 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation of the role of surface nitrate species in the oxidation of propene on a Pt-based diesel oxidation catalyst†
Sarayute
Chansai
*a,
Robbie
Burch
a,
Christopher
Hardacre
a,
Harry
Oh
b and
William S.
Epling
c
aCenTACat, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, N. Ireland, UK. E-mail: s.chansai@qub.ac.uk
bDepartment of Chemical Engineering, University of Waterloo, 200 University Ave W, Waterloo, ON, Canada
cDepartment of Chemical and Biomolecular Engineering, University of Houston, 4800 Calhoun Road, Houston, TX 77204, USA
First published on 21st June 2013
Abstract
Enhancing the low temperature activity of diesel oxidation catalysts is important for cold-start conditions and the possible importance of nitrate species in oxidation reactions has been proposed although definitive evidence has not been reported. To investigate the possible role of surface nitrates, their adsorption and reactivity on a Pt-based diesel oxidation catalyst have been investigated using the Short Time on Stream (STOS) transient kinetic technique. The results provide for the first time definitive evidence for the oxidation of propene by some of these nitrate-type species.
1. Introduction
Diesel engine aftertreatment has received considerable attention over the last several years. Commercial systems include a diesel oxidation catalyst (DOC) upstream of a selective catalytic reduction (SCR) of NOx catalyst and/or a NOx storage/reduction (NSR) catalyst.1,2 The role of the DOC is to oxidize CO and hydrocarbon species exiting the combustion process, to oxidize NO to NO2 and to periodically provide heat for the downstream components via exothermic hydrocarbon oxidation. The NO oxidation functionality is important for the NOx reduction catalysts, as the SCR reaction rate is highest with an equimolar mixture of NO and NO2, the so-called fast SCR reaction,3,4 and NSR catalysts trap NO2 more readily than NO,5–9 thereby improving NOx storage performance10–13 as the engine out ratios of NO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NO2 are typically high. The formed NO2 can also oxidize soot at significantly lower temperatures than O2.1 DOCs are typically comprised of Pt, Pd or Pt–Pd blends, due to their oxidation ability, supported on a high surface area oxide.14–21
:NO2 are typically high. The formed NO2 can also oxidize soot at significantly lower temperatures than O2.1 DOCs are typically comprised of Pt, Pd or Pt–Pd blends, due to their oxidation ability, supported on a high surface area oxide.14–21
Recent evidence14,15 has pointed to the possibility that NO2 is also involved in the oxidation of CO and hydrocarbons over the DOC. Under certain circumstances, NO2 has been shown to be preferentially consumed relative to O2 in the oxidation reaction.14,15 This leads to no NO2 observed until all, or most, of the CO and hydrocarbons are oxidized and it is only after they are combusted is NO2 observed. Katare and co-workers22 also showed that as a DOC is thermally aged, this effect becomes more pronounced since the activity of the catalyst for the oxidation of CO and hydrocarbon drops. The definitive elucidation of the role of species such as surface nitrates for alumina-supported catalysts lies in the difficulty in differentiating between active and spectator surface species that build up during catalyst exposure to reactants.23–25 As an example, Chansai et al.24 have observed nitrates species of different reactivities during H2-enhanced hydrocarbon SCR of NOx over Ag-based catalysts. Upon exposure to NOx, a significant amount of nitrate species can form on Pt/Al2O3 catalysts, and as mentioned above, some of these surface nitrates may participate in hydrocarbon oxidation. To confirm the involvement of some types of surface nitrates, the Short Time on Stream (STOS) DRIFTS technique23–25 was used to study the mechanistic details of the reaction between surface nitrates and C3H6 during the oxidation of the hydrocarbon.
2. Experimental details
The 1 wt% Pt/Al2O3 powder sample used was purchased from Alfa Aesar. Pt dispersion was measured using H2 chemisorption in a Hiden Catlab micro-reactor. The catalyst was heated to 500 °C in 5% H2 in He for 30 minutes and then 100 μL of 5% H2 in He was pulsed into a 50 mL min−1 of He flow every 30 seconds, with a total of 26 pulses used. A stoichiometric ratio of two Pt sites per H2 molecule was used for calculation purposes.26–28 The Pt/Al2O3 catalyst Pt dispersion was 23.1%.In situ DRIFTS measurements were performed with a Bruker Vertex 70 FTIR spectrometer equipped with a liquid N2-cooled detector. 25 mg of the 1.0% Pt/Al2O3 catalyst sample was placed in a ceramic crucible in the DRIFTS cell. The exit lines were connected to a Hiden Analytical HPR20 quadrupole mass spectrometer in order to monitor the changes of gas phase species.
Prior to the experiments, the catalyst was pre-treated by heating in Ar with a total flow rate of 50 cm3 min−1 up to 300 °C for 1 h and then cooled down in flowing Ar to 250 °C. The IR spectrum of the Pt/Al2O3 catalyst at 250 °C under flowing Ar was taken as a background. Two 4-way VICI valves were installed to allow us to switch between two gas mixtures. The first was used to switch between Ar and the gas feed and the second was used for switching between other gas components, for example, switching between NO + O2 and C3H6. Kr was used a tracer for the switching experiments. The concentrations of the reactants used were: 500 ppm NO, 5% O2, 500 ppm C3H6, 0.5% Kr (when added) and Ar balance. The total flow rate was 50 cm3 min−1.
Three different sets of transient switching experiments were performed. The first two sets of experiments were carried out using the short time on stream (STOS) technique.24,25 In these experiments the catalyst is only exposed to reactants for the shortest time possible (typically tens of seconds) so that any adsorption on the support is minimised and it is easier to distinguish between potential reactant intermediates, at or near the active metal, from those, mostly inactive, species which have similar infrared spectra, and are located at a larger distance from the active metal. In these STOS experiments we switched between NO–O2 and C3H6–Kr, or between NO–O2 and C3H6–O2–Kr. The catalyst was exposed to the NO–O2 gas feed for 1 min at 250 °C before performing fast cycling transient switches from one gas mixture to another every 1 min. For comparison, in a final set of experiments, we show a conventional long time on stream procedure using an NO–O2 feed for 90 min, after which Ar was used to purge for 5 min before replacing this with the C3H6 feed. This allows us to compare the results from a conventional long exposure experiment with those from a STOS experiment.
In all cases, the in situ DRIFTS spectra were recorded with a resolution of 4 cm−1 and with the accumulation of 16 scans every 10 s during transient switches. The DRIFTS spectra were analyzed by the OPUS software. For the gas phase analysis, the following mass-to-charge (m/z) ratios were monitored as a function of time: 27 (C3H6), 28 (N2 and CO), 30 (NO), 32 (O2), 44 (N2O and CO2), 46 (NO2) and 82 (Kr). The results for gaseous CO, N2 (m/z = 28), NO2 (m/z = 46) are not reported because; (1) the MS signal of m/z = 46 was not observed as NO2 nearly 100% fragments to m/z = 30 and therefore, the MS signal at m/z = 30 is assigned to either NO or NOx; and (2) because CO2 (m/z = 44) is formed throughout the experiments and can be fragmented into m/z = 28 which then severely overlaps with the signals for CO and N2.
3. Results and discussion
3.1 STOS experiments when switching between NO–O2 and C3H6 [in the absence of O2]
Fig. 1 shows the results obtained during the first 2 min cycle when the catalyst was exposed to the NO–O2 mixture for just 1 min before switching to C3H6 for 1 min.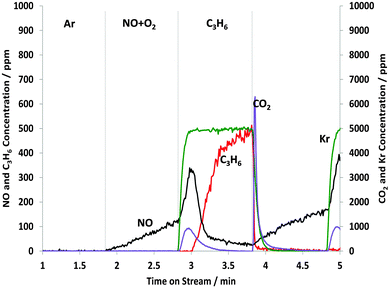 | ||
| Fig. 1 Changes in the gas phase species obtained from the STOS experiment when switching from NO + O2 to C3H6: outlet 14NOx and C3H6 concentrations as a function of time on stream during the 1st cycle of 60 s switches between NO–O2 and C3H6 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
The NO signal gradually increased over the course of the first minute and the total amount of NOx adsorbed was calculated to be 40.9 μmol gcat−1 (see Table 1). When the C3H6 was introduced, even in the absence of gaseous O2, it was found that the C3H6 was completely consumed for at least 10 s. The total amount of C3H6 consumed was calculated to be 18.9 μmol gcat−1 over the course of 1 min. At the same time, CO2 was formed and NO was released. The amount of NOx released and CO2 produced is 9.8 and 23.3 μmol gcat−1, respectively. The amount of CO2 production was significantly smaller than that calculated from the C3H6 consumption using the stoichiometric reaction of 3 moles of CO2 (56 μmol gcat−1) being formed from 1 mole of C3H6. As we shall see later from the DRIFTS experiments, some of the “missing” carbon can be accounted for by formation of CO and by carboxylate and formate species adsorbed on the support.
| Cycle no. | NO + O2 | C3H6 | NO + O2 | C3H6 + O2 | ||||
|---|---|---|---|---|---|---|---|---|
| NOx adsorption | NOx desorption | C3H6 consumption | CO2 formation | NOx adsorption | NOx desorption | C3H6 consumption | CO2 formation | |
| a All the calculated values are reported in μmol gcat−1. | ||||||||
| 1 | 40.9 | 9.8 | 18.9 | 23.3 | 39.8 | 7.2 | 46.4 | 127.9 |
| 10 | 32.8 | 10.1 | 18.6 | 23.9 | 26.2 | 7.7 | 46.4 | 129.4 |
These data indicate that some of the C3H6 or an intermediate species was adsorbed on the catalyst. Further evidence for this can be seen in Fig. 1 when replacing C3H6 with NO–O2. A sharp peak in CO2 production (31.5 μmol gcat−1) was observed which is due to the O2(g) + CxHy(ads) reaction, and the oxidation of CO(ads) and NCO(ads). In combination with the amount of CO2 formed during the NO–O2 pulse, this now accounts for all the carbon introduced during the C3H6 pulse.
The amount of NOx adsorbed in the second cycle (see Fig. 1) was less than the amount adsorbed in the first cycle, which indicates that not all of the NO that was adsorbed in the first cycle was removed by the propene. Nevertheless, a significant amount of NO still adsorbed in the second cycle. Therefore under these STOS conditions, the results indicate that C3H6 was indeed reacting with adsorbed NOx species. As well as reaction with the adsorbed NOx, some of the propene can react with PtOx formed on exposure to the NO–O2 mixture. It should be noted that in this switching apparatus, we can monitor the Kr switch to show that there is very little leakage of gaseous oxygen from the NO–O2 pulse to the C3H6 pulse and so very little of the C3H6 removed will be due to the O2 + C3H6 reaction. Also, Fig. 1 shows that CO2 formation took place for at least 40 s, compared with the disappearance of O2 which is rapid (<12 s (not shown)) on removal of the NO–O2 from the feed. Overall, we can confidently attribute some propene oxidation to a reaction with previously adsorbed species, of which a nitrate-type species is the most probable.
Clear confirmation that adsorbed NOx was removed when C3H6 was added is seen in Fig. 2, which shows the corresponding DRIFTS spectra for these STOS experiments. When the NO–O2 mixture was introduced to the catalyst for the first minute, the bands at 1230 cm−1 (nitrites) and at 1308 and 1545 cm−1 (nitrates) increased steadily.29–35 On the other hand, when the NO–O2 stream was replaced by C3H6 these bands decreased in intensity and essentially disappeared after 1 min of exposure to C3H6. It is observed that nitrates (1308 cm−1) react with C3H6 slightly faster than nitrites (1230 cm−1). This is probably due to the fact that in a short period of 1 min a small amount of nitrates are adsorbed weakly and very close to the Pt interfaces and then can react quickly when replacing the NO–O2 mixture with C3H6, whereas nitrites are formed quickly under the NO–O2 feed, allowing some to be adsorbed slightly further away from the Pt on the support.34,35 However, there are no observable changes in the band at 1545 cm−1 due to the fact that it is swamped with bands due to carboxylate-type species forming in the same region.33–36 New IR bands also appeared at 2016 and 2097, which are assigned to CO adsorbed on Pt,37–39 and at 2246 cm−1, which is assigned to adsorbed NCO species.40–42 Overall, it seems clear that C3H6 reacted with adsorbed NOx, which was probably in the form of a surface nitrate or nitrite.
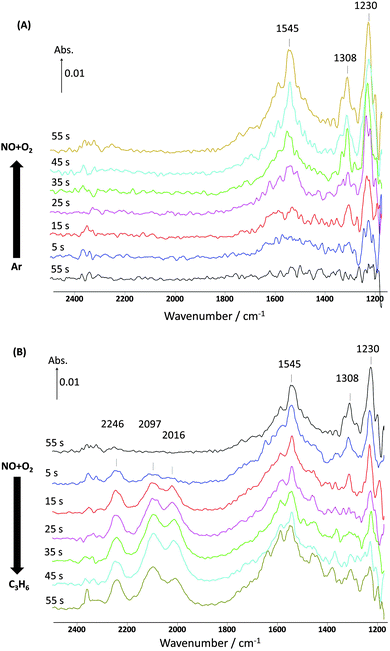 | ||
| Fig. 2 Changes in the surface species corresponding to Fig. 1 obtained from the DRIFTS-MS system as a function of time on stream during the 1st cycle of 60 s switches. (A) Switch from Ar to NO + O2 and (B) switch from NO + O2 to C3H6 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
Repeating the STOS experiment for more of the two-minute cycles, the results in the data for the tenth cycle are shown in Fig. 3.
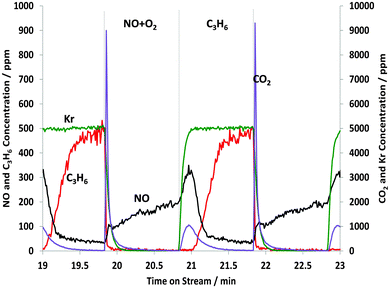 | ||
| Fig. 3 Changes in the gas phase species obtained from the STOS experiment when switching from NO + O2 to C3H6: outlet 14NOx and C3H6 concentrations as a function of time on stream during the 10th cycle of 60 s switches between NO–O2 and C3H6 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
Qualitatively, these kinetic results look similar to those seen for the first cycle, and almost identical to the results for the second cycle, as shown in Fig. 1, so again it appears that there is reaction between gaseous propene and adsorbed nitrate-type species. However, see Table 1, the calculated amount of NOx adsorbed from the NO–O2 feed is 32.8 μmol gcat−1 during the 10th cycle, compared with 40.9 μmol gcat−1 from the first cycle. Thus, as we increase the number of cycles we find that less nitrates are formed so presumably only a fraction of the adsorbed nitrates is being reduced by propene and the total amount of residual nitrate-type species has increased with each cycle. The amount of NOx released and the amount of C3H6 consumed remained about the same at 10.1 and 18.6 μmol gcat−1, respectively. However, the DRIFTS experiments presented in Fig. 4 for the 10th cycle appear to be contrast with this information.
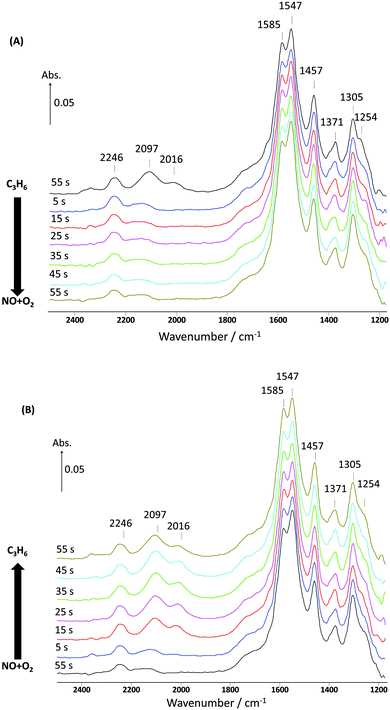 | ||
| Fig. 4 Changes in the surface species corresponding to Fig. 3 and obtained from the DRIFTS-MS system as a function of time on stream during the 10th cycle of 60 s switches. (A) Switch from C3H6 to NO + O2 and (B) switch from NO + O2 to C3H6 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
The DRIFTS results in Fig. 4 show how the amount of retained surface nitrates changed as we increased the number of two-minute cycles. By the tenth two-minute cycle, there was essentially no change in the DRIFTS spectra. No significant changes in the nitrate bands were observed either on exposure to the NO–O2 mixture or to the C3H6. The surface had apparently become saturated with essentially inactive nitrate and nitrite species. The band at 1457 cm−1 and the unresolved bands between 1397 and 1370 cm−1, that are attributed to acetate-type species and formate-type species, respectively,33–36 showed a similar lack of change when the gas atmosphere was changed. Note, however, that the bands attributed to CO (2097 and 2016 cm−1) were removed by the NO–O2 pulse which is consistent with oxidation of intermediate CO adsorbed on the Pt and the reoxidation of the Pt during this pulse.
It is clear that exposure of the catalyst to the reaction mixtures for as little as 20 minutes in total took the experiment beyond the point where active surface species can be detected and their concentrations monitored. Already the active nitrates, as previously seen to be reactive under the STOS conditions and shown in Fig. 2, were swamped by spectroscopically similar inactive species, presumably due to nitrates that formed on the support but which are not close enough to the metal to be catalytically important. These species were inactive, or at least had an activity that was much lower than that found for the “active” intermediates observed in the first cycle. These STOS experiments showed that there was a reaction between adsorbed nitrate-type species and gaseous propene. However, the ability to detect and monitor these surface reactions was lost after only a few two-minute cycles of the reactants.
3.2 STOS experiments with switching between NO–O2 and C3H6–O2 [in the presence of O2]
The effect of including oxygen with the propene in the STOS switching experiments is shown in Fig. 5. In contrast to the experiments in the absence of oxygen, complete combustion of C3H6 (46.4 μmol gcat−1) was observed and summarised in Table 1. However, during the first 1 minute cycle under C3H6–O2 feed, 127.9 μmol gcat−1 of gas phase CO2 was produced which is less than the 139.2 μmol gcat−1, calculated using the stoichiometric reaction of 3 moles of CO2 formed from 1 mole of C3H6. This is due to the formation and subsequent adsorption of acetate and carbonate species (see Fig. 6). On the other hand, the calculated amount of NOx adsorbed was 39.8 μmol gcat−1 which is essentially identical to the amount found in the absence of oxygen, although comparison of Fig. 1 and 5 shows that the NOx profile was quite different when NO was replaced by C3H6–O2 rather than C3H6 alone. However, once again, it seems that some of the surface NOx species can react with propene even when excess oxygen was present in the C3H6–O2 mixture.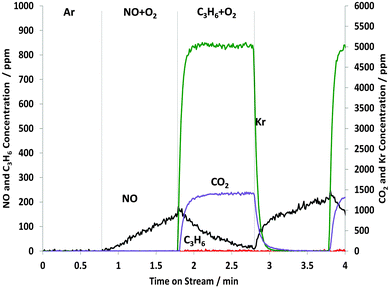 | ||
| Fig. 5 Changes in the gas phase species obtained from the STOS experiment when switching from NO + O2 to C3H6 + O2: outlet 14NOx and C3H6 concentrations as a function of time on stream during the 1st cycle of 60 s switches between NO–O2 and C3H6–O2 over Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
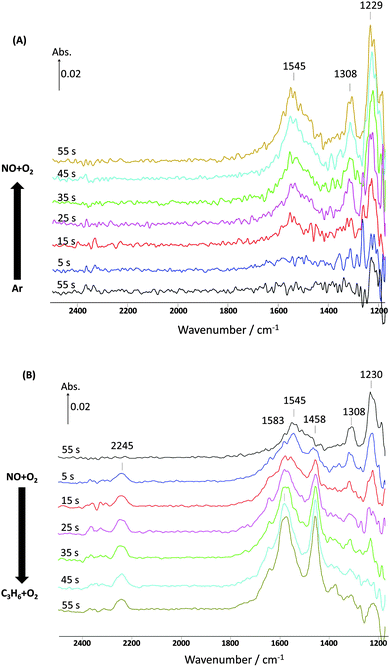 | ||
| Fig. 6 Changes in the surface species corresponding to Fig. 5 and obtained from the DRIFTS-MS system as a function of time on stream during the 1st cycle of 60 s switches. (A) Switch from Ar to NO + O2 and (B) switch from NO + O2 to C3H6 + O2 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
Importantly, as shown in Table 1, the amount of NOx desorbed under the C3H6 + O2 feed in the first cycle when O2 was present with the C3H6 is 7.2 μmol gcat−1 which contrasts with the 9.8 μmol gcat−1 of NOx desorbed when O2 was not added to the C3H6. This would seem to indicate that there is less direct reaction between adsorbed NO and the C3H6 in the case where there is O2 in the gas phase. If correct, this would indicate that either the reoxidation of the Pt by O2, followed by the reaction between PtOx and C3H6 is faster than the direct reaction between C3H6 and adsorbed NOx, or that the nitrate species are more stable in the presence of O2. The latter is consistent with literature describing NOx adsorption on Pt/Ba/Al2O3 materials.43,44
Further evidence that there is still some reaction with adsorbed NOx can be seen in the DRIFTS spectra shown in Fig. 6. These results for the first two-minute cycle show that when replacing NO–O2 with C3H6–O2, surface nitrites (1230 cm−1) and nitrates (1308 cm−1) were removed. It was also observed that carboxylate peaks (1458 and 1583 cm−1) appeared from the partial oxidation of C3H6 by O2 or NOx. This demonstrates that nitrate stability is not limiting the reaction.
During the 10th cycle, Fig. 7 and Table 1 show that the amount of NOx adsorption (26.2 μmol gcat−1) under the NO–O2 feed becomes smaller due to more adsorbed NO being retained from the earlier cycles (compare cycle 1 in Fig. 5), and/or because of some residual oxidation of the Pt blocking NOx uptake.
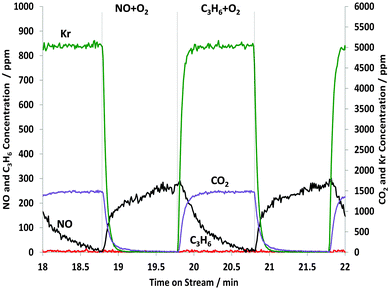 | ||
| Fig. 7 Changes in the gas phase species obtained from the STOS experiment when switching from NO + O2 to C3H6 + O2: outlet 14NOx and C3H6 concentrations as a function of time on stream during the 10th cycle of 60 s switches between NO + O2 and C3H6 + O2 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
Again, it is seen that, when replacing with C3H6–O2, the NO signal slowly decreased and the amount of NOx desorbed was 7.7 μmol gcat−1, which is the same within experimental error to the amount seen in the first cycle. However, there was no significant change in the infrared bands for the nitrates as can be seen in Fig. 8.
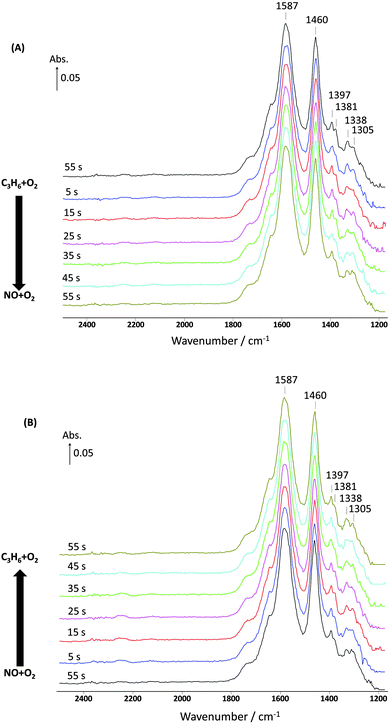 | ||
| Fig. 8 Changes in the surface species corresponding to Fig. 7 and obtained from the DRIFTS-MS system as a function of time on stream during the 10th cycle of 60 s switches. (A) Switch from C3H6 + O2 to NO + O2 and (B) switch from NO + O2 to C3H6 + O2 over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
Once again, in this case with O2 added to the C3H6, we find that even after as few as ten two-minute exposures to the reaction mixtures it becomes impossible to obtain any useful information from the DRIFTS results. On the other hand, using the STOS technique we can clearly demonstrate the removal of adsorbed nitrates, and the production of gaseous NOx, when a C3H6–O2 mixture was passed over a Pt catalyst that had previously been exposed to a NO–O2 mixture. These results also help clarify an apparent discrepancy in previous data.45 These showed for propene oxidation that the activation energy associated with nitrates as oxidant is significantly lower than that for O2 as oxidant but there were only slight improvements in the rate of propene oxidation when nitrates were present.45 These STOS data indicate that this is due to the relatively small amount of reactive nitrates (likely residing near the Pt) compared to the abundant nitrate species that form over the support.
3.3 Conventional long time on stream experiments with switching from NO–O2 to Ar to C3H6
The critical role of the surface nitrates in the oxidation of the hydrocarbon is not obvious from an examination of “conventional” long time on stream experiments. In this case, the Pt catalyst was exposed to the NO + O2 feed for 90 min, followed by Ar for 5 min and then by C3H6. Over a period of 90 min the surface nitrate and nitrite bands developed continuously (Fig. S1, ESI†). Using Ar as a purge will clarify whether or not a fraction of adsorbed NOx can be decomposed or desorbed into gas phase. After switching to inert Ar to flush the catalyst (Fig. S2, ESI†), it is shown that there was essentially no change in the surface nitrate or nitrite bands as well as gas phase NOx (not shown).In addition, Fig. 9A clearly shows that after switching to C3H6 both gas phase NOx and CO2 were rapidly released due to the interaction between adsorbed NOx and C3H6. However, it is important to note that although changes in the gas phase species were observed, which demonstrate that some adsorbed nitrate-type species were reacting with C3H6, the DRIFT spectra in Fig. 9B were unchanged even after 30 min exposure to C3H6. Clearly, under these “traditional” transient experimental conditions virtually all the infrared detectable nitrates and nitrites were inactive.
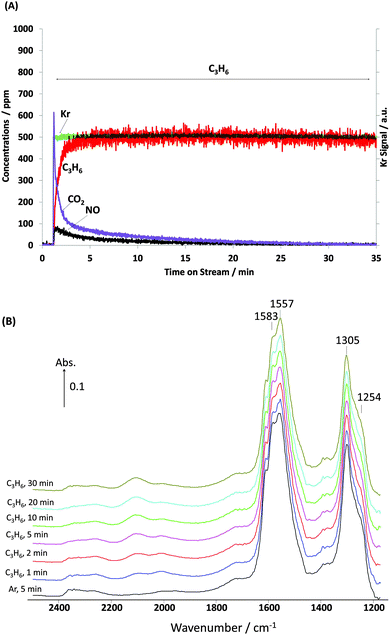 | ||
| Fig. 9 Changes in (A) gas phase and (B) surface species obtained from a “conventional” steady state (long time on stream) transient experiment as a function of time on stream over the course of 30 min under C3H6 flow over 1% Pt/Al2O3 at 250 °C. Feed conditions: 500 ppm NO, 500 ppm C3H6, 5% O2, Kr (when added) and Ar balance. | ||
4. Conclusions
The contribution of surface nitrate-type species to the oxidation of propene over a Pt diesel oxidation catalyst has been demonstrated unequivocally for the first time both for propene-only and propene + oxygen reaction mixtures. The use of the Short Time on Stream (STOS) transient kinetic technique has allowed the identification of a surface nitrate-type species that can react with propene to produce CO2 and NOx. The STOS results show that after long times on stream most of the nitrate-type species that are detectable by DRIFTS are either unreactive or have a very low reactivity. The benefit of using the STOS transient technique is the ability to clearly differentiate between unreactive spectator species and reactive intermediates.Acknowledgements
We are grateful to the EPSRC for providing the funds to develop the STOS technique through the CASTech project (EP/G012156/1).References
- L. Castoldi, R. Matarrese, L. Lietti and P. Forzatti, Appl. Catal., B, 2006, 64, 25 CrossRef CAS.
- M. Koebel, M. Elsener and G. Madia, Ind. Eng. Chem. Res., 2001, 40, 52 CrossRef CAS.
- A. Kato, S. Matsuda, T. Kamo, F. Nakajima, H. Kuroda and T. Narita, J. Phys. Chem., 1981, 85, 4099 CrossRef CAS.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312 CrossRef CAS.
- S. Erkfeld, E. Jonson and M. Larsson, Top. Catal., 2001, 16–17, 127 CrossRef.
- F. Rodrigues, L. Juste, C. Potvin, J. F. Tempere, G. Blanchard and G. Djega-Mariadassou, Catal. Lett., 2001, 72, 59 CrossRef CAS.
- N. W. Cant and M. J. Patterson, Catal. Today, 2002, 73, 271 CrossRef CAS.
- S. Hodjati, P. Bernhardt, C. Petit, V. Pitchon and A. Kiennemann, Appl. Catal., B, 1998, 19, 2098 Search PubMed.
- S. Hodjati, K. Vaezzadeh, C. Petit, V. Pitchon and A. Kiennemann, Catal. Today, 2000, 59, 323 CrossRef CAS.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks, Catal. Rev., 2004, 46, 163 Search PubMed.
- S. Salasc, M. Skoglundh and E. Fridell, Appl. Catal., B, 2202, 36, 145 Search PubMed.
- H. Mahzoul, J. F. Brilhac and P. Gilot, Appl. Catal., B, 1999, 20, 47 CrossRef CAS.
- F. Pronetto, G. Ghiotti, I. Nova, L. Lietti, E. Tronconi and P. Forzatti, J. Phys. Chem. B, 2001, 105, 12732 CrossRef.
- K. Irani, W. S. Epling and R. Blint, Appl. Catal., B, 2009, 92, 422 CrossRef CAS.
- H. Oh, J. Luo and W. S. Epling, Catal. Lett., 2011, 141, 1746 CrossRef CAS.
- J.-H. Lee and H. H. Kung, Catal. Lett., 1998, 51, 1 CrossRef CAS.
- E. Xue, K. Seshan and J. R. H. Ross, Appl. Catal., B, 1996, 11, 65 CrossRef CAS.
- P. Denton, A. Gioir-Fendler, H. Praliaud and M. Primet, J. Catal., 2000, 189, 410 CrossRef CAS.
- L. Olsson and E. Fridell, J. Catal., 2002, 210, 340 CrossRef CAS.
- S. Bernard, L. Retailleau, F. Gaillard, P. Vernoux and A. Giroir-Fendler, Appl. Catal., B, 2005, 55, 11 CrossRef.
- L. Olsson, M. Abul-Milh, H. Karlsson, E. Jobson, P. Thormaehlen and A. Hinz, Top. Catal., 2004, 30/31, 85 CrossRef CAS.
- S. R. Katare, J. E. Patterson and P. M. Laing, SAE paper 2007-01-3984, 2009.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2011, 281, 98 CrossRef CAS.
- S. Chansai, R. Burch and C. Hardacre, J. Catal., 2012, 295, 223 CrossRef CAS.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2010, 276, 49 CrossRef CAS.
- J. Freel, J. Catal., 1971, 25, 139 CrossRef.
- H. L. Gruber, J. Phys. Chem., 1962, 66, 54 CrossRef.
- J. Dawody, L. Eurenius, H. Abdulhamid, M. Skoglundh, E. Olsson and E. Fridell, Appl. Catal., A, 2005, 296, 157 CrossRef CAS.
- Y. Ji, T. J. Toops, J. A. Pihl and M. Crocker, Appl. Catal., B, 2009, 91, 329 CrossRef CAS.
- T. J. Toops, D. B. Smith and W. P. Partridge, Catal. Today, 2006, 114, 112 CrossRef CAS.
- B. Westerberg and E. Fridell, J. Mol. Catal. A: Chem., 2001, 165, 249 CrossRef CAS.
- C. Sedlmair, K. Seshan, A. Jentys and J. A. Lercher, J. Catal., 2003, 214, 308 CrossRef CAS.
- W. S. Epling, C. H. F. Peden and J. Szanyi, J. Phys. Chem. C, 2008, 112, 10952 CAS.
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493 CrossRef CAS.
- F. C. Meunier, V. Zuzaniuk, J. P. Breen, M. Olsson and J. R. H. Ross, Catal. Today, 2000, 59, 287 CrossRef CAS.
- K. Shimizu, H. Kawabata, A. Satsuma and T. Hattori, J. Phys. Chem. B, 1999, 103, 5240 CrossRef CAS.
- M. Haneda, M. Sasaki, H. Hamada and M. Ozawa, Catal. Lett., 2001, 141, 1262 CrossRef.
- G. Busca, E. Finocchio and V. S. Escribana, Appl. Catal., B, 2012, 113–114, 172 CrossRef CAS.
- M. M. Schubert, M. J. Khalich, H. A. Gasteiger and R. J. Behm, J. Power Sources, 1999, 84, 175 CrossRef CAS.
- N. Bion, J. Saussey, C. Hedouin, T. Seguelong and M. Daturi, Phys. Chem. Chem. Phys., 2001, 3, 4811 RSC.
- N. Bion, J. Saussey, M. Haneda and M. Daturi, J. Catal., 2003, 217, 47 CAS.
- B. Wichterlová, P. Sazama, J. P. Breen, R. Burch, C. J. Hill, L. Čapek and Z. Sobalík, J. Catal., 2005, 235, 195 CrossRef.
- J. Anderson, A. Paterson and M. Fernandez-Garcia, Stud. Surf. Sci. Catal., 2000, 130, 1331 CrossRef.
- K. Eguchi and T. Hayashi, Catal. Today, 1998, 45, 109 CrossRef CAS.
- H. Oh, I. S. Pieta, J. Luo and W. S. Epling, Reaction kinetics of C3H6 oxidation for various reaction pathways over diesel oxidation catalysts, Top. Catal., 2013 Search PubMed , in press.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00349c |
| This journal is © The Royal Society of Chemistry 2013 |