 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards acid MOFs – catalytic performance of sulfonic acid functionalized architectures†
Jana
Juan-Alcañiz
*a,
Robin
Gielisse
a,
Ana B.
Lago
b,
Enrique V.
Ramos-Fernandez
a,
Pablo
Serra-Crespo
a,
Thomas
Devic
b,
Nathalie
Guillou
b,
Christian
Serre
b,
Freek
Kapteijn
a and
Jorge
Gascon
*a
aCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 136, 2628 BL, Delft, The Netherlands. E-mail: j.gascon@tudelft.nl
bInstitut Lavoisier, UMR 8180 CNRS Université de Versailles St Quentin en Yvelines, 45 avenue des Etats-Unis, 78035 Versailles, France
First published on 4th June 2013
Abstract
In this work, the inclusion of free sulfonic acid groups in highly stable MOFs is explored. The synthesized catalysts have been applied in a model esterification reaction. Two metal organic frameworks bearing sulfonic acid moieties are investigated: HSO3-MIL-101(Cr) synthesized following different approaches and a new structure based on HSO3-bdc and Zr. The acidic properties, catalytic performance, deactivation and stability of the different structures are critically evaluated. In the case of MIL-101(Cr), deactivation of the sulfonic groups via formation of butanol sulfonic esters has been observed. Due to the strong interaction between –SO3− and the Cr open metal site where usually fluorine (F−) is located in the structure, the HSO3-MIL-101(Cr) catalysts are not stable under acidic regeneration conditions. When using Zr as a metal node, a new and stable sulfonic acid containing porous structure was synthesized. This structure showed high activity and full re-usability in the esterification of n-butanol with acetic acid. In this case, deactivation of the catalyst due to sulfonic ester formation could be reversed by reactivation under acidic conditions.
Introduction
During the last few decades, much effort has been put into the development of acidic solid materials such as ion-exchange resins based on sulfonic acid groups,1 sulfated oxides based on zirconia, silica, or alumina2 and activated carbons with different surface functional groups.3–5 However, the application window of such solids is limited: some exhibit swelling in solvents (polymers) or strong leaching issues (sulfated oxides and activated carbons), while none of them possess a well-defined porosity. The development of strongly acidic nano-structured catalysts is a long-standing challenge for materials’ researchers and is only partially solved by organic functionalized mesoporous silicas.6In the field of synthetic nano-structured materials, metal–organic frameworks, hereafter MOFs, are attracting a great deal of attention. Next to a high surface area and pore volume, the chemical environment in MOFs can be fine-tuned by selecting the appropriate building blocks and/or by post-synthetic treatment, resulting in nearly infinite design possibilities.7 When it comes to catalytic applications, as recently highlighted in several reviews,8,9 MOFs hold many promises: the inorganic connectors may hold certain functional sites by themselves, as shown for MOFs possessing coordinatively unsaturated sites (CUS),10 or may display photoactivity.11 Besides the intrinsic activity of such CUS, they can also be utilized as anchoring points for molecules to create catalytically active or sorption centers.12 Regarding the organic linkers, it is generally anticipated that MOFs may bridge the gap between organic and inorganic chemists,13,14 since almost any organic moiety of choice could be immobilized. The formation of guest-accessible functional organic sites (FOS) may be achieved following two different strategies:15 direct incorporation of functionality during the synthesis16 or post-functionalization.17
Different approaches have been followed for the introduction of strong Brønsted acidity in MOFs, these include mainly post-synthetic and encapsulation routes, like controlled framework destruction to increase the number and acidity of CUS,18 post-synthetic framework sulfation,19 post-functionalization of amine groups with sultones20 and encapsulation of heteropolyacids.21 To date, Kitagawa and co-workers have reported the direct synthesis of free sulfonic acid functionalized MOFs, namely MIL-101(Cr) and doped UiO-66(Zr), via direct synthesis.22–24 Very recently Biswas et al. presented a new direct synthesis procedure for SO3H-UiO-66.25 Several works describe the benefits of sulfonic MOFs for applications like catalysis,26 proton conductivity or adsorption.23–25 In this work, we explore two synthetic routes for the production of sulfonic structures like MIL-101(Cr) and a Zr based MOF, along with a thorough assessment of their acid catalytic activity, deactivation and influence of synthesis conditions on framework stability and catalyst reusability.
Experimental section
All the chemicals and reference catalysts were obtained from Sigma-Aldrich and used without further purification, unless otherwise stated.HSO3-MIL-101(Cr)HCl
HSO3-MIL-101(Cr)HCl has been synthesized according to the procedure reported by Kitagawa and co-workers, a mixture of monosodium 2-sulfoterephthalic acid (2 g, 7.5 mmol), CrO3 (0.75 g, 7.5 mmol), concentrated aqueous hydrochloric acid (12 N, 0.546 g) was dissolved in water (30 g), and it was hydrothermally treated at 453 K for 168 h.22HSO3-MIL-101(Cr)HF
HSO3-MIL-101(Cr)HF has been synthesized from a mixture of chromium(III) nitrate nonahydrate (2.00 g, 5 mmol), monosodium 2-sulfoterephthalic acid (Tokyo Chemical Company, 2.70 g, 10 mmol), deionized water (30 g) and hydrofluoric acid (47–51 wt%, 0.3 g) by heating at 463 K for 24 h. The as-synthesized solid was washed with deionized water and dried in air at 393 K.KSO3-bdc
25 g (0.134 mol) of 2,5-dimethylbenzenesulfonic acid dehydrated (Acros Organics, 99%) was dissolved in 100 ml of water and KMnO4 (106 g, 0.67 mol) was slowly added. The reaction mixture was heated under reflux for 48 h, and followed by filtration to remove the residual MnO2. The solution was concentrated until approx. 80%, and 12 N hydrochloric acid was added to the filtrate (approx. 200 ml). Immediately, a precipitation of a mixture of 2-sulfoterephthalic acid and the monopotassium salt is formed. The white precipitate is washed with EtOH and dried at 100 °C. 1H NMR (dmso-d6): δ 8.34 (s, 1H), 7.99 (dd, 1H, J = 2 Hz, J = 8.6 Hz), 7.69 (d, 1H, J = 8.5 Hz). MS-ESI: m/z (%) = 245 (100) [HSO3-bdc]+. EDS analysis: (K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S/0.6:1). IR (cm−1): 1710 vs, 1487 m, 1260 m, 1208 vs, 1120 m, 1386 m, 1200 w
:S/0.6:1). IR (cm−1): 1710 vs, 1487 m, 1260 m, 1208 vs, 1120 m, 1386 m, 1200 w
HSO3-ZrMOF
Zirconium tetrachloride (1.17 g, 5 mmol) and KSO3-bdc (2.50 g, 8.5 mmol) were reacted in deionized water (25 g) at 423 K for 24 h. The resulting white solid was washed with hot deionized water and dried in air under ambient conditions.Scanning electron microscopy
SEM was measured in a JEOL JSM 6500F setup coupled to an Energy Dispersive Spectrometer (EDS) for micro elemental analysis.Nitrogen adsorption at 77 K
Nitrogen adsorption at 77 K was measured in a Quantachrome Autosorb-6B unit gas adsorption analyzer.Carbon dioxide adsorption at 273 K
Low-pressure adsorption was measured in a Micromeritics TriStar II 3020 based on the volumetric technique. The samples were pre-treated under vacuum for 16 h at different temperatures depending on the sample.X-ray diffraction
X-ray diffraction was performed using a Bruker-AXS D5005 with CoKα radiation. For HSO3-ZrMOF, high-resolution synchrotron data were collected at ID31, ESRF, France. Subsequent pattern indexing and Le Bail structure-independent fits were performed using Topas software.ICP-OES
Samples were digested in duplo in a mixture of 8 ml aqua regia and 2 ml HF using microwave irradiation. After digestion, the samples were diluted to 50 ml with MQ, and analyzed using an ICP-OES Perkin Elmer Optima 5300dv.Diffuse reflectance IR spectroscopy
DRIFT spectra were recorded on a Nicolet model 8700 spectrometer, equipped with a high-temperature DRIFT cell (KBr windows), DTGS-TEC detector and a 633 nm laser. The spectra were registered from 4000 to 600 cm−1 after accumulation of 128 scans and a resolution of 4 cm−1. A flow of helium at 20 ml min−1 was maintained during the measurements. Before collecting the spectra, the different samples were pre-treated in the same helium flow at 393 K for 30 min. KBr was used to perform background experiments.X-ray photoelectron spectroscopy (XPS)
Measurements were made using VG-Microtech Multilab equipment, MgKα (hν: 1253.6 eV) radiation and a pass energy of 50 eV. The XPS system analysis pressure was kept at 5 × 10−10 mbar. The binding energy (BE) and the kinetic energy (KE) scales were adjusted by setting the C1s transition to 284.6 eV. BE and KE values were determined with the peak-fit software of the spectrometer. The intensities were estimated by calculating the integral of each peak, after subtraction of the S-shaped background, and by fitting the experimental curve to a combination of Lorentzian (30%) and Gaussian (70%) peak shapes. The surface atomic ratios were estimated from the integrated intensities corrected by the atomic sensitivity factors (D. A. Shirley, Phys. Rev. B., 1972, 5, 4709).Acid–base titration
Volumes of known solutions of two different bases (NaOH and bicycloguanidine) were added to 100 mg MOF dispersed in 20 ml water, while analyzed using a pH-meter, at RT. The diffusion of the bases inside of the MOF was in agreement with their size, and in both cases long times were needed for pH stabilization (1 ml needs at least one hour).Esterification of acetic acid and n-butanol
The reaction was performed without solvent, using a molar ratio mixture of acetic acid:n-butanol = 1:1. The reaction mixture (25 ml) was introduced into a round bottom flask, while being stirred under reflux the temperature was increased to 343 K. A ratio of 3 g catalyst per mole of acetic acid was used. After recovering the catalyst, it was filtered, and treated with an acidic solution before use in consecutive runs. The acidic treatment was performed by putting the MOF catalyst in contact with a 0.2 M solution of HCl or pellets of an acidic ion exchange resins 1:1 mass ratio (DOWEX® 50W X8 hydrogen form) in water under stirring conditions for 30 minutes at room temperature.
Results and discussion
HSO3-MIL-101(Cr)
HSO3-MIL-101(Cr)HCl has been synthesized according to the procedure reported by Kitagawa and co-workers, while a new route has been developed for the synthesis of HSO3-MIL-101(Cr)HF. Both catalysts exhibit the PXRD pattern expected for MIL-101 structure (Fig. S1, ESI†). N2 adsorption isotherms measured at 77 K (Fig. S2, ESI†) show the two characteristic steps of MIL-101, related to the filling of the mesoporous cavities. At very low relative pressures (P/P0 < 0.05) only the supertetrahedra are filled. As pressure increases, the medium (P/P0 = 0.15) and later the large cavities (P/P0 = 0.20) are filled. Samples synthesized in the presence of HCl and at long synthesis times display a lower surface area (circa 1200 m2 g−1, calculated between 0.05 and 0.10 relative pressure) than samples synthesized in shorter times and in the presence of HF (circa 1800 m2 g−1). In both cases, the surface area is smaller than for pure MIL-101(Cr)27 but in good agreement with other functionalized MIL-101s.28SEM pictures (Fig. S3, ESI†) reveal a smaller particle size for HSO3-MIL-101(Cr)HCl (≈200 nm) than for HSO3-MIL-101(Cr)HF (≈700 nm). Elemental analysis of the evacuated samples demonstrates that ion exchange occurs during the synthesis, resulting in an exchange of Na+ from the linker with protons, yielding a partial protonation of the sulfonic acid groups. This ion exchange is more effective for HSO3-MIL-101(Cr)HCl (1.28 wt% Na resulting in 0.3 Na+/SO3−) in contrast with HSO3-MIL-101(Cr)HF (2.75 wt% Na resulting in 0.41 Na+/SO3−), probably due to the longer synthesis times, the stronger acidity of the synthesis medium and the smaller particle size.
Thermo-gravimetric analysis of both samples (Fig. S4, ESI†) reveals a significantly higher amount of inorganic residue in the case of the HSO3-MIL-101(Cr)HCl. While the remaining 20 wt% in the case of HSO3-MIL-101(Cr)HF is in good agreement with the expected molecular formula (Cr3OF(H2O)2[(O2C)-C6H3(SO3NaxHy)-(CO2)]3), and the amount of water found in the sample (15 wt%), as indicated also by EDS with Cr/S of 1. The residue for HSO3-MIL-101(Cr)HCl is higher than expected and can be attributed to the presence of Cr2O3 in the material.
Infrared spectroscopy confirms the successful functionalization. Fig. 1 shows a comparison between the pristine MIL-101(Cr) and both functionalized HSO3-MIL-101(Cr). Clear differences are already revealed when observing the whole spectrum (4000–1000 cm−1). The ν(OH) region (4000–3000 cm−1) exhibits a broad band centered at 3400 cm−1 for the sulfonic materials, absent in the bare samples. This vibration corresponds to water molecules retained in the pores by strong hydrogen bonding with the sulfonic acid even after in situ treatment at 423 K for one hour. Specific sulfonic acid stretching can be observed in the fingerprint region (between 1800 and 1000 cm−1).
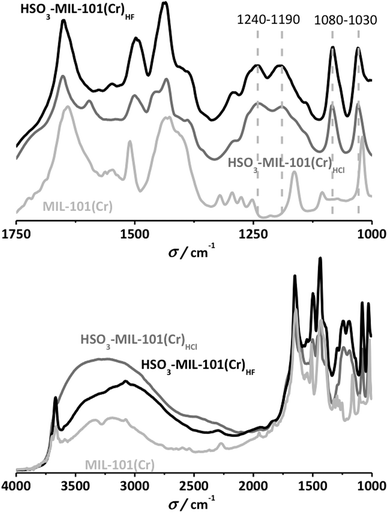 | ||
| Fig. 1 FTIR spectra of the different HSO3-MIL-101(Cr) structures compared to that of non-functionalized MIL-101(Cr). Top: full spectral range. Bottom: details of the 1000–1750 cm−1 region. | ||
New bands appear at 1190 and 1240 cm−1, attributed to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO symmetric and asymmetric stretching modes. The peak at 1080 cm−1 corresponds to the S–O stretching vibration, with a slight shift of the band at 1020 to 1030 cm−1, possibly attributed to the influence of the SO3 substituted aromatic ring.29
SO symmetric and asymmetric stretching modes. The peak at 1080 cm−1 corresponds to the S–O stretching vibration, with a slight shift of the band at 1020 to 1030 cm−1, possibly attributed to the influence of the SO3 substituted aromatic ring.29
The catalytic activity of the HSO3-MIL-101(Cr) catalysts has been benchmarked against Amberlyst®-15 by the liquid-phase stoichiometric esterification of n-butanol and acetic acid at low temperatures (343 K). Experiments were performed using the same ratio of 3 g mol−1 reactant for all catalysts. The evolution of the butyl acetate yield (the only reaction product next to water) with time for all studied catalysts is plotted in Fig. 2.
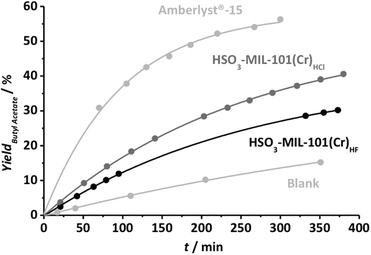 | ||
| Fig. 2 Esterification of n-butanol and acetic acid (1:1 molar fraction) using 3 grams of the catalyst (specified in the graph) per mole of reactant at 343 K. | ||
The commercial catalyst (Amberlyst®-15) displays a higher activity on a weight basis, while a clear difference in activity between both HSO3-MIL-101(Cr) samples can be noticed. The latter can be explained based on the amount of protonated HSO3 groups in the HSO3-MIL-101(Cr)HCl catalyst. On the other hand, the difference in activity found between commercial and MOF based catalysts can be explained when the density of acid sites is considered: the amount of H+ in Amberlyst®-15 is 4.2 mmol g−1, higher than 1.9 and 2.2 mmol g−1 for HSO3-MIL-101(Cr)HF and HSO3-MIL-101(Cr)HCl, respectively (Table 1).
:1 molar ratio) using 3 gram of the catalyst per mole of reactant at 343 K. TOF obtained for several catalytic runs
| Catalyst | Acidity [mmol g−1] | TOF/min−1 | |||
|---|---|---|---|---|---|
| 1st Run | 2nd Run | 3rd Run | 4th Run | ||
| a Obtained from ref. 30. b Obtained by molecular formula. c Obtained by the theoretical molecular formula, and confirmed accessibility of all protons by acid–base titration with NaOH and 1,5,7-triazabicyclo[4.4.0.]dec-5-ene. | |||||
| Amberlyst®-15 | 4.2a | 0.36 | 0.31 | 0.20 | |
| HSO3-MIL-101(Cr)HF | 1.9b | 0.23 | 0.12 | 0.11 | |
| HSO3-MIL-101(Cr)HCl | 2.2b | 0.30 | 0.22 | 0.13 | |
| HSO3-ZrMOF | 2.6c | 0.29 | 0.23 | 0.20 | 0.33 |
TOF values of 0.36, 0.23 and 0.30 min−1 were calculated based on the amount of protonated sulfonic groups for Amberlyst®-15, HSO3-MIL-101(Cr)HCl and HSO3-MIL-101(Cr)HF, according to the theoretical molecular formula. The TOF values for the MOFs are systematically 15–30% lower, although equal values could be expected, since all three catalysts are based on benzene-sulfonic moieties and therefore their acidic strength should not differ much. On the other hand, it has to be noted that TOF values obtained for the MOF catalysts are even higher than most heterogeneous catalysts reported in the literature, including sulfonic functionalized mesoporous silicas.31,32 Recycling tests were performed to assess the heterogeneous character of the samples. These experiments suggest a strong deactivation of both HSO3-MIL-101(Cr) samples, in contrast to a less pronounced deactivation found for Amberlyst®-15. A large decrease in activity is observed for the MOF catalysts. Indeed, after 3 cycles, similar conversion levels as those in the blank run (in the absence of catalyst) were observed, demonstrating the complete deactivation of the HSO3-MIL-101(Cr) samples.
To unravel the nature of the deactivation, the different HSO3-MIL-101(Cr) catalysts were analyzed after several reaction cycles: PXRD confirmed the integrity of the crystalline structure (Fig. S5, ESI†) while SEM did not show any strong attrition of the catalyst particles, both discounting framework collapse as a reason for the deactivation (Fig. S6, ESI†).
On the other hand, BET measurements (Fig. S7, ESI†) show a large decrease in surface area after every reaction cycle, even after pre-treatment at 433 K under vacuum. This decrease in accessible pore volume and the absence of particle disintegration point at chemisorbed reactants and products as possible causes for deactivation.
In order to gain insight into the specific interactions between both reactants and the HSO3-MIL-101(Cr) framework, DRIFT spectroscopy has been performed during the adsorption and thermal desorption of n-butanol and acetic acid. Fig. 3 shows the collected IR spectra at different temperatures. Impregnation with acetic acid does not result in the appearance of new vibrations other than those representative of the carboxylic acid group at 2650, 2575 and 1725 cm−1. All bands disappear after heat treatment of the sample, demonstrating that the framework–acetic acid interaction is rather mild. In contrast, when the HSO3-MIL-101(Cr) catalysts are contacted with n-butanol, in addition to the CH2 and CH3, symmetric and asymmetric stretching modes, between 2960 and 2880 cm−1, several new vibrations can be observed after heat treatment, namely a doublet at 2650 and 2575 cm−1 and a broadening of the 1725 cm−1 and SO bands (1260 and 1180 cm−1). Both the doublet and the broadening of the 1725 cm−1 bands can be ascribed to carbonyl vibrations resulting from the partial hydrolysis of terephthalate–Cr bonds, while the broadening of the SO bands and the shoulder formed at 1065 cm−1 are attributed to a strong SO3–n-butanol interaction.
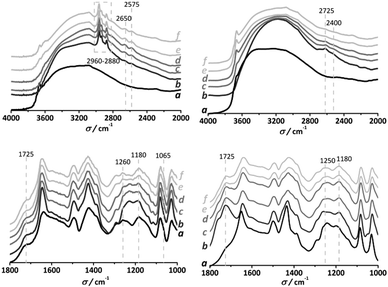 | ||
| Fig. 3 DRIFTS (a) HSO3-MIL-101(Cr)HF (also observed for HCl samples) upon contact with an esterification reactant at RT (b) and desorption under helium at increasing temperature steps: (c) 30 min at 323 K, (d) 30 min at 373 K, (e) 30 min at 423 K, (f) 120 min at 443 K. Left: n-butanol. Right: acetic acid. | ||
Very recently, Fraile and co-workers3 demonstrated that in the case of sulfonic functionalized microporous carbons the formation of sulfonate esters accounts for the deactivation of the catalysts in the presence of an alcohol. In our work, the infrared and adsorption results are in line with this formation of sulfonate esters.4 In view of such a strong chemical interaction, we re-activated the HSO3-MIL-101(Cr) catalysts using either acidic ion exchange resins (DOWEX® 50W X8 hydrogen form) or very diluted HCl–water mixtures. Infrared spectra of the reactivated samples show the disappearance of all n-butanol related bands, demonstrating the successful exchange. However, XRD and adsorptive characterization after several ion exchanges show a clear loss of crystallinity along with a reduced N2 uptake, inferring the partial collapse of the framework. These results demonstrate that although active during the first catalytic runs, the application window of HSO3-MIL-101(Cr) is rather limited.
Kitagawa et al. claimed that electroneutrality in the HSO3-MIL-101(Cr) framework is achieved by partial deprotonation of SO3H groups, instead of halide or hydroxide inclusion in the Cr trimers, resulting in one out of three SO3− moieties in anionic form.22 If this is the case, it is easy to envisage that due to a strong interaction of these anionic moieties with n-butanol and during the ion exchange with the acidic resin, charge neutrality may be compromised, resulting in a partial collapse of the framework. Two different approaches were followed to improve stability of the Cr3+ trimers and to achieve charge neutrality in the framework without using the SO3− functional groups: in the first approach, the as-synthesized catalysts were ion exchanged with NH4F with the idea that F− would stabilize the Cr3+ trimers and that NH4+ would act as counter ion for the SO3−, but this approach resulted in the collapse of the framework. In a second approach, we synthesized mixed linker MIL-101(Cr) using a 50:50 mixture of monosodium 2-sulfoterephthalic acid and terephthalic acid and the same synthesis conditions as in the case of HSO3-MIL-101(Cr)HF (Fig. S8, ESI†).
Elemental analysis (XPS) on the resulting solids demonstrates that both linkers are present and that F− incorporation in the framework took place during synthesis (Fig. S9, ESI†). However, the resulting solids did not display any activity in the esterification of n-butanol and acetic acid, inferring that most SO3− moieties are used to stabilize the framework Cr trimers and confirming the rather limited availability of protons in HSO3-MIL-101(Cr) for catalysis.
XPS analysis of the S 2p shows one peak centered at 168 eV, indicating the presence of sulfonic groups.33 These peaks decrease significantly for the mix-linker system, suggesting the rather low inclusion of sulfonic linkers compared to the 100% functionalized linker sample. For both structures, contributions from two different S 2p peaks are observed after precise deconvolution: 169 eV and 167.8 eV. Although these binding energies are typically attributed to the S 2p3/2 and S 2p1/2 transitions of sulfonic groups (R–SO3H),34 several publications partially attribute this transition to sulfone (R–SO3–R′) groups,35 suggesting a possible interaction of sulfonic groups with another part of the framework. This hypothesis is emphasized when looking at the F 1s spectra. When full sulfonic linker is present in the synthesis, a very small amount of fluor is found in the final solid, with a binding energy of 684.2 eV. Interestingly, in the case of pristine MIL-101(Cr) (no sulfonic groups) and when only half of the linkers are sulfonated, a much larger amount of fluor, with a binding energy of 689 eV, is observed.36,37 These results demonstrate that in the fully sulfonic structure, SO3− plays the same role as F− in stabilizing the chromium trimers and that full proton exchange results in the collapse of the structure.
It is indeed well known that fluorine is involved in the terminal bond of the trimeric chromium species and partly substitutes the terminal –OH molecules attached to chromium in MIL-100 and MIL-101. Although it is not fully clear yet, it seems that the use of fluorine provides a strong interaction with the chromium octahedral motif and enhances the formation of MIL-101 during the hydrothermal reaction.38,39 In our case, when a similar stabilization role is taken by the sulfonic groups, the final structure is more likely to collapse due to the weaker interaction with the sulfonic groups.40
HSO3-Zr-MOF
Zr-carboxylate MOFs like UiO-66(Zr) and others have been shown to be among the most stable MOFs.41 Functionalization of UiO-66(Zr) has been extensively studied, both by direct synthesis for groups like –NH2, –NO2 or –Br,42 and by post-synthetic treatment for more complex groups.43–45 Several theoretical studies already proposed the benefits of sulfonic acid functionalization on the UiO-66(Zr)46 framework and very recently Kitagawa and co-workers and Biswas et al. described the first synthetic route towards fully sulfonated and mixed-linker UiO-66(Zr) and the effect of linker functionalization on adsorption properties.24,25 More precisely, whereas the mixed bdc/bdc-SO3H linker based compounds were shown to present a rather high specific surface area, the fully substituted compound loses its crystallinity upon activation. Exploring the reactivity towards a MOF formation of zirconium- and potassium 2-sulfoterephthalic in water rather than DMF, a new crystalline porous solid could be isolated, denoted HSO3-ZrMOF (see Experimental section).The XRD powder pattern of the new structure could be indexed (Fig. S10, ESI†) within a I cubic unit-cell (Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 41.5331(2) Å), first indicating that the product is a single crystalline phase. Although this cell presents similarities with the one of UiO-66 (cubic, SG: Fmm, a = 20.7004(2) Å) with a doubling of the cell parameter, preliminary structural analysis suggests that it does not represent a UiO-66 type topology built up from 12-connected Zr6 clusters solely, but is rather based on 8-connected Zr6 clusters, similar to the ones observed in some very recently reported Zr-MOFs.47–49 This is furthermore in accordance with the Zr/ligand ratio extracted from EDS analysis (Zr/S ∼ 1.5, repeated at least 10 times with equal results), which suggests a Zr6(bdc-SO3H)4 formula rather than a Zr6(bdc-SO3H)6 one. Using state of the art structure solution methods (charge flipping), some Zr atoms could be allocated. This unambiguously confirms the presence of Zr6 clusters similar to those found in many Zr-based MOFs (Fig. S11, ESI†); one can nevertheless not preclude that some clusters are still missing in our model. Moreover, the ligands could not be localized yet. Nevertheless, the examination of the intercluster distances gives information about their connection. The shortest intercluster Zr–Zr distances (11.4 Å) are indeed almost identical to those found in UiO-66, indicating that at least one part of the cluster is connected in a similar way to that in the UiO-66 solid, i.e. through Zr–terephthalate–Zr bridges. This proves that at least one part of the sulfonic groups is not connected to Zr ions and is thus available for catalysis.
m, a = 41.5331(2) Å), first indicating that the product is a single crystalline phase. Although this cell presents similarities with the one of UiO-66 (cubic, SG: Fmm, a = 20.7004(2) Å) with a doubling of the cell parameter, preliminary structural analysis suggests that it does not represent a UiO-66 type topology built up from 12-connected Zr6 clusters solely, but is rather based on 8-connected Zr6 clusters, similar to the ones observed in some very recently reported Zr-MOFs.47–49 This is furthermore in accordance with the Zr/ligand ratio extracted from EDS analysis (Zr/S ∼ 1.5, repeated at least 10 times with equal results), which suggests a Zr6(bdc-SO3H)4 formula rather than a Zr6(bdc-SO3H)6 one. Using state of the art structure solution methods (charge flipping), some Zr atoms could be allocated. This unambiguously confirms the presence of Zr6 clusters similar to those found in many Zr-based MOFs (Fig. S11, ESI†); one can nevertheless not preclude that some clusters are still missing in our model. Moreover, the ligands could not be localized yet. Nevertheless, the examination of the intercluster distances gives information about their connection. The shortest intercluster Zr–Zr distances (11.4 Å) are indeed almost identical to those found in UiO-66, indicating that at least one part of the cluster is connected in a similar way to that in the UiO-66 solid, i.e. through Zr–terephthalate–Zr bridges. This proves that at least one part of the sulfonic groups is not connected to Zr ions and is thus available for catalysis.
Nitrogen adsorption at 77 K proved not to be the best method to characterize porosity of the new Zr MOF: N2 uptakes depended on the pre-treatment prior to the measurement. For instance, after pre-treatment at room temperature under vacuum, samples presented a decent adsorption capacity (400 cm3 g−1), whereas when pre-treated at 353 K under vacuum, samples did not show any N2 uptake. In contrast, adsorption of carbon dioxide at 273 K demonstrated sample micro-porosity, independent of the pre-treatment used (2 mmol CO2 per g at 100 kPa, see Fig. 4).
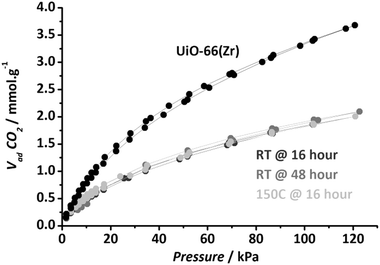 | ||
| Fig. 4 Carbon dioxide adsorption isotherms (273 K) of the HSO3-Zr-MOF pre-treated under different conditions compared to UIO-66(Zr) pretreated at 427 K for 16 hours. | ||
Compared to the pristine UiO-66(Zr), the accessible surface area is lower: 496 m2 g−1 for UiO-66(Zr) vs. 339 m2 g−1 for Zr6(bdc-SO3H)4, as calculated from CO2 adsorption data using the Dubinin Radushkevich model. This adsorption capacity is however higher than the values shown by fully sulfonated UiO-66(Zr) presented by Kitagawa et al.24 In addition, since the synthesis takes place in the presence of the highly acidic precursor ZrCl4, almost full K+ exchange takes place during synthesis (∼0.36 wt% residual K compared to the 2.5 wt% Na in the MIL-101 samples). In fact, after one single use of the catalyst in reaction all the K+ is exchanged (<0.01 wt% K). DRIFT spectroscopy demonstrates the presence of sulfonic groups, with the same new vibrations related to Caromatic–SO3 bonds (Fig. S12, ESI†) as observed for the MIL-101 sulfonic samples.
In order to demonstrate that all sulfonic groups present in the new Zr structure are accessible for catalysis, titration experiments were performed using two different bases: sodium hydroxide and 1,5,7-triazabicyclo[4.4.0.]dec-5-ene; differing very much in size. Titration results indicated a similar consumption of both bases, and were in agreement with the amount of sulfonic groups in the theoretical molecular formula (∼3 mmol H+ per g). These results demonstrate that even though pores might not be accessible to N2 adsorption after sample activation, in the liquid phase much bulkier molecules like triazabicyclo[4.4.0.]dec-5-ene can enter the pores and titrate all sulfonic groups.
The acidic performance of the new Zr MOF has been assessed in the esterification of acetic acid and n-butanol. Fig. 5 shows a very high initial catalytic activity, well in comparison with the benchmark catalyst Amberlyst®-15.
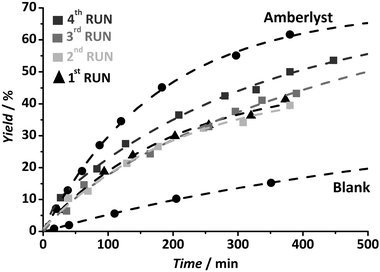 | ||
| Fig. 5 Esterification of n-butanol and acetic acid (1:1 molar ratio) using 3 grams of the catalyst per mole of reactant at 343 K. Before every use the catalyst has been pre-treated with 0.2 M HCl solution for 1 h at room temperature and thoroughly washed with H2O until neutral pH. | ||
Reusability of the catalyst was tested. Similar to that for MIL-101(Cr) samples, formation of sulfonic esters resulted in a strong decrease in catalytic activity. In addition, both the Na+ and K+ salts of the sulfonic linker were tested in blank experiments, and did not show any catalytic activity. In order to regenerate the spent catalyst, acid treatments were applied after each reaction cycle with a 0.2 M HCl solution at room temperature for one hour followed by thorough cleaning with water until pH 7 was reached. In the case of the Zr-MOF, re-usability and full recovery of the catalytic activity upon regeneration is possible (Fig. 5), with crystallinity and porosity being intact after 4 catalytic-regeneration cycles, demonstrating the much higher chemical stability of the new MOF. Possible dissolution of the framework could be discarded by 13C, 1H NMR and infrared analysis of the reaction solution: the presence of a sulfonic linker in solution could not be detected using any of the above-mentioned techniques.
Conclusions
In this work the inclusion of free sulfonic groups is explored in, a priori, highly stable MOFs. When applied in a model esterification reaction, deactivation of the sulfonic groups via formation of butanol sulfonic esters has been observed. In the case of MIL-101(Cr), due to the strong interaction between –SO3− and the Cr open metal site where usually fluorine (F−) is located in the structure, the catalysts are not stable under acidic regeneration conditions. When using Zr as a metal node, a new and stable sulfonic acid containing porous structure was synthesized. This structure showed high activity and full reusability in the esterification reaction of n-butanol with acetic acid. In this case, deactivation of the catalyst due to the formation of sulfonic esters could be tackled by reactivation under acidic conditions.Notes and references
- M. A. Harmer and Q. Sun, Appl. Catal., A, 2001, 221, 45–62 CrossRef CAS.
- T. A. Peters, N. E. Benes, A. Holmen and J. T. F. Keurentjes, Appl. Catal., A, 2006, 297, 182–188 CrossRef CAS.
- J. M. Fraile, E. García-Bordejé and L. Roldán, J. Catal., 2012, 289, 73–79 CrossRef CAS.
- L. Peng, A. Philippaerts, X. Ke, J. Van Noyen, F. De Clippel, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, Catal. Today, 2010, 150, 140–146 CrossRef CAS.
- J. H. Clark, V. Budarin, T. Dugmore, R. Luque, D. J. Macquarrie and V. Strelko, Catal. Commun., 2008, 9, 1709–1714 CrossRef CAS.
- G. Morales, R. van Grieken, A. Martín and F. Martínez, Chem. Eng. J., 2010, 161, 388–396 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- A. Corma, H. García and F. X. L. I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- H. Khajavi, J. Gascon, J. M. Schins, L. D. A. Siebbeles and F. Kapteijn, J. Phys. Chem. C, 2011, 115, 12487–12493 CAS.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS.
- I. Luz, F. X. L. I. Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS.
- I. Luz, F. X. L. I. Xamena and A. Corma, J. Catal., 2010, 276, 134–140 CrossRef CAS.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- C. Volkringer and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 4644–4648 CrossRef CAS.
- F. Vermoortele, R. Ameloot, L. Alaerts, R. Matthessen, B. Carlier, E. V. R. Fernandez, J. Gascon, F. Kapteijn and D. E. De Vos, J. Mater. Chem., 2012, 22, 10313–10321 RSC.
- M. G. Goesten, J. Juan-Alcañiz, E. V. Ramos-Fernandez, K. B. S. S. Gupta, E. Stavitski, H. Van Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177–187 CrossRef CAS.
- D. Britt, C. Lee, F. J. Uribe-Romo, H. Furukawa and O. M. Yaghi, Inorg. Chem., 2010, 49, 6387–6389 CrossRef CAS.
- J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS.
- G. Akiyama, R. Matsuda, H. Sato, A. Hori, M. Takata and S. Kitagawa, Microporous Mesoporous Mater., 2012, 157, 89–93 CrossRef CAS.
- M. L. Foo, S. Horike, T. Fukushima, Y. Hijikata, Y. Kubota, M. Takata and S. Kitagawa, Dalton Trans., 2012, 41, 13791–13794 RSC.
- S. Biswas, J. Zhang, Z. Li, Y.-Y. Liu, M. Grzywa, L. Sun, D. Volkmer and P. Van Der Voort, Dalton Trans., 2013, 42, 4730–4737 RSC.
- Y. Zang, J. Shi, F. Zhang, Y. Zhong and W. Zhu, Catal. Sci. Technol, 2013 10.1039/C3CY00044C.
- C. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef.
- P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, Chem. Mater., 2011, 23, 2565–2572 CrossRef CAS.
- P. Atorngitjawat, R. J. Klein and J. Runt, Macromolecules, 2006, 39, 1815–1820 CrossRef CAS.
- A. S. Singh, B. M. Bhanage and J. M. Nagarkar, Green Chem. Lett. Rev., 2011, 5, 27–32 Search PubMed.
- I. Díaz, F. Mohino, J. Pérez-Pariente and E. Sastre, Appl. Catal., A, 2001, 205, 19–30 CrossRef.
- E. Cano-Serrano, G. Blanco-Brieva, J. M. Campos-Martin and J. L. G. Fierro, Langmuir, 2003, 19, 7621–7627 CrossRef CAS.
- W. Wang, X. Zhuang, Q. Zhao and Y. Wan, J. Mater. Chem., 2012, 22, 15874–15886 RSC.
- B. Brunetti, E. De Giglio, D. Cafagna and E. Desimoni, Surf. Interface Anal., 2012, 44, 491–496 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, M. Y. Lee, S. G. Jang, J. A. Dumesic and S. L. Scott, ACS Catal., 2011, 1, 719–728 CrossRef CAS.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97–A101 CrossRef CAS.
- I. Minami, T. Inada, R. Sasaki and H. Nanao, Tribol. Lett., 2010, 40, 225–235 CrossRef CAS.
- T. Loiseau and G. Férey, J. Fluorine Chem., 2007, 128, 413–422 CrossRef CAS.
- D. Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J. S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- A. Vimont, J. M. Goupil, J. C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS.
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC.
- Q. Yang, A. D. Wiersum, P. L. Llewellyn, V. Guillerm, C. Serre and G. Maurin, Chem. Commun., 2011, 47, 9603–9605 RSC.
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS.
- V. Bon, V. Senkovskyy, I. Senkovska and S. Kaskel, Chem. Commun., 2012, 48, 8407–8409 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00272a |
| This journal is © The Royal Society of Chemistry 2013 |