 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A silver-free system for the direct C–H auration of arenes and heteroarenes from gold chloride complexes†
Nanna Ahlsten,
Gregory J. P. Perry,
Xacobe C. Cambeiro,
Tanya C. Boorman and
Igor Larrosa*
School of Biological and Chemical Sciences, Queen Mary University of London, Joseph Priestley Building, Mile End Road, E1 4NS, London, UK. E-mail: i.larrosa@qmul.ac.uk; Web: http://webspace.qmul.ac.uk/ilarrosa/index.html
First published on 14th June 2013
Abstract
A new methodology for the direct C–H auration of electron-deficient arenes and heteroarenes with simple bases and readily available [Au(PR3)Cl] complexes is described. This system allows the preparation of a wide scope of aryl–Au(I) compounds without the need for using Ag(I) additives or preparing and isolating basic Au(I) hydroxide complexes.
Introduction
Direct functionalisation of arenes by metal-catalysed C–H activation is among the most active topics in organic synthesis. Although dominated by Pd, other metals such as Cu, Rh, Ru, Ir or Co have been shown in recent years to mediate this type of reaction.1 Au(III) salts have also been used in C–H activation of electron-rich arenes under mild conditions.2,3 In contrast to Au(III), Au(I)-promoted C–H activation to afford (hetero)aryl–Au(I) compounds has been demonstrated only recently and is specific for electron-poor arenes.4Studies on the chemistry of (hetero)aryl–Au(I) compounds (Scheme 1) have shown them to be suitable organometallic partners for Pd- or Ni-catalysed cross-coupling reactions,5 although systems catalytic in Au are scarce.6 These species have also been shown to react with appropriate electrophiles such as CO2,7 electrophilic halogen sources8 or strong acids.6a,8b Finally, oxidation with I(III) reagents has allowed their use in the direct C–H arylation of electron-rich arenes exploiting the orthogonal selectivity of both Au oxidation states towards C–H activation, which opens a path for the development of Au(I/III)-catalysed double C–H activation cross-coupling reactions.9
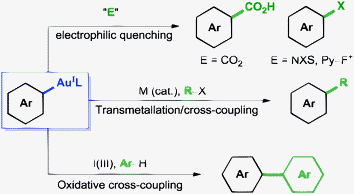 | ||
| Scheme 1 Transformations of aryl–Au(I) compounds. | ||
Aryl–Au(I) compounds are remarkably stable towards common decomposition pathways that affect related organometallic compounds.10,11 Thus, in addition to being air and moisture-stable, they do not participate in oxidative addition reactions,12 undergo protodeauration only in the presence of relatively strong acids6a,8b,13 and, although one-electron reduction to Au(0) is possible with mild reductants such as Pd(0), this reaction is sufficiently slow to be outcompeted by other processes.6,14
This combination of rich reactivity and stability towards undesired decomposition pathways makes aryl–Au(I) compounds extremely interesting intermediates in the design of metal-catalysed transformations.
Two C–H auration systems have been reported to date by Nolan's group4b,c (eqn (1)) and our group4a (eqn (2)) based on the use of a [Au(IPr)(OH)] or a [Au(PR3)Cl]–Ag2O–K2CO3–PivOH reagent system, respectively.
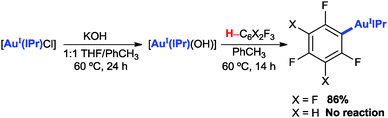 | (1) |
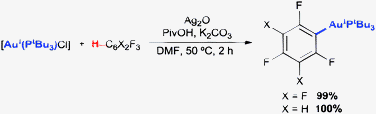 | (2) |
We considered that the development of a system using a readily available [Au(PR3)Cl] starting complex and a simple base, without other additives, would not only be synthetically useful but also help us determine which are the real differences between the reactivity of phosphine- and NHC-ligated gold complexes towards C–H activation of electron-deficient arenes.
Results and discussion
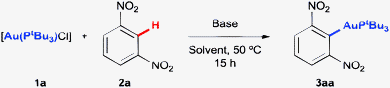
We took as a starting point for this investigation our previously reported conditions for the direct auration of electron-deficient arenes ([Au(PtBu3)Cl], PivOH, Ag2O and K2CO3, eqn (2)) and studied the possibility of performing the reaction with only the base, in the absence of PivOH or Ag+ salts. Unsurprisingly, K2CO3 alone was not able to promote the reaction by itself when applied to [Au(PtBu3)Cl] (1a) and 1,3-dinitrobenzene (2a) in DMF (Table 1, entry 1). However, shifting to the stronger, but still simple, bases KOH (entry 2) or KOtBu (entry 3) allowed us to obtain product 3aa in 47 and 80% yields, respectively. Interestingly, use of the more soluble LiOtBu (entry 4) resulted in a decreased yield, while the less soluble NaOtBu (entry 5) gave a result comparable to that obtained with KOtBu. Consistently, NaOH (entry 6) afforded a somewhat higher yield than KOH, although still lower than the tert-butoxide salts. The use of 4 equiv. of 2a was necessary for optimal performance of the reaction, lower yields being obtained with smaller amounts of 2a (entry 7).17 Less polar solvents, shown to be effective in direct auration with [Au(IPr)(OH)], turned out to be detrimental in this case (entries 8 and 9), while dioxane (entries 10 and 11) afforded a yield very similar to that obtained in DMF. Finally, increasing the temperature to 75 °C in DMF with NaOtBu (entry 12) allowed us to obtain the aurated product in 82% yield after purification (90% NMR yield).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yield (%) |
| a Reactions carried out on the 0.03 mmol scale with 1a as the limiting reagent, 4 equiv. of 2a, 4 equiv. of base and 0.15 mL of solvent (0.2 M), at 50 °C for 18 h. Yields determined using 1H NMR with an internal standard.b Reaction performed with 2 equiv. of 2a.c Reaction at the 0.1 mmol scale, at 75 °C for 5 h.d Isolated yield. | |||
| 1 | K2CO3 | DMF | 0 |
| 2 | KOH | DMF | 47 |
| 3 | KOtBu | DMF | 80 |
| 4 | LiOtBu | DMF | 57 |
| 5 | NaOtBu | DMF | 81 |
| 6 | NaOH | DMF | 64 |
| 7b | NaOtBu | DMF | 31 |
| 8 | NaOtBu | THF | 45 |
| 9 | NaOtBu | PhCH3 | 30 |
| 10 | NaOtBu | 1,4-Dioxane | 80 |
| 11 | NaOH | 1,4-Dioxane | 74 |
| 12c | NaOtBu | DMF | 90 (82)d |
With these optimised conditions, we set out to explore the scope of the reaction, aiming to determine the reactivity of this system towards electron-poor arenes (Scheme 2). A variety of polyfluorinated benzenes 2b–e, as well as 1-fluoro-3-nitrobenzene 2f, reacted smoothly to afford the corresponding aryl–Au(I) compounds 3ab–3af in excellent yields with only slight variations in the general procedure in some cases (use of NaOH as the base in 1,4-dioxane). Remarkably, even 2d reacted at 50 °C without any additive. This contrasts with previous observations where [Au(IPr)(OH)] was not able to activate this substrate under the standard conditions (eqn (1)), requiring the use of a silver salt as an additive.16 A higher basicity of the active phosphine–Au(I) complex compared to that of [Au(IPr)(OH)] could explain this difference (vide infra). Benzene derivatives bearing two F atoms and another halogen such as Br or I (2g-h) reacted preferentially with the C–H bond ortho to the two F atoms, with virtually complete regioselectivity at nearly room temperature. These substrates are particularly interesting since, after functionalisation of the C–Au bond, they would give place to products bearing a useful functional group for further reactions via other metal-catalysed coupling processes.
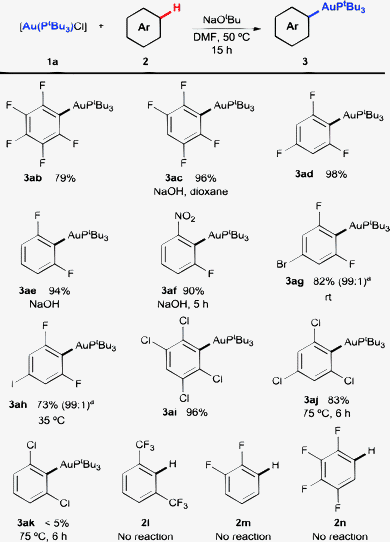 | ||
Scheme 2 Scope of the direct C–H auration of arenes. Reactions carried out between the 0.06 and 0.1 mmol scale with 1a as the limiting reagent, 4 equiv. of 2 and 4 equiv. of NaOtBu in DMF (0.2 M) at 50 °C for 15 h, unless stated otherwise. Yields are of the pure isolated product. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Regioisomer ratios determined using 1H NMR of the crude mixture. Regioisomer ratios determined using 1H NMR of the crude mixture. | ||
Polychlorinated benzenes 2i–k were also reactive, although in this case at least three chlorine atoms were necessary for good yields. 1,3-Di(trifluoromethyl)benzene 2l failed to afford any aurated product, as well as benzenes without a C–H bond ortho to two electron-withdrawing groups (2m-n).
A wide scope was also observed for electron-deficient heteroarenes (Scheme 3), with a clear dependence of reactivity on the acidity of the C–H bond to be activated. A brief re-examination of the reaction conditions showed that NaOH in 1,4-dioxane consistently afforded higher yields in the case of heteroarenes. Under these conditions, oxazole (4a), thiazole (4b), benzoxazole (4c) and benzothiazole (4d) all reacted to afford the corresponding auration products 5aa–5ad in good yields. In the case of thiazole, displaying two roughly equally acidic protons (pKa = 29.5 for the C2 position and 29.6 for C4),15 selective C2-auration (>99:1) to give 5ab could be achieved at 35 °C. Additionally, blocking the C2 position with a Br substituent allowed preparation of the C4-aurated product 5ae, keeping the Br intact and available for further functionalisation. The less acidic thiophene (4f, pKa = 32.5)15 was found not to be active in the reaction, thus determining an upper limit for the activity of our system. However, the presence of just one halogen substituent lowered the pKa sufficiently for the corresponding aurated products 5ag–5ai to be obtained in excellent yields and regioselectivities, with auration occurring selectively α to the sulfur. Similarly, just one strong electron-withdrawing group allowed C2-auration of indole-type substrates (pKa of N-methylindole = 37.3)15 to obtain 5aj, and two electron-withdrawing groups allowed preparation of pyridyl–Au(I) compounds 5ak–5al. Finally, a stable, electron-deficient imidazole-type substrate such as caffeine (4m) could be used, affording product 5am in good yield and using only 1.1 equiv. of the parent heteroarene.
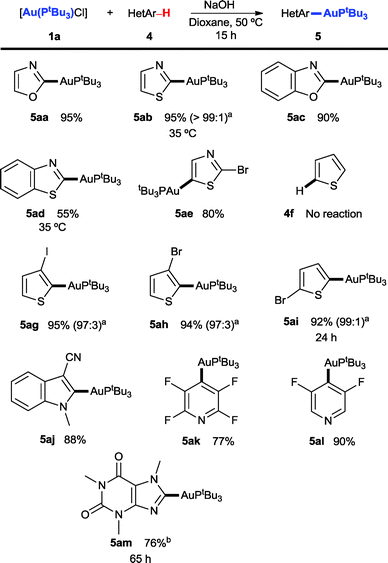 | ||
| Scheme 3 Scope of the direct C–H auration of heteroarenes. Reactions carried out between the 0.06 and 0.1 mmol scale with 1a as the limiting reagent, 4 equiv. of the corresponding heteroarene 4 and 4 equiv. of NaOH in 1,4-dioxane (0.2 M) at 50 °C for 15 h, unless stated otherwise. Yields are of the pure isolated product. aRegioisomer ratios determined using 1H NMR of the crude product. bReaction carried out with 1.1 equiv. of caffeine. | ||
Phosphine ligands other than PtBu3 were also explored (Scheme 4). For the highly electron-deficient tetrachlorobenzene, both alkyl and aryl phosphines (3ai–3ci) afforded high yields, and only with the very bulky XPhos (3di) was the yield significantly reduced. Reaction of 1,3,5-trichloro- and 1,3-difluorobenzene with [Au(PEt3)Cl] also led to excellent yields, comparable to those obtained with the PtBu3 ligand (compare 3bj, 3be and 3bd in Scheme 4 with 3aj, 3ae and 3ad in Scheme 2). More interestingly, in some cases where [Au(PtBu3)Cl] had offered limited or no success, the use of a PEt3 ligand allowed efficient preparation of the corresponding aryl–Au(I) compounds. This allowed efficient auration of the previously unreactive 1,3-dichlorobenzene (3bk), and even satisfactory reaction with 1,3-dibromobenzene (3bo).
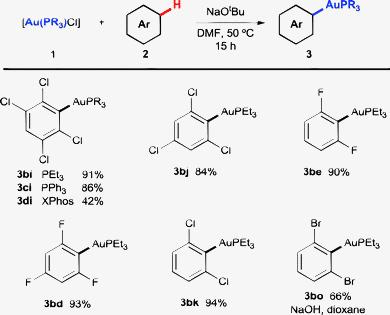 | ||
| Scheme 4 Application of different phosphines. Reactions carried out between the 0.06 and 0.1 mmol scale with 1a as the limiting reagent, 4 equiv. of 2 and 4 equiv. of NaOtBu in DMF (0.2 M) at 50 °C for 15 h, unless stated otherwise. Yields are of the pure isolated product. | ||
In order to determine the origin of this system's ability to perform the auration of less acidic substrates, such as 1,3,5-trifluorobenzene, in contrast to the [Au(IPr)(OH)] complex, we tested the auration in our conditions with [Au(IPr)Cl] (6, Scheme 5). Treatment of 6 with 2b and NaOH or NaOtBu cleanly provided the aryl–Au(I) complex 7b in 95 and 93% yields, respectively, showing that the auration process with NHC ligands does not require isolation of the gold hydroxide complex.18 Reaction with 2d under the same conditions, however, did not afford the corresponding aryl–Au(I) compound. This shows that, at least under these reaction conditions, phosphine-ligated Au(I) complexes are intrinsically more active in the auration reaction than the corresponding NHC-ligated ones.19
![Direct auration with [Au(IPr)Cl].](/image/article/2013/CY/c3cy00240c/c3cy00240c-s5.gif) | ||
| Scheme 5 Direct auration with [Au(IPr)Cl]. | ||
The exact nature of the species effecting the C–H activation proved elusive. Titration of 1a with NaOtBu or NaOH, monitored by in situ 31P NMR, showed complete disappearance of the signal corresponding to the starting material (90.7 ppm), giving place to a new peak at 83.2 ppm with 1.0 equiv. of NaOtBu or at 83.5 ppm with 1.5 equiv. of NaOH. Further addition of base did not give place to any other observable product. The product of reaction between 1a and NaOtBu, unfortunately, resisted all attempts to isolate it, but it has been reported previously that treatment of [Au(PR3)Cl] complexes with NaOtBu in THF gives place to the corresponding [Au(PtBu3)OtBu] complexes20 (Scheme 6). Higher order complexes of the type {[Au(PR3)]2OR}+ have also been described,21 but their preparation usually involves the use of a “cationic” gold complex (such as [Au(PR3)(BF4)]), which makes it unlikely in our case. Thus, we hypothesise that the product of 1a + NaOtBu could be 8. Regarding the reaction between 1a and NaOH, the NMR signals of the product matched those previously described for {[Au(PtBu3)]3O}BF4 (9′).22 Addition of NaBF4 into the mixture of 1a + NaOH allowed isolation of 9′ as a white solid in 92% yield, leading us to conclude that the product obtained is {[Au(PtBu3)]3O}Cl (9).
![Reactions of [Au(PtBu3)Cl] (1a) with NaOtBu and NaOH.](/image/article/2013/CY/c3cy00240c/c3cy00240c-s6.gif) | ||
| Scheme 6 Reactions of [Au(PtBu3)Cl] (1a) with NaOtBu and NaOH. | ||
However, the reaction of 1a, 2a and either 1.0 equiv. of NaOtBu or 1.5 equiv. of NaOH afforded the auration product 3aa in only 45 and 15% yield, respectively. These results seem to indicate different behaviour of compounds 8 and 9: in the case of 8 the yield obtained was moderate, still lower than that obtained under the standard conditions but indicative of some activity. On the other hand, 9 was less reactive and required the presence of a large excess of base (only 15% yield was obtained, despite the presence of 1.5 equiv. of base, more than theoretically needed for formation of 9).
Trinuclear μ3-oxo Au(I) species with biphenyl(dialkyl)phosphines (12, Scheme 7) have recently been shown to be related to 10, 11 and 13 by an equilibrium which depends on the stoichiometry with respect to the base, as well as on the steric properties of the phosphine ligand.23 The analogous compound 1d (PR3 = XPhos) was effective in the auration process under the conditions reported here (product 3di in Scheme 4), leading us to hypothesise that a qualitatively similar situation could take place also with the other phosphines. Thus, in situ formation of a Au(I) hydroxide species, even as a minor component in the equilibrium mixture, can be hypothesised. Alternatively, direct deprotonation of the arene by the base followed by attack of the resulting aryl anion on an electrophilic gold(I) complex cannot be discarded either.24
![Previously reported products obtained from basic treatment of [Au(L)(NCMe)]BF4 complex 10. L = XPhos or JohnPhos.23](/image/article/2013/CY/c3cy00240c/c3cy00240c-s7.gif) | ||
| Scheme 7 Previously reported products obtained from basic treatment of [Au(L)(NCMe)]BF4 complex 10. L = XPhos or JohnPhos.23 | ||
Conclusions
In summary, we have developed a simplified methodology for efficiently performing direct C–H auration of electron-deficient arenes and heteroarenes, combining readily available [Au(PR3)Cl] complexes and simple bases. This system shows excellent activity for a wide range of substrates without requiring the use of silver salts as additives or the preparation and isolation of Au(I) hydroxide complexes.Acknowledgements
We gratefully acknowledge the Engineering and Physical Sciences Research Council for generous funding, the European Research Council for a Starting Research Grant (to I.L.), the Marie Curie Foundation for an Intra-European Fellowship (to X.C.C.) and the EPSRC National Mass Spectrometry Service (Swansea).Notes and references
- For recent reviews on C–H activation, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS; (d) Modern Arylation Methods, ed. L. Ackermann, 1st edn, Wiley-VCH, Weinheim, 2009 Search PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (g) C–H Activationin Topics in Current Chemistry, ed. J.-Q. Yu and Z. Shi, Springer, Heidelberg, 1st edn, 2010, vol. 292 Search PubMed; (h) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- (a) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053–3059 CrossRef CAS; (b) M. S. Kharasch and T. M. Beck, J. Am. Chem. Soc., 1934, 56, 2057–2060 CrossRef CAS; (c) K. S. Liddle and C. Parkin, J. Chem. Soc., Chem. Commun., 1972, 26a RSC; (d) F. Calderazzo and D. B. Dell'Amico, J. Organomet. Chem., 1974, 76, C59–C60 CrossRef CAS; (e) P. W. J. de Graaf, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1976, 105, 399–406 CrossRef CAS.
- For reviews on Au-catalysed C–H activation: (a) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (b) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS; (c) T. De Haro and C. Nevado, Synthesis, 2011, 2530–2539 CAS; (d) R. Skouta and C.-J. Li, Tetrahedron, 2008, 64, 4917–4938 CrossRef CAS.
- (a) P. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580–5581 CrossRef CAS; (b) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC; (c) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS.
- Metal-catalysed coupling reactions with stoichiometric organo-Au(I) complexes: (a) Y. Shi, S. D. Ramgren and S. A. Blum, Organometallics, 2009, 28, 1275–1277 CrossRef CAS; (b) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243–8246 CrossRef CAS; (c) A. S. K. Hashmi, R. Döpp, C. Lothschütz, M. Rudolph, D. Riedel and F. Rominger, Adv. Synth. Catal., 2010, 352, 1307–1314 CrossRef CAS; (d) M. Peña-López, M. Ayán-Varela, L. A. Sarandeses and J. Pérez-Sestelo, Chem.–Eur. J., 2010, 16, 9905–9909 CrossRef; (e) J. J. Hirner, Y. Shi and S. A. Blum, Acc. Chem. Res., 2011, 44, 603–613 CrossRef CAS; (f) J. J. Hirner and S. A. Blum, Organometallics, 2011, 30, 1299–1302 CrossRef CAS.
- For a Pd/Au dual-catalysed Stille coupling proceeding through an aryl–Au(I) intermediate, see: (a) J. Delpozo, D. Carrasco, M. H. Pérez-Temprano, M. García-Melchor, R. Alvarez, J. A. Casares and P. Espinet, Angew. Chem., Int. Ed., 2013, 52, 2189–2193 CrossRef CAS . For other dual Au/TM catalytic systems, see: ; (b) L. A. Jones, S. Sanz and M. Laguna, Catal. Today, 2007, 122, 403–406 CrossRef CAS; (c) Y. Shi, S. M. Peterson, W. W. Haberaecker and S. A. Blum, J. Am. Chem. Soc., 2008, 130, 2168–2169 CrossRef CAS; (d) Y. Shi, K. E. Roth, S. D. Ramgren and S. A. Blum, J. Am. Chem. Soc., 2009, 131, 18022–18023 CrossRef CAS; (e) B. Panda and T. K. Sarkar, Tetrahedron Lett., 2010, 51, 301–305 CrossRef CAS; (f) J. J. Hirner, K. E. Roth, Y. Shi and S. A. Blum, Organometallics, 2012, 31, 6843–6850 CrossRef CAS; (g) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. De Buck Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133–147 CrossRef CAS; (h) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS.
- I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858–8859 CrossRef CAS.
- (a) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592–597 CrossRef CAS; (b) J. Cornella, M. Rosillo-Lopez and I. Larrosa, Adv. Synth. Catal., 2011, 353, 1359–1366 CrossRef CAS.
- X. C. Cambeiro, T. C. Boorman, P. Lu and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 1781–1784 CrossRef CAS.
- Some of the aryl–Au(I) compounds described herein have been stored under bench conditions for up to two years without any observable decomposition.
- K. E. Roth and S. A. Blum, Organometallics, 2010, 29, 1712–1716 CrossRef CAS.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS.
- For other preparation processes and properties of aryl–Au(I) compounds, see: (a) J. M. Forward, J. P. Fackler and R. J. Staples, Organometallics, 1995, 14, 4194–4198 CrossRef CAS; (b) A. Sladek, S. Hofreiter, M. Paul and H. Schmidbaur, J. Organomet. Chem., 1995, 501, 47–51 CrossRef CAS; (c) E. J. Fernández, A. Laguna and M. E. Olmos, Adv. Organomet. Chem., 2004, 52, 77–141 CrossRef; (d) H. Schmidbaur and A. Schier, in Comprehensive Organometallic Chemistry III, ed. R. Crabtree and M. Migos, Elsevier, New York, 2006, vol. 2, Section 2.05 Search PubMed; (e) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188–8191 CrossRef CAS.
- D. Weber and M. R. Gagné, Chem. Commun., 2011, 47, 5172–5174 RSC.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568–1576 CrossRef CAS.
- Nolan and coworkers have reported that the addition of Ag(I) salts to their system allows the auration of 1,3,5-trifluorobenzene: S. R. Patrick, I. I. F. Boogaerts, S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2011, 7, 892–896 CrossRef CAS.
- This is likely due to side reactions between the arene and the base competing with the auration process.
- Similar behaviour has been recently reported in solid-phase reactions with IPrAuCl. J. D. Egbert, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 2271–2274 CrossRef CAS.
- The exact nature of the active Au species is, however, not clear. In the case of NHC-ligated complexes with NaOH as the base, the similarity between our reaction conditions and the ones employed by Nolan suggests that [Au(IPr)(OH)] could be formed, while when NaOtBu was used as the base, [Au(IPr)(OtBu)] or [Au(IPr)(OH)] could be present.
- (a) B. R. Sutherland, K. Folting, W. E. Streib, D. M. Ho, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1987, 109, 3489–3490 Search PubMed; (b) S. Komiya, M. Iwata, T. Sone and A. Fukuoka, J. Chem. Soc., Chem. Commun., 1992, 1109–1110 Search PubMed.
- A. Kolb, P. Bissiier and H. Schmidbaur, Inorg. Chem., 1993, 32, 5132–5135 Search PubMed.
- (a) H. Schmidbaur, A. Kolb, E. Zeller, A. Schier and H. Beruda, Z. Anorg. Allg. Chem., 1993, 619, 1575–1579 Search PubMed; (b) K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1994, 474, 1–21 Search PubMed.
- A. Zhdanko, M. Ströbele and M. E. Maier, Chem.–Eur. J., 2012, 18, 14732–14744 Search PubMed.
- Both hypotheses would explain the need for excess base, due to its influence on the equilibrium between 12 and 13 in one case, or between arene and aryl anions in the other.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data and spectra of new compounds. See DOI: 10.1039/c3cy00240c |
| This journal is © The Royal Society of Chemistry 2013 |