent internal atoms of the core display disorder which was refined with a model based on site occupancies of 50% W(10) and 50% Co(2) which are in agreement with elemental analysis and overall charge balance.
(Table S1†). The two identical lacunary β-{BiW9O33} units of the [{Co(H2O)3}2{CoBi2W19O66(OH)4}]10− polyanion are connected via two disordered {WO2|CoO2} groups W(10)|Co(2) and two {Co(H2O)3} (Co(1)) moieties which form the tetranuclear metal core. The two crystallographically equivalent internal atoms of the core display disorder which was refined with a model based on site occupancies of 50% W(10) and 50% Co(2) which are in agreement with elemental analysis and overall charge balance.
The central Bi atoms of Co/Bi-POM (1) exhibit pyramidal environments consisting of a basal plane with three Bi–O bonds (2.130(6) to 2.154(6) Å) and a lone pair at the top of the pyramid. All of the non-disordered tungsten atoms are octahedrally coordinated with W–O bond distance ranges of 1.766(7)–2.083(7) Å for equatorial W⋯O bonds. The longer internal W⋯O bond distances range from 2.209(7) to 2.320(7) Å, whereas the external W⋯O bonds are characteristically short (1.726(8) and 1.771(7) Å). Intermetallic W⋯W distances range from 3.2848(6) to 3.3880(6) Å for the edge-sharing WO6 octahedra and from 3.6708(7) to 3.7097(9) Å for the corner-sharing WO6 polyhedra, respectively.
The disordered W(10)|Co(2) sites are octahedrally coordinated by six oxygen atoms with metal-oxygen distances in the 1.893(9)–2.112(8) Å range and intermetallic distances between 3.5461(8) and 3.8728(10) Å for corner-sharing W|Co⋯W octahedra. The remaining two terminal {Co(H2O)3} (Co(1)) units of the tetranuclear core show longer Co⋯OH2 bonds (2.088(8)–2.211(7) Å) than Co⋯O bonds (2.030(7)–2.070(7) Å). Interatomic distances between corner-sharing Co⋯W octahedra are in the expected range (3.5435(18) to 3.7862(15) Å).
Five crystallographically independent and fully occupied Na positions were located in the course of the refinement (Na(1), Na(2), Na(3), Na(4), Na(5)). They are coordinated by oxygen atoms of the polyoxoanion or by crystal water molecules. Their coordination numbers increase from 3 (Na(5)) over 6 (Na(1), Na(2), Na(3)) to 7 (Na(4)) with Na⋯O bond lengths ranging from 2.18(2) to 2.88(1) Å. Several aqualigands complete the environment of Co/Bi-POM (1).
Bond valence sum (BVS) values26 for all tungsten atoms (5.8–6.1) agree well with the common W oxidation state of +6 in POMs. Bi is present in the +3 oxidation state according to its BVS of 3.05. The BVS value of 1.7 for Co(1) indicates the presence of Co2+. Most oxygen atoms of Co/Bi-POM (1) have BVS values corresponding to an oxidation state of −2 (between 1.4 and 2.1), except for O(33), O(34), O(35), O(36) and O(37). BVS values of 0.28, 0.32 and 0.34 for O(33), O(34) and O(35), respectively, identify them as terminal H2O molecules. According to their medium BVS values (0.8 and 0.7), O(36) and O(37) are bridging hydroxo groups.
Structure of Na8[Co2.5(H2O)6{Bi2W19.5O66(OH)4}]·32H2O (Co/Bi-POM (2))
Co/Bi-POMs (1) and (2) have the same POM architecture in common, i.e. they both consist of two identical lacunary β-{BiW9O33} shells which are connected through a Co|W tetranuclear metal core. Co/Bi-POM (2), however, crystallizes in the monoclinic space group P21/n (Table S1†). Analogous to Co/Bi-POM (1), the tetranuclear metal core of (2) displays disorder as well. Other than in (1), both crystallographically independent core metal positions of Co/Bi-POM (2) are disordered. Refinement of this disordered model resulted in occupancies of 50% W(10)|Co(1) and 75/25% Co(2)|W(11), respectively. Bismuth displays the same pyramidal oxygen coordination environment with related bond distances (2.122(8), 2.135(8) and 2.148(8) Å) as described for (1) above. Likewise, all tungsten centers of (2) are octahedrally coordinated with medium bond lengths (1.780(9)–2.068(8) Å) for the equatorial W⋯O bonds, long internal W⋯O bonds (2.198(7)–2.301(8) Å) and short external W⋯O distances (1.714(8)–1.756(8) Å). W⋯W contacts range from 3.2770(9) to 3.3561(11) Å for edge-sharing WO6 moieties and from 3.6610(10) to 3.7095(10) Å for the corner-sharing WO6 octahedra.The disordered core metal sites W(10)|Co(1) and Co(2)|W(11) are both octahedrally coordinated with oxygen atoms which exhibit bond distance ranges of 1.863(1)–2.113(9) Å and 1.967(1)–2.108(8) Å, respectively. All intermetallic distances (W(10)|Co(1), Co(2)|W(11) and fully occupied tungsten atoms) cover a range from 3.498(19) to 3.913(8) Å.
The surroundings of Co/Bi-POM (2) are constituted by crystal water molecules, three fully occupied Na positions and two 50% occupied Na positions. Refinements revealed an increase of Na coordination numbers from 3 (Na(5)) over 4 (Na(4)) to 6 (Na(1), Na(2), Na(3)) with Na⋯O bond distances between 2.310(19) and 2.825(16) Å.
BVS values between 5.83 and 6.09 for all tungsten atoms clearly point to an oxidation state of +6 throughout, and the BVS value of 3.05 for Bi corresponds well with the expected oxidation state of +3. All oxygen atoms exhibit BVS values between 1.4 and 2.1 suggesting an oxidation state of −2, except for lower values for O(30), O(33), O(35), O(36) and O(37) (0.9, 0.9, 0.7, 0.5 and 0.6, respectively). The two values of 0.9 for O(30) and O(33) indicate the presence of hydroxo groups, while the values of 0.5, 0.6 and 0.7 for O(36), O(37) and O(35) are characteristic for water molecules.
Structure of Na6[Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]·36H2O (Mn/Bi-POM (3))
Mn/Bi-POM (3) is isostructural with the Co/Bi-POMs (1) and (2), and it crystallizes in the monoclinic space group P21/n (Table S1†). The β-{BiW9O33} building blocks sandwich a tetranuclear Mn|W core, and both of its crystallographically independent positions are disordered. Other than in compounds (1) and (2), the terminal metal position displays more pronounced disorder than the internal sites: occupancies were refined to 90|10% for W(10)|Mn(1) and to 35|65% for W(11)|Mn(2) (Fig. 1). Nine different oxygen atoms of (3) form terminal W⋯O bonds of the octahedral {WO6} building blocks (distances between 1.704(16) and 1.740(13) Å), and three oxygen positions form connecting internal bonds with notably longer distances (2.214(12) to 2.299(13) Å). The octahedral coordination of the nine fully occupied tungsten positions is completed by oxygen atoms in equatorial positions with bond lengths ranging from 1.784(15) to 2.056(16) Å. The W⋯W distances within the lacunary β-{BiW9O33} units range from 3.6654(11) to 3.6998(11) Å for corner-sharing and from 3.2712(11) to 3.3514(11) Å for edge-sharing {WO6} octahedra, respectively.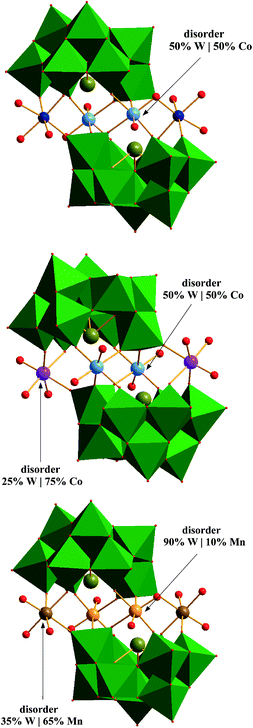 | ||
| Fig. 1 Polyhedra and ball-and-stick representations (WO6 = green, O = red) of Co/Bi-POM (1) (top; Co = dark blue, W|Co = light blue), Co/Bi-POM (2) (middle; 50W|50Co = light blue, 25W|75Co = pink) and Mn/Bi-POM (3) (bottom; 90W|10Mn = orange, 35W|65Mn = brown). | ||
The tungsten centers of the disordered W(10)|Mn(1) and W(11)|Mn(2) sites are octahedrally coordinated with W⋯O bond lengths ranging between 1.76(2) to 2.186(13) Å, associated to interatomic metal cation distances between 3.5518(13) and 3.9239(13) Å for corner-sharing WO6|MnO6⋯WO6 octahedra.
As observed for the Co/Bi-POMs (1) and (2), the pyramidal coordination of the Bi heteroatoms is constituted by a lone electron pair oriented towards the tetranuclear metal core and three oxygen atoms with Bi⋯O distances of 2.131(13), 2.142(13) and 2.147(12) Å.
Mn/Bi-POM (3) is surrounded by three Na positions which are all fully occupied and octahedrally coordinated with Na⋯O bond lengths ranging between 2.32(3) and 2.64(5) Å. Crystal water molecules complete the surroundings of the [Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]6− polyanion. All tungsten BVS values (5.88 to 6.15) clearly indicate an oxidation state of +6, and the BVS value of 3.16 for Bi is in line with Bi3+.
Oxygen BVS values of (3) are in the range for −2 oxidation states (between 1.5 and 2.1), except for O(34), O(35), O(36), O(37) and O(38). Lower BVS values (0.61, 0.45 and 0.49) identify O(36), O(37) and O(38) as terminal water molecules, whereas medium BVS values of 1.4 and 1.3 for O(34) and O(35), respectively, suggest the presence of hydroxo groups.
Phase purity of representative Co/Bi-POM (1), Co/Bi-POM (2) and Mn/Bi-POM (3) samples was investigated with PXRD measurements (Fig. S1–S3†). The related PXRD patterns are in line with the isostructural POM series, and the experimental data agree with the calculated theoretical patterns.
Our results on Co/Bi-POMs (1) and (2) confirm the strong tendency towards Co|W core disorder which has previously been observed for the isostructural POM Na(10−4x)[Bi2W(20+x)Co(2−x)O70(H2O)6]·41H2O (x = 0.28) with intermediate cobalt content compared to (1) and (2).25 This may indicate a certain phase width for Co|W cores with variable compositions, and further studies into the synthetic parameters are under way. Furthermore, high quality single crystal growth of Na10[Bi2W20Co2O70(H2O)6]·30H2O and Na10[Bi2W20Mn2O70(H2O)6]·26H2O, respectively, was not possible via a related protocol, so that no detailed crystallographic data were reported for these compounds.24 Furthermore, ICSD data for the Na/K-containing analogue of Mn/Bi-POM (3) with 68% Mn on the terminal sites (100% W on the internal sites) are related to the above mentioned disorder pattern of the Mn sites.27 Nevertheless, crystallographic evidence for [Bi2W20M2O70(H2O)6]10− (M = Mn, Co) with fully occupied M sites is missing to date, and the present data clearly suggest that metal core disorder is a characteristic property of this sandwich-type tungstobismutate type.
Cyclic voltammetry of POMs (1)–(3)
Generally, electrochemical characterization of sandwich-type polyoxotungstates in aqueous solutions by cyclic voltammetry (CV) is strongly pH dependent. In the negative potential range, 2–4 processes involving tungsten centers can be observed which are particularly evident in acidic media (0.4 M NaOAc/HOAc buffer at pH 3, cf. Fig. S6†). All compounds were furthermore studied under catalytic conditions at pH 5.8 in silicate buffer (20 mM Na2SiF6/NaHCO3) to enhance the otherwise weakly developed redox waves of CoII. For stability tests of Co/Bi-POM (1), combined CV and UV/Vis measurements were performed on aged solutions which were stored for 24 h under ambient conditions. No significant changes in cyclic voltammograms and UV/Vis spectra were observed after the ageing process.Cyclic voltammograms of Co/Bi-POM (1) in 0.4 M H2SO4 (pH 3) (Fig. S6†) display defined oxidation peaks of the W6+/W5+ couple at −0.060 V, −0.104 V and −0.240 V together with an associated but less defined reduction peak at −0.670 V. The Co2+/Co3+ redox couple shows a quasi-reversible wave with an oxidation peak at +0.965 V and a rather undefined reduction peak. The observed peak broadening may point to a sum of a pairs of bands which are undergoing a two electron oxidation process.
It is well known from literature that only monosubstituted POMs and [CoxIIW11−xO40]6− display an electroactive Co center.28 CV measurements in protic solvents may afford asymmetric electrochemical patterns with much larger anodic than cathodic branches. These phenomena are often related to water oxidation processes occurring at the glassy carbon surface,29 and they are reinforced at pH values of the supporting electrolyte which are close to catalytic conditions, thereby excluding reliable assignment of electrons to different redox waves of the metal centers.
The Co2+/Co3+ redox couple of Co/Bi-POM (1) in catalytic buffer medium (20 mM Na2SiF6/NaHCO3, pH 5.8, cf.Fig. 2) displays less defined oxidation peaks in the positive region at +0.623 V (I) and +0.934 V (II) together with associated reduction peaks at +0.743 V (II′) and +0.607 V (I′). The redox processes can be assigned to the two different intermediates 2(CoII3)/2(CoII2CoIII).30 The negative domain (Fig. S7†) displays two characteristic wave pairs of the W6+ centers with different intensities at −0.135 V and −0.328 V for the oxidation process and at −0.160 V and −1.009 V for the reduction process, respectively. These intensity differences are due to the different numbers of involved electrons, i.e. 2 electrons for the −1.009 /−0.328 V couple and one electron for the −0.160/−0.135 V couple.28a
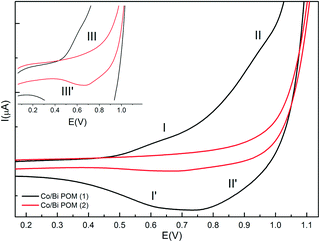 | ||
| Fig. 2 Cyclic voltammograms of Co/Bi-POM (1) (15.8 mM – black line) and Co/Bi-POM (2) (15.8 mM – red line) in Na2SiF6/NaHCO3 buffer (20 mM) at pH 5.8; scan rate: 10 mV s−1, (vs. Ag/AgCl). Insert: magnification of Co/Bi-POM (2). | ||
In comparison, Co/Bi-POM (2) shows less defined and convoluted W oxidation waves in the negative domain at −0.546 V, −0.361 V and −0.271 V which are accompanied by reduction waves located at −0.718 V and −0.595 V (Fig. S7†). The observed oxidation processes of CoII are in line with literature:31 a broadened and rather undefined oxidation wave at +0.852 V (III) corresponds to the respective reduction at +0.648 V (III′) (Fig. S7†). We would like to point out that Co centers in POMs are generally difficult to monitor with CV methods which renders their observed response in (1) and (2) under catalytic conditions truly significant and selective.32
Cyclic voltammograms of Mn/Bi-POM (3) (Fig. 3) showed a broad reduction wave of the tungsten centers in the negative range at −0.962 V which can be interpreted as a convolution of two processes. One re-oxidation peak at −0.256 V is associated with the first reduction wave and the 70 mV peak potential difference corresponds to a reversible one electron redox transfer.28a However, the re-oxidation wave corresponding to the second reduction process displays a more complex splitting pattern with another wave located at −0.186 V. The Mn2+/Mn3+ couple in the positive scan range displays a potential difference of +0.409 V (oxidation: +0.536 V, reduction: +0.127 V). There is also indication of a second Mn2+/Mn3+ redox couple with weak peaks appearing at +0.427 V and at +0.295 V with a more defined shoulder (I–I′,II′), respectively. In line with literature data for Mn-POMs, these redox couples are related to one-electron Mn2+/Mn3+ transfer processes between different Mn sites of the POM moiety.33 Computational studies and more detailed electrochemical investigations (controlled-potential electrolysis and coulometry) are currently in progress.
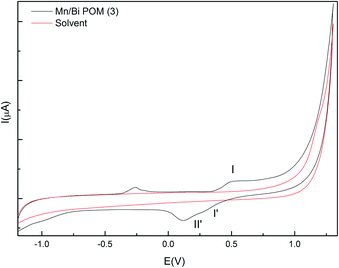 | ||
| Fig. 3 Cyclic voltammograms of Mn/Bi-POM (3) (30 mM) in Na2SiF6/NaHCO3 (20 mM) buffer at pH 5.8; scan rate: 10 mV s−1, (vs. Ag/AgCl). | ||
Spectroscopic and stability investigations
Stability tests of POMs (1)–(3) in catalytic media (Na2SiF6 buffer at pH 5.8 and NaPi buffer at pH 8) were conducted with UV/Vis spectroscopy. Co/Bi-POM (1) shows two characteristic bands in NaSiF6 media assigned to Od–W charge transfer (< 450 nm) and to the 4T1g(P)→4T1g(F) transition of octahedral Co2+ at 543 nm, respectively.32b,34 Most importantly, spectra of (1) recorded after 3 h of ageing in catalytic media did not show significant intensity decreases of the above Co absorption bands and a slight intensity loss (<5%) was only observed after 12 h (Fig. 4). Based on this evidence, we conclude that POM-WOC (1) enters the photocatalytic cycle as a structurally intact active species with no substantial decomposition or side product formation processes involved.32b Similar to (1), UV/Vis spectra of Co/Bi-POM (2) display a O–W charge transfer band in the higher energy region (above 370 nm) as well as the Co2+ related 4T1g(P) → 4T1g(F) transition at 543 nm. A third absorption band around 490 nm might indicate Jahn–Teller distortion of the octahedrally coordinated Co centers.46a Likewise, Co/Bi-POM (2) remained stable after 2 h of exposure to catalytic conditions with Na2SiF6 buffer.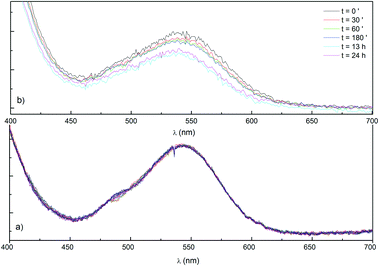 | ||
| Fig. 4 Time-dependent UV/Vis absorption spectra of (a) Co/Bi-POM (2) (70 μM) and (b) Co/Bi-POM (1) (50 μM) (final recording after 24 h) in Na2SiF6/NaHCO3 buffer (30 mM, pH 5.8). | ||
Concerning stability tests in NaPi media, Co/Bi-POM (1) did not show any intense absorption bands, whereas spectra of (2) are virtually identical to those recorded in Na2SiF6 buffer (Fig. S8†).
UV/Vis spectra of Mn/Bi-POM (3) in catalytic buffer media exhibit an intense O–W charge transfer peak below 293 nm. Another broad band around 390–400 nm indicates the presence of Mn2+,47 whereas the typical bands of Mn3+ and Mn4+ in the 450–500 nm range were absent.35 Given that no significant intensity loss after 24 h of ageing was observed, the Mn-containing sandwich type POM (3) is assumed to be stable under catalytic test conditions.
Visible light-driven water oxidation
Visible-light-driven water oxidation activity of POMs (1)–(3) was investigated according to standard literature protocols13,36 (for details cf. Experimental) with [Ru(bpy)3]2+ as photosensitizer (PS) and S2O82− as a sacrificial electron acceptor in different buffer media: NaOAc/HOAc (40 mM, pH 4.7), NaPi (40 mM, pH 7 and 8), and Na2SiF6/NaHCO3 buffer (20 mM, pH 5.8). Na2SiF6 buffer offers the advantage of reducing the radical decomposition of the Ru-PS.37Catalytic performance was evaluated with special emphasis on (a) turnover number (TON) expressing the ratio between total molar amount of O2 produced and number of Co centers involved in the catalytic cycle, and (b) yield (per S2O82−), based on a maximum oxygen yield corresponding to half of the initial molar S2O82− concentration. A precise description of the applied calibration and Clark electrode techniques can be found in the ESI† (cf. also Fig. S10–11).
Interestingly, only Co/Bi-POM (1) exhibited catalytic water oxidation activity in Na2SiF6 media, whereas compounds (2) and (3) remained inactive under the applied test conditions. The catalytic performance parameters of (1) were investigated in detail.
Co/Bi-POM (1) exhibits water oxidation activity in the concentration range from 62 μM to 400 μM, and maximum TON (21) with 97% oxygen yield were observed for 115 μM. This oxygen yield is rather high in comparison with comparable Co-POM WOCs under related catalytic conditions.38 At 166 μM, POM-WOC (1) afforded the highest molar O2 production (28.7 μmol, 97% yield; cf.Fig. 5). This is in stark contrast to the observed catalytic inactivity of (2) over the same concentration range (62 μM–400 μM, cf. Fig. S11 and S14†), because both compounds differ only with respect to the disorder on the W|Co sites (Fig. 1). For the optimal concentration (166 μM) of the active POM-WOC (1), the initial turnover frequency (TOF) was determined as 0.9 min−1.
![Kinetic monitoring (GC) of O2 formation from photocatalytic water oxidation with Co/Bi-POM (1). Conditions: LED lamp, 470 nm; 1 mM [Ru(bpy)3]Cl2, 5 mM Na2S2O8, Na2SiF6 buffer pH 5.8 (20 mM) [cyan star: 400 μM, black square: 62 μM, blue prism: 255 μM, green triangle: 166 μM, red circle: 115 μM].](/image/article/2013/CY/c3cy00475a/c3cy00475a-f5.gif) | ||
| Fig. 5 Kinetic monitoring (GC) of O2 formation from photocatalytic water oxidation with Co/Bi-POM (1). Conditions: LED lamp, 470 nm; 1 mM [Ru(bpy)3]Cl2, 5 mM Na2S2O8, Na2SiF6 buffer pH 5.8 (20 mM) [cyan star: 400 μM, black square: 62 μM, blue prism: 255 μM, green triangle: 166 μM, red circle: 115 μM]. | ||
Concentration screening of Co/Bi-POM WOC (1) is in line with previously reported trends.11,36 Gradual increase of the POM concentration leads to maximum POM-WOC efficiency at 166 μM for Co/Bi-POM WOC (1). However, higher POM concentrations above this optimal working point induce a significant decline in catalytic activity (Fig. 5, Table 1), e.g. O2 yields decrease from 76% (16 μmol) for 255 μM of (1) to 26% (5.6 μmol for 400 μM (1)), respectively. This trends clearly shows that POM-WOC concentration is a key optimization parameter.
| (1) (μM) | O2 (μmol) | Yield (%) | TON |
|---|---|---|---|
| 62 | 6.8 | 36 | 10 |
| 115 | 26.3 | 97 | 21 |
| 166 | 28.7 | 97 | 16 |
| 255 | 16.5 | 77 | 6 |
| 400 | 5.7 | 26 | 2 |
Ion pairing between the highly negatively charged Co/Bi-POM (1) and the positively charged [Ru(bpy)3]2+ PS is occurring over the entire POM concentration range from 62 μM onwards and can be observed as formation of a dark orange colloidal suspension of the POM-PS complex as reported in our previous work.13 POM-PS complex formation becomes more evident for higher POM concentrations and seems to be an integral part of the catalytic process. However, the precise structure of these ion associates in solution remains to be further elucidated.
The POM-PS complex of (1) has been studied with various analytical techniques, including FT-IR (Fig. 7–8 and S15†) and TG (Fig. 9), as outlined in the next sections. Furthermore, the influence of different catalytic parameters on the WOC efficiency of (1) was investigated. First, the S2O82− concentration was varied between 2.5 and 9 mM and all other parameters were kept constant. We observed that the standard 5 mM concentration for catalytic tests leads to maximum O2 production rates. Higher and lower persulfate concentrations decrease the O2 yields and no O2 evolution was detected in the absence of S2O82− (cf. also Table S8†).
Analogous tests were performed to investigate the influence of the [Ru(bpy)3]Cl2 concentration parameter in the range from 0 mM to 2 mM. Again, the standard 1 mM PS concentration applied for catalytic tests afforded maximum oxygen yield, and no O2 evolution occurred in the absence of PS (Table S8†). POM-PS complex formation was observed over the entire PS concentration range, and the amounts formed were determined to be approx. the same for (1) and (2) after centrifugation and subsequent drying at room temperature under N2 atmosphere. Obviously, stoichiometric complex formation is reached at 0.5 mM PS concentration, and further investigations are under way to determine if excess PS would accelerate WOC oxidation.
Detailed kinetic evaluations of Co/Bi-POM (1) are difficult, because the correlation between WOC concentration and O2 evolution appears to be rather complex. Whereas oxygen formation reaches its maximum for medium POM-WOC concentrations (around 166 μM), the reaction rate apparently declines again for higher WOC concentrations (255 μM and 400 μM). A follow-up study is in progress to establish a coherent kinetic model correlating POM-PS complex formation with the concentration-dependent reaction rates.
Catalyst stability tests
Stability of POM-WOC catalysts against leaching of metal cations which might form secondary – and possibly heterogeneous – active species is a key design criterion for artificial photosynthesis processes.18,39 UV/Vis experiments (cf. above) indicate stability of the Bi-POMs (1)–(3) under the applied catalytic conditions.The formation of free Co2+ species was furthermore excluded on the basis of CV measurements of Co(NO3)2 solutions at different concentrations (5–20% of the above WOC concentrations, i.e. 15.8–50 mM under standard conditions with Na2SiF6 buffer). These reference solutions show Co redox waves at 0.652 V/0.464 V and 0.973 V/0.774 V (see Fig. S7†), i.e. they display entirely different electrochemical behavior than observed for the Co/Bi-POMs (1) and (2) (Fig. 6).
![Cyclic voltammograms of Co/Bi-POMs (1) (black) and (2) (dashed black), and of Mn/Bi-POM (3) (blue); conditions: 1 mM [Ru(bpy)3]Cl2 in Na2SiF6/NaHCO3 (20 mM) buffer at pH 5.8; scan rate: 10 mV s−1 (vs. Ag/AgCl). Main picture: water oxidation current flow for Co/Bi-POM (1) (inset: magnification 0–1.2 V range).](/image/article/2013/CY/c3cy00475a/c3cy00475a-f6.gif) | ||
| Fig. 6 Cyclic voltammograms of Co/Bi-POMs (1) (black) and (2) (dashed black), and of Mn/Bi-POM (3) (blue); conditions: 1 mM [Ru(bpy)3]Cl2 in Na2SiF6/NaHCO3 (20 mM) buffer at pH 5.8; scan rate: 10 mV s−1 (vs. Ag/AgCl). Main picture: water oxidation current flow for Co/Bi-POM (1) (inset: magnification 0–1.2 V range). | ||
CV has furthermore proven useful to predict possible WOC activity of POMs in the presence of [Ru(bpy)2]3+ photosensitizer: if the electrochemical potential of the [Ru(bpy)2]2+/3+ couple is higher than that of the respective metal couple in the POM under consideration, the water oxidation process is thermodynamically favored.40 In the present POM series, the [Ru(bpy)2]2+/3+ couple is capable of oxidizing the active cobalt sites in Co/Bi-POM (1), whereas activation of the cobalt centers in compound (2) or the Mn2+/Mn3+ redox couple in (3) is not possible according to CV results (Fig. 6). Furthermore, the rapidly ascending wave in the anodic part of the voltammogram of (1) is consistent with the observed WOC activity, whereas this phenomenon is absent for the catalytically inactive POMs (2) and (3).
After catalyst stability and suitable electrochemical properties of Co/Bi-POM (1) had been established, its role as unique WOC was further investigated with a series of stability tests (Table 2). The results show that the simultaneous presence of (1), Na2S2O8 and [Ru(bpy)3]2+ under light irradiation exclusively leads to O2 evolution, which is not observed if any of these four prerequisites is absent. Next, the POM-PS complex of [Ru(bpy)3]2+ and Co/Bi-POM (1) was recovered by centrifugation and characterized. Reference POM-PS complex of (1) was obtained from the reaction of both components in buffer media or distilled water, and FT-IR spectra confirm the identity of recovered and as-synthesized complex (Fig. 7–8).
| Catalyst | e− acceptor | PS | Irradiation | WOC activity |
|---|---|---|---|---|
| — | S2O82− | — | yes | no |
| — | — | Ru(bpy)32+ | yes | no |
| — | S2O82− | Ru(bpy)32+ | no | no |
| 1 | — | — | yes | no |
| 1 | S2O82− | — | yes | no |
| 1 | — | Ru(bpy)32+ | yes | no |
| 1 | S2O82− | Ru(bpy)32+ | no | no |
| 1 | S2O82− | Ru(bpy)32+ | yes | yes |
![FT-IR spectra of pristine (1) (black), POM-PS (1) complex recovered from Na2SiF6 buffer media after catalytic tests (red) and [Ru(bpy)3]Cl2 (blue).](/image/article/2013/CY/c3cy00475a/c3cy00475a-f7.gif) | ||
| Fig. 7 FT-IR spectra of pristine (1) (black), POM-PS (1) complex recovered from Na2SiF6 buffer media after catalytic tests (red) and [Ru(bpy)3]Cl2 (blue). | ||
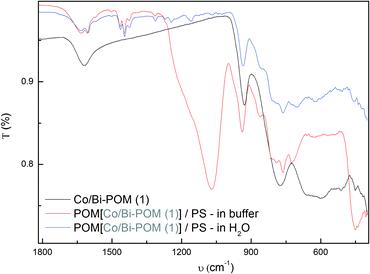 | ||
| Fig. 8 FT-IR spectra of pristine (1) (black), POM-PS (1) complex synthesized from Na2SiF6 buffer medium (red) and POM-PS (1) complex obtained from H2O (blue). | ||
A comparison of FT-IR spectra of recovered POM-PS (1) complex with pristine PS and POM (Fig. 7) shows characteristic bands in the 1600–1400 cm−1 region (1635, 1604, 1500, 1467, 1446, 1428 cm−1) arising from the [Ru(bpy)3]2+ countercation. The intense and broad band around 1067 cm−1 (s) with a shoulder at 1184 cm−1 (w) is attributed to Si–O vibrations emerging from hydrolysis products of the buffer solution, which are convoluted with the sharp peak of the pristine [Ru(bpy)3]2+ sensitizer at 1161 cm−1 (s). In the 900–400 cm−1 region, characteristic POM bands are observed (942, 869, 798, 763, 550, 449 cm−1) which underwent an average shift of +20 cm−1 compared to pristine POM, probably due to the electrostatic POM-PS interactions. Overlap between POM bands of POM-PS (1) complex (620 cm−1, 514 cm−1, 453 cm−1, 418 cm−1) synthesized in water and pristine POM (617 cm−1, 515 cm−1, 453 cm−1, 416 cm−1) is even more accurate (Fig. 8).
Catalytic tests on recycled POM-PS (1) complex showed WOC activity only in freshly prepared standard media under irradiation (Table 3). The complex can be recycled twice for O2 evolution and its activity dropped after further cycles, although FT-IR spectra did not point to significant structural changes (cf. Fig. S15†).
| Catalyst | e− acceptor | PS | Light | Activity |
|---|---|---|---|---|
| Recovered 1 | — | — | yes | no |
| Recovered 1 | S2O82− | — | yes | no |
| Recovered 1 | — | Fresh Ru(bpy)32+ | yes | no |
| Recovered 1 | S2O82− | Fresh Ru(bpy)32+ | no | no |
| Recovered 1 | S2O82− | Fresh Ru(bpy)32+ | yes | yes |
The POM-PS (1) complex was furthermore characterized with TG measurements which are compared to results for pristine (1) in Fig. 9. The initial mass loss of ca. 4.7% for both compounds can be ascribed to the associated 10 aqua ligands of the POM moiety. The POM-PS (1) complex undergoes a mass loss of 15.3% which is probably due to decomposition of the 2,2′-bipyridine ligands into CO2 and NOx and points to the electrostatic attachment of 2/3[Ru(bpy)3]2+ molecules per POM polyanion.
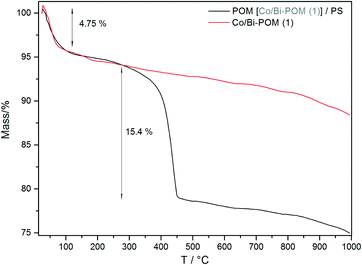 | ||
| Fig. 9 TG curves of (1) (red) and POM-PS (1) complex recovered from Na2SiF6 media after catalytic tests (air, Al2O3 crucible, heating rate 10 K min−1). | ||
Catalytic activity among the POM series
The most striking result emerging from the present study is the drastic difference in WOC activity between the isostructural Co/Bi-POMs (1) and (2). Given that no indications of substantial instability were observed for either compound, reduction of the Co occupancy of the terminal core positions by 25% in (2) is detrimental for its catalytic activity. As far as we know, such a notable influence of subtle disorder differences on the catalytic performance has never been reported for POM-based WOCs. Furthermore, this result is in contrast to recent reports on the WOC activity of single-site [CoIIICoII(H2O)W11O39]7− with a disordered catalytic center.11 Comparison of (1) and (2) suggests at first glance that the terminal {Co(H2O)3} sites are decisive for WOC activity, whereas the internal {M(H2O)2} (M = 50% Co, 50% W) sites are less active. As outlined above, the well-defined Co-rich POM [Bi2W20Co2O70(H2O)6]10− remains difficult to access for reference experiments,24,25 so that theoretical calculations and isotope labeling experiments are scheduled to assign the precise roles of the different core sites in the water oxidation mechanism.Meanwhile, electrochemical measurements are a promising pre-screening method for the targeted selection of WOC candidates among the manifold POM motifs as illustrated by predictive CV measurements for (1) and (2) (Fig. 6).
Concerning Mn/Bi-POM (3), its lack of WOC activity does not come as a comparable surprise, because ongoing work on bio-inspired Mn-containing WOC complexes demonstrated that their construction is demanding due to the manifold key criteria for electron transfer and flexibility, which limit the number of known examples.41,42 Selected molecular Mn-WOCs include [(bpy)2Mn(μ-O)2Mn(bpy)2]3+ immobilized on clay or mica43 and a bio-inspired dinuclear manganese complex with imidazole and carboxylate containing ligands.44 Both compounds already show strong tendencies towards aggregation into higher nuclearities, and consequently, Ca-Mn-oxides have recently emerged as promising solid state Mn-WOCs.45 Nevertheless, POMs as a “missing link” between molecules and oxide networks remain an attractive target for WOC construction so that the general lack of active Mn-WOCs merits further clarification.
Conclusions
An isostructural series of sandwich-type Co- and Mn-POMs, namely Na10[{Co(H2O)3}2{CoBi2W19O66(OH)4}]·37H2O (Co/Bi-POM (1)), Na8[Co2.5(H2O)6{Bi2W19.5O66(OH)4}]·32H2O (Co/Bi-POM (2) and their Mn-containing analogue Na6[Mn1.5(H2O)6{Bi2W20.5O68(OH)2}]·36H2O (Mn/Bi-POM (3)) were obtained from systematic parameter variations of a solution-based synthetic protocol. All POMs were characterized with a variety of analytical methods, and their performance as visible-light-driven water oxidation catalysts (WOCs) was investigated. Extensive parameter screening indicated that a certain extent of M/W-disorder (M = Mn, Co) on the internal and terminal positions, respectively, of the four-membered M2+|W core is strongly favored over the formation of pure M2+ metal cores which have, to the best of our knowledge, not yet been reported for this tungstobismutate sandwich-type. Co/Bi-POM (1) displays (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) Co/W disorder only on the internal sites of the metal core, whereas the external {Co(H2O)3} sites are fully occupied. In comparison, Co/Bi-POM (2) displays the same disorder pattern on the internal sites together with a (3:1) Co/W ratio on the terminal core positions. Mn/Bi-POM (3) also exhibits disorder over the entire core region: the internal positions are mainly occupied with W (90%), whereas the terminal sites are Mn-rich (65%).
:1) Co/W disorder only on the internal sites of the metal core, whereas the external {Co(H2O)3} sites are fully occupied. In comparison, Co/Bi-POM (2) displays the same disorder pattern on the internal sites together with a (3:1) Co/W ratio on the terminal core positions. Mn/Bi-POM (3) also exhibits disorder over the entire core region: the internal positions are mainly occupied with W (90%), whereas the terminal sites are Mn-rich (65%).
The core composition exerts a tremendous influence on the WOC performance, and only Co/Bi-POM (1) exhibits catalytic activity among the series under standard test conditions ([Ru(bpy)3]2+ photosensitizer with S2O82− as sacrificial electron acceptor). Co/Bi-POM (1) performance ranks among the most prominent POM-WOCs reported to date with respect to oxygen yield (96% for 115 μM). Moreover, Co/Bi-POM (1) forms a POM/PS complex with [Ru(bpy)3]2+ which can be recycled for further oxygen evolution runs. Thermogravimetric analyses of the Co/Bi-POM (1)/PS associate indicate that its PS:POM ratio is below values around 4:1 as reported by other groups. Further XAS studies on the precise structure of the POM/PS complex are in progress. However, development of neutral or positively charged PS systems would be a more sustainable long-term alternative.
Detailed reference experiments and stability tests indicated that Co/Bi-POM (1) is stable on the timescale of WOC experiments, and no indications for nanoparticle formation through Co2+ leaching from the metal core were found. Cyclovoltammetric measurements confirmed the integrity of the newly found POM-WOC, and the redox potentials of all three title POMs in comparison with the electrochemical potential of the [Ru(bpy)2]2+/3+ couple agree well with their experimentally determined WOC activities. This renders CV techniques a useful tool for the selective screening of potential POM-WOC candidates.
Most importantly, the results shed new light on the structure–activity relationships for POM-WOC construction. Comparison of the active WOC Co/Bi-POM (1) with its inactive analogue Co/Bi-POM (2) shows that a subtle difference in the Co occupancy of the terminal positions decides about WOC activity or inactivity. This result opens up new perspectives for investigations into the minimum requirements for POM-WOC construction. If the only terminal core sites were essential for WOC activity, economic single-site POMs would be attractive design targets. As the two Co/Bi-POM end members of the series with (a) fully occupied Co core and (b) Co on terminal sites and W on internal sites could not be preparatively accessed to date, we pursue computational and labeling strategies in parallel to assign the precise roles of the core sites in the water oxidation mechanism. We have furthermore started to investigate the influence of the heteroatom within the lacunary POM shells, and our present results indicate that the presence of a lone pair as in Bi3+ exerts a productive influence on the WOC performance.
Our extensive screenings in search of Mn-POMs with WOC activity further underscore the need for SAR. The present study shows a representative example for their elusive character, and additional results will be reported in due course. In the case of Mn/Bi-POM (3), WOC inactivity might be due to the lower Mn occupancy of the terminal sites. However, the general contrast between reported WOC activity of various Mn-based molecular and nano-oxide catalysts on the one hand and the ongoing lack of Mn-POM-WOCs on the other hand might point to underlying drawbacks associated with the POM shell architecture. Further theoretical and synthetic studies are currently in progress to establish reliable rules for POM-WOC core construction.
Experimental section
Materials and methods
Na9[α-BiW9O33]·19.5H2O was prepared and purified according to literature protocols.25 Tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate ([Ru(bpy)3]2+) was purchased (Sigma Aldrich, reagent grade) and recrystallized before use. All other chemicals and salts were commercially purchased with maximum purity levels from commercial suppliers (Sigma Aldrich, TCI).Synthesis of compounds (1–3)
Co/Bi-POM (1) was synthesized according to a literature protocol from the dissolution of freshly prepared Na9[α-BiW9O33]·19.5H2O in sodium acetate buffer, followed by the addition of CoCl2·6H2O dissolved in a minimum of distilled water.24 After heating to 80 °C under reflux, hot filtering and cooling, Co/Bi-POM (1) was obtained in phase pure and single crystalline form as violet needle-shaped crystals (yield: 65% based on W). Elemental analysis (%), calc. (found): Bi, 6.72 (7.35); Co, 2.84 (2.77); W, 56.23 (56.0); Na, 3.70 (4.4). Thermogravimetric analysis: one weight loss step in the range of 30–100 °C was observed, corresponding to the loss of hydrating and coordinated water molecules (Fig. 9). IR (ATR-FTIR, cm−1): 930 cm−1 (s), 775 cm−1 (s), 830 cm−1 (w), 664 cm−1 (w), 589 cm−1 (w), 425 cm−1 (w) (Fig. S4†). Intense bands around 450 and 535 nm in the UV/Vis absorption spectrum (Fig. 4) of Co/Bi-POM (1) in solution (50 μM) are in a good agreement with other CoII polyoxometalates.46Co/Bi-POM (2) was obtained through a slight modification of the above procedure:24 Na9[α-BiW9O33]·19.5H2O was dissolved in distilled water instead of sodium acetate buffer, followed by longer stirring after addition of CoCl2·6H2O, heating to 70–80 °C for 60 min and cooling to room temperature. (2) was isolated as pink needle-shaped crystals (yield: 82% based on W). Elemental analysis (%), calc. (found): Bi, 6.53 (5.65); Co, 1.84 (1.95); W, 57.5 (60.2); Na, 3.6 (3.87). Thermogravimetric analysis: one weight loss step in the range of 30–100 °C was observed, corresponding to the loss of hydrating and coordinated water molecules (Fig. S16†). IR (ATR-FTIR, cm−1): 938 (s), 953 (w), 823 (s), 789 (w), 733 (w) (Fig. S4†). Intense bands around 450 and 535 nm in the UV/Vis absorption spectrum (Fig. 4) of Co/Bi-POM (2) in solution (70 μM) are in a good agreement with other CoII polyoxometalates.46
Mn/Bi-POM (3) was obtained from Na9[α-BiW9O33]·19.5H2O and MnSO4·2H2O in sodium acetate buffer via related literature procedures24 as yellow needle-shaped crystals (yield: 70% based on W). Elemental analysis (%), calc. (found): Bi, 6.65 (6.52); Mn, 1.31 (1.28); W, 59.96 (59.21); Na, 2.20 (3.12). Thermogravimetric analysis: one weight loss step in the range of 30–100 °C was observed, corresponding to the loss of hydrating and coordinated water molecules (Fig. S5†). IR (ATR-FTIR, cm−1): 926 (s), 870 (w), 759 (s), 749 (s), 578 (w) (Fig. S4†). Intense bands lower than 350 nm and around 400 nm in the UV/Vis absorption spectrum (Fig. S9†) of Mn/Bi-POM (3) in solution (50 μM) are in a good agreement with other MnII polyoxometalates.47 Slight discrepancies in the heavy atom contents found by elemental analysis can be explained in terms of cation disorder as has been frequently observed in polyoxometalate chemistry.
Analytical characterization
Cyclic voltammetry (CV) was performed on a Metrohm 797 VA Computrace instrument with glassy carbon electrode (Metrohm AG, 3 mm diameter) as working electrode and an Ag/AgCl reference electrode. Prior to all measurements, solutions were deaerated with pure N2. Cyclic voltammograms were obtained in 0.4 M H2SO4 at pH 3 and 20 mM Na2SiF6/NaHCO3 buffer at pH 5.8 containing the respective polyoxoanion (2.0 mM) at room temperature (scan rate 10–100 mV s−1), to investigate the according redox waves of different metallic centers. The potential domain of the voltammograms is divided into a positive and a negative section (vs. Ag/AgCl) for their sequential analysis.FT-IR spectra were recorded on a Perkin Elmer BXII spectrometer with KBr pellets. Attenuated total reflectance Fourier-transform (ATR-FTIR) spectra were recorded on a Bruker Vertex 70 spectrometer equipped with a Platinum ATR accessory with a diamond crystal. Elemental analyses were performed by Mikroanalytisches Labor Pascher, Remagen, Germany. Thermogravimetric analyses (TGA) were performed on a Netzsch STA 409 CD instrument between 30 and 1000 °C with a heating rate of 10 K min−1. UV/Vis spectra were recorded on a Lambda 650 S Perkin Elmer UV-Visible spectrometer in the range 300–800 nm using a Quartz SUPRASIL precision cell (10 mm). TG measurements were performed on a Netzsch STA 449 C between 25 and 800 °C with a heating rate of 10 K min−1 in nitrogen atmosphere.
X-ray crystallography
Suitable crystals of (1)–(3) for X-ray analysis were all coated with Paratone-N oil and mounted inside a small fiber loop placed in a cooled N2 gas flow at 183 K. Data collections of (1), (2) and (3) were performed on an Oxford Xcalibur Ruby CCD single-crystal diffractometer (Mo Kα radiation, λ = 0.71073 Å). Routine Lorentz and polarization corrections were applied, and an absorption correction was applied using the program CrysAlis (multi-scan).48 Structural analysis was performed with the Win-GX software package.49 Direct methods were used to locate heavy metal atoms (SHELXS-97) and the remaining atoms were located from successive Fourier maps (SHELXL-97).50Further details on the crystal structure investigations may be obtained from the Fachinformationszentrum Karlsruhe, D-76344 Eggenstein-Leopoldshafen, Germany (Fax: +49 7247 808 666; email: E-mail: crysdata@fiz-karlsruhe.de), on quoting the depository numbers CSD-426302 (for Co/Bi-(1)), CSD-426303 (for Co/Bi-(2)), and CSD-426304 (for Mn/Bi-(3)).
Visible-light-driven water oxidation
Water oxidation experiments were performed in a 10 mL headspace vial sealed with an aluminum crimp cap with a rubber septum (PTFE). Reaction mixtures were prepared under dark conditions as follows: 8 mL of a solution containing 1 mM [Ru(bpy)3]Cl2, different concentrations of Na2S2O8 (2 to 7 mM), 20 mM Na2SiF6/NaHCO3 buffer at pH 5.8 (freshly prepared), and the according concentration of catalyst concentration were added to a glass vessel. The solution was deaerated by purging with helium (6.0 purity) for 30 min.Oxygen evolution was measured both in solution and in the headspace using an Oxygen Sensor (OX-N) Clark electrode (Unisense). Constant temperature was maintained with a mineral insulated thermosensor (2 mm tip diameter, TP2000, Unisense).
After purging an initial 100 μL sample of the headspace was injected into a gas chromatograph as background reference for the GC calibration. Gas chromatograms were recorded using an Agilent Technologies 7820A gas chromatograph with helium as the carrier gas and a 3 m × 2 mm packed molecular sieve 13X 80–100 column to separate O2 and N2. The oven was operated isothermally at 80 °C. Analysis of the headspace was performed by taking 100 μL samples with a Hamilton (1825 RN) gas-tight microliter syringe. Gases were detected using a thermal conductivity detector (Varian) operated at 200 °C. Contamination of the headspace by air was constantly quantified by measuring the N2 peak on GC chromatograms. Calibration was performed by injection of known quantities of pure oxygen diluted in the same headspace vial containing the same volume and concentration of Na2SiF6 as used for the measurements.
After background calibration of the GC, the Clark-type O2 probe was introduced into the catalytic vessel, first in the headspace until the signal remained constant, and afterwards below the liquid surface accordingly.
Signals were first calibrated using the normal procedure supplied for the applied electrode model. After complete aeration of the water (5 min of vigorous bubbling) in the Unisense calibration chamber CAL300, the gas stream was turned off and the saturation point for the calibration line for atmospheric partial pressure was added. Background calibration was performed with an anoxic solution prepared from sodium ascorbate and NaOH. Controlled stirring (500 rpm) was applied throughout all experiments.
Data collection was performed with the SensorTrace software from Unisense, with a frequency of 1 data point per sec for both probes (T and O2). After a constant signal for the O2 sensor was recorded, the catalytic reaction was initiated by exposing the reaction vessel to the light of a 470 nm high flux LED from Rhopoint Components LTD. The LED power was determined as 26.1 mW cm−2.
Catalyst recycling
Catalysts were recovered from reaction mixtures containing 1 mM [Ru(bpy)3]Cl2, 5 mM Na2S2O8 and the given concentration of WOC in Na2SiF6 aqueous media (20 mM, pH 5.8). The solution was kept under LED irradiation for the catalytic reaction time and a red precipitate was obtained after centrifugation. The obtained powder was washed twice with distilled water, dried in N2 atmosphere and characterized with FT-IR spectroscopy, TG and XRD.Acknowledgements
This work was supported by the Swiss National Science Foundation (SNSF Professorship PP00P2_133483/1 and Sinergia Grant No. CRSII2_136205/1) and by the University of Zurich (Institute of Inorganic Chemistry and UFSP LightChEC). We thank PD Dr. Bernhard Spingler for discussions on the crystal structures.Notes and references
- (a) Y. Tachinaba, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef; (b) R. J. Cogdell, A. T. Gardiner and L. Cronin, Philos. Trans. R. Soc. London, Ser. A, 2012, 370, 3819–3826 CrossRef CAS PubMed; (c) J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC; (d) D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed; (e) K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- (a) G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed; (b) E. S. Andreiadis, M. Charavot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed; (c) A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC; (d) I. Rivalta, G. W. Brudvig and V. S. Batista, Curr. Opin. Chem. Biol., 2012, 16, 11–18 CrossRef CAS PubMed.
- (a) Polyoxometalates: from Platonic Solids to Antiretroviral Activity ed. M. T. Pope and A. Müller), Kluwer, Dordrecht, 1994 Search PubMed; (b) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 19–20 CrossRef PubMed; (c) Chem. Rev., Special Issue on Polyoxometalates, ed. L. Hill, 1998, 98, pp. 1–389 Search PubMed; (d) L. Cronin, Angew. Chem., Int. Ed., 2006, 45, 3576–3578 CrossRef CAS PubMed; (e) Polyoxometalate Molecular Science, ed. J. J. Borras-Almenar, E. Coronado, A. Müller and M. T. Pope, Kluwer, Dordrecht, The Netherlands, 2004 Search PubMed; (f) D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (g) E. Cadot, M. N. Sokolov, V. P. Fedin, C. Simonnet-Jegat, S. Floquet and F. Secheresse, Chem. Soc. Rev., 2012, 41, 7335–7353 RSC.
- (a) N. D. Morris and T. E. Mallouk, J. Am. Chem. Soc., 2002, 124, 11114–11121 CrossRef CAS PubMed; (b) Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed; (c) Y. V. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quinonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360–17370 CrossRef CAS PubMed; (d) C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 46, 2784–2786 RSC; (e) M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC; (f) F. Puntoriero, G. La Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725–4727 RSC; (g) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed; (h) Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS PubMed; (i) A. Sartorel, M. Truccolo, S. Berardi, M. Gardan, M. Carraro, F. M. Toma, G. Scorrano, M. Prato and M. Bonchio, Chem. Commun., 2011, 47, 1716–1718 RSC; (j) R. N. Biboum, C. P. N. Njiki, G. Zhang, U. Kortz, P. Mialane, A. Dolbecq, I. M. Mbomekalle, L. Nadjo and B. Keita, J. Mater. Chem., 2011, 21, 645–650 RSC; (k) M. Murakami, D. Hong, T. Suenobu, S. Yamaguchi, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605–11613 CrossRef CAS PubMed.
- (a) H. Lv, Y. V. Geletii, C. Zhao, J. G. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC; (b) A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC; (c) R. Sivakumar, J. Thomas and M. Yoon, J. Photochem. Photobiol., C, 2012, 13, 277–298 CrossRef CAS PubMed.
- (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed; (b) A. Savini, P. Belanzoni, G. Bellachioma, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Green Chem., 2011, 13, 3360–3374 RSC; (c) A. Singh, R. K. Hocking, S. L.-Y. Chang, B. M. George, M. Fehr, K. Lips, A. Schnegg and L. Spiccia, Chem. Mater., 2013, 25, 1098–1108 CrossRef CAS.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
- (a) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed; (b) M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC.
- (a) G. Zhu, Y. V. Geletii, J. Song, C. Zhao, E. N. Glass, J. Bacsa and C. L. Hill, Inorg. Chem., 2013, 52, 1018–1024 CrossRef CAS PubMed; (b) H. Lv, J. Song, Y. V. Geletii, W. Guo, J. Bacsa and C. L. Hill, Eur. J. Inorg. Chem., 2013, 1720–1725 CrossRef CAS.
- D. Guo, S. Teng, Z. Liu, W. You and L. Zhang, J. Cluster Sci., 2013, 34, 549–558 CrossRef.
- F. Song, Y. Ding, B. Ma, C. Wang, Q. Wang, X. Du, S. Fu and J. Song, Energy Environ. Sci., 2013, 6, 1170–1184 CAS.
- S. Tanaka, M. Annaka and K. Sakai, Chem. Commun., 2012, 48, 1653–1655 RSC.
- P. E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680–1688 RSC.
- J. Song, Z. Luo, H. Zhu, Z. Huang, T. Lian, A. L. Kaledin, D. G. Musaev, S. Lense, K. I. Hardcastle and C. L. Hill, Inorg. Chim. Acta, 2010, 363, 4381–4386 CrossRef CAS PubMed.
- K. Heussner, K. Peuntinger, N. Rockstroh, L. C. Nye, I. Ivanovic-Burmazovic, S. Rau and C. Streb, Chem. Commun., 2011, 47, 6852–6854 RSC.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324–7331 CrossRef CAS PubMed.
- J. Gao, S. Cao, Q. Tay, Y. Liu, L. Yu, K. Ye, P. C. S. Mun, Y. Li, G. Rakesh, S. C. J. Loo, Z. Chen, Y. Zhao, C. Xue and Q. Zhang, Sci. Rep., 1853, 2013(3), 1–5 Search PubMed.
- J. Stracke and R. J. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808–8810 RSC.
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209–1219 CrossRef CAS.
- J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753–4755 CrossRef PubMed.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- C.-Y. Sun, S. X. Liu, C. L. Wang, L. H. Xie, C.-D. Zhang, B. Gao and E.-B. Wang, J. Coord. Chem., 2007, 60, 567–579 CrossRef CAS.
- I. Loose, E. Droste, M. Bösing, H. Pohlmann, M. H. Dickman, C. Rosu, M. T. Pope and B. Krebs, Inorg. Chem., 1999, 38, 2688–2694 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- M. Piepenbrink, D. Drewes and B. Krebs, ICSD 391152, private communication, 2002 Search PubMed.
- (a) L. Ruhlmann, L. Nadjo, J. Canny, R. Contant and R. Thouvenot, Eur. J. Inorg. Chem., 2002, 975–986 CrossRef CAS; (b) G. G. Gao, L. Xu, W. Wang, X. Shu Qu, X. Liu and Y. Y. Yang, Inorg. Chem., 2008, 47, 2325–2333 CrossRef CAS PubMed.
- S. Goberna-Ferron, L. Vigara, J. Soriano-Lopez and R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707–11715 CrossRef CAS PubMed.
- (a) Y. Z. Voloshina, O. A. Varzatskii, A. S. Belova, Z. A. Starikovaa, A. V. Dolganova, V. V. Novikova and Y. N. Bubnov, Inorg. Chim. Acta, 2011, 370, 322–332 CrossRef PubMed; (b) J. B. Serra, I. Todorut, N. C. Pastor, J. P. Server-Carrió, L. C. W. Baker and R. Accrete, Synth. React. Inorg. Met.-Org. Chem., 1995, 25(6), 869–882 CrossRef.
- (a) B. S. Bassil, M. Ibrahim, S. S. Mal, A. Suchopar, R. N. Biboum, B. Keita, L. Nadjo, S. Nellutla, J. van Tol, N. S. Dalal and U. Kortz, Inorg. Chem., 2010, 49, 4949–4959 CrossRef CAS PubMed; (b) D. J. Wasylenko, C. Ganesamoorthy, J. Borau Garcia and J. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- (a) K. Nomita, M. Miwa, R. Kobayashi and M. Aiso, Bull. Chem. Soc. Jpn., 1981, 54, 2983–2987 CrossRef; (b) N. K. Kala Raja, V. G. Puranikb, C. Gopinathana and A. V. Ramaswamy, Appl. Catal., A, 2003, 256, 265–273 CrossRef.
- M. Lebrini, I. M. Mbomekalle, A. Dolbecq, J. Marrot, P. Berthet, J. Ntienoue, F. S. Secheresse, J. Vigneron and A. Etcheberry, Inorg. Chem., 2011, 50, 6437–6448 CrossRef CAS PubMed.
- Y. Liu, B. Liu, G. Xue, H. Hu, F. Fu and J. Wang, Dalton Trans., 2007, 3634–3639 RSC.
- (a) X. Y. Zhang, M. T. Pope, M. R. Chancet and J. B. Jameson, Polyhedron, 1995, 14, 1381–1392 CrossRef CAS; (b) I. Bar-Nahum, H. Cohen and R. Neumann, Inorg. Chem., 2003, 42, 3677–3684 CrossRef CAS PubMed.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. J. Sutin, J. Am. Chem. Soc., 1984, 106, 4772–4779 CrossRef CAS.
- (a) Y. V. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quinonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360–17370 CrossRef CAS PubMed; (b) G. S. Fink, Angew. Chem., Int. Ed., 2008, 47, 5888–5890 CrossRef PubMed; (c) Z. Huang, Z. Luo, Y. V. Geletii, J. V. Vickers, Q. Yin, Y. W. Wu, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS PubMed; (d) A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- A. Sartorel, M. Truccolo, S. Berardi, M. Gardan, M. Carraro, F. M. Toma, G. Scorrano, M. Prato and M. Bonchio, Chem. Commun., 2011, 47, 1716–1718 RSC.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808–8810 RSC.
- M. Wiechen, H.-M. Berends and P. Kurz, Dalton Trans., 2012, 41, 21–31 RSC.
- (a) L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC; (b) P. Kurz, G. Berggren, M. Anderlund and S. Styring, Dalton Trans., 2007, 4258–4261 RSC.
- M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef CAS PubMed.
- E. A. Karlsson, B.-L. Tee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233–2237 CrossRef CAS PubMed.
- (a) B. J. Johnson and A. Stein, Inorg. Chem., 2001, 40, 801–808 CrossRef CAS; (b) N. K. Kala Raj, V. G. Puranik, C. Gopinathan and A. V. Ramaswamy, Appl. Catal., A, 2003, 256, 265–273 CrossRef.
- K. Patel, P. Shringarpure and A. Patel, Transition Met. Chem., 2010, 36, 171–177 CrossRef PubMed.
- Oxford Diffraction CrysAlis CCD and CrysAlis RED, Oxford Diffraction Ltd., Abingdon, UK, 2005 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–851 CrossRef CAS.
- G. M. Sheldrick, SHELX97, Programs for Crystal Structure Analysis; Release 97–2, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data (ICSD 426302, 426303 and 426304) and BVS calculations for all compounds, powder X-ray diffraction patterns, FT-IR and UV/Vis spectra, TG measurements and cyclic voltammograms, experimental details for catalytic water oxidation, FT-IR and TG data for POM/PS complexes. See DOI: 10.1039/c3cy00475a |
| This journal is © The Royal Society of Chemistry 2013 |