 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How metallic is gold in the direct epoxidation of propene: an FTIR study
Jiaqi Chena, Evgeny A. Pidkob, Vitaly V. Ordomskya, Tiny Verhoevenb, Emiel J. M. Hensenb, Jaap C. Schoutena and T. Alexander Nijhuis*a
aLaboratory of Chemical Reactor Engineering, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: t.a.nijhuis@tue.nl; Fax: +31 40 2446653; Tel: +31 40 2473671
bSchuit Institute of Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 28th August 2013
Abstract
Unraveling the oxidation state of gold is important to understand the role of gold in direct propene epoxidation on gold–titania catalysts. A Fourier transform infrared study of low-temperature carbon monoxide adsorption was performed over Au/TiO2 and Au/Ti–SiO2 under an atmosphere of the reaction mixture of oxygen, hydrogen, and propene. Data reveals that the active gold sites treated by the reaction mixture are fully covered by reaction intermediates and deactivating species. Oxidation at 573 K removes these carbonaceous species on gold. Oxygen adsorption at the reaction temperature leads to positively charged gold, which can be reduced to metallic gold in the presence of hydrogen. Propene acts as an electron donor to the gold atoms resulting in negatively charged gold with the carbonyl band at 2079 cm−1. The results in this study may provide a general scheme of electron transfer via gold on the gold–titania catalysts for direct propene epoxidation.
1. Introduction
Gold nanoparticles in the direct propene epoxidation over gold–titania catalysts are considered to have two main functions. The first is the formation of the active oxidizing species, the hydro-peroxy species (HOOH, OOH), which has been spectroscopically confirmed via an inelastic neutron scattering (INS) study.1 From a combination of in situ UV-vis and XANES study,2 it was suggested that the hydro-epoxidation of propene over gold–titania catalysts involves the formation of HOOH on gold and a sequential transfer of HOOH to Ti4+ forming the real active intermediate Ti–OOH. This route resembles the chemistry of propene epoxidation by H2O2 over TS-1 in the HPPO process.3 The second function of gold in the epoxidation of propene, which is less prominent, is that propene may be activated on gold nanoparticles,4 or, in another sense, that the adsorption of propene on gold strongly affects the activity of gold in the formation of hydro-peroxy species, since the inhibiting effect of the alkene in hydrogen oxidation on gold catalysts is generally observed.5The rate-determining step in the hydro-epoxidation of propene on gold–titania catalysts is considered to be the dissociative adsorption of hydrogen.6–8 The study by Boronat et al.9 suggests that the hydrogen dissociation over Au/TiO2 occurs only on low coordinated and neutral gold atoms at corner or edge sites of gold nanoparticles but not directly bonded to the support. The H2–D2 exchange reaction studied by Fujitani et al.10 on model Au/TiO2(110) catalysts with gold nanoparticles of controlled sizes has shown a constant turnover frequency of HD formation when based on the perimeter length of gold nanoparticles, indicating that hydrogen dissociation on gold–titania catalysts may very likely be an interfacial phenomenon. Green et al.11 studied low-temperature hydrogen oxidation over Au/TiO2 by infrared (IR) spectroscopy and density functional theory (DFT) calculations. Their results suggest a pathway of hydrogen dissociation at the perimeter sites at the interface between Au and TiO2, in which oxygen molecules adsorb on Ti5c perimeter sites and hydrogen dissociates into Au–H and Ti–OOH in the first step. Yang et al.12 performed a DFT study of hydrogen dissociation and diffusion at the perimeter sites of Au/TiO2(110) and depicted two ways of hydrogen dissociation. In their study, the heterolytic dissociation of hydrogen with one H atom on gold and another on the bridging oxygen atom in the support is energetically more favourable, while the homolytic hydrogen dissociation mainly occurs after all Au–O–Ti sites are passivated into Au–O(H)–Ti. Using coadsorbed CO in an IR study of hydrogen dissociation over Au/TiO2 at room temperature, Panayotov et al.13 revealed that the most active sites for hydrogen dissociation are defect Au0 sites away from the Au–O–Ti interface. Passivation of Au–O–Ti to Au–O(H)–Ti was also observed. The rate of hydrogen atom spillover to the support was determined to be proportional to PH21/2. The coadsorption (or competitive adsorption) of CO does not change the chemistry of the hydrogen dissociation and spillover, but it can suppress the initial rate of hydrogen dissociation by a factor of 2.6 as demonstrated by Panayotov et al.13 In summary, hydrogen dissociation occurs on low-coordinated Au0 atoms at edges and corners and the role of the support cannot be neglected over gold–titania catalysts.
Another interesting phenomenon in the hydro-epoxidation of propene over gold–titania catalysts is that gold can catalyze the propene hydrogenation.14–17 Although in our previous study18 it was demonstrated that the support itself can contribute significantly to the propene hydrogenation and that the presence of a small amount of CO (10 ppm to 1000 ppm) can switch off the propene hydrogenation, we cannot exclude a contribution from gold. Gold-catalyzed alkene hydrogenation has long been known.19 Specifically, over the gold–titania system, the explanation to this side reaction can be that propene hydrogenation is sensitive to the structures of gold nanoparticles or clusters.14,16,20 It could also be that Au+ exists and functions as proposed by Oyama et al.17 The Au+ in their study is in the form of Au(CN)2−1 resulting from NaCN leaching of Au/TS-1 and has no activity in propene epoxidation but is active in its hydrogenation. Bond and Thompson21 suggested that gold atoms with a low coordination number are more prone to be oxidized than those with a high coordination number. The findings by Oyama and coworkers17 thus provide another mechanistic view towards the hydrogenation of propene, in which small gold clusters have probably oxidized interphase atoms. Nevertheless, Au+ should be neither easy to produce nor stable in the reaction conditions for the direct propene epoxidation where dihydrogen and propene are also present. Dekkers et al.22 observed Au+ by pre-oxidizing Au/TiO2 in pure dioxygen at 300 °C for one hour and found that Au+ was gradually reduced to Au0 in a CO–O2 mixture at room temperature. Venkov et al.23 could not obtain Au+ by treating Au/TiO2 under oxygen at 300–500 °C and only observed Au+ after oxidation in NO + O2 at 500 °C. Thus it is doubtful that Au+ would exist under a H2/O2/C3H6 mixture at 50–200 °C.
The most informative technique used in gold catalysis to determine the gold oxidation state is using CO as a probe molecule to observe the frequency of ν(CO) bands by the IR spectrum. The sites on gold nanoparticles where CO adsorbs are also those gold atoms with low coordination numbers at the edges and corners, where all the chemistry is supposed to happen. The spectral regions of surface carbonyls on gold with different status are summarized well in reviews by Freund et al.24 and Mihaylov et al.25 Though some of the bands overlap with CO interacting with Ti4+ and hydroxyls, they can be distinguished by progressive adsorption/desorption.
The aim of this work is to have a general image of the oxidation state of gold in Au/Ti–SiO2 and Au/TiO2, which are active in direct propene epoxidation. The catalysts were investigated by low-temperature CO adsorption after different pretreatments or in different atmospheres. We also have the two following questions left from previous studies.5,18 The first is regarding the reason for the suppressing effect of propene adsorption on water formation over gold. The second is to what extent can we observe the competitive adsorption of CO and H2 if the inhibiting effect of CO on propene hydrogenation is due to CO occupying gold. Co-adsorption of CO and H2/C3H6 was thus investigated and discussed in this study.
2. Experimental
2.1. Catalyst preparation and testing
The catalyst consisting of 1 wt% gold on titania dispersed on silica, which has been fully investigated in our earlier study,8 was used in this study and denoted as Au/Ti–SiO2 (SiO2, Davisil 643, Aldrich, 300 m2 g−1, pore size 150 Å, pore volume 1.15 cm3 g−1). The preparation of 1 wt% Au/SiO2 (Davisil 643, Aldrich) and 1 wt% Au/TiO2 (P25, Degussa, 70% anatase, 30% rutile, 45 m2 g−1) followed the deposition–precipitation method using ammonia described in our earlier study.4 Safety concerns and suggestions on the possible formation of explosive fulminating gold when using ammonia have been addressed earlier.4 Due to the limited amount of gold and ammonia, the risks are very minor in this study. A total of 2 g of the support was dispersed in 100 mL of water. The pH of the slurry was adjusted to 9.5 by dropwise adding ammonia (2.5 wt%), a total of 115 mg of an acidic 30 wt% aqueous ammonia. After the addition of the gold solution, the slurry was stirred for one hour. The slurry was filtered and washed 3 times using 200 mL of water. The catalyst was dried overnight at 80 °C and calcined first at 120 °C (5 °C min−1 heating) for 2 h and afterwards at 400 °C (10 °C min−1 heating) for 4 h. Drying and calcination of the support and the catalyst were performed under atmospheric pressure in stationary air.Catalytic tests were performed in a flow setup equipped with a fast Interscience Compact GC system (3 min analysis time) containing a Porabond Q column and a Molsieve 5A column in two separate channels, each with a thermal conductivity detector. 300 mg of catalyst was loaded into the tubular quartz reactor (6 mm inner diameter) and tested with a gas feed rate of 50 mL min−1 in total, consisting of 10 vol% each of hydrogen, oxygen, and propene with helium as the balance. The term ‘spent’ or ‘after reaction’ hereinafter means that the catalyst had been tested in the reaction mixture for epoxidation for at least 2 hours. The term ‘regenerated’ hereinafter means that the catalyst or sample had been calcined in 5 vol% or 10 vol% oxygen diluted in helium at 573 K for at least 30 min. When mentioned in an IR experiment, the regeneration was always conducted in situ in 5 vol% oxygen at 573 K for 30 min.
2.2. Characterization techniques
Transmission electron microscope (TEM) images were recorded with a FEI Tecnai G2 Sphera transmission electron microscope at an acceleration voltage of 200 kV. Loadings of gold and titanium were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Spectra CirosCCD system. In ICP analyses, gold was dissolved with aqua regia and grafted titanium was etched by 5 mol L−1 H2SO4 solution. The H2SO4 solution containing dissolved titanium was then diluted to 2.9 mol H2SO4 L−1 for analyses. The X-ray photoelectron spectroscopy (XPS) measurements were carried out with a Kratos AXIS Ultra spectrometer, equipped with a monochromatic X-ray source and a delay-line detector (DLD). Spectra were obtained using the aluminum anode (Al Kα = 1486.6 eV) operating at 150W. A detachable U-shape Pyrex tubular reactor (6 mm inner diameter) with valves on both ends was used for post-reaction catalysts. The U-shape reactor can be heated using an aluminum heating jacket up to 300 °C. The post-reaction catalysts were then flushed in helium and afterwards sealed in the U-shape reactor. The reactor was transferred into a nitrogen glove box free from oxygen (<2 ppm) and moisture (<0.5 ppm). The post-reaction samples for XPS measurements were prepared in the glove box. Transport of the samples from the glove box to the spectrometer was performed in an inert atmosphere by using a small nitrogen-purged chamber equipped with a magnetic arm. In this way, the post-reaction catalysts were kept ‘in situ’. XPS spectra were referenced to the C 1s line at 284.9 eV.Infrared spectra were recorded with a Bruker IFS 113v spectrometer at a 2 cm−1 optical resolution and accumulation of 128 scans (ca. 2 min of acquisition time per spectrum). The samples were pressed in self-supporting discs at 0.5 MPa (diameter: 12.7 mm, ca. 7.9 mg cm−2). The sample wafer was placed in a homemade double-walled IR cell.26 The high-vacuum (below 10−5 mbar) transmission IR cell was in-house made and can be cooled by liquid nitrogen and heated by electric wire. The IR cell is connected to a gas dosing system, through which the progressive adsorption of adsorbates can be performed via a 5 μL or 50 μL sample loop on a Valco six-port valve. In a typical CO dosing experiment, the CO pulse was automatically carried out upon the acquisition of each spectrum (128 scans) finished. The stabilization of one spectrum was fast and normally within a few scans. An extra pair of inlet and outlet is located on top of the IR cell, through which in situ calcination or pretreatment under different atmospheres can be performed. The gas for sample pretreatment can be dried by liquid nitrogen trap when necessary. The catalysts were tested in the tube reactor confirming the activity before IR experiments. The catalyst pellets were generally used for 3–4 sequential IR measurements. In situ calcination in 5 vol% oxygen diluted by helium at 573 K for 30 min followed by evacuation at 573 K for 30 min was carried out between each IR experiment.
3. Results
3.1. Catalyst performance and characterization
The 1 wt% Au/TiO2 and Au/Ti–SiO2 catalysts were tested for propene epoxidation in hydrogen and oxygen in the tubular reactor. Their catalytic performance was listed in Table 1. The conversion of propene and the selectivity to PO were in the typical range for these two catalysts at these conditions.4 The side products were mainly propionaldehyde, acetone, acetaldehyde and carbon dioxide. No propane formation was found. The size distribution of gold nanoparticles was narrow for both catalysts (spent, i.e. after more than 2 hours of propene epoxidation), centered at 4 nm for Au/TiO2 and 4.5 nm for Au/Ti–SiO2. The catalyst stability was investigated for these two catalysts by repeatedly performing catalytic testing followed by 1 hour of regeneration at 573 K in 10 vol% oxygen. The results are shown in Fig. 1. The time-on-stream performance of both catalysts was highly reproducible. The breakthrough curve at the beginning of the reaction cycles in particular for Au/TiO2 is due to the adsorption of PO and water on the catalyst surface until the adsorption capacity was reached.27 Adsorbed bidentate propoxy species and consecutive oxidized species thereof lead to the catalyst deactivation.27–29 The reason for these stability tests was to exclude a possible catalyst change in the IR experiments since each pellet was re-used 3–4 times. The effect of high vacuum at 573 K after calcination in oxygen in the IR cell on possible gold sintering was examined by repeatedly treating one Au/TiO2 sample under such conditions before cooling down to 323 K in vacuum 9 times. The average size of gold particles after such treatment increased slightly, by less than 0.5 nm, but it was still well below 5 nm as shown in Fig. 2 and the size distribution remained narrow. Thus, sintering of gold nanoparticles in IR experiments for each pellet was not supposed to occur or had very limited effect on CO adsorption.| Sample ID | Au loading (wt%) | Ti loading (wt%) | Au size (nm) | Catalytic performancea | |||
|---|---|---|---|---|---|---|---|
| Temperature (K) | C3H6 conversion (%) | PO selectivity (%) | H2 Efficiencyb (%) | ||||
a No activity in propene hydrogenation observed, activity taken at 2 h, H2:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) O2:C3H6:He = 1:1:1:7, GHSV 10000 mL gcat−1 h−1b Determined as rPO/rH2O O2:C3H6:He = 1:1:1:7, GHSV 10000 mL gcat−1 h−1b Determined as rPO/rH2O | |||||||
| Au/Ti–SiO2 | 0.91 | 1.29 | 4.5 ± 1.1 | 423 | 1.32 | 85.5 | 7.1 |
| Au/TiO2 | 0.93 | — | 4.0 ± 1.2 | 333 | 0.20 | 98.0 | 15.9 |
| Au/SiO2 | 0.95 | — | 3.1 ± 0.8 | — | |||
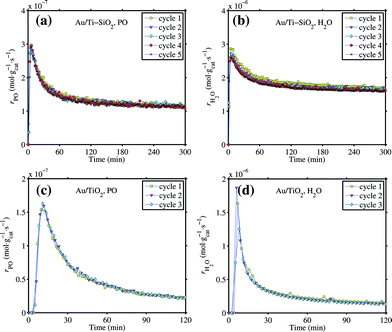 | ||
| Fig. 1 Tests of activity stability for the 1 wt% Au/Ti–SiO2 (top, a and b) and Au/TiO2 (bottom, c and d) catalysts in 10/10/10/70 H2/O2/C3H6/He mixture. GHSV 10000 mL gcat−1 h−1, 423 K for Au/Ti–SiO2, 333 K for Au/TiO2. A few hours of continuous testing in the reaction mixture followed by 1 h calcination in 10 vol% oxygen at 573 K constitutes one ‘cycle’. | ||
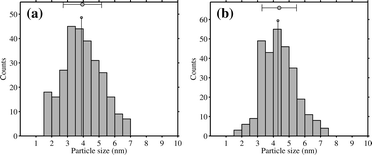 | ||
| Fig. 2 Change in the size distribution of gold nanoparticles on the 1 wt% Au/TiO2 catalyst after repeating 1 h calcination in vacuum at 573 K in the IR cell and cooling down to 323 K 9 times: (a) before treatment and (b) after treatment. | ||
3.2. CO adsorption on catalysts after epoxidation and regeneration
Fig. 3 shows the changes in IR spectra during progressive CO adsorption on a clean Au/SiO2 sample at 90 K. The spectrum of the dehydrated sample before CO dosing is quite simple and similar to what can be observed for SiO2. In the OH stretching region, the band at 3743 cm−1 is assigned to unassociated Si–OH. The band at 3715 cm−1 is assigned to the terminal silanol in a pair or chain of hydrogen-bonded hydroxyls (hydrogen perturbed OH), while the broad band centered at 3552 cm−1 is assigned to those within the hydrogen-bonded hydroxyl groups (oxygen perturbed OH).30 The adsorption at 3653 cm−1 is from inaccessible Si–OH. The adsorption at 1980 cm−1 and 1875 cm−1 is attributed to the skeleton vibrations of silica.31 The 1640 cm−1 band is most likely to be assigned to the bending mode of some remaining adsorbed water. Upon adsorption, the CO molecules interact with terminal OH groups leading to a rise of the perturbed band at ca. 3550 cm−1, which gradually shifted to lower wavelengths when CO coverage increased. CO also interacted with unassociated OH groups giving the decreased band at ca. 3746 cm−1 (shifted from 3743 cm−1 before CO was introduced) and lowered transparency at ca. 3610 cm−1. The isosbestic point located at ca. 3680 cm−1. Three carbonyl bands were observed after introducing CO. The band at 2158 cm−1 is known for CO interacting with OH groups. The band at 2136 cm−1 can be assigned to physisorbed CO.32,33 The 2136 cm−1 band is unlikely to be assigned to P-branch adsorption of gas phase CO. The contribution from gas phase CO at 1 mbar and 90 K was found to be very minor in our study. Besides, the R-branch component at higher frequencies is lacking as seen for Au/SiO2. The band at 2099 cm−1 is assigned to Au0–CO, which shifted from 2105 cm−1 at low CO coverage. A red shift of ν(CO/Au) with increased coverage of CO was generally observed and can be explained by the balancing between dipole–dipole coupling, which leads to a blue shift, and the chemical shift, which leads to a wave number decrease since the donation from the weakly antibonding 5σ orbital dominates in the Au–CO interaction whereas the backbonding from gold atoms to the 2π* CO orbital is lacking.34–36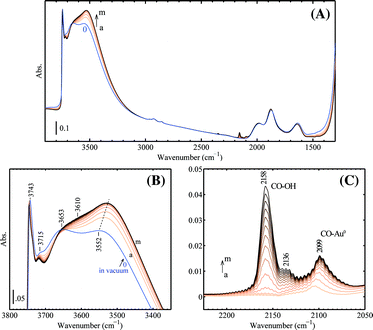 | ||
| Fig. 3 The development of IR spectra during progressive CO adsorption on the 1 wt% Au/SiO2 sample at 90 K: (A) wide-frequency range, (B) range of ν(OH) and (C) difference spectra in the range of ν(CO), subtracted from the spectrum ‘0’ before CO dosing. The following CO pressures after each dose were used (in mbar, spectra a–m): 0.06, 0.14, 0.23, 0.30, 0.36, 0.43, 0.50, 0.58, 0.66, 0.73, 0.81, 0.90, 0.99. Prior to low-temperature CO adsorption, the catalyst was calcined in 10 vol% O2 at 573 K and then tested in 2 vol% H2 and 2 vol% O2 at 423 K in the flowing reactor. Afterwards the pellet was prepared ex situ and transferred into the IR cell for evacuation at 473 K for 1 h. | ||
Progressive adsorption of CO on the spent and regenerated Au/Ti–SiO2 sample is shown in Fig. 4. Comparing to CO adsorption on Au/SiO2, a new band above 2180 cm−1 appeared as seen from Fig. 4, which can be assigned to CO adsorbed on Ti4+ ions.32 The spent catalyst showed very weak adsorption of CO on both Ti4+ and Au. The weak band of linear carbonyl on Au0 at 2095 cm−1 was blue shifted by 12 cm−1 to 2107 cm−1 on the regenerated sample, on which propoxy species adsorbed at the Au–Ti interface are supposed to be fully removed. After regeneration, the sample showed a much stronger adsorption of CO on gold indicating that during the reaction the gold sites active for epoxidation were fully covered by strongly adsorbed species, though the water formation rate remained high as shown in Fig. 1. Upon dosing CO on the regenerated sample as shown in Fig. 4B, Auδ+ can be detected by the shoulder of ν(CO) at 2125 cm−1.23,37,38
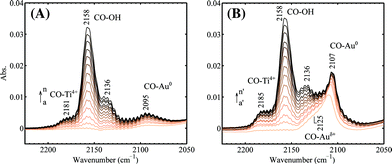 | ||
| Fig. 4 Progressive CO adsorption on the (A) spent and (B) regenerated 1 wt% Au/Ti–SiO2 catalyst. The spectra were taken at 90 K and the following CO pressures after each dose were used (in mbar): 0.06, 0.12, 0.19, 0.26, 0.33, 0.40, 0.46, 0.53, 0.61, 0.67, 0.74, 0.82, 0.89, 0.96 for a–n; 0.06, 0.12, 0.18, 0.25, 0.32, 0.39, 0.45, 0.53, 0.62, 0.68, 0.76, 0.83, 0.90, 0.97 for a′–n′. The sample in vacuum was used as the background. | ||
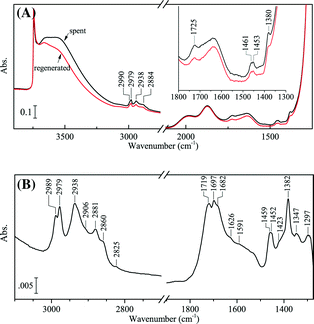
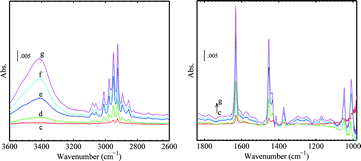
The spectra of the spent and regenerated sample before CO dosing are given and compared in Fig. 5. The difference spectrum given in Fig. 5 confirmed the presence of strongly adsorbed propoxy on the catalyst surface. In Fig. 5A, it is clear that alkoxy adsorbed on the support cannot be fully removed by oxidation at 573 K as indicated by the remaining bands in the CH stretching region after regeneration. The difference spectrum between the spent and regenerated sample given in Fig. 5B did show increased transparency by regeneration in the CH stretching and bending regions, which was due to the removed propoxy species similar to what was observed by Mul et al..28 In the CH stretching region, the bands at 2989 and 2979 cm−1 are assigned to νas(CH3), while the bands at 2938, 2906, 2881, and 2860 cm−1 can be assigned to νas(CH2), ν(CH), νs(CH3), and νs(CH2), respectively.4,28,29 A weak band at 2825 cm−1 was observed, which may be assigned to the ν(CH) of surface formate.39 In the CH bending region, the bands at 1459/1452, 1382, and 1347 cm−1 can be assigned to δs(CH3)/δ(CH2), δs(CH3), and δ(CH), respectively. The bands at 1719 and 1697 cm−1 are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations from surface species containing a carbonyl group. Since the sample of the spent catalyst was already evacuated at 573 K, the carbonyl group is mostly likely from dehydrogenation of intermediates such as Ti–O–CH2CH(OH)CH3 or Ti–O–CH(CH3)CH2OH (preferred, however, there is no obvious adsorption of ν(CH) at 2720–2750 cm−1 for aldehydes) by the ring opening of propene oxide on the surface.28,40,41 Less intensive bands at 1682, 1626, 1591, and 1423 cm−1 may be attributed to surface carbonate and carboxylate groups.42
O stretching vibrations from surface species containing a carbonyl group. Since the sample of the spent catalyst was already evacuated at 573 K, the carbonyl group is mostly likely from dehydrogenation of intermediates such as Ti–O–CH2CH(OH)CH3 or Ti–O–CH(CH3)CH2OH (preferred, however, there is no obvious adsorption of ν(CH) at 2720–2750 cm−1 for aldehydes) by the ring opening of propene oxide on the surface.28,40,41 Less intensive bands at 1682, 1626, 1591, and 1423 cm−1 may be attributed to surface carbonate and carboxylate groups.42
 | ||
| Fig. 5 IR spectra of the 1 wt% Au/Ti–SiO2 sample after reaction and regeneration: (A) wide-frequency range and (B) difference spectrum of the spent subtracted from the regenerated. Both spectra were taken at 90 K in vacuum. The sample of the spent catalyst (2 h in 10/10/10/70 H2/O2/C3H6/He mixture at 423 K, GHSV 10000 mL gcat−1 h−1, ex situ) was evacuated in vacuum at 573 K in the IR cell for 1 h and then cooled down to 90 K. After CO adsorption was performed, the same sample was then calcined in flowing 5 vol% O2 at 573 K in situ for 0.5 h to clean the carbonaceous surface, evacuated at 573 K for 0.5 h and then cooled down to 90 K. | ||
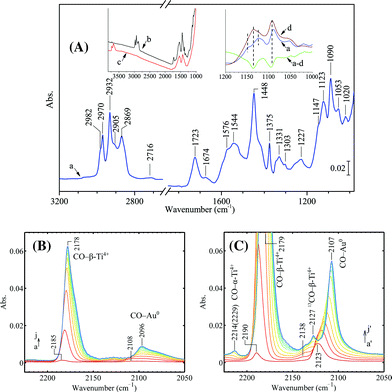
The spectrum given in Fig. 6A provides information on species adsorbed on the spent Au/TiO2 catalyst. Fig. 6B and C show CO adsorption on the spent and regenerated Au/TiO2 catalyst. The left inset in Fig. 6A compares the IR spectra of the spent Au/TiO2 sample after evacuation at 573 K (spectrum b) and the same sample after regeneration (spectrum c). In the left inset, the regenerated sample clearly shows OH stretching bands above 3500 cm−1. However, there are hardly any OH stretching bands observed on the spent catalyst but when evacuated at 573 K. These OH groups on a clean or regenerated catalyst sample can hardly be completely removed by evacuation at even 673 K. As for the spent catalyst, evacuation at 573 K eliminated all the surface hydroxyl groups indicating that probably some adsorbed monodentate propoxy species reacted with remaining Ti–OH during heating. The right inset in Fig. 6A shows the change of the spent sample in the Ti–O–C/C–C stretching region during heating. After heating the spent sample in vacuum at 573 K, the intensity decrease of two bands at 1135 and 1090 cm−1 was observed. As seen from Fig. 6A (spectrum a), the spent catalyst was fully covered by bidentate propoxy species, carboxylates as well as aldehyde adsorbed on the surface. The presence of a bidentate propoxy species on the spent catalyst is indicated by the pattern of bands in the CH stretching region at 2970 [νas(CH3)], 2932 [νas(CH2)], 2905 [ν(CH)], and 2869 cm−1 [νs(CH3)] together with two sharp bands in the Ti–O–C and C–C stretching regions at 1123 and 1090 cm−1.4,29 The band at 1123 cm−1 can be assigned to Ti–O–C stretching, while for PO or 1,2-propanediol adsorbed on Au/TiO2 the frequency of this band is ca. 20 cm−1 higher.28,29 The assignment for the 1090 cm−1 band can be ν(Ti–O–C),4 or ν(C–C).28,43 Aldehyde remaining on the surface is evidenced by the peak at 1723 cm−1 for CO vibration and the weak peak at 2716 cm−1 for H–CO, while the peak at ca. 2820 cm−1 is superimposed by bands of νs(CH3) and νs(CH2). Originally, the band at 1723 cm−1 was superimposed by a much stronger band at 1685 cm−1 of the spent sample evacuated at 323 K (not shown). However, the band at 1685 cm−1 disappeared after evacuation at 573 K, while the band at 1723 cm−1 remained intact. The strong and broad bands at 1435 cm−1 (overlapped with the sharp band at 1448 cm−1 for δas(CH3)/δ(CH2)) and 1544 cm−1 are assigned to νs(COO) and νas(COO) of surface acetate.44–46 Additional bands at 1576 cm−1 and 1331 cm−1 are tentatively assigned to surface formate.46,47 The surface acetate and formate species are mainly from the oxidation of the strongly adsorbed bidentate propoxy species as proposed in the literature.4,29 IR adsorption by acetate/formate is less pronounced on the spent catalyst evacuated at 323 K. After heating the sample to 573 K in vacuum, the increase of bands at 1435 cm−1 and 1544 cm−1 corresponded to the decrease of bands in the Ti–O–C/C–C stretching region between 1050 and 1200 cm−1 (the right inset in Fig. 6A). It is more likely to assign the bands at 1140 and 1090 cm−1 to ν(Ti–O–C) since these two bands turned into COO vibrations and have the same width and reduced intensity. Probably, lattice oxygen took part in the breakage of C–C bonds and consequent oxidation while heating up to 573 K in vacuum. After regeneration, a small amount of carbonate or carboxylate groups remained on the surface as indicated by strong adsorption at ca. 1540 and 1440 cm−1 (spectrum c), which were merely spectators on the surface since the catalyst activity was fully restored after regeneration.
 | ||
| Fig. 6 (A) Difference spectrum of the spent and regenerated sample of Au/TiO2. Progressive CO adsorption on (B) the spent (pre-evacuated at 573 K) and (C) the regenerated sample. The left inset in (A) compares the IR spectra of the spent and regenerated sample. The spent catalyst was under the reaction conditions ex situ at 333 K for 1 h after a 5 h catalytic testing and regeneration. The sample was then evacuated at 323 K and 573 K in the IR cell. All spectra were taken at 90 K. The following CO pressures after each dose were used (in mbar): 0.01, 0.03, 0.05, 0.07, 0.09, 0.11, 0.13, 0.16, 0.18, 0.20 for a–j in (B) and 0.01, 0.03, 0.05, 0.06, 0.08, 0.10, 0.13, 0.16, 0.18, 0.21 for a′–j′ in (C). In (A), spectrum a is the difference spectrum between the sample evacuated at 573 K (spectrum b) and the regenerated sample (spectrum c) and spectrum d is the difference spectrum between the sample evacuated at 323 K (not shown) and the regenerated sample (spectrum c). The green spectra ‘a−d’ in the right inset is the subtraction of ‘a’ by ‘d’. | ||
As shown in Fig. 6, on the spent Au/TiO2 catalyst there was limited adsorption of CO on both gold and Ti4+, similar to what was observed for the spent Au/Ti–SiO2 catalyst. The position of the weak CO band located at 2096 cm−1 (shifted from 2108 cm−1 at low coverage) indicates that the accessible gold atoms are typical Au0, although 2096 cm−1 is at the lower side for CO adsorption on Au0.24,25 On the regenerated sample, CO adsorption on metallic gold is clearly featured by the sharp band at 2107 cm−1, which was originally at 2123 cm−1 at low CO coverage. The red shift of ν(CO/Au0) is very typical for supported gold catalysts due to the fact that the backbonding from the gold atoms to the 2π* CO orbital is lacking.34–36 A weak peak located at 2138 cm−1 upon CO adsorption can also be identified and it gradually evolved to lower frequencies at higher CO coverages, seemingly saturated as the small band at 2127 cm−1. The small band at 2127 cm−1 can be assigned to vibrations of 13CO arising from natural abundance, which interacts with 5-fold coordinated Ti atoms (Ti5c4+, or β-Ti4+).23,32,48 This assignment can also be supported by Fig. 13, which shows CO adsorption on bare TiO2 (P25). However, it is not unambiguous to distinguish the contribution from CO adsorbed on Auδ+, which also gives a peak located between 2140 and 2125 cm−1. The band of ν(CO) adsorbed on Ti5c4+ located at 2179 cm−1, which shifted from 2190 cm−1 at low CO coverage on the clean and dehydrated Au/TiO2, while on the spent catalyst this band was at a bit lower frequency (2185 cm−1) at low CO coverage. On the regenerated Au/TiO2 the peak at 2214 cm−1 (shifted from 2229 cm−1) is assigned to ν(CO) on the stronger Lewis acid site Ti4c4+, or α-Ti4+.23,32,48
The XPS survey spectrum given in Fig. 7 also indicates the carbonaceous surface after the reaction. The O 1s, Ti 2p, and Au 4f XPS peaks of the spent catalyst are weaker than their counterparts after regeneration, while the C 1s peak is much stronger. Since the samples were kept in situ, the surface carbon was most likely not from contamination. Peak fitting of the O 1s XPS spectra shows five components on the catalyst surface, which are located at BE 529.9, 531.2, 532.0, 532.8, and 533.6 eV, respectively. The components at 529.9 and 531.2 eV are assigned to oxygen from Ti–O–Ti and Ti–OH, respectively.49 Combining the IR data in Fig. 6 and their intensity change after regeneration, the components of the O 1s line at 532.0, 532.8, and 533.6 eV can be assigned to CO32− or OC–O in carboxylates, CO in ketone/aldehyde, and C–O–Ti species, respectively.50,51 The Au 4f7/2 line is located at BE 83.4 eV for both the spent and regenerated catalyst and the spin doublet separation is 3.6 eV for the Au 4f line. The surface gold is simply in its metallic form.15,52 The difference in the intensity of the Au 4f lines from the spent and regenerated catalyst is much less profound than the difference observed from CO adsorption by IR. It can be concluded that XPS is not sensitive for detecting the active gold sites in our reaction since this method counts all the gold atoms at the surface rather than those specifically at edges and corners.
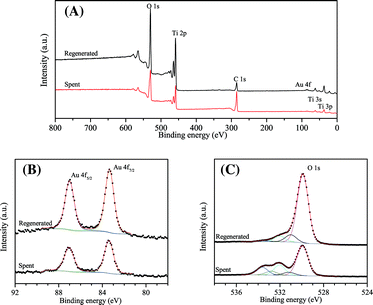 | ||
| Fig. 7 (A) XPS survey spectrum, (B) Au 4f photoelectron lines, and (C) O 1s photoelectron lines of the spent and regenerated Au/TiO2 catalyst. The spent catalyst was dehydrated in flowing helium at 473 K. Spectra were referenced to the C 1s line at 284.9 eV. | ||
3.3. CO adsorption on Au/Ti–SiO2 treated by O2 and H2
The effect of O2/H2 pretreatment on the gold oxidation state is shown in Fig. 8. On the regenerated Au/Ti–SiO2 sample (Fig. 8A), two bands can be distinguished: the sharp one at 2108 cm−1 and a shoulder at 2128 cm−1. They are assigned to CO adsorbed on Au0 and Auδ+, respectively. After pre-oxidation at 423 K (Fig. 8B), the ν(CO/Au0) band became much weaker while the ν(CO/Auδ+) almost remained the same. By further treating the pre-oxidized sample in H2 (Fig. 8C), the ν(CO/Auδ+) band could not be clearly observed. The incremental adsorption of CO between the second and first dose (Fig. 8D) indicates that on the reduced Au/Ti–SiO2 there were mainly Au0 sites and that the remaining small amount of Auδ+ may come from the interfacial Au–O–Ti. As for the regenerated sample, there may be more Au–O–O–Ti sites at the perimeter as proposed in the literature.38 On the pre-oxidized sample, it seems that Au0 atoms were covered by molecular oxygen even after evacuation at 423 K, which is in accordance with the high desorption temperature of O2 on small gold nanoparticles.53 However, when Au/TiO2 was used, the disturbance of ν(13CO) on Ti4+ to ν(CO) on Auδ+ obscured the band assignment and thus is not discussed. On the other hand, treating Au/TiO2 in the same way as in Fig. 8 but at 323 K (the typical epoxidation temperature for Au/TiO2) it showed no significant change in the adsorption intensity of CO on Auδ+ and Au0 when comparing to the literature,23,38 probably due to the low pre-oxidation temperature and low gold loading.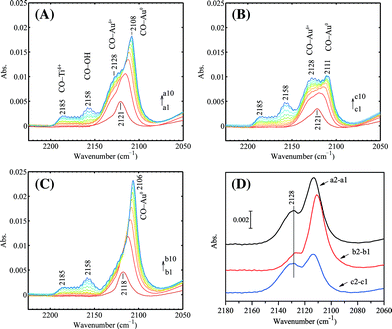 | ||
| Fig. 8 Progressive CO adsorption on the Au/Ti–SiO2 sample after different pretreatment: (A) regeneration at 573 K followed by 0.5 h in vacuum at 573 K and quenching to 90 K, (B) regeneration at 573 K followed by 0.5 h in vacuum at 573 K, cooling down to 423 K within 15 min, then 0.5 h in 5 vol% O2 at 423 K, afterwards 0.5 h in vacuum at 423 K and finally quenching to 90 K, (C) regeneration at 573 K followed by 0.5 h in vacuum at 573 K, cooling down to 423 K within 15 min, then 0.5 h in 5 vol% O2 at 423 K, 0.5 h in vacuum at 423 K, afterwards 0.5 h in 5 vol% H2 at 423 K, 0.5 h in vacuum at 423 K and finally quenching to 90 K. (D) The incremental adsorption between the second and first dose in (A), (B) and (C). The treated sample in vacuum at 90 K was used as the background. CO pressure increased from 0.01 mbar to 0.20 mbar after 10 doses with increments of ca. 0.02 mbar per dose. | ||
3.4. CO adsorption on Au/TiO2 in the presence of H2
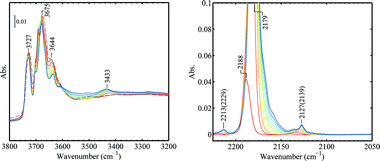
Fig. 9 shows the progressive CO adsorption on Au/TiO2 in the presence of H2 at 90 K. In the OH stretching region before CO dosing, three sharp bands at 3734, 3675, and 3420 cm−1 together with several weak shoulders at 3715, 3690, 3658, and 3648 cm−1 can be observed. During the H2 treatment at 323 K, trace amounts of H2O were formed as evidenced by the weak adsorption of δ(H2O) at 1617 cm−1. Upon CO dosing, a sharp band in the OH stretching region at 3648 cm−1 appeared together with a band for CO2 at ca. 2350 cm−1. Surprisingly, the pressure in the IR cell continued to decrease till the 3rd or 4th dose. CO adsorbed on gold from the first 3 doses (via the 5 μL loop) was almost fully and instantly converted to CO2. The CO2 vibration band reached its peak intensity after the 3rd dose and gradually decreased after subsequent CO dosage, very likely due to the replacement of CO on the surface. The change in the OH stretching region after CO dosing was more difficult to explain. The band at 3734 cm−1 remained at its original position, which is generally assigned to impurities such as Si–OH.28,48 The weak shoulder at 3715 cm−1 shifted to 3706 cm−1 upon CO dosing and can be assigned to isolated Ti–OH of the anatase phase.54,55 The major band centered at 3679 cm−1 decreased its intensity as the CO coverage increased and shifted to the broad band at 3547 cm−1 due to the interaction with CO. This major band can be assigned to Ti–OH of anatase.48,56 The assignment of the 3425 cm−1 band was proposed to be from water molecules or hydroxy groups on TiO2 (rutile),28,56,57 but this may need further investigation.57 The sharp band at 3648 cm−1 developed into the band at ca. 3500 cm−1 with a shift of 150 cm−1 as the CO coverage increased. A clear assignment of the 3648 cm−1 band is difficult in our case. This band was very weak before CO dosing. Upon CO dosing in the presence of H2, this band became very strong and sharp. Considering the decrease of cell pressure after CO dosing and the CO2 formation, it is clear that CO reacted with lattice oxygen in the presence of H2, forming CO2 and hydroxy groups and/or water. Water formation can be evidenced by the weak band at 3492 cm−1 upon CO dosing (red spectrum, dose 1 in Fig. 9B), which can be assigned as ν(OH) bound to water. On the other hand, from analysis of the difference spectra in the OH stretching region (not shown), the perturbation of a component at 3658 cm−1 upon CO dosing also contributed to the band increase at 3648 cm−1 but to a much lesser extent. The contribution of pre-adsorbed water to the bands at 3648 cm−1, either by the reaction with CO or by the dissociation of water on coordination unsaturated Ti4+ sites, was excluded by the experiment shown in Fig. 10.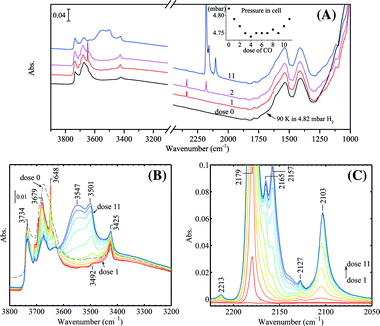 | ||
| Fig. 9 Progressive CO adsorption on Au/TiO2 in the presence of H2: (A) wide frequency range, the inset shows the pressures in the IR cell after each dose of CO, (B) spectra in the ν(OH) region, (C) difference spectra in the ν(CO) region using the spectrum of dose 0 as the background. The regenerated Au/TiO2 sample was kept in 50 mbar H2 (pre-dried with liquid nitrogen trap) at 323 K for 10 min followed by quenching down to 90 K and adjusting the H2 pressure by evacuation to 4.82 mbar before CO dosing. All spectra were taken at 90 K. | ||
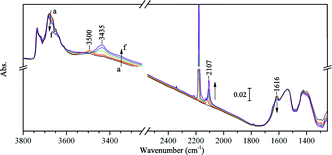 | ||
| Fig. 10 Progressive CO adsorption on Au/TiO2 pre-covered by a trace amount of H2O: (a) in vacuum, (b–e) dose 1 to 4, in 0.01, 0.03, 0.05, 0.07 mbar CO, respectively, (f) dose 10, in 0.21 mbar CO. The sample was pre-treated by partially dried H2 at 323 K and evacuated at 323 K for 1 h. All spectra were taken at 90 K. | ||
In Fig. 10, the coverage of water before CO adsorption was evidenced by the relatively stronger band of δ(H2O) at 1616 cm−1 and a band of ν(OH) perturbed by H2O at 3500 cm−1. After CO adsorption, no significant change occurred to the 3648 cm−1 band. With the increased coverage of CO, the band for δ(H2O) gradually shifted to 1645 cm−1. The band at 3500 cm−1 gradually shifted to 3435 cm−1. The band for ν(CO) on Au0 shifted from 2119 cm−1 at the low CO coverage and saturated at 2107 cm−1. It seems that water is not necessary for the band at 3648 cm−1, although its decrease in intensity was observed concomitantly with outgassing of water molecules on the TiO2 surface in other studies.57
Fig. 9 shows that the bands of ν(CO) at 2213 (α-Ti4+), 2179 (β-Ti4+), 2165 (γ-Ti4+), and 2103 (Au0) cm−1 did not shift with the CO coverage in the presence of H2. The band at 2157 cm−1 is assigned to ν(CO) interacting with OH groups. The small band at 2127 cm−1 is assigned to vibration of 13CO (arising from natural abundance) interacting with β-Ti4+.23 The bands at 2165 and 2157 cm−1 would only appear at very high CO pressures if there were no H2 presence. After the experiment performed as shown in Fig. 9, the IR cell was immediately evacuated at 90 K and the progressive CO adsorption on this sample was repeated but without H2 presence. The results are given in Fig. 11. The pressure in the IR cell increased monotonously by each dose of CO with an average increment of ca. 0.02 mbar, which is significantly different from what was observed in Fig. 9A, where CO and H2 reacted leading to a pressure decrease. The band at 3648 cm−1 remained. The bands at 2165 and 2157 cm−1 were not observed. It can be concluded that the presence of H2 significantly enhanced the adsorption of CO on TiO2 at low temperature.
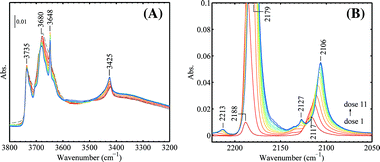 | ||
| Fig. 11 CO adsorption on Au/TiO2 after removing hydrogen (to be compared with Fig. 9, see text). The following CO pressures after each dose were used (in mbar): 0.00, 0.01, 0.03, 0.05, 0.07, 0.09, 0.11, 0.13, 0.16, 0.18, 0.20 for dose 1 to dose 11 (red to blue). All spectra were taken at 90 K. | ||
The same experiments were performed on the bare TiO2(P25) sample for reference. The results are shown in Fig. 12 and 13. The pressure in the IR cell increased monotonously after each CO dose when H2 was present, as shown in Fig. 12, which in turn confirmed that CO and H2 reacted on Au/TiO2 with lattice oxygen as indicated in Fig. 9. The results shown in this study are consistent with those that Widmann and Behm58 revealed. They showed that only for Au on TiO2 can TiO2 be reduced by CO. The presence of H2 simply improved the CO coverage at low CO pressures, featured by the stronger interaction of CO with hydroxy groups (3400–3600 cm−1, 2159 cm−1) and the weak acidic γ-Ti4+(2165 cm−1) as shown in Fig. 12B and C. After the experiment in Fig. 12 was done, the cell was fully evacuated and CO adsorption on TiO2 without H2 present was performed at the same temperature (Fig. 13). The only and obvious difference in the ν(CO) region as compared to Fig. 11 is that no CO on gold was observed. The remaining peaks were exactly the same, which also confirms the assignment of the 2127 cm−1 band to 13CO. In the OH stretching region no obvious sharp bands at 3648 and ca. 3420 cm−1 were seen.
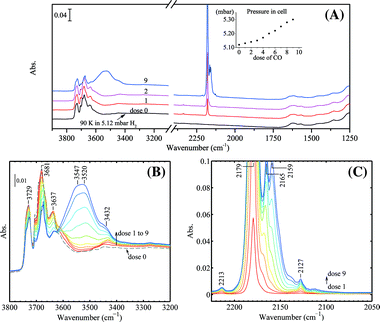 | ||
| Fig. 12 Progressive CO adsorption on TiO2(P25) in the presence of H2: (A) wide frequency range, the inset shows the pressures in the IR cell after each dose of CO, (B) spectra in the ν(OH) region, (C) difference spectra in the ν(CO) region using the spectrum of dose 0 as the background. The TiO2 sample was calcined in O2 at 573 K and treated in 50 mbar H2 (pre-dried with liquid nitrogen trap) at 323 K for 20 min followed by quenching down to 90 K and adjusting the H2 pressure by evacuation to 5.12 mbar before CO dosing. All spectra were taken at 90 K. | ||
 | ||
| Fig. 13 CO adsorption on TiO2 after removing hydrogen. The following CO pressures after each dose were used (in mbar): 0.02, 0.04, 0.07, 0.11, 0.14, 0.18, 0.22, 0.26, 0.30 for dose 1 to dose 9 (red to blue). All spectra were taken at 90 K. | ||
3.5. C3H6 adsorption on Au/TiO2 in the presence of CO
Fig. 14 shows the IR spectra of C3H6 adsorption on Au/TiO2 in the presence of CO at 230 K (above the boiling point of C3H6, 226 K at atmospheric pressure). The main adsorption band of ν(CO) on gold in 10 mbar CO was located at 2108 cm−1. Two weak bands at 2132 and 2069 cm−1 were also observed. The band at 2132 cm−1 can be assigned to CO on Auδ+. The broad band at 2060–2070 cm−1 was also observed in several other studies when at relatively high CO pressures (e.g., 20 mbar) and temperatures (e.g., at room temperature).59,60 In our experiment, this band was observed after a few minutes after the sample contacted with CO. It stabilized without further evolution. As proposed in the literature, this weak band is more likely due to morphology change of gold nanoparticles (lowered coordination number of gold atoms) in CO, but to a small extent in our case. The progressively dosed C3H6 gradually replaced CO on the surface interacting with both Ti4+ and Au as indicated by the weakened CO bands in Fig. 14A. It is known that C3H6 forms a stronger π-complex with Ti4+ than CO,61 thus almost all CO adsorbed on Ti4+ was removed. It seems that C3H6 has also a stronger interaction with gold than CO does. When there was 2.5 mbar C3H6 present, the band of CO on gold red-shifted by 30 cm−1 to 2079 cm−1. Interaction with C3H6 does not lead to the reconstruction of gold nanoparticles. This was confirmed by low temperature CO adsorption on the Au/TiO2 sample, which had contacted with 50 mbar C3H6 at 323 K for 30 min followed by evacuation. Thus, the red shift of ν(CO/Au) was more likely an effect of change in the electron density of gold atoms. The band at 2079 cm−1 may be assigned to ν(CO/Auδ−).62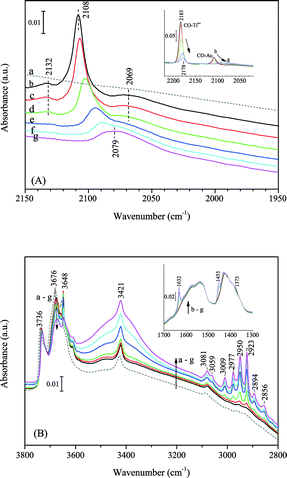 | ||
| Fig. 14 Progressive C3H6 adsorption on Au/TiO2 in the presence of CO at 230 K: (A) the CO stretching region; the spectra were shifted for clarity, spectra in the inset were not shifted. (B) The OH and CH stretching region; the inset shows the CC stretching and CH bending region. Spectra (gas phase compensated): (a) in vacuum before CO and C3H6 adsorption, (b) in 10.3 mbar CO after 20 min, (c) in 10.3 mbar CO + 0.5 mbar C3H6, (d) in 10.3 mbar CO + 1.0 mbar C3H6, (e) in 10.3 mbar CO + 1.5 mbar C3H6, (f) in 10.3 mbar CO + 2.0 mbar C3H6 and (g) in 10.3 mbar CO + 2.5 mbar C3H6. | ||
The presence of C3H6 on the catalyst surface was confirmed by the IR spectra in the CH stretching and bending regions as given in Fig. 14B. The details and assignment of bands from C3H6 adsorbed on the Au/TiO2 sample are given in Fig. 15 and Table 3. The decrease of the main bands at 3676 and 3648 cm−1 and increase of the broad band centred at 3410 cm−1 is attributed to the formation of hydrogen bonds between the allylic hydrogen of C3H6 and oxygen in Ti–OH.63 The sharp band of ν(CC) at 1632 cm−1 is slightly lower than the gas-phase C3H6, indicating a weak π-bonding to Ti4+ and Au. Three bands above 3000 cm−1 were observed. The bands at 3081 and 3009 cm−1 can be assigned to νas(CH2) and ν(CH), respectively. The origin of the 3059 cm−1 band is unclear. This band was not observed for C3H6 adsorbed on TiO2 (anatase).63 We tentatively assign this 3059 cm−1 band as νas(CH2) of C3H6 adsorbed at the Au–Ti interface or on gold. However, further investigation may be needed. After evacuation, no bands in the CH stretching region and for CC could be observed. The weak bands at 1576 and 1252 cm−1 (Fig. 15) can be assigned to carbonates due to the presence of CO.42
| Au/SiO2 | Au/Ti–SiO2 | Au/TiO2 | Assignment | Ref. | ||
|---|---|---|---|---|---|---|
| S | R | S | R | |||
| a S, spent; R, regenerated. | ||||||
| 2181 | 2185 | On Ti4+ of Ti–SiO2 | 32 | |||
| 2229 | On α-Ti4+ of TiO2, at low CO coverage | 23,32,48 | ||||
| 2214 | On α-Ti4+ of TiO2, at high CO coverage | |||||
| 2185 | 2190 | On β-Ti4+ of TiO2, at low CO coverage | 23,32,48 | |||
| 2178 | 2179 | On β-Ti4+ of TiO2, at high CO coverage | ||||
| 2165 | On γ-Ti4+ of TiO2 | 23 | ||||
| 2157 | With isolated OH groups on TiO2 | 23 | ||||
| 2158 | 2158 | 2158 | With OH groups on SiO2 and/or Ti–SiO2 | |||
| 2136 | 2136 | 2136 | Physisorbed CO | 32,33 | ||
| 2138 | Not fully resolved, in Fig. 6C, tentatively 13CO on β-Ti4+ at low CO coverage | |||||
| 2127 | 13CO on β-Ti4+ | 23,32,48 | ||||
| 2125, 2128 | On Auδ+ | 23,37,38 | ||||
| 2105 | 2118, 2121 | 2108 | 2123 | On Au0, low CO coverage | 24,25 | |
| 2099 | 2095 | 2107 | 2096 | 2107 | On Au0, high CO coverage | |
| 2079 | On Auδ−, C3H6 present | 62 | ||||
 | ||
| Fig. 15 Difference spectra of progressive C3H6 adsorption on Au/TiO2 in the presence of CO at 230 K (see Fig. 14, spectrum b as the background). | ||
| Vibrations | Gas phase64 | In polyethylene65 | On Ir4/Al2O366 | On TiO263 | This work |
|---|---|---|---|---|---|
| νas(CH2) | 3090 | 3075 | 3077 | 3090 | 3081, 3059 |
| ν(CH) | 3013 | 3008 | 3014 | 3010 | 3009 |
| νs(CH2) | 2992 | 2977 | 2979 | 2990 | 2977 |
| νas(CH3) | 2954, 2933 | 2958, 2933 | 2968 | 2960 | 2950, 2923 |
| νs(CH3) | 2870 | 2912 | 2859 | 2930, 2870 | 2894, 2856 |
| ν(CC) | 1652 | 1645 | 1640 | 1635 | 1632 |
| δas(CH3) | 1474, 1443 | 1449, 1443 | 1458, 1442 | 1460, 1440 | 1453, 1435 |
| δ(CH) | 1419, 990 | 1411, 988 | 1296 | 1415, 1010 | |
| δs(CH3) | 1378 | 1370 | 1376 | 1380 | 1373 |
| δ(CH2) | 1298 | 1293 | 1415 | 1295 | |
| ρ(CH2) | 1229 | 1169 | 1170 | 1171 | |
| ρ(CH3) | 1171, 1045 | 1040 | 1049, 1007 | 1047 |
Since the co-adsorption of CO and C3H6 was performed at a low temperature of 230 K, it is interesting to know how C3H6 would compete with CO at temperatures where the epoxidation occurs. Previously we have demonstrated that CO can switch off propene hydrogenation over the gold–titania catalysts.18 However, it is not clear if the role of CO is on gold or the support or both. If the effect of CO is on gold, propene should not have much effect on CO oxidation if we assume all the chemistry happens on the low coordinated gold atoms close to the Au–Ti interface.
The effect of C3H6 on CO oxidation over Au/TiO2 is shown in Fig. 16. It can be seen that CO oxidation was suppressed by C3H6 at either 303 K or 373 K. No significant deactivation was observed in the first 8 hours. However, after flushing in C3H6 (period III) and further flushing in helium till the C3H6 concentration was lower than 10 ppm, the activity in CO oxidation was severely reduced. Co-feeding of C3H6 completely suppressed CO oxidation. At 373 K after C3H6 flushing (11.5–13.5 h), the activity in CO oxidation gradually restored, which is very likely due to desorption of adsorbed propene. However, after period IV when C3H6 was removed from the CO/ceO2 stream (the last 1 hour), the activity could not be restored. It seems that propene formed a strongly adsorbed species at the Au–Ti interface when CO oxidation was proceeding (O adatoms available) and thus deactivated the catalytic site. Competition for oxygen by C3H6 is unlikely to explain the suppression of CO oxidation, since no reaction was proceeding at 303 K and no oxygenates were found at 373 K during C3H6 co-feeding. A trace amount of water was released at the beginning of C3H6 co-feeding at 373 K, but soon died away. The likelihood of full combustion of C3H6 at 373 K is small. Otherwise, there should be continuous water and CO2 formation. The deactivated catalyst in CO oxidation with C3H6 co-feeding can be regenerated by treatment in 10 vol% O2 at 573 K. The Au/TiO2 catalyst, after being tested at 303 K as shown in Fig. 16a, was then heated to 333 K and tested for the direct propene epoxidation. The epoxidation activity at 333 K is given in Fig. 17. There was only ca. 30% loss in the maximum PO formation rate. The trajectory of rH2O against rPO was still on par with the original performance of the catalyst as indicated by Fig. 17c. It can be concluded that the strongly adsorbed species formed from C3H6 during CO oxidation can be removed by hydro-epoxidation.
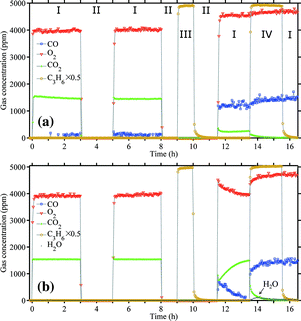 | ||
| Fig. 16 Effect of C3H6 on CO oxidation over Au/TiO2: (a) at 303 K and (b) at 373 K. (I) 1500/4750 ppm of CO/O2, helium balance; (II) helium flush; (III) 10000 ppm C3H6 flush; (IV) 1500/4750 ppm of CO/O2, co-feeding 10000 ppm C3H6. 150 mg of catalyst, GHSV 20000 mL gcat−1 h−1. | ||
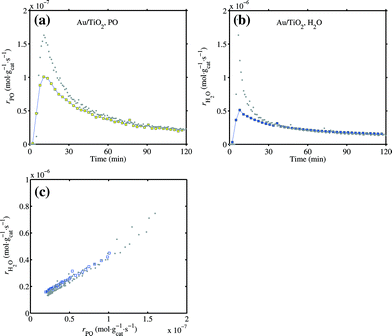 | ||
| Fig. 17 The activity of Au/TiO2 in the direct propene epoxidation at 333 K after testing the effect of C3H6 on CO oxidation as shown in Fig. 16a. GHSV 10000 mL gcat−1 h−1, 10:10:10:70 H2:O2:C3H6:He mixture. (a) PO formation rate, (b) water formation rate and (c) relation between the PO and water formation rates. Grey dots are data of three reaction cycles from Fig. 1 for reference. | ||
4. Discussion
The oxidation state of gold in the direct propene epoxidation can be summarized in the scheme given in Fig. 18. Our data of propene adsorption on the Au/TiO2 catalyst suggests that the low-coordinated gold atoms become negatively charged in the presence of propene, which leads to a broad carbonyl band at a low frequency of 2079 cm−1. The negatively charged gold has been also recently described well by Chakarova et al. on Au/SiO2 system by CO reduction.62 Combining with our previous work of propene adsorption on Au/SiO2,5 we would not expect much difference for Au/Ti–SiO2 in terms of charge transfer over gold in different atmospheres. In our case for Au/TiO2, contributions from propene adsorbed on gold and the support near the interface may both count to increase the electron density on gold. But for Au/Ti–SiO2, the contribution of charge transfer from the support and the role of the support oxygen may be less pronounced.67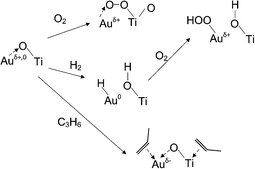 | ||
| Fig. 18 Schematic of gold oxidation state under conditions for propene epoxidation. | ||
Propene adsorption on gold in this study was performed at a relatively low temperature (230 K) and in an atmosphere where oxygen was absent. However, under the real reaction conditions, the species adsorbed on gold are much more complicated. Oxygen molecules are activated on gold by charge transfer from reduced gold forming a gold–oxygen complex.68,69 When hydrogen is present, OOH is formed.1 The study by Bravo-Suárez et al. on propane oxidation in hydrogen and oxygen over gold catalysts suggests that even propane can be oxidized by Ti–OOH forming isopropoxy species.70 The real role of propene modifying the reactivity of gold may be complicated. On one hand, propene can pi-bond to gold and reduce the water formation as shown by Nijhuis et al. in their XANES study.5 The spectra shown in Fig. 14 provide another evidence of the electron-donating ability from propene to gold, which seems to be even stronger than CO. On the other hand, oxidants adsorbed on gold may also react with propene forming strongly adsorbed species as proposed by Nijhuis et al.5 and as suggested by the CO oxidation experiment co-fed with propene in this study.
Although propene adsorption on either gold single crystal surfaces or a model Au/TiO2 catalyst is found to be weak,71,72 it may be not the case for a real catalyst. On a model Au/TiO2(110) catalyst in the work by Ajo et al.,72 the most stable sites for propene adsorption were those at the edge of gold islands with a TPD peak at ca. 240 K, while for 2D Au islands the temperature was higher till 310 K. For a real Au/TiO2 catalyst that is active for the direct propene epoxidation, the TPD peak of propene desorption on gold and/or at the perimeter was found to be at 323 K.14 TPD data over Au/Ti–SiO2 by Bravo-Suárez et al. suggests that the presence of gold and titania can significantly enhance the adsorption of C3H6 and a desorption temperature higher than 450 K was reported by them.73 However, it would be more interesting to know if propene and oxygen can cooperatively adsorb on gold, just as CO and oxygen do on gold clusters.74,75 In the coadsorption of CO and oxygen on gold clusters, it is proposed that CO acts as the electron donor and oxygen as the acceptor, both not competing for adsorption sites but cooperating.74,75 Our finding on the stronger electron-donating ability of propene than CO on gold might provide a starting point to this question.
The desorption temperature for O2 on gold is generally high, as reported in the literature. In the early work by Hayashi et al.,14 the TPD curve of Au/TiO2, which had been treated at 573 K in O2, showed that O2 desorbed at above ca. 500 K. The desorption of recombined O adatoms was reported to be at 470 K on Au(100) and above 500 K on Au(111),71 and even higher for thin gold nanoparticles supported on TiO2.53 The results in Fig. 8, of Au/Ti–SiO2 treated by O2 and H2, also indicate that the removal of O2 needs a relatively high temperature. Evacuation at 573 K seems not enough for us to fully remove oxygen on gold for Au/Ti–SiO2. The presence of O2 on gold, or preferably at the interface,76 makes the gold atoms electron deficient. The Auδ+ sites can be reduced by hydrogen to Au0 as shown in Fig. 8.
The CO adsorption on spent catalysts, as shown in Fig. 4 and 6, indicates that the gold sites during reaction are fully covered by deactivating species and reaction intermediates. Very limited gold sites were accessible for the probe molecule CO, even if the Au/Ti–SiO2 catalyst still had half its activity in PO formation after deactivation. CO adsorbed on these remaining gold sites shows the typical band location for Au0. However, these CO vibration bands show a long tailing shape towards lower wavenumbers, indicating a certain degree of reduction of the supports due to the adsorbed organic species and thus less electron transfer from gold to the support. IR using CO as the probe molecule may not be the preferred method to detect gold oxidation state under the real reaction conditions due to the fully covered surface. When using other bulk methods like XANES or XAFS one may also need to pay attention to the small contribution from the low-coordinated gold atoms if the gold particle size is not small enough.
A very interesting phenomenon observed in this study is that the presence of H2 greatly increased the amount of CO adsorbed on the TiO2 support at low temperature, as shown in Fig. 9 and 12. This may provide one explanation for the preferential oxidation (PROX) of CO over Au/TiO2 with the aid of H2. On Au/TiO2, the metal–support interface is considered to be the active site for O2 dissociation.76,77 It is also found that the diffusion of CO adsorbed on TiO2 to the Au–Ti interface contributes more to CO oxidation over Au/TiO2 at low temperature.77 Thus, besides a facile dissociation of O2 in the presence of H2, H2 may also promote the adsorption of CO on the support and consequently increase the reaction rate.
5. Conclusions
On both the Au/TiO2 and Au/Ti–SiO2 catalysts investigated after use in the direct propene epoxidation, the active gold sites can hardly be detected by CO molecules. The low-coordinated gold atoms are most likely occupied by propoxy species and carboxylates, which are most abundant on the catalyst surface after the reaction. Those gold atoms that can still be detected by CO show a very weak band at 2095 cm−1, indicating their metallic state. XPS spectra show that the oxidation state of surface gold atoms is metallic before and after reaction. However, XPS is not a sensitive tool to pinpoint those low-coordinated atoms at the perimeter of gold nanoparticles.Calcination in oxygen at 573 K restores the catalyst activity by removing the deactivating species and carbonaceous spectators. Oxygen can strongly adsorb on gold or at the perimeter of gold nanoparticles over the Au/Ti–SiO2 catalyst at the reaction temperature for propene epoxidation (e.g. 423 K), leading to positively charged gold atoms as evidenced by the carbonyl band on gold at ca. 2125 cm−1. Reduction in hydrogen at the reaction temperature removes adsorbed oxygen on the catalyst and restores the low-coordinated gold atoms to electron-neutral state, which give carbonyl bands at 2100–2110 cm−1. Over the Au/TiO2 catalyst, it has been observed that the presence of hydrogen at low temperatures can significantly enhance CO adsorption on the support, which may help explain the role of hydrogen in the preferential oxidation of CO.
The low-coordinated gold atoms become negatively charged in the presence of propene. Propene shows a stronger chemisorption than CO on both gold and Ti4+. Progressive propene adsorption leads to the weakening and broadening of the CO band on gold with a red shift of ca. 30 cm−1 towards 2079 cm−1. This implies a strong interaction between propene and gold through electron donation, which may explain the suppressed water formation over the gold catalysts when propene is present.
Acknowledgments
The Netherlands Organization for Scientific Research (NWO) is kindly acknowledged for providing an ECHO grant (700.57.044).References
- C. Sivadinarayana, T. V. Choudhary, L. L. Daemen, J. Eckert and D. W. Goodman, J. Am. Chem. Soc., 2004, 126, 38–39 CrossRef CAS PubMed.
- J. J. Bravo-Suárez, K. K. Bando, J. Lu, M. Haruta, T. Fujitani and S. T. Oyama, J. Phys. Chem. C, 2008, 112, 1115–1123 Search PubMed.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71–83 CrossRef CAS.
- T. A. Nijhuis, T. Visser and B. M. Weckhuysen, J. Phys. Chem. B, 2005, 109, 19309–19319 CrossRef CAS PubMed.
- T. A. Nijhuis, E. Sacaliuc, A. M. Beale, A. M. J. van der Eerden, J. C. Schouten and B. M. Weckhuysen, J. Catal., 2008, 258, 256–264 CrossRef CAS PubMed.
- B. Taylor, J. Lauterbach, G. E. Blau and W. N. Delgass, J. Catal., 2006, 242, 142–152 CrossRef CAS PubMed.
- J. Lu, X. Zhang, J. J. Bravo-Suárez, S. Tsubota, J. Gaudet and S. T. Oyama, Catal. Today, 2007, 123, 189–197. Search PubMed.
- J. Chen, S. J. A. Halin, J. C. Schouten and T. A. Nijhuis, Faraday Discuss., 2011, 152, 321–336 RSC.
- M. Boronat, F. Illas and A. Corma, J. Phys. Chem. A, 2009, 113, 3750–3757 CrossRef CAS PubMed.
- T. Fujitani, I. Nakamura, T. Akita, M. Okumura and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 9515–9518 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates Jr., Angew. Chem., Int. Ed., 2011, 50, 10186–10189 CrossRef CAS PubMed.
- B. Yang, X.-M. Cao, X.-Q. Gong and P. Hu, Phys. Chem. Chem. Phys., 2012, 14, 3741–3745 RSC.
- D. A. Panayotov, S. P. Burrows, J. T. Yates and J. R. Morris, J. Phys. Chem. C, 2011, 115, 22400–22408 CAS.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- A. Zwijnenburg, A. Goossens, W. G. Sloof, M. W. J. Crajé, A. M. Van der Kraan, L. J. De Jongh, M. Makkee and J. A. Moulijn, J. Phys. Chem. B, 2002, 106, 9853–9862 CrossRef CAS.
- C. Qi, J. Huang, S. Bao, H. Su, T. Akita and M. Haruta, J. Catal., 2011, 281, 12–20 CrossRef CAS PubMed.
- J. Gaudet, K. K. Bando, Z. Song, T. Fujitani, W. Zhang, D. S. Su and S. T. Oyama, J. Catal., 2011, 280, 40–49 CrossRef CAS PubMed.
- J. Chen, S. J. A. Halin, D. M. Perez Ferrandez, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 285, 324–327 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- J. Chou, N. R. Franklin, S.-H. Baeck, T. F. Jaramillo and E. W. McFarland, Catal. Lett., 2004, 95, 107–111 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41–50 CrossRef CAS.
- M. A. P. Dekkers, M. J. Lippits and B. E. Nieuwenhuys, Catal. Lett., 1998, 56, 195–197 CrossRef CAS.
- Tz. Venkov, K. Fajerwerg, L. Delannoy, Hr. Klimev, K. Hadjiivanov and C. Louis, Appl. Catal., A, 2006, 301, 106–114 CrossRef CAS PubMed.
- R. Meyer, C. Lemire, Sh. K. Shaikhutdinov and H.-J. Freund, Gold Bull., 2004, 37, 72–124 CrossRef CAS.
- M. Mihaylov, H. Knözinger, K. Hadjiivanov and B. C. Gates, Chem. Ing. Tech., 2007, 79, 795–806 CrossRef CAS.
- E. J. M. Hensen, D. G. Poduval, D. A. J. M. Ligthart, J. A. R. van Veen and M. S. Rigutto, J. Phys. Chem. C, 2010, 114, 8363–8374 CAS.
- T. A. Nijhuis, T. Q. Gardner and B. M. Weckhuysen, J. Catal., 2005, 236, 153–163 CrossRef CAS PubMed.
- G. Mul, A. Zwijnenburg, B. Van der Linden, M. Makkee and J. A. Moulijn, J. Catal., 2001, 201, 128–137 CrossRef CAS.
- A. Ruiz, B. van der Linden, M. Makkee and G. Mul, J. Catal., 2009, 266, 286–290 CrossRef CAS PubMed.
- Characterization and Chemical Modification of the Silica Surface, ed. E. F. Vansant, P. Van Der Voort and K. C. Vrancken, Elsevier, 1995, pp. 59–77. Search PubMed.
- A. Burneau and J. P. Gallas, in The Surface Properties of Silicas, ed. A. P. Legrand, John Wiley and Sons, 1998, ch. 3, Hydroxyl Groups on Silica Surfaces, pp. 147–234 Search PubMed.
- K. Hadjiivanov, B. M. Reddy and H. Knözinger, Appl. Catal., A, 1999, 188, 355–360 CrossRef CAS.
- D. A. Boyd, F. M. Hess and G. B. Hess, Surf. Sci., 2002, 519, 125–138 CrossRef CAS.
- J. France and P. Hollins, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 251–258 CrossRef CAS.
- F. Boccuzzi, S. Tsubota and M. Haruta, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 241–250 CrossRef CAS.
- C. Ruggiero and P. Hollins, J. Chem. Soc., Faraday Trans., 1996, 92, 4829–4834 RSC.
- F. Boccuzzi, A. Chiorino and M. Manzoli, Mater. Sci. Eng., C, 2001, 15, 215–217 CrossRef.
- I. X. Green, W. Tang, M. McEntee, M. Neurock and J. T. Yates, J. Am. Chem. Soc., 2012, 134, 12717–12723 CrossRef CAS PubMed.
- F. Meunier, D. Reid, A. Goguet, S. Shekhtman, C. Hardacre, R. Burch, W. Deng and M. Flytzani-Stephanopoulos, J. Catal., 2007, 247, 277–287 CrossRef CAS PubMed.
- J. Gong, D. W. Flaherty, T. Yan and C. B. Mullins, ChemPhysChem, 2008, 9, 2461–2466 CrossRef CAS PubMed.
- S. Y. Liu and S. M. Yang, Appl. Catal., A, 2008, 334, 92–99 CrossRef CAS PubMed.
- M. A. Bollinger and M. A. Vannice, Appl. Catal., B, 1996, 8, 417–443 CAS.
- W.-C. Wu, C.-C. Chuang and J.-L. Lin, J. Phys. Chem. B, 2000, 104, 8719–8724 CrossRef CAS.
- G. A. M. Hussein, N. Sheppard, M. I. Zaki and R. B. Fahim, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 1723–1741 RSC.
- V. Sanchez Escribano, G. Busca and V. Lorenzelli, J. Phys. Chem., 1990, 94, 8939–8945 CrossRef CAS.
- F. P. Rotzinger, J. M. Kesselman-Truttmann, S. J. Hug, V. Shklover and M. Grätzel, J. Phys. Chem. B, 2004, 108, 5004–5017 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- G. Busca, H. Saussey, O. Saur, J. C. Lavalley and V. Lorenzelli, Appl. Catal., 1985, 14, 245–260 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- G. P. López, D. G. Castner and B. D. Ratner, Surf. Interface Anal., 1991, 17, 267–272 CrossRef.
- J. Stoch and J. Gablankowska-Kukucz, Surf. Interface Anal., 1991, 17, 165–167. Search PubMed.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- V. A. Bondzie, S. C. Parker and C. T. Campbell, Catal. Lett., 1999, 63, 143–151 CrossRef CAS.
- M. Primet, P. Pichat and M. V. Mathieu, J. Phys. Chem., 1971, 75, 1216–1220 CrossRef CAS.
- H. Lin, J. Long, Q. Gu, W. Zhang, R. Ruan, Z. Li and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 9468–9474 RSC.
- P. Du, A. Bueno-López, M. Verbaas, A. R. Almeida, M. Makkee, J. A. Moulijn and G. Mul, J. Catal., 2008, 260, 75–80 CrossRef CAS PubMed.
- C. Deiana, E. Fois, S. Coluccia and G. Martra, J. Phys. Chem. C, 2010, 114, 21531–21538 CAS.
- D. Widmann and R. J. Behm, Angew. Chem., Int. Ed., 2011, 50, 10241–10245 CrossRef CAS PubMed.
- T. Diemant, Z. Zhao, H. Rauscher, J. Bansmann and R. J. Behm, Top. Catal., 2007, 44, 83–93 CrossRef CAS PubMed.
- M. Boronat, P. Concepción and A. Corma, J. Phys. Chem. C, 2009, 113, 16772–16784 CAS.
- A. A. Efremov, Yu. D. Pankratiev, A. A. Davydov and G. K. Boreskov, React. Kinet. Catal. Lett., 1982, 20, 87–91 CrossRef CAS.
- K. Chakarova, M. Mihaylov, S. Ivanova, M. A. Centeno and K. Hadjiivanov, J. Phys. Chem. C, 2011, 115, 21273–21282 CAS.
- A. A. Efremov and A. A. Davydov, React. Kinet. Catal. Lett., 1980, 15, 327–331 CrossRef CAS.
- R. C. Lord and P. Venkateswarlu, J. Opt. Soc. Am., 1953, 43, 1079–1085 CrossRef CAS.
- J. G. Radziszewski, J. W. Downing, M. S. Gudipati, V. Balaji, E. W. Thulstrup and J. Michl, J. Am. Chem. Soc., 1996, 118, 10275–10284 CrossRef CAS.
- A. M. Argo, J. F. Goellner, B. L. Phillips, G. A. Panjabi and B. C. Gates, J. Am. Chem. Soc., 2001, 123, 2275–2283 CrossRef CAS PubMed.
- T. A. Nijhuis, E. Sacaliuc-Parvulescu, N. S. Govender, J. C. Schouten and B. M. Weckhuysen, J. Catal., 2009, 265, 161–169 CrossRef CAS PubMed.
- J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651–4654 CrossRef CAS PubMed.
- N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. M. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240–2241 CrossRef CAS PubMed.
- J. J. Bravo-Suárez, K. K. Bando, J. Lu, T. Fujitani and S. T. Oyama, J. Catal., 2008, 255, 114–126 CrossRef PubMed.
- K. A. Davis and D. W. Goodman, J. Phys. Chem. B, 2000, 104, 8557–8562 CrossRef CAS.
- H. M. Ajo, V. A. Bondzie and C. T. Campbell, Catal. Lett., 2002, 78, 359–368 CrossRef CAS.
- J. J. Bravo-Suárez, J. Lu, C. G. Dallos, T. Fujitani and S. T. Oyama, J. Phys. Chem. C, 2007, 111, 17427–17436 Search PubMed.
- W. T. Wallace and R. L. Whetten, J. Am. Chem. Soc., 2002, 124, 7499–7505 CrossRef CAS PubMed.
- A. Prestianni, A. Martorana, F. Labat, I. Ciofini and C. Adamo, J. Phys. Chem. B, 2006, 110, 12240–12248 CrossRef CAS PubMed.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538–8546 RSC.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |