New trends in the synthesis of crystalline microporous materials
Giuseppe Bellussi, Angela Carati, Caterina Rizzo and Roberto Millini*
eni s.p.a., Refining & Marketing Division, San Donato Milanese Research Center, via F. Maritano 26, I-20097 San Donato Milanese, Italy. E-mail: roberto.millini@eni.com; Fax: +39 02 52036347; Tel: +39 02 52056543
First published on 7th November 2012
Abstract
Zeolites and related microporous materials form the most important class of microporous solids due to their practical importance in different technological areas. In spite of the extensive research carried out in previous decades, it is surprising to realize that there is still space for innovation in this area, making microporous crystalline solids special among all classes of materials known today. We illustrate here the most recent advances in the field, focusing on the research topics that, in our opinion, are most likely to provide results of practical interest. Three main topics are discussed: (1) the synthesis of new framework topologies, with particular attention to those having extra-large and/or multidimensional pore systems; (2) the modification of the morphological and textural properties of known zeolites, including the discussion on two-dimensional structures and on the synthesis of nanocrystals and of the hierarchical porous structures; (3) the still poorly explored field of silica-based hybrid organic–inorganic porous crystalline materials (hybrid zeolites). For each of these topics, a selection of the most relevant results reported in the literature is provided together with some considerations on the potentialities and future perspectives.
 Giuseppe Bellussi | Giuseppe Bellussi is Senior Vice President R&D in the Refining & Marketing Division of eni company. He holds a University degree in Chemistry cum-laude from University of Parma-I. In 1981 he moved from Montedison to eni and since then he has been involved in research activities related to process technologies. He is the co-author of 90 patents and 100 publications. For his activities he has been recognized with several awards among which the 1992 D. Breck Award from IZA, the 2003 Johnson-Matthey EFCATS International Award, the 2007 IZA Award from IZA, the 2008 P. Pino Gold medal from Italian Chemical Society and the 2013 Houdry Award from NACS. Since 2010 he has been the President of International Zeolite Association. |
 Angela Carati | Angela Carati received the University degree in Industrial Chemistry in 1985 from the University of Bologna. In 1986 she joined the research centre of eni s.p.a. in San Donato Milanese. She has been engaged in research activities concerning synthesis and characterization of heterogeneous catalysts, with particular attention to the field of nanoporous ordered materials. She has skills in the development of synthetic catalysts from the laboratory to the pilot scale. Actually she is responsible for the activity related to material development. She is a co-author of more than 100 papers and conference contributions and 36 patents. |
 Caterina Rizzo | Caterina Rizzo received the University degree in Chemistry in 1986 from the University of Calabria. After an Education Grant at the University of Trento, she joined the research centre of eni s.p.a. in Monterotondo (Rome) where she has been engaged in research on advanced ceramic materials. In 1996 she moved to the research centre in San Donato Milanese where she is presently Senior Project Manager of the Downstream Process Technology Department. Her main research interests are focused on synthesis, textural and adsorption properties of several porous solids. She is a co-author of 90 papers and conference contributions and 19 patents. |
 Roberto Millini | Roberto Millini received the University degree in Chemistry cum-laude from the University of Pavia in 1983. After gaining experience in important research institutions in Germany (Max Planck Institut für Strahlenchemie – Mulheim/Ruhr, 1983–1984, and Institut für Anorganische Chemie – Erlangen/Nürnberg Universität, 1984–1985), in 1986 he joined the corporate research centre of eni s.p.a. in San Donato Milanese, where he is currently Manager of the Physical Chemistry Department. His research interests focus on the structural, morphological and spectroscopic characterization of heterogeneous catalysts, in particular zeolites and related porous solids. He has been president of the Italian Zeolite Association and is a member of the Synthesis Commission of the International Zeolite Association. He holds 18 patents and has authored more than 100 scientific papers. |
Introduction
The availability of new materials with novel advanced properties is the basis of the progress in all fields of industry and technology. In this regard, porous materials, in particular the microporous ones that exhibit permanent and accessible pores with dimensions below 20 Å, have attracted the attention of chemists and material scientists owing to the commercial interest in their applications. Speaking about crystalline porous solids, the thought turns immediately to zeolites. They, in fact, constitute one of the most extensively studied classes of materials and are applied in the industry as catalysts, molecular sieves and ion-exchangers.Known since 1756, when the Swedish mineralogist and chemist A. F. Cronstedt coined the term “zeolite”,1 for two centuries, these materials represented a subject of study for mineralogists only. The modern era of zeolite science began in the late 1940's, when Barrer and Milton carried out their pioneering work on the synthesis of zeolites, while the first industrial applications of synthetic zeolites (Ca-exchanged zeolite A for the separation of n- and i-paraffins, zeolite Y as a component of the catalyst for the catalytic cracking of hydrocarbons) followed just 10 years later.2,3 A milestone in the history of zeolites was the introduction of organic molecules in the synthesis, which enabled the disengaging from the limitations imposed by the synthesis carried out in the fully inorganic system (low Si/Al ratios with detrimental consequences on the stability and acid properties of the materials), favouring at the same time the discovery of new crystalline porous structures.4 Since then, there has been literally an explosion of studies dedicated to the preparation of new zeolite structures with different pore architectures through the use of organic molecules (structure directing agents, SDAs) of increasing complexity and of advanced synthesis procedures. The actual portfolio of crystalline microporous structures consists of 201 framework types and 22 disordered zeolite frameworks (i.e. intergrowth of two or more different but structurally related frameworks) officially recognized by the Structure Commission of the International Zeolite Association,5 and several other microporous phases with still unknown (or not yet officially recognized) structures. Moreover, if one considers that each framework-type has several compositional variants, not only in terms of the Si/Al ratio but also of the chemical nature of the elements constituting the framework itself, a huge number of microporous materials are available today.
This area seems to have reached such a level of maturity to make new breakthroughs unlikely. As the history of materials science of the past twenty years demonstrates, however, referring to zeolites as the only class of crystalline microporous solids is not correct. In fact, several other classes of materials, having in common with zeolites just the crystalline porous structure but far away from them in terms of chemical composition, coordination of the metals etc., have been discovered. We are referring, for instance, to the octahedral-pentahedral-tetrahedral framework metallosilicates (OPT),6 the oxide molecular sieves (e.g. manganese octahedral molecular sieves, OMS),7 and most importantly, constituting by themselves a specific research area, the metal organic frameworks (MOFs).8
In spite of the variety of different classes of materials available today, the scientific community is wondering itself how to meet the growing demand for new crystalline porous materials.
Examining the present and the future synthesis challenges for zeolites, J. Coronas has recently identified the morphology control (crystal size and growth habit), the synthesis of hierarchical porous systems, of extra-large pore structures, of chiral zeolites and membranes and the preparation and modification of layered zeolites as the most important fields of advancement.9 R. E. Morris, on the other hand, has expressed his own opinion on what are the most difficult challenges in the synthesis of nanoporous (a term that is better than the classical “microporous” reflecting the characteristics of the zeolites and related phases) materials, consisting in the subtle control of the structure and morphology of zeolites and MOFs.10 With regard to the catalytic applications of zeolites, J. Čejka et al. have recently identified five areas on which attention should be focused: synthesis of new zeolite structures, chemical modification of existing zeolites, T-atoms siting, crystal size and morphology, defect sites in the structure.11
In general, the different authors agree on the main areas of action and on the consequent challenges in the synthesis of crystalline porous materials. While agreeing with this view, we believe that the most promising research areas on which efforts should be concentrated are those shown in Fig. 1. Therefore, we will discuss the synthesis of new zeolite structures (in particular with extra-large porous systems and/or with multidimensional pore architectures) and, in a functional way to this objective, the modification of the composition of the framework through the use of heteroatoms. This process, known as the isomorphous substitution, in the past, led to results of high interest, being aimed at modifying the catalytic properties of known materials by incorporating trivalent elements (e.g. B, Ga) for modulating the acidic strength of the zeolite or tetravalent elements (e.g. Ti) for giving red-ox catalytic properties to the microporous material. Currently, the use of elements other than Si and Al is conversely considered as a means to stabilize particular secondary building units (SBUs) and, therefore, to synthesize new framework topologies, otherwise difficult to obtain using the classic Si–Al system. Special attention will also be devoted to 2D zeolites, namely to microporous materials which are (or can be) synthesized in the form of layered precursors. This topic is interesting because it provides the possibility of designing and synthesizing new materials with different porous characteristics.
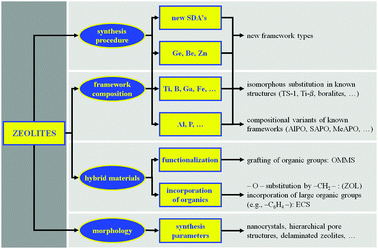 | ||
| Fig. 1 Main areas of innovation in zeolite science. | ||
An important topic of research is aimed at changing the morphological and textural properties of zeolites. The development of synthesis procedures able to produce solids in the form of nanocrystals or to generate a second level of porosity in the crystals or in the aggregates of nanocrystals (hierarchical porous systems) is expected to give a response to the important problem related to the diffusion limitations that often affect the efficiency and the overall life of the zeolite catalysts.
Finally, we will treat an area potentially of high interest, related to the synthesis of crystalline silica-based hybrid organic–inorganic porous materials (Fig. 1). In fact, although there is a strong interest in the field of mesostructured hybrid organic–inorganic materials (in particular, periodic mesoporous organosilicas, PMOs),12,13 the same cannot be said for crystalline microporous hybrid materials, which are still relatively unexplored. Starting from recent results obtained in the eni's laboratories related to the synthesis of Eni Carbon Silicates (ECSs), we will draw an overall picture of this subject still little explored, but certainly full of perspectives.
Before getting to the heart of the arguments, it is necessary to point out that some of the materials discussed in this review strictly speaking do not fall within the definition of zeolites. We refer, in particular, to some phases derived from the precursors of layered zeolites (2D zeolites) and to the ECSs, in which the inorganic (zeolitic) component contributes only partly to define the overall properties of the material. However, we have considered it useful to include them in this review because of the close relationship they have with the classical zeolites, well aware that they do not fall within any of the classes of materials known so far.
Synthesis of new zeolites
The current activity in the synthesis of zeolites has as objectives the increase of the complexity of the pore architecture (from 1D to 3D porous systems) and the preparation of large and extra-large pore structures. To reach these objectives it is necessary to know in detail the phenomena occurring during the complex hydrothermal process that leads to the crystallization of a zeolite. In this regard, it is worth stressing what Cundy and Cox reported in their excellent review on the hydrothermal synthesis of zeolites: “It is unfortunately fairly common to see in the scientific literature statements to the effect that this process is still at an empirical stage, or poorly understood, or even steeped in some form of alchemical mystery. There is also the tendency to evoke special explanations for some of the phenomena observed, as if they were somewhat outside the legitimate realm of classical orthodoxy. Such implications are misleading…”.2 In fact, although we do not have yet a complete understanding of this process, we are nevertheless able to draw detailed mechanisms for the formation of zeolites. Instead, the problem is that we are still far away from having a full control of the system and, hence, from the possibility of predicting its behaviour. Historically, the strategies followed for crystallizing new structures were based on the screening of the some critical synthesis parameters (e.g. crystallization temperature and time, stirring, Si/Al ratio, H2O/SiO2, nature and concentration of the alkali metal ions, pH, etc.). In the last period, however, emphasis is given mainly on three different but closely related aspects: the organic additives (SDAs), the synthesis approaches and the heteroatoms.Most of the zeolite structures known so far were synthesized thanks to use of organic additives, whose role in the crystallization of zeolites was, for a long time, a matter of discussion. According to Davis and Lobo, the organic additives can play three different roles.14 Small organic molecules, for instance, are believed to act as void fillers, stabilizing the inorganic structure towards successive transformation into thermodynamically more stable systems (Ostwald rule of successive transformations). They can also act as “templates”, thereby expressing a dream of the material scientists, i.e. the possibility of designing the characteristics of the porous system by appropriately modifying the dimensions and shape of the organic molecule. Unfortunately, the dream has remained as such because the desired correspondence (zeolite structure ↔ organic additive) rarely occurs, the cases of the triquaternary ammonium cation (triquat) for ZSM-18 (MEI)15 and [18]-crown-[6] for hexagonal faujasite (EMT)16 being the well known exceptions. In contrast, it is commonly observed that, depending on the synthesis conditions adopted and on the composition of the reaction mixture, a given SDA may favour the crystallization of two or more different zeolite phases. The lack of specificity that normally characterizes the SDAs implies that a large number of syntheses should be run to explore the influence of the main parameters on the nature of the products. It is true that some porous systems reflect the structure of the SDA (e.g. the tetrapropylammonium cations in the intersecting 10-ring channels of ZSM-5 (MFI)17 or the complex triquaternary ammonium cation (triquat) in ZSM-1815) but in most cases this correspondence is less satisfactory and other factors should be considered for explaining the structure directing actions of the SDAs. Detailed experimental and computational studies were performed during the 1990's for defining the properties that an organic molecule should have for acting as an SDA. Examining the results of the syntheses of silica clathrasils, Gies and Marler defined the conditions that an organic molecule should fulfil for being a suitable SDA.18,19 Besides the obvious stability of the molecule during the hydrothermal treatment, the intrinsic characteristics of the molecule itself (shape, flexibility, basicity, polarizability, tendency to interact with the solvent) and its interactions with the host framework (maximization of the van der Waals contacts with low deformation from the equilibrium conformation) constitute the most important parameters. Successive studies demonstrated that most of these characteristics (i.e. hydrophobicity, size, shape, charge, flexibility) are of general validity.
Kubota et al. found that the hydrophobicity and rigidity of the SDA, evaluated in terms of phase transfer behaviour from water to chloroform and of a number of tertiary and quaternary C atoms, respectively, are important for predicting its structure-direction abiltiy in zeolite synthesis.20 By studying the effect of the C/N+ ratio of the organic additive on the crystallization of zeolites, they found that the optimum value lies between 11 and 16, which corresponds to molecules with moderate hydrophobicity. When the SDA has a strong hydrophobic character, crystallization of a zeolite is difficult, while rigid, bulky and relatively short (<10 Å for the longest axis) molecules with moderate hydrophobicity are the best candidates for acting as SDAs.20 Even the size and shape of the molecule are important because it is expected that they influence the characteristics of the porous phase. A well known example is that reported by Wagner et al., who demonstrated that the porous characteristics of the zeolite phases strongly depend on the size and shape of the SDAs (Fig. 2).21 The systematic modification of the structure of a parent organic molecule is the concept at the base of several interesting works. Some significant examples include: quaternary imidazolidinium compounds,22 substituted [5.2.1.02.6]-tricyclodecane derivatives,23 ring-substituted piperidinium and spiro-piperidinium derivatives.24–28
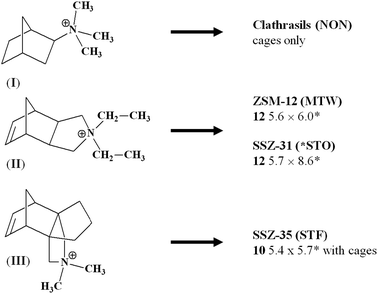 | ||
| Fig. 2 Phases obtained with three structurally related SDAs with increasing sizes and different shapes. The small norbornyl derivative (I) favours the crystallization of clathrasils only; upon expanding the length of the molecules as in the case of the tricyclic derivative (II), zeolites with monodimensional linear channel systems (MTW and *STO) are produced. When bulky molecules, such as the pseudo-propellane SDA (III) are used, zeolite SSZ-35 (STF; with 1D channel system with cages) crystallizes.21 | ||
The use of positively charged SDAs (or, at least, of neutral amines that can be found in protonated form when occluded in the zeolite pores) is important because, apart from the steric effects, they also compensate the negative framework charges. This is one of the most important concepts in zeolite science, since the partial replacement of alkali metal ions by charged organic molecules has allowed the synthesis of materials with a higher Si/Al ratio.4 This is because, for a given zeolite structure, the larger dimensions of the SDA compared to the alkali metal ion imply a lower number of charged species in the pores and, consequently, a lower incorporation of Al. SDAs are also known to favour the crystallisation of pure-silica zeolite phases. When these phases are crystallized in basic media (OH−), the charge of the SDA is compensated by the presence of siloxy groups (Si–O−, stabilized by H-bonds).29–31 Alternatively, when the syntheses are carried out in fluoride-containing media, the charge of the SDA is compensated by F− ions encapsulated in the zeolite structure and/or located in the pores. Irrespective of that, it is clear that for a given zeolite, the framework charge density is the result of a compromise between the characteristics of the SDA (size and charge) and the presence of F− or Si–O− and Al (or other trivalent metal ions). Since each SDA has a well-defined size/charge ratio, the number of SDA molecules hosted in a zeolite will depend on the framework charge density. An example is given by the systematic study on the structure-direction ability of N,N-dimethylpiperidinium (DPM), which favours the crystallization of the various zeolite phases (including the new ERS-7 (ESV) zeolite), whose framework density decreases upon decreasing the SiO2/Al2O3 molar ratio in the reaction mixture (Fig. 3).32
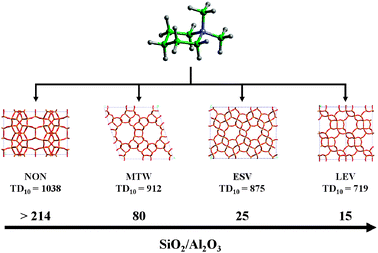 | ||
| Fig. 3 Change in phase selectivity obtained by varying the SiO2/Al2O3 molar ratio in the reaction mixture containing N,N-dimethylpiperidiunium as a SDA. The topological density TD10 of each phase is also reported (syntheses performed at 443 K with crystallization time >5 days).32 | ||
As proposed by Gies and Marler, an important condition that a SDA should fulfil is the maximization of the van der Waals contacts with the framework atoms without significant deviation from the equilibrium conformation.18,19 Taking advantages of the impressive advancements in computational science occurred from the early 1990's and, in particular, of the development methodologies for determining the location and energetics of organic molecules within porous solids,33 it was possible to confirm computationally the plausibility of this hypothesis, otherwise impossible to verify experimentally. Lewis et al. performed molecular mechanics calculations for evaluating the relative stabilization of the MFI and MEL structures when docked with one and two adjacent TPA and TBA ions, concluding that an organic molecule is able to favour the crystallization of a zeolite only when the favourable non-bonding interactions with the framework are maximized along with efficient packing of the SDA molecules within the pores.34 On the basis of these criteria, successively confirmed by several other works, the authors claimed the possibility of predicting suitable SDA molecules for a given (even hypothetical) framework. To do that, they developed the ZEBEDDE (ZEolites by Evolutionary De novo Design) code,35,36 which proved to be efficient in the selection of new SDAs, namely 4-piperidinopiperidine for DAF-5 (CHA)37 and 2-methycyclohexylamine for DAF-4 (LEV)38 (Fig. 4). This code may find wide application in the selection of SDA for the crystallization of new zeolites selected, for example, among those reported in the databases of hypothetical zeolite structures.39–44
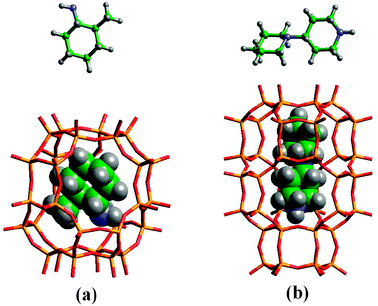 | ||
| Fig. 4 New SDA molecules designed by the ZEBEDDE code: (a) 4-piperidinopiperidine for DAF-5 (CHA); (b) 2-methycyclohexylamine for DAF-4 (LEV). | ||
Coming back to the synthesis of new zeolite structures, besides the SiO2/Al2O3 molar ratio, there are also other parameters that may determine the nature of the crystalline products. Among them, the crystallization conditions (temperature and time), the use of F− as a mineralizing agent and the concentration of the reaction mixture are often key factors for obtaining new crystalline phases. As far as the crystallization conditions are concerned, several cases demonstrate the importance of the crystallization temperature and time in zeolite synthesis. A representative example is given by the previously mentioned study on the structure directing ability of DMP, in particular for what concerns the determination of the optimal conditions for the crystallization of ERS-7, which gave the results reported in Table 1.32 The synthesis of ERS-7 demonstrates that temperature and time play a fundamental role, since this zeolite was obtained only at relatively high temperature (≥428 K) with the consumption of other zeolite phases which are formed first (ANA or MOR, Table 1). In other words, it is a clear example of Ostwald's step rule, i.e. that crystallization from a solution occurs in steps in such a way that thermodynamically unstable phases occur first, followed by the recrystallization to thermodynamically more stable phases.45
| Time (days) | Crystallization temperature | ||
|---|---|---|---|
| 403 K | 428 K | 443 K | |
| a AM = amorphous, ANA = analcime, ESV = ERS-7, MOR = mordenite. | |||
| 3 | AM | ANA | MOR |
| 5 | AM | ANA | ESV |
| 7 | AM | ANA + ESV | ESV |
| 14 | AM + ANA | ESV | — |
The use of F− as a mineralizing agent opened new perspectives in the synthesis of zeolites. In fact, compared to the conventional syntheses performed in strongly basic aqueous media containing OH− ions, the use of F− allows the crystallization of zeolites at lower pH values with some undoubted advantages. First, F− ions compensate the positive charges of the SDA ions occluded in the pores, favouring the formation of defect-free high-silica zeolites. Moreover, the neutral to slightly acidic pH values may allow the framework incorporation of heteroatoms that are insoluble at high pH values as well as the use of SDAs that are not stable in basic media. Finally, fluoride ions may also stabilize small cages (in particular the double four ring, D4R), in which F− ions are often occluded. The use of F− gives also the opportunity to perform the syntheses with concentrated gels, as firstly demonstrated by Camblor et al. who were able to crystallize the pure silica form of low-density zeolites.46,47 More specific studies confirmed that, in the presence of the same SDA, the gel concentration (expressed as the H2O/SiO2 molar ratio) can influence the nature of the crystalline products and that, in general, lower H2O/SiO2 molar ratios in the reaction mixture produce zeolites with lower framework density. Moreover, under concentrated conditions, the nucleation rate increases, facilitating the incorporation of F− ions and of SDA molecules, favouring the formation of zeolite structures with larger micropore volume.28,48–52 On the other hand, when working under basic OH− conditions, it is the extent of the trivalent element (e.g. Al) substitution that determines the nature of the products, in the sense that zeolites with lower framework density are produced by lowering the SiO2/Al2O3 molar ratio.23,28
The review of the main factors that affect the synthesis of zeolites cannot be concluded without considering the role of the heteroatoms. The attempts to incorporate elements different from Si and Al in the microporous framework of zeolites date back to the early 1980's when this approach was taken into great consideration for modifying the catalytic properties of zeolites. Known as isomorphous substitution, this approach gave very interesting results with, e.g., the incorporation of Ti in the pure silica MFI framework,53 producing the well-known TS-1 oxidation catalyst, and B in several known zeolite structures.54 Among the heteroatoms used in zeolite synthesis, B is one of the most versatile since several interesting new zeolite structures have been recently prepared in the form of borosilicates.55–60 This trivalent element can be incorporated into a large variety of zeolite frameworks because the average Si–O–B angle is smaller than the Si–O–Si and Si–O–Al ones. In contrast, the possibility that B stabilizes specific secondary building units (SBUs) is excluded, an effect attributed to other heteroatoms. For instance, zeolites containing 3-rings are preferentially formed in the presence of divalent ions. In this way, the naturally occurring Lovdarite (LOV),61 Nabesite (NAB)62 and Roggianite (–RON)63 as well as the synthetic OSB-1 (OSO)64 and OSB-2 (OBW)64 crystallize in the presence of Be2+, while VPI-7 (VSV),65 VPI-8 (VET),66 VPI-9 (VNI)67 and RUB-17 (RSN)68 with Zn2+.
Germanium, on the other side, favours the crystallization of zeolites containing the SBU D4R (double 4-ring), as demonstrated by the synthesis of a series of pure microporous germanates69–71 and successively of zeolites. Corma et al. found that the addition of small amounts of Ge (together with F− as a mineralizing agent) to the gel for the synthesis of ITQ-7 (previously obtained in pure silica form)72 reduces the crystallization time from 7 days to only 12 hours (Fig. 5).73,74 Moreover, quantum mechanical calculations indicated that the incorporation of up to three Ge atoms stabilizes the D4R unit.75 This stabilization can be better explained by comparing the geometrical features of the SBU D4R in a pure germanate structure and in its silicon-rich counterpart, an opportunity given by the germanate synthesized by Conradsson et al.70 and by ITQ-17 (with Si/Ge = 1.8),75 both possessing the BEC framework topology. As shown in Fig. 6, the Ge–O–Ge angles are on average smaller than the corresponding values measured in Si-containing D4R units; in other words, the presence of Ge reduces the strain due to the spatial arrangement of the T atoms, with all the T–T–T angles of 90°. The stabilization of the D4R units by Ge is the key parameter in the crystallization of zeolites having the UTL-type topology, as recently demonstrated by Shvets et al., who reported that germanosilicates with this topology can be synthesized in the presence of 13 different SDAs (all with the proper structure, rigidity, hydrophilicity/hydrophobicity balance and pKa), provided that the Si/Ge ratio in the reaction mixture is close to 2. Since zeolites with the UTL-type topology were not obtained from a reaction mixture with a Si/Ge ratio less than 1 and higher than 5, the authors concluded that the role of Ge prevails over that of the SDA.76
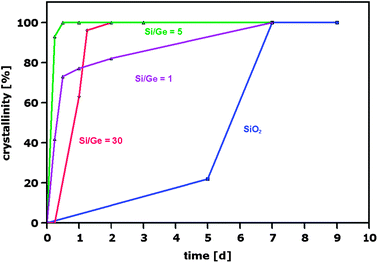 | ||
| Fig. 5 Crystallization curves of ITQ-7 with different Ge content compared to that of the pure silica parent structure.73,75 | ||
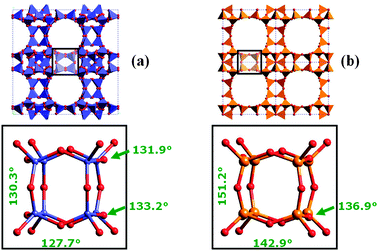 | ||
| Fig. 6 T–O–T angles in the D4R unit contained in (a) the pure germanate structure70 and (b) in ITQ-17.75 | ||
These successful results opened the possibility to synthesize several other new framework structures; the researchers, however, are well aware of the drawbacks resulting from the use of Ge: its cost and the low hydrothermal stability of the crystallized products. For these reasons, once obtained a new Ge-containing zeolite, the efforts are generally driven toward the strong reduction if not the elimination of this expensive element and toward the incorporation of catalytically active components (e.g. Al, B). In most cases, this goal was reached, leading to the preparation of new materials of high potential interest.
Improving the screening capabilities
What reported above demonstrates the complexity of the synthesis of the zeolites, which is the result of the proper combination of parameters and conditions, only apparently independent of each other. In general, a synthesis parameter does not prevail over the others in a clear manner; therefore, the careful inspection of a given system requires an extensive screening activity, consisting in a huge number of synthesis runs. This is time consuming and expensive in terms both of manpower and of raw materials: each synthesis run requires the preparation of the reaction mixture, the hydrothermal treatment, the separation, washing and drying of the solid product and its preliminary characterization. On the other hand, the raw materials may be expensive, particularly when complex SDAs and high-purity reactants are employed. An important question therefore arises: is it possible to perform rapidly the screening of a given system by saving at the same time the raw materials? To answer to this question we have to go back to the end of 1990's, when researchers from Sintef (Norway) announced the development of a new multiautoclave system properly designed for applying the combinatorial approach to the synthesis of zeolites.77,78 The combinatorial approach originated from other fields of the organic (in particular from the pharmaceutical) and inorganic chemistry where it is widely applied for the systematic production and evaluation of molecules and materials.79 The multiautoclave system consists of Teflon blocks containing 100 reaction chambers each with 1 ml volume (500 μl capacity corresponding to 10–50 mg of the solid); up to ten different blocks can be stacked so that, in each experiment, 1000 syntheses with different gel compositions can be run at the maximum temperature of 473 K. Clearly, the multiautoclave is only one of the key components of the entire system, which includes also the tools for the experimental design, an automated reagent transfer and mixing, the automated logging of all relevant reaction conditions, the parallel work-up and isolation of the solid products and, last but not least, a tool for their automated XRD analysis.78 The effectiveness of this approach was assessed by inspecting the Na2O·Al2O3·SiO2·H2O and R2O·Al2O3·SiO2·H2O (R = Li, Na, Cs, TMA) systems;78 successively, it was employed for the screening of the structure-directing capabilities of ethyltrimethylammonium in zeolite synthesis,80 in the synthesis of zinc titanium silicates81 and zinc phosphates.82 Choi et al. used a similar system but with smaller reaction volumes (400 or 700 μl) accommodating 100–300 μl of reactant gel, hence producing up to 30–35 mg of the solid.83 Almost at the same time, Klein et al. reported another multiautoclave system composed of a small Teflon disk with up to 100 reaction chambers hosting up to 2 μl of gel (i.e. 250 times less than the previous one), producing 50 to 150 μg of the product.84 Such a small amount of solid requires special equipment for the XRD analysis; as a matter of fact, the authors made use of a microdiffractometer for collecting the XRD patterns of the solid products sintered on the Si wafer used as the reactor base.84 Though the efficiency of the system was verified by performing the synthesis of TS-1 with different raw materials and compositions of the reaction mixture, Newsam et al. highlighted some potential complications that may arise when the reaction volume is reduced to less than 10 μl.85Corma et al. proposed an in-house developed high-throughput system consisting of an array of 15 autoclaves supported by a robotic system for the automatic loading of the reagents.86 Differently from the previous systems, a factorial design was used for selecting the syntheses to be performed; moreover, the analysis of the results by artificial neural networks resulted in a non-linear model able to predict the occurrence and crystallinity of the zeolite under study better than the other methods did.86 The robustness of the approach was demonstrated by the synthesis of new zeolites including ITQ-24,87 ITQ-30,88 ITQ-33,89 ITQ-4390 and ITQ-44.91
It is easy to realize that these combinatorial or high-throughput approaches provide some obvious advantages related to the possibility of exploring, at the same time, a large number of synthesis parameters (combinatorial methods) or the rational design of the experiments to be run (high-throughput approach), to the saving of expensive raw materials and to the shortening of the time required for the screening of a given system. These advantages surely compensate the high costs of investment related to the in-house fabrication or to acquisition of the complex integrated experimental tools, but do not assure the facile scaling-up of the most interesting results eventually obtained.
Extra-large pore zeolites
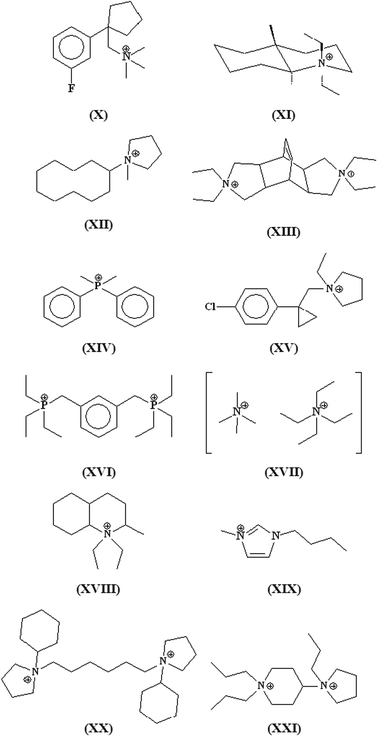
As stated above, one of the main goals in the synthesis of zeolites concerns the crystallization of frameworks having porous systems with at least 14-ring openings. This subject has recently been reviewed by Jiang et al.,92 but some interesting results obtained after the publication of this review deserve the attention.Table 2 lists the extra-large pore zeolites synthesized in the last decade, using the SDAs shown in Fig. 7. As a general comment, it should be noted that none of these phases has been obtained in the classical aluminosilicate system, since their crystallization requires the presence of a heteroatom (Ge, B and, in the case of ECR-34, Ga), combined with the use of the proper SDA. By inspecting the structure of the organic additives reported in Fig. 7, one realizes that the use of large and rigid SDAs is not mandatory for synthesizing extra-large pore zeolites. It is true that some of them were obtained with bulky organic molecules (e.g. the 1D 14-ring channel systems of the borosilicates SSZ-53 and SSZ-59 with SDAs I and II)55 but even that, sometimes, there is a clear role of the type of heteroatoms and of their concentration in the framework, favouring the crystallization of low-density phases even in the presence of relatively small SDAs. The 3D 18-8-8-ring channel system of ECR-34, for instance, was obtained as gallosilicate (with only traces of Al) by using TEA+ (III), Na+ and K+.93 The high micropore volume of this zeolite is generated by the large amounts of extra-framework cations required to compensate the high concentration of Ga3+ and Al3+ in the framework. All the other zeolites listed in Table 2 were obtained by using Ge as a heteroatom. It is worth noting that all are low-density phases characterized by multidimensional channel systems. Besides the two zeolites with the UTL topology (IM-1294 and ITQ-1595 prepared with the SDAs IV and V) having a 2D 14-12-ring channel system, the synthesis of ITQ-33 was quite interesting because it was obtained by using the relatively small and unselective hexamethonium dication (VI).89 The framework of ITQ-33 is formed by SBUs 3 and D4R stabilized by Ge and is characterized by a 3D 18-10-10-ring porous system; thanks to the presence of Al in the framework (Table 2), ITQ-33 could be an ideal acid catalyst for reactions involving large substrates.
| Material (code) | Pore structurea | Framework composition | FDb | SDA | Ref. |
|---|---|---|---|---|---|
| a Following the formalism of the Atlas of Zeolite Framework Types.5b Framework density (T atoms 1000 Å−3). | |||||
| SSZ-53 (SFH) | [001] 14 6.4 × 8.7* | B1.6Si62,4O128 | 17.9 | I | 55 |
| SSZ-59 (SFN) | [001] 14 6.2 × 8.5* | B0.35Si15.65O32 | 17.8 | II | 55 |
| ECR-34 (ETR) | [001] 18 10.1* ↔⊥ [001] 8 2.5 × 6.0** | Ga11.6Al0.3Si36.1O96 | 14.7 | III | 93 |
| IM-12 (UTL) | [001] 14 7.1 × 9.5* ↔ [010] 12 5.5 × 8.5* | Ge13.8Si62.2O152 | 15.2 | IV | 94 |
| ITQ-15 (UTL) | [001] 14 6.7 × 10.0* ↔ [010] 12 5.8 × 8.4* | Ge8Si68O152 | 15.3 | V | 95 |
| ITQ-33 (—) | [001] 18 12.2* ↔ 〈100〉 10 4.3 × 6.1** | Ge13.8Al1.8Si30.4O92 | 12.3 | VI | 89 |
| ITQ-37 (-ITV) | 〈100〉 30 4.3 × 19.3*** | Ge80Si112O368(OH)32 | 10.3 | VII | 96 |
| ITQ-40 (—) | 〈100〉 16 9.4 × 10.4** ↔ [001] 15 9.9* | Ge32.4Si43.6O150(OH)4 | 10.1 | VIII | 97 |
| ITQ-43 (—) | [001] 28 9.7 × 21.9* ↔ [100] 12 6.1 × 6.8* ↔ 〈110〉 12 5.7 × 7.8** | Ge49.6Si110.4O320 | 11.4 | IX | 90 |
| ITQ-44 (IRR) | [001] 18 12.5* ↔ 〈100〉 12 6.0 × 8.2** | Ge16.6Al1.6Si33.8O104 | 10.9 | IX | 91 |
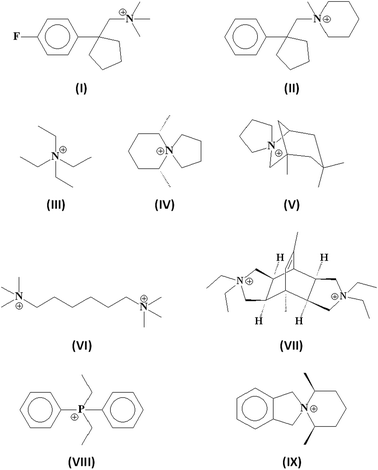 | ||
| Fig. 7 SDAs used in the syntheses of extra-large pore zeolites. | ||
A breakthrough in the synthesis of zeolites was achieved in 2009 with the discovery of ITQ-37, synthesized in the concentrated gel containing Ge and F− and the rigid and bulky SDA VII.96 ITQ-37 is a very low density phase (FD = 10.3) with a rather complex interrupted framework containing two large cavities per unit cell, each of them being connected to other three cavities through 30-ring windows of 4.3 × 19.3 Å, i.e. at the upper border of the microporous region (Fig. 8). Another interesting feature of ITQ-37 is the chirality of the framework, a very rare feature among the zeolites known so far. It is interesting to note that, in spite of the very low density, ITQ-37 is stable up to 813 K, a necessary condition for its use as a catalyst. As a matter of fact, when small amounts of Al were incorporated in the framework, ITQ-37 proved to be more active than zeolite beta in the acetalisation of bulky aldehydes with triethyl-orthoformate.96
 | ||
| Fig. 8 Polyhedral representation of the interrupted framework of ITQ-37, showing the 30-MR channels.96 | ||
Diethyldiphenylphosphonium (VIII) was the SDA employed in the synthesis of ITQ-40, performed in the concentrated gel containing Ge and NH4F.97 This phase has the lowest framework density (FD = 10.1) among the O-containing zeolites and it is the first structure containing the SBU D3R (double 3-ring) together with the D4R. The stabilization of the SBU 3DR by Ge was predicted by theoretical calculations and confirmed by examining the geometry of the SBU. The unprecedented 3D porous system of ITQ-40 is formed by 16-ring channels running parallel along the hexagonal a and b axes and 15-ring channels running along the c axis (Table 2). Due to its low density, ITQ-40 is not thermally stable: it can be calcined up to 723 K without loss of crystallinity but, at this temperature, only partial removal of the SDA (75%) is achieved.
Two new interesting extra-large pore zeolites, ITQ-4390 and ITQ-44,91 were recently synthesized by using the rigid and bulky SDA IX. ITQ-43 is a germanosilicate possessing a complex 3D porous structure, including 28-ring cloverleaf-like channels running along [001] (Fig. 9), similar to that found in cloverite (–CLO)98 but with dimensions of 19.6 × 21.9 Å in their longest axes in the range of the mesopores. The presence of other interesting 12-ring channels allowed us to define ITQ-43 as the first hierarchical micro-mesoporous zeolite structure synthesized so far. ITQ-43 can incorporate also small amounts of B and Al but, as for other extra-large pore zeolites, it is characterized by a low thermal stability due to the low framework density (FD = 11.4) and even by the high Ge content of the framework. That renders impossible the application of this zeolite as a heterogeneous catalyst. The crystallization of ITQ-44, on the other hand, was achieved by increasing the Al concentration in the gel.91 It is another low density phase (FD = 10.9) whose framework contains D3R and D4R secondary building units, both stabilized by Ge. Its structure, closely related to ITQ-33, contains a 3D 18–12–12-ring porous system and is thermally stable after calcination at 823 K.
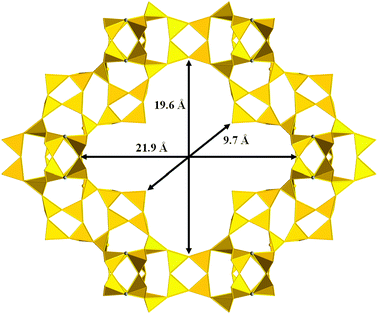 | ||
| Fig. 9 Polyhedral representation of the interrupted framework of ITQ-43, showing the 28-MR channels.90 | ||
Large pore zeolites
A number of large pore zeolites have been synthesized in the last decade (Table 3), by using the SDAs shown in Fig. 10. With few exceptions (SSZ-5599 and SSZ-48100), they are characterized by multidimensional (2D or 3D) porous systems, with framework densities ranging from 14.3 (ITQ-26) to 18.7 (SSZ-48) (Table 3). The presence of multidimensional channel systems is quite interesting, providing new opportunities for the use of these phases as heterogeneous catalysts. Differently from extra-large pore zeolites, whose crystallization mandatorily requires the use of Ge/F− or B (Table 2), some phases were obtained as aluminosilicates or aluminogermanosilicates and, often, in strong basic media, i.e. without F−. A first interesting example is ITQ-24 (IWR). This zeolite, having a 3D 12-10-10-ring porous system, was crystallized in OH−-containing gel in the presence of both Ge and Al and by using the hexamethonium dication (VI, Fig. 7) as a SDA.101 Interestingly, Ge stabilizes the D4R units within the framework, without the necessity of using F−. ITQ-24 proved to be active in acid catalyzed reaction (alkylation of benzene with propylene to cumene) as a consequence of the presence of Al in framework position.101 MCM-68 (MSE), on the other hand, was obtained as aluminosilicate from a classical synthesis approach (Si/Al, OH−-containing gel), by using the rigid and bulky SDA XIII and K+.102 The alkali metal ion plays a fundamental role in driving the crystallization of MCM-68, since zeolite beta was obtained upon replacing K+ by Na+. Its framework, containing a 3D 12-10-10-ring porous system, is composed of an unusual combination of SBUs 6, 4 and 5.102 ITQ-27 (IWV)103 and ITQ-26 (IWS)104 provided two interesting cases of zeolites synthesized in the presence of phosphonium-based SDAs: diphenyl-dimethyl-phosphonium (XIV) and 1,3-bis-(triethyl-phosphonium-methyl)-benzene (XVI), respectively. Both zeolites contain D4R units but only in ITQ-26 they are stabilized by Ge and F− contemporarily present in the gel; in the case of ITQ-27, F− ions only were used and the phase was obtained as aluminosilicate, a feature that renders it intrinsically more hydrothermally stable than ITQ-26.| Material (code) | Pore structurea | Framework composition | FDb | SDA | Ref. |
|---|---|---|---|---|---|
| a Following the formalism of the Atlas of zeolite framework types.5b Framework density (T atoms 1000 Å−3).c See Fig. 7. | |||||
| SSZ-55 (ATS) | [001] 12 6.5 × 7.2* | B1.5Si22.5O48 | 17.2 | X | 99 |
| SSZ-48 (SFE) | [001] 12 5.4 × 7.6* | AlxSi14−xO28 | 18.7 | XI | 100 |
| ITQ-24 (IWR) | [001] 12 5.8 × 6.8* ↔ [110] 10 4.6 × 5.3* ↔ [010] 10 4.6 × 5.3* | Ge5.1Al2.6Si48.3O112 | 15.5 | VIc | 101 |
| SSZ-63 (Beta) | Ordered zeolite beta | Si/B = 39 | n.d. | XII | 57 |
| MCM-68 (MSE) | {[001] 12 6.4 × 6.8 ↔ [100] 10 5.2 × 5.8 ↔ [110] 10 5.2 × 5.2}*** | Al11.4Si100.6O224 | 16.6 | XIII | 102 |
| ITQ-27 (IWV) | {[001] 12 6.2 × 6.9 ↔ [011] 12 6.2 × 6.9}** | Al5Si147O304 | 15.7 | XIV | 103 |
| SSZ-65 (SSF) | 〈100〉 12 5.9 × 6.9** | B1.5Si52.5O108 | 17.5 | XV | 58 |
| ITQ-26 (IWS) | [100] 12 7.1* ↔ 〈110〉 12 7.0×7.3** | Ge27.2Si108.8O272 | 14.3 | XVI | 104 |
| LZ-135 (LTF) | [001] 12 7.1*| [001] 12 6.5* | Al32Si76O216 | 16.5 | XVII | 105 |
| SSZ-56 (SFS) | [010] 12 5.9×8.4* ↔ [001] 10 4.8 × 5.5* | B1.3Si54.7O112 | 17.0 | XVIII | 59 |
| IM-20 (UWY) | {[001] 12 6.1 × 7.7* | [001] 10 4.2 × 5.5*} ↔ {[010] 10 5.0 × 5.9* | [010] 10 4.2×5.0*} ↔ [100] 10 5.0×5.8* | Ge17.8Si42.2O120 | 16.2 | XIX | 107 |
| SSZ-82 (—) | [010] 12 5.2 × 8.0* ↔ [100] 10 4.9 × 5.5* | B4.7Si61.3O132 | 16.8 | XX | 60 |
| ITQ-39 (—) | Intermediate between MFI and Beta | Si/Al = 9–11 | n.d. | XXI | 108 |
 | ||
| Fig. 10 SDAs used in the synthesis of large pore zeolites. | ||
Continuing with the large pore aluminosilicate phases, the case of LZ-135 (LTF)105 deserves attention. Though known since 1989,106 only recently its structure was solved thanks to availability of new powerful methods for determining complex structures from powder diffraction data.105 This phase crystallizes in the presence of Na+ and mixtures of TMA and TEA (XVII), the latter being necessary for the nucleation of LC-135 but not for its growth, since it was not detected in the final product. Its porous structure is unique, being composed by two types of 12-ring and two types of 8-ring channels, not interconnected, running parallel along [001]. The material is stable upon calcination but it is reported that TMA cations occluded in the pores are not completely eliminated.
Ge and F− are necessary for the crystallization of IM-20 (UWY), a zeolite characterized by a complex pore system, synthesized in the presence of 3-butyl-1-methyl-3H-imidazol-1-ium (XIX).107 Its 3D channel system is composed of linear interconnected 12-ring and 10-ring channels running parallel to the three directions of the orthorhombic unit cell. Along the c-axis run 12-ring and 10-ring channels, two types of 10-ring channels are parallel to the b-axis and a third kind of 10-ring channels lie along the a-axis. Similarly to the other germanosilicates, IM-20 is thermally stable under a dry atmosphere, the Ge–O bonds undergoing hydrolysis when exposed to moisture.
Also the use of B proved to be a key factor for the synthesis of large pore structures; however, only zeolites with 1D (SSZ-5599) or 2D (SSZ-65,58 SSZ-5659 and SSZ-8260) porous systems were obtained, probably as a consequence of the lack of stabilization of structural units such as the 3-ring and the double 4-ring that are at the origin of less dense frameworks.
Zeolite synthesis with diquaternary cations (diquats)
Among the several organic additives used in zeolite synthesis, those derived from the quaternarization of linear α,ω-diamines are of high interest. In this way, it is possible to modulate the dimensions of the polar heads and to modify their distance by appropriately choosing the length of the alkyl chain. Systematic works were performed by using a series of (H3C)3N–(CH2)n–N(CH3)3 with n = 5–12109 or 3–10.110 Moini et al. reported the synthesis of EU-1 (EUO, with n = 5 and 6), ZSM-23 (MTT with n = 7, 8, 11 and 12), ZSM-12 (MTW with n = 9), NU-87 (NES with n = 10),109 while Lee et al. found that the phase selectivity of these SDAs is sensitive to the Al and alkali content in the synthesis mixtures as well as to the length of the alkyl chain;110 they also observed the highest phase selectivity with n = 5, which yielded EU-1, ZSM-48, ZSM-12, MCM-22 (MWW) and Mordenite (MOR). Using additives of general formula (H5C2)3N–(CH2)n–N(C2H5)3 with n = 3–10, the same authors found similar results;111 again, the highest phase selectivity was achieved with n = 5, since it produced Na-P1 (GIS), SSZ-16 (AFX), SUZ-4 (SZR), ZSM-57 (MFS) and Mordenite. These results suggested that the structure directing ability of these SDAs is not strong enough to dominate over the inorganic gel chemistry.110 As a matter of fact, ITQ-13 (ITH), possessing an unusual 3D 10-10-9-ring porous system, was obtained when F− was added to the pure silica or B-containing reaction gel and with (H3C)3N–(CH2)6–N(CH3)3 as a SDA.112 Two other zeolites, ITQ-24101 and ITQ-33,89 were obtained with the same dication from Ge and F− containing gels.A number of studies focused on the modification of the size and shape of the positively charged heads. Three linear diquats consisting of two quinuclidinium groups linked by a chain of 4, 6 and 8 methylene units were employed as SDAs in the presence of F− ions.113 The most interesting results were obtained with the shortest diquat which, besides the layered ITQ-8 phase, gave ITQ-10 and ITQ-14 both disordered phases related to zeolite beta. Interestingly, ITQ-14 was found to consist of crystallites of polymorph C overgrown on standard zeolite beta crystals.114
Interesting is the case of the α,ω-bis-(N-methylpyrrolidinium)-alkanes XXII–XXIV shown in Fig. 11. 1,4-Bis-(N-methylpyrrolidinium)-butane (XXII) was used for the synthesis of TNU-9, a medium pore zeolite characterized by a 3D interconnected system of 10-MR channels.115,116 This zeolite can be synthesized in a very narrow range of SiO2/Al2O3 and NaOH/SiO2 ratios; moving out from the optimal ratios, other zeolites are obtained. When the length of the alkyl chain is increased by another methylene group (XXIII), the SDA favours the crystallization (again in a very narrow range of SiO2/Al2O3 and NaOH/SiO2 ratios) of the quite complex IM-5 (IMF) zeolite, a medium pore zeolite with a 3D porous system consisting of six different 10-MR channels.117 It is interesting to note that Corma et al. employed the same SDA XXIII in fluoride-free media and the presence of Ge obtaining, in a wide range of composition, the ITQ-22 (IWW) zeolite, characterized by a 3D 12-10-8-ring porous system.118 Finally, when 1,6-bis-(N-methylpyrrolidinium)-hexane (XXIV) was used, the medium pore zeolite SSZ-74 (-SVR), characterized by an interrupted framework, was obtained.119 The 3D porous system is constituted by undulating channels running along the [110] direction interconnected to other 10-ring channels along the [001] direction.
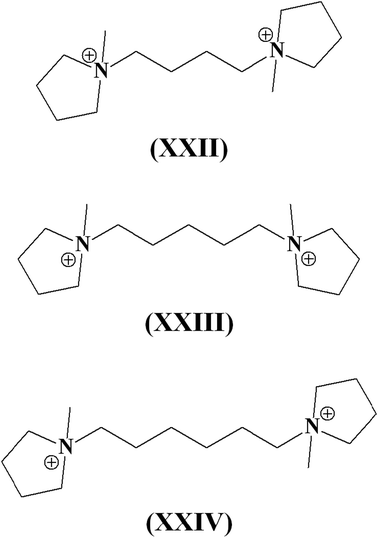 | ||
| Fig. 11 α,ω-Bis-(N-methylpyrrolidinium)-alkanes used as SDAs in zeolite synthesis. | ||
These interesting results prompted Jackowski et al. to carry out a systematic study on the structure directing ability of diquats, examining the effect of the charge separation by the methylene chain length from C4 to C6, of the size and geometric constraints of the heterocyclic end groups containing the charged nitrogen and of the inorganic context in which the syntheses are performed.120 Eighteen different zeolite structures were obtained including the already cited SSZ-74 and the new SSZ-75, and the data collected led to a better understanding of the role of diquats in the crystallization of zeolites.120
Two-dimensional (2D) zeolites
Most of the synthetic zeolites crystallize with their complete 3D four-connected framework. In some cases, however, the product recovered from the hydrothermal treatment consists of a layered phase. The layers, 2–3 nm thick, can be considered as 2D periodic building units (PerBU) and the 3D framework forms upon calcination through the condensation of the silanol groups present on the surface. The first example of a zeolite formed by this pathway was EU-20121 obtained by topotactic condensation of the layered precursor EU-19 constituted by hydrated silica layers intercalated by piperazinium cations.122 The structure of EU-20 remained unsolved for a long time and only recently Marler et al. confirmed that it is a disordered framework formed by CAS-type of stacking (88% probability) and NSI-type of stacking (12% probability).123The system at the base of the interest of two-dimensional zeolites is certainly the MWW-type structure. In 1995, we demonstrated for the first time that the precursor of ERB-1 (the borosilicate analogue of MCM-22) possesses a layered structure and that it is able to intercalate polar molecules.124 The 3D MWW structure is obtained by calcination at 543 K through the condensation of the silanol groups present on the layer surface. Later, this finding was confirmed on the aluminosilicate analogue, MCM-22.125
The discovery of this 2D zeolite precursor opened new opportunities in the synthesis of materials and stimulated the search of new systems with similar behaviour. As reported by Roth and Čejka in a very recent perspective paper, eleven different zeolite frameworks have been recognized to have a layered precursor.126 Only part of them, namely NU-6(2) (NSI),127 CDS-1/MCM-65 (CAS),128,129 RUB-24 (RWR),130 and RUB-41 (RRO)131 are characterized by a formation pathway that, as in the case of MWW, naturally involves a layered precursor; in the other cases (ALPO-41 (AFO),132 Ferrierite (FER),133,134 Sodalite (SOD),135 ZSM-5 (MFI)136), in fact, the original recipe leads directly to the formation of the 3D structure and special conditions and/or different SDAs are required for isolating the layered phase.
The availability of materials composed of zeolite layers is not merely a scientific curiosity but should be considered as an opportunity for designing new materials with enhanced properties. Fig. 12 summarizes the different type of materials that, besides the conventional 3D structure produced by calcination of the layered precursor, can be prepared. We are referring to:
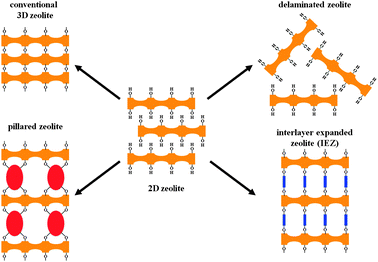 | ||
| Fig. 12 The different types of materials that can be prepared from the layered precursor of zeolites. | ||
- the possibility of expanding permanently the layers by adding pillars constituted by silica oligomers or other thermally stable inorganic components.
- the synthesis of the so-called interlayer expanded zeolites (IEZ) through the treatment of the layered precursor with specific monomeric species (e.g. silylating agents), which react with the surface silanol groups, expanding the interlayer distance and generating a 3D structure with pore system larger than that of the conventional parent zeolite.
- the full delamination of the 2D zeolite with the formation of materials constituted by single layers randomly arranged and connected by hydrogen bonds.
The MCM-22 (MWW) family of materials gives a well known example of the opportunities offered by a 2D zeolite.137 As reported above, this zeolite is normally obtained in the form of a layered precursor, MCM-22(P), and only under well defined conditions the synthesis pathway does not involve the formation of a 2D intermediate. In particular, it was reported that, by using the same SDA (e.g. hexamethyleneimine), MCM-22(P) is obtained when the mole ratio of the organic additive to the inorganic cations in the gel is greater than 2.0; below this value, the 3D zeolite, MCM-49, is obtained.125 This indicates that an excess of SDA in the gel is necessary for blocking the growth of the 3D structure and for stabilising the layers; as a matter of fact, the SDA molecules in the interlayer region can be easily removed by mild treatment with ammonium acetate solution or eliminated at relatively low temperature (543 K) with the contemporary condensation of the surface silanol groups.124
Pillaring of MCM-22(P) was also achieved with a variety of different oligomeric species. The procedure required first the swelling of the layers with a mixture of a surfactant (cetyltrimethylammonium, CTMA) and tetrapropylammonium, followed by the addition of the source of inorganic pillars. In this way, the mesoporous MCM-36 material was obtained firstly with tetraethylorthosilicate (TEOS)138 and silica oligomers,139 successively with other components (e.g. Al2O3, MgO, BaO and mixtures of them).140,141 Successful results were also obtained with NU-6(1), the layered precursor of NSI, which was pillared with silica oligomers obtaining the material called MCM-39(Si).142
Following the same concept, the treatment of the layered zeolite precursor with monomeric species able to condensate with the silanol groups present on the surfaces of adjacent layers may lead to the preparation of 3D crystalline structures with expanded interlayer spacing (IEZ) and larger pore size. Layered precursors of different zeolites (MWW, FER, CDO and MCM-47) were treated with diethoxydimethylsilane [(CH3)2Si(C2H5O)2] in 2 M HNO3 under reflux obtaining the corresponding IEZs, which are successively calcined to remove the organic groups. 3D ordered structures were formed as a result of the insertion of O–Si–O linkages between the layers.143 Corma et al. treated the CTMA-swollen MCM-22(P) precursor with 1,4-bis-(triethoxysilyl)benzene (BTEB); the structural evidence obtained was consistent with the formation of pillars constituted by two condensed BTEB molecules.144 These long pillars (∼15 Å long) generate a hierarchical pore structure stable up to 673 K; notably, the phenylene groups can be functionalised with amino groups generating an acid–base bifunctional catalyst which proved to be very active in one-pot acetal hydrolysis–Knoevenagel condensation cascade reactions.144 This family of materials includes also: IEZ-FER, obtained by treating PREFER with diethoxydimethylsilane,145 while dichlorodimethylsilane was the silylating agent employed in the preparation of COE-1 and -2 (IEZ-RRO) from RUB-39146 and COE-3 and -4 (IEZ-CDO) from RUB-36.147 It is worth noting that COE-2 and -4 are the calcined forms of the pristine silylated materials COE-1 and -3 in which the methyl groups are substituted by –OH. All these materials possess 3D ordered structures, with enhanced accessibility of the internal porosity.
What is reported above demonstrates that in 2D zeolites the layers can be swollen and successively re-assembled in the presence of pillars, to form 3D structures with enhanced pore openings both in the micro- and mesopore ranges. Another opportunity is given by the possibility of exfoliating the layered precursor leading to the preparation of the so-called delaminated zeolites. Corma et al. were the first to report the successful delamination of MCM-22(P) by treating it with large excess of CTMA-Br and TPA-OH at 353 K followed by a treatment in an ultrasound bath. After acidification at pH = 2, the solid is separated and calcined giving the fully delaminated ITQ-2 material.148,149 The rather drastic conditions (high pH values and temperature) used for exfoliating MCM-22(P) do not assure the structural integrity of the MWW layers. Therefore, milder conditions were proposed, including the careful control of the amount of organic base (e.g. TPA-OH) used in conjunction with the surfactant for swelling the layered structure,150 the decrease to 298 K of the reaction temperature,151 the treatment of the MCM-22(P) at 353 K using CTMA-Br, TBA-F and TBA-Cl at pH = 9 (UCB-1).152 Other materials were obtained by the delamination of PREFER (ITQ-6,153,154 UCB-2,155) and NU-6(2) (ITQ-18).156
The materials reported so far are related to zeolite frameworks in which a clear alternation of high and low density regions exists. However, two interesting cases involve zeolite frameworks that apparently are not suitable candidates for being obtained in the form of layered structures. The first one concerns the cubic sodalite (SOD) structure. Depending on the cutting direction of the 3D structure, different materials can be obtained. The layered silicate hydrated RUB-15 crystallised at moderate temperature (393–413 K) from a gel containing the silica source and TMA-OH.135 Syntheses performed at higher temperature (453 K) gave Dodecasil-3C, while conventional zeolites crystallised in the presence of heteroatoms: sodalite, gismondine and NU-1 with Al, AlPO-20 and AlPO-12 with Al and P. Interestingly, RUB-15 did not convert to an ordered phase when calcined as such135 but it did when TMA+ was firstly removed by exchange with acetic acid.157 The silicate layers of RUB-15, derived from clipping the SOD structure along the [110] direction, were found also in other materials: RUB-51, synthesized in the Si/B system in the presence of benzyltrimethylammonium (BTMA) as a SDA,158and DLS-2, prepared by hydrothermal treatment of a clear solution of octakis(tetramethylammonium) octasilsequioxane.159 Other materials are formed by silicate layers obtained by clipping the SOD cages along the [100] direction: the helix layered silicate (HLS), obtained by using TMA+ and 1,4-dioxane,160 and the Hiroshima University Silicate-1 (HUS-1) layered phase prepared by the so-called interzeolite conversion method, consisting in the hydrothermal treatment of a mixture of two primary gels obtained by decomposition of dealuminated FAU and beta zeolites by hydrothermal treatment with, respectively, TMA-OH and BTMA-OH.161 A very recent paper demonstrated that it is possible to treat the acid-exchanged HUS-1 phase with dimethyldichlorosilane producing an IEZ material, which proved to effectively adsorb TMA+ from aqueous solution.162
The other intriguing example concerns the synthesis of MFI nanosheets.136 In this case, the key factor for obtaining such kind of nanoparticles was the design of the bifunctional SDA, which contains, in the same molecule, the quaternary ammonium cations and a long alkyl surfactant chain: H3C(CH2)21–N(CH3)2–(CH2)6–N(CH3)2–(CH2)5CH3 (designated as C22-6-6).136 The MFI nanosheets synthesized in this way are 2 nm thick in the crystallographic b directions, i.e. that along which the straight 10-MR channel system run. It is interesting to note that the proposed structural models for the single MFI nanosheet consist of surfactant molecules aligned along the straight channels, with the quaternary ammonium sites located at the channel intersections, one inside the channel and the other on the external surface. The nanosheets can either stack along b or assemble in a random way. The removal of the surfactant would result, in the first case, in the condensation of the nanosheets to form MFI crystals, in the second in the partial condensation of the nanosheets with the formation of materials similar to those obtained by delamination of 2D zeolites. The experimental evidence supports the second model, which accounts for the presence of a hierarchical micro-mesoporous structure.136 This approach is reported to be effective even in the synthesis of Ti-containing MFI nanosheets with single unit cell thickness, displaying high selectivities in the epoxidation of bulk molecules with both H2O2 and t-butylhydroperoxide as oxidazing agents.163,164 Similarly to 2D zeolites, MFI nanosheets can be assembled by using amorphous silica oligomers,165 following the method proposed by Maheshwari et al.151
More recently, Roth et al. reported the first case of a 3D → 2D conversion of a zeolite structure.166 In particular, they found that calcined germanosilicate with UTL topology containing or not B in the framework is converted to a lamellar structure by mild hydrolysis; the layered structure obtained can be treated similarly to the other lamellar precursors. This finding is important because it opens the possibility of extending the 3D → 2D conversion to other materials with similar structural features.
What is reported above demonstrates that new materials with different characteristics can be prepared starting from the layered precursors of zeolites. This field of research is far from being fully explored, but the promising results obtained so far clearly indicate the opportunities given by the different families of materials produced. These opportunities can be better understood when considering the applications of zeolites, in particular as heterogeneous catalysts. The concept at the base of the different synthesis pathways described in Fig. 12 is the increase of the accessibility of the active sites located on the internal surface of the layers through the expansion of the pore openings (IEZ materials), the creation of hierarchical micro-mesoporous systems (pillared zeolites) or the delamination of the layers with the (nearly) complete exposure of the surface of the layers (delaminated zeolites). To exemplify the concept, it is useful to come back to the MWW-type of materials. The conventional 3D phases (e.g. MCM-22, ERB-1) proved to be effective catalysts in the alkylation of aromatics. For instance, it has been reported that MCM-22 behaves similarly to other 12-MR zeolites (e.g. MOR and beta) in the alkylation of toluene with methanol and ethanol167 and in the isopropylation of benzene to cumene.168,169 This is quite unusual when considering the peculiar pore structure of this zeolite, composed of two independent channel systems with 10-ring apertures: one possessing large supercages (a sort of cylinder 18 Å long and 7.1 Å wide) interconnected with slightly elliptical apertures, the other characterized by 2D sinusoidal channels.170 Detailed computational studies predicted that the diffusion of even relatively small molecules such as cumene and benzene is hampered by the narrow pore openings, leading to the conclusion that the reaction occurs on the external crystal surface where a high concentration of cavities (corresponding to half of the supercage) is present (Fig. 13).168,171 Under these conditions, the reaction occurs on the crystal surface under steric control but without any diffusion limitation. Therefore, significant benefits are expected if the external surface is maximized and this can be done either by decreasing the dimensions of the crystallites (particularly along the stacking direction of the layers) or by exfoliating the layered precursor to give the delaminated zeolite. Though the enhanced catalytic performances of delaminated zeolites with respect to the 3D conventional parent structure have been demonstrated, e.g.,148,149,172,173 it is evident that the quality of the materials should be improved. In fact, the small dimensions of the layers combined with the drastic conditions necessary for the delamination and the high calcination temperature necessarily induce the collapse of the structure. The new synthesis approaches seem to have a lower impact on the structure of the layers but the lack of catalytic data renders it impossible to verify their effectiveness.
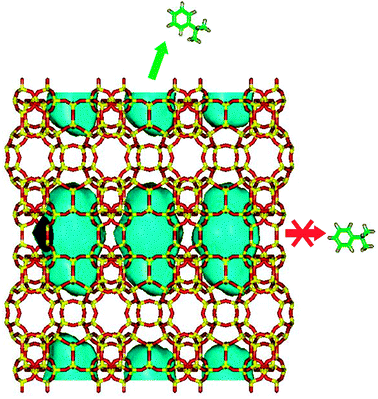 | ||
| Fig. 13 Model of the MWW-type zeolites highlighting supercages present in the interior of the crystal and the hemisupercages on the surface. Cumene molecules can form within the supercages but cannot diffuse outside the crystal. In contrast, they can be formed in the hemisupercages under steric control but without any diffusion limitation.168 | ||
Another question concerns the possible increase of the number of zeolites that can be synthesised in lamellar form. Apparently, this is limited to a few structures, which, in their 3D forms, show a regular alternation of regions of high and low density even if the discovery of the layered form of SOD (RUB-15) and of the MFI nanosheets demonstrates that this possibility cannot be ruled out. The case of MFI nanosheets is particularly important because it represents the first material obtained by using a SDA specifically designed for this purpose. In the other cases, the lamellar forms were obtained either as a naturally occurring step in the formation of the 3D structure (e.g. MWW) or accidentally during exploratory works on the structure directing ability of new SDAs (e.g. PREFER). A better understanding of the phenomena that lead to the formation of 2D zeolites is therefore necessary to create the basis of a rational design of lamellar phases, which can be used as building blocks for preparing new materials according the synthesis routes depicted in Fig. 12. This is quite important when considering that the increased accessibility of the active sites located on the surface of the layers may have a positive impact on the catalytic activity, avoiding the diffusion limitations imposed by the parent 3D structure. This is the basic concept of an important research line which aims at preparing materials with hierarchical porous structure, i.e. with interconnected micro-mesoporous systems, to which the materials derived from 2D zeolites belong. However, some negative aspects justify further investigations of 2D zeolites. First, the costs of the materials derived from 2D zeolites are significantly higher than those of conventional zeolites due to the increase of the number of unit operations necessary for preparing them. Without the possibility of reducing them, the only reason that could make these materials economically attractive would be the outstanding catalytic performances. Moreover, the intrinsic thermal/hydrothermal stability of these materials is low. Delaminated zeolites, for instance, are in principle very interesting but it is easy to understand that the nanoparticles, which assemble themselves in the form of a “house of cards”, are nearly “all surface” and, though ordered, they lack stabilization given by an extended 3D crystalline structure. Finally, most of the 2D zeolites known so far are highly siliceous, therefore, unattractive for catalytic applications. Obviously, an increase of the Al content is highly desirable, otherwise the risk that these materials remain a scientific curiosity is high.
Nanocrystals and hierarchical porous structures
In the previous section, we have already introduced an important concept in the modern zeolite science: the enhancement of the catalytic performances of zeolites through the increasing accessibility of the active sites located inside the crystals, bypassing the diffusion limitations imposed by the pore sizes. This would be very important not only for reactions involving bulky molecules occurring on the pore mouths but even for the exploitation of the active sites located in the interior of relatively large zeolite crystals (and by large we mean on the micrometer level). In other words, the degree of utilization of a zeolite catalyst is defined by the effectiveness factor η, i.e. the ratio between the observed and intrinsic reaction rates, which, in turn, depends on the extent to which the diffusional transport limits the rate of conversion defined by the Thiele modulus ϕ. A low value of ϕ means that the process occurs without diffusional constraints and the whole utilisation of the catalyst particle (ϕ = 0 → η = 1); on the other hand, as the Thiele modulus increases, the degree of utilisation of the catalyst particle progressively decreases (e.g. ϕ = 10 → η = 0.1, which means that only 10% of the catalyst particle is effectively used). It is intuitive that high values of Thiele modulus have negative impacts on the process: the low utilisation of the catalyst requires high reactor volumes, while the diffusional constraints may influence the selectivity and the life of the catalyst. Since the intrinsic rate coefficient is constant for a given reaction, to increase the effectiveness factor it is necessary to shorten the length of the diffusion path by decreasing the dimensions of the crystals or to enhance the effective diffusivity of the molecules by employing catalysts with larger pore dimensions. In the first case, suitable synthesis conditions should be designed for obtaining the zeolite in the form of small crystals having however in mind that their separation from the mother liquor could be difficult.174,175 Other more specific approaches involve, for instance, the synthesis in confined space in which the crystallization occurs inside the mesopores of a rigid matrix (e.g. carbon black), producing nanosized zeolite crystals with narrow size distribution,176–178 or addition of organosilanes in the synthesis. For instance, Choi et al. added the rationally designed amphiphilic organosilanes surfactant ([3-(trimethoxysilyl)-propyl]hexadecyldimethylammonium chloride) to the reaction mixture to generate mesoporosity in ZSM-5 and zeolite A particles,179 while the group of Serrano added phenylaminopropyl-trimethoxysilane to the preformed suspension of zeolite seeds to prevent crystal growth, stabilizing in this way zeolite nanoparticles. In this way, ultrasmall crystals of ZSM-5,180,181 beta182 and mordenite183 were prepared.On the other hand, the discovery of the M41S family of ordered mesoporous materials by using quaternary alkylammonium surfactants (e.g. CTMA-Br), announced 20 years ago by scientists of Mobil,184,185 opened great expectations in the scientific community because they were considered an extension of zeolites in the mesoporous region. In reality, it was soon clear that mesostructured materials as such are not suitable for being used as heterogeneous catalysts because they are characterized by lower acidic strength and thermal/hydrothermal stability with respect to zeolites.186 These severe drawbacks hamper the industrial application of mesostructured materials and, with the exception of the positive case announced by a Chinese group related to the use of MCM-41 in the formulation of a FCC catalyst (containing 30 wt% of RHUSY and 5 wt% MCM41) tested on a commercial FCC riser unit of 60 kton y−1 capacity with 6 ton catalyst inventory,187 no industrial applications have been realized so far.
In order to fill the gap between the characteristics of mesostructured materials and zeolites, the ideal situation is to confer to the materials the same properties of the zeolites, while maintaining the characteristics of the mesoporous structure. Ideally, the materials will be constituted by a system of mesopores (not necessarily ordered) with crystalline microporous walls. These materials would encompass the efficient mass transport assured by the mesopores with the thermal/hydrothermal stability and the better control of the characteristics of the active sites given by the crystalline zeolite structure. In other words, this is the definition of a system with a hierarchical porous structure. The strategies proposed for preparing hierarchical porous structures are divided into destructive and constructive methods. The former is based on the generation of mesopores in a preformed zeolite crystal by selective extraction of framework atoms (demetallation), mainly Al and Si but even Ti and B. Dealumination is surely the most important demetallation method since it is employed for the preparation of industrial catalysts.188 The selective extraction of Al from low silica zeolites was originally applied for increasing the acid strength but it assumed higher importance once realized that the process is accompanied by the formation of intra-crystalline mesoporosity. As reviewed by van Donk et al.,188 dealumination can be achieved through calcination, steaming, acid leaching and chemical treatments with chelating agents (e.g. EDTA). The extraction of Si atoms (desilication) is also deeply studied but it is performed only through alkaline leaching with strong mineral (NaOH) or mild organic bases (e.g. TPAOH, TBAOH).189–191
Though effective, demetallation methods do not permit an accurate control of the characteristics of the mesoporosity, which is randomly generated within the zeolite crystals. On the other hand, better control of the hierarchical porous structure can be achieved by the so-called constructive methods.190,191
The first, we would say, obvious strategy proposed was aimed to crystallize the amorphous walls of preformed mesostructured materials, including MCM-41,192,193 HMS192 and SBA-15.194–196 The preformed material is firstly impregnated with an SDA (usually TPA+) and then hydrothermally treated under the desired conditions. In the case of MCM-41 and HMS the local organization of the thin amorphous walls was evidenced spectroscopically (appearance of a weak absorption in the FT-IR spectra at 550 cm−1, attributed to the asymmetric stretching of 5-rings in ZSM-5, transformation of strongly anisotropic Al into an isotropic tetrahedral environment, a situation similar to that usually observed in the 27Al MAS NMR spectra of zeolites), but no extended 3D organization of the structure was detected.192,193 Better results were claimed in the crystallization of the thicker walls of SBA-15, eventually stabilized by the presence of organic material derived from the carbonization of the surfactant or of a polymer generated inside of mesopores.194–196 In general, the characteristics of the resulting material strongly depend on the conditions used in the hydrothermal treatment. Mild treatments generally maintain the ordered mesoporous structure while producing embryonal zeolite crystals, while more severe conditions produce the progressive collapse of the mesoporous structure and the growth of zeolite crystals separated from the mesostructured phase. In other words, even under the best conditions, the materials obtained should be considered composites rather than mesostructured phase with crystalline walls.197
An alternative strategy consists of the deposition of zeolite seeds or nanocrystals, obtained by interrupting the synthesis in an early stage, on a preformed mesostructured phase. Due to the average dimensions of the zeolite seeds (2–5 nm), this approach was successfully applied to substrates with very large mesopores (e.g. SBA-15, mesocellular foams)198–200 but not to materials with narrow pore dimensions (MCM-41, MCM-48). In fact, the seeds are too large for coating the internal surface of the mesopores, avoiding at the same time the blocking of the pores as a consequence of the deposition of the particles on the external surface.
More attractive strategies are based on the so-called dual templating approaches, which involve the use of a mixture of a micro-template (i.e. a conventional SDA used for the crystallization of zeolites) and a meso-template able to generate the mesoporous structure. Meso-templates, in turn, are defined soft or hard when they are constituted, respectively, by organized systems of molecules (micelles, polymers) or solid particles of different nature (e.g. carbon particles, nanotubes, etc.).
The use of a soft meso-template implies that the formation of the micro-mesoporous material occurs through the aggregation of zeolite seeds or nanocrystals around the surfactant micelles and, depending on whether the reaction mixture contains at the same time the micro- and the meso-templates or that the zeolite nanoparticles are formed separately and then organized in the presence of a meso-template, the syntheses are defined as one-pot or two-pot.
The one pot approach has essentially failed the expectations because the materials obtained were composites of intimately mixed individual microporous crystals and mesoporous aggregates, rather than true hierarchical porous systems. The reason is probably that the processes of formation of zeolite units and the organization of the mesoporous structure come into competition, due to the simultaneous presence of micro- and meso-templates.201–207 To avoid that, attempts were made to isolate the two processes, preparing first the zeolite seeds and then organizing them in the presence of a surfactant in two separate vessels (two-pot synthesis). The advantage of this approach is that there are no limitations in the type of zeolite structure used for assembling the mesostructural phase as firstly demonstrated by Liu et al. in the cases of zeolite Y,208 beta and ZSM-5.209 Several other hierarchical porous materials were successively synthesized, including, for instance, the highly ordered hexagonal mesoporous aluminosilicate MAS-5, assembled with zeolite beta seeds, and CTMA-Br,210,211 a series of materials related to SBA-15 derived from the assembly of zeolite beta (MAS-7),212,213 TS-1 (MTS-9),212,213 silicalite-1 (MPS-9)214 and Fe-ZSM-5 (MFS-9),215 cubic MCM-48 mesoporous silicas and aluminosilicates assembled by ZSM-5, beta and silicalite-1 seeds,216 wormhole mesostructured aluminosilicates (TUD-1 analogues) obtained by assembly ZSM-5 seeds,217 MCM-41 assembled by Ti,Al-beta218 and by TS-1 seeds.219
The large number of materials synthesized by the two-pot approach testifies to its effectiveness. A distinctive character of most of the materials reported so far is the presence, in the XRD pattern, of the low angle reflections associated to the ordered array of mesopores but even the lack of reflections in the wide-angle region. In the few cases in which the reflections associated to a zeolite phase were clearly observed, the materials showed the typical characteristics of the composite.218,219
The zeolitic character of the walls was demonstrated by the presence of the typical bands of zeolite structure in the FT-IR spectra (e.g., the already cited band at 550–600 cm−1, assigned to the asymmetric stretching mode of 5-ring blocks, was always reported when MFI-type and zeolite beta seeds were used),220 the tetrahedral coordination of Al (resonance peak at 55–60 ppm in the 27Al MAS NMR spectra), and the presence of microporosity. Albeit partial, the ordering of the walls confers thermal/hydrothermal stability to these materials representing, therefore, a distinctive feature of the product synthesized by the two-pot approach.209
A further evolution of the characteristics of the materials towards a real hierarchical porous structure was given by the introduction of the hard templates in the synthesis. The concept at the base of this approach directly derives from the synthesis performed in confined space, where the crystallization occurs within the mesoporosity of a carbon black rigid matrix.176–178 Successively, it was found that by working with an excess of reactant gel with respect to the carbon particles (carbon black,221 multiwall nanotubes222,223 or nanofibers224), these were embedded in the μm-sized zeolite crystals of ZSM-5, leaving a regular mesoporosity once removed by calcination. Selected area electron diffraction analysis of several particles demonstrated that they are single crystals rather than agglomerates of nanocrystals.221 The approach was successively extended to mesoporous TS-1 crystals prepared either by conventional225 or microwave226 heating, MEL,227 MTW228 and beta229 zeolites, even in fluoride media.230 Hierarchical ZSM-5 crystals with up to 0.82 cm3 g−1 mesopore volume were synthesized by Park et al. by using carbon black under microwave heating.231 From the catalytic point of view, these materials are more active than the conventional ones, at least than those formed by large μm-sized well formed crystals. In general, the higher activity is attributed to the more efficient mass transport assured by the extended mesoporous system and by the shorter length of the micropores. Significant examples were reported for cracking of n-tridecane and 1,3-dimethylcyclohexane with mesoporous ZSM-12,228 for the alkylation of benzene with ethylene232 and dealkylation of ethylbenzene233 with mesoporous ZSM-5, and for the bulky bimolecular reaction of 2′,4′-dimethoxyacetophenone with 4-methoxybenzaldehyde to give vesidryl.231
A beneficial effect of the generation of mesoporosity within the crystals was also reported for the TS-1 oxidation catalyst. For instance, by comparing the catalytic activity of a mesoporous TS-1 catalyst with that of a conventional sample in the epoxidation of 1-octene and cyclohexene, Schmidt et al. found that the enhanced mass transport and easier accessibility of the mesoporous catalyst affect only the conversion of the bulkier substrate (cyclohexene), while the epoxidation of 1-octene was substantially similar for the two catalysts.225 On the other hand, Ok et al. observed that the epoxidation of cyclooctene and cyclododecene proceeds with higher activity and selectivity on a mesoporous TS-1 catalyst with respect to a conventional sample, while no difference was reported in the case of cyclohexene.226 Due to the large dimensions of cyclooctene and cyclododecene, most likely the improved catalytic performance of the mesoporous TS-1 catalyst is derived from the greater availability of active sites on the external surface of the crystallites, which are much larger in the mesoporous catalyst than in the conventional one.
The examples here reported, representative of the many available in the literature, do not prove the effective advantages of the hierarchical porous structure. First, what is defined “conventional catalyst” is generally a sample constituted by large (catalytically speaking) single crystals. In this case, the advantages are evident and expected but, certainly, the reference sample does not have the characteristics of the “state of the art” catalysts, generally constituted by mesoporous aggregates of crystals of small dimensions. Moreover, from the data reported in the literature, we do not know exactly what is the effect of the combustion process of large amounts of hard templates on the structural characteristics of the zeolite and, in particular, of the active sites. In our opinion, these aspects should be further investigated for defining the effectiveness of this approach, even for justifying the higher costs for the production of hierarchical porous structures compared to conventional catalysts.
In the mean time, the search for new synthesis approaches is continued, producing results of considerable interest. In particular, these new strategies are finalized to the control of the extent and the organization of the mesoporous system and are based on the use of carbon replicas. An example is represented by CMK (Carbon Mesostructured at KAIST) materials, derived from the mild carbonization of sucrose impregnated into the mesopores of 3D mesostructured materials (e.g. MCM-48, SBA-15), followed by the dissolution of the inorganic phase.234–236 These carbon replicas were successively used as templates for preparing RMM-1 and RMM-3 amorphous mesoporous materials analogous to MCM-48 and SBA-15, possessing microporous characteristics due to the presence of zeolite secondary building units in the framework.237 Highly crystalline ZSM-5 zeolites with disordered mesopores can also produced by applying the appropriate hydrothermal conditions.238 More recently, 3DOm carbon replicas of regular assemblies of size-tunable (10–40 nm) silica nanoparticles were employed for synthesizing silicalite-1,239 LTA, FAU, BEA and LTL zeolites.240 Confined crystal growth within the mesopores led to uniformly shaped zeolite nanocrystals regularly organized in large domains with regular mesoporosity, easily tuned by varying the mesopores size and structure of the carbon template. These methods, especially the second, are excellent examples of materials engineering, but, despite their apparent simplicity, they are expensive and time-consuming. For these reasons, as admitted also by the authors, their main destination is not heterogeneous catalysis, but rather the preparation of advanced devices (e.g. sensors, thin films, membranes) with high added value.
Finally, there is another approach that deserves attention because it may have practical implications: the reorganization of pre-formed zeolite crystals in the presence of a surfactant. The concept is simple: the zeolite crystals are dissolved generating fragments in solution which successively re-organize themselves around the micellar mesophase, producing an ordered mesostructured material and at least partial ordering of the walls. Ultimately, this approach is similar to the dual template methods based on the use of soft templates, except that the fragments are produced by the dissolution of a pre-formed zeolite and not generated by the presence of a micro-template. The characteristics of the materials strongly depend on the synthesis procedure adopted. The early reports refer to the preparation of micro/meso-composites rather than single phases with a hierarchical porous system.241–244 Successively, mesostructured materials with partially ordered walls were obtained by introducing the filtration of the solution resulting from the zeolite degradation step.245–247 The most interesting result, however, was that claimed by Ying and Garcia-Martinez, consisting in the short-range reorganization of pre-formed zeolite crystals (Y, mordenite, ZSM-5) in the presence of a surfactant to accommodate mesoporosity without losing zeolite crystallinity.248–250 Though the only data known so far are those reported in the patents, it seems clear that the materials claimed by the inventors really possess a hierarchical porous structure formed by mesoporous zeolite crystals. Among them, the mesoporous zeolite Y is surely the most important because it shows outstanding performances in the MicroActivity Test (MAT), the main technique used for evaluating the performances of FCC catalysts: using vacuum gas–oil as a feed, the test showed that, with respect to the parent zeolite, mesostructured zeolite Y produces an increase of gasoline (+43%), propylene (+75%) and butenes (+110%), and a decrease of coke (−32%), dry gas (−23%) and LPG (−12%).248 To demonstrate the interest in these results, a partnership between Rive Technology Inc. (the proprietary of the technology) and Grace Davison (one of the largest FCC catalyst manufacturers) for commercializing advanced FCC catalysts based on the use of the mesostructured Y zeolite has been announced.251 If successful, this will be the first example of mesostructured material for operation in an industrial process.
Hybrid organic–inorganic zeolites
The last section of this perspective review is devoted to a topic still largely unexplored but that may have important implications in several technological areas: the synthesis of hybrid organic–inorganic zeolites. The incorporation of organic groups in zeolites, in fact, would open up new opportunities for modulating hydrophilic/hydrophobic character useful for adsorption and catalysis. In this way, the lower affinity of the internal surface towards polar molecules could control more efficiently host–guest interactions and facilitate, for instance, the rapid elution of polar products limiting side reactions. In addition, new functionalities and active sites would be useful in acid–base catalysis, separation and even for optical applications. Different approaches for functionalizing microporous materials with organosilanes of different nature were proposed, most of them, however, with limited or no success at all.The preparation of organic–inorganic hybrid zeolites was firstly approached via post-synthesis treatment of preformed zeolites with organosilanes, a well-known process usually applied for the functionalization of amorphous silica and periodic mesoporous materials (e.g., MCM-41) for chromatographic and catalytic purposes.252 If the functionalisation of these materials is facilitated by the high concentration of surface silanol groups, the same is not true for zeolites. In fact, Corma et al.253–255 reported that the functionalisation of ultra-stable Y zeolites with organosilanes (used for anchoring metal complex catalysts)255 occurs in the mesoporous region. Cauvel et al. investigated the functionalisation of a series of Y zeolites with increasing content of mesopores produced by steaming, thus confirming this hypothesis.256 In this case, the efficiency of functionalisation increased with the mesopores (regions rich in silanol groups) content. These results led to the conclusion that the functionalisation does not occur within the zeolite micropores, but only where the silanols are mainly located, i.e., on the external crystal surface and in the mesoporous region. Though these materials can be classified as organic–inorganic hybrids, they do not possess the desired characteristics (i.e., the presence of new organic functions in the microporous region).
More promising is the direct synthesis route. Maeda et al. reported the synthesis of a series of microporous alumino-methylphosphonates, named AlMepO.257–260 Formally, these materials cannot be classified as zeolites because of the octahedral coordination of Al ions in the framework. Nevertheless, they are interesting since their preparation demonstrated the possibility of obtaining organic-functionalized crystalline microporous solids. Two phases, AlMepO-α259 and AlMepO-β,257,258 were obtained by using methylphosphonic acid as phosphorus source; both phases are characterized by monodimensional 18-ring linear channel systems, with the methyl groups located in the channels. These materials display interesting gas adsorption properties but, because of the presence of methyl groups only, they do not possess either any catalytic activity nor can be further functionalized for such a purpose.
With regard to the AlPO system, functionalization of AlPO4-H1 (VFI) with phenylphosphonic acid was reported recently.261 The synthesis route of the hybrid material is the same used for the pure phase with the exception that phenylphosphonic acid was added to the reaction mixture. The functionalisation is rather limited since a maximum of 2 mol% was achieved, as estimated by 31P MAS NMR.
Jones et al. reported the first silica-based organic functionalized molecular sieve (OFMS), demonstrating the possibility of synthesizing directly hybrid organic–inorganic zeolites.262–265 To avoid any high-temperature thermal treatment, zeolite beta was chosen because the SDA (TEA+) can be removed by extraction. Pure silica zeolite beta was synthesized in a conventional way by replacing a small fraction of TEOS by phenylethyltrimethoxysilane. After removal of the SDA with acetic acid solution, the phenylethyl groups were sulphonated by reaction with SO3 vapour at room temperature, obtaining a catalyst active in the reaction of ethylene glycol with cyclohexanone to pentamethylene-1,3-dioxolane.262 Other OFMSs with polar (e.g., aminopropyl-trimethoxysilane, mercaptopropyl-trimethoxysilane)263 or non-polar (e.g., 3-butenyl-triethoxysilane, 2-(3-cyclohexenylethyl)-trimethoxysilane))264 organic groups were successfully synthesized, demonstrating that this methodology may effectively lead to the discovery of new shape selective catalysts. However, no other communications followed the original papers probably because zeolites with extractable SDAs are very uncommon and there is therefore a lack of possible candidates for such kind of functionalisation.
The other approach consists in the incorporation of bivalent organic groups in the framework, the simplest being the replacement of the framework oxygen atoms by methylene (–CH2–) groups. A new family of hybrid organic–inorganic materials named ZOL (Zeolite with Organic groups as Lattice) comprising different known zeolite structures (MFI, LTA and Beta) were synthesized by using bis-(triethoxysilyl)-methane (BTEM) as the only silica source.266–268 The crystallization of ZOLs requires much more time than the fully inorganic counterpart and the C content in the crystalline product is generally well below the expected value. For instance, ZOL-A (the hybrid counterpart of zeolite A) crystallized in 14 days instead of 1 day required by zeolite A under the same conditions and the C content was ca. the half of the expected (2.2 vs. 4.3 wt%).
The syntheses of hybrid zeolites with the ITQ-21, MFI and beta structures and with an unprecedented high organic content were claimed by Díaz et al.269 These authors employed a mixture of conventional (TEOS) and unconventional (BTEM and bis-(triethoxysilyl)-ethane, BTEE) silica sources at different molar ratios. No crystalline products were obtained by using BTEE and this is probably expected because the substitution of –O– by –CH2CH2– would introduce an intolerable strain in the structure and can be predicted unsuitable from the energetic point of view. In contrast, successful results were reported for the syntheses performed with BTEM, particularly for fully crystalline ZSM-5 and beta, which showed C contents of 3.6 and 4.9 wt%, respectively. The methylene groups incorporated in the framework are stable up to 673 K, a temperature which allows the elimination of the SDA molecules.269
More recently, Su et al. reported the synthesis of ZOF-X, a hybrid material with the FAU structure containing methylene groups in the framework.270 The elemental analysis of the best samples indicated a relatively low C content (1.3–1.5 wt%) while 29Si MAS NMR and FTIR analysis confirmed the presence of Si–C and –CH2– moieties, respectively. However, the same authors admitted that “…none of the characterization techniques can unambiguously ascertain carbon incorporation into the zeolite framework, due to the difficulty to exclude the presence of amorphous impurities in amounts of a few percent.”,270 highlighting in this way the still debated question on the real possibility to incorporate methylene groups in the zeolite framework. It is true that this process was predicted to be feasible from the energetic point of view,271 but direct and unambiguous proof is necessary for definitely assessing it. In all cases, it is evident that the incorporation of methylene in the framework is difficult as demonstrated by the following evidence:
- when the bis-silylated organic precursor only is used as silica source, the crystallization rate is very low compared to the conventional syntheses performed with, e.g., TEOS.
- the crystallization does not take place under nearly neutral conditions.
- for all the crystalline phases reported so far, the C content is much lower than the theoretical one, namely the maximum C content corresponding to the condensation of the disilane only.
All these lines of evidence indicate that the crystallization takes place only when [SiO4] groups are available and these groups can be added in the form of the conventional silica source or produced via the hydrolysis of the Si–C bonds, a very slow reaction occurring only in strong basic media.
In spite of this, bis-silylated organic precursors are the most promising route for preparing crystalline microporous hybrid materials. As a matter of fact, these compounds are the silica sources for the synthesis of hybrid organic–inorganic mesostructured materials known as periodic mesoporous organosilicas (PMOs).12,13 The pioneering work of Inagaki et al. with 1,4-bis-(triethoxysilyl)-benzene (BTEB) is interesting because it yielded a pseudo-ordered material in which the hydrolyzed BTEB units organized themselves into alternating organic and inorganic layers, with regular spacing of ca. 7.6 Å, lacking 3D order.272 Obviously, organization is imposed by silanol group condensation which, under some conditions, may lead to truly crystalline structures.273 Condensation of bis-silylated precursors inevitably leads to compact structures and additional components such as [SiO4] or [AlO4] tetrahedra are required to create open crystalline frameworks. In fact, the use of sodium aluminate gave a new class of crystalline microporous hybrid organic–inorganic aluminosilicates called Eni Carbon Silicates (ECS) with integral Si–C bonds and [AlO4] tetrahedra acting as spacers between the [CSiO3] groups.274 For the crystallization of ECSs, an accurate control of the hydrothermal conditions is necessary, in particular of the temperature which should be set at ca. 373 K in order to have a reasonable crystallization rate, avoiding the hydrolysis of the Si–C bonds. This reaction, in fact, prevails at higher temperatures leading to the formation of [SiO4] units which, in the presence of high concentration of Al and alkali metal ions (Na and/or K), leads to the crystallization of conventional, low silica zeolites (e.g. sodalite, cancrinite, Na-P1).274 A number of different crystalline phases have been synthesised so far using bis-silylated precursors of general formula (EtO)3Si–R–Si(OEt)3 with R = phenylene (ECS-1, ECS-2, ECS-3, ECS-14), biphenyl (ECS-5), p-diethylbenzene (ECS-6), trimethylene (ECS-7). For three of them (ECS-2,274 ECS-3275 and ECS-14276) the crystal structure has been determined (Fig. 14), allowing a better definition of the characteristics of this class of materials. As a common feature, the ECSs are formed by organic and inorganic layers, which alternate along the stacking direction. The aluminosilicate layer consists of [AlO4] tetrahedra bound to four different [CSiO3] (three in the case of ECS-14, being the fourth coordinated to a Na ion) tetrahedra. The [AlO4] tetrahedra do really act as spacers of the bis-silylated units, leading to the formation of low-density phases with different pore systems. In the case of ECS-2, the arrangement of these units forms cages surrounded by six phenylene rings not open to the exterior (Fig. 14b). By analogy with zeolites, ECS-2 can be regarded as a chlatrasil-type structure; in fact, in the cage there are ethanol molecules (well identified by 13C MAS NMR and by the structural analysis) that cannot be removed without leading to the collapse of the structure.274 ECS-3, on the other hand, possesses open porosity in the organic layer (Fig. 14c) and also two 8MR openings in the inorganic layer (Fig. 14d).275 The most interesting result has recently been achieved with the synthesis of ECS-14, whose structure resembles that of the AFI framework type (i.e. that of the well known AlPO-5277 and of silica counterpart SSZ-24278) with a porous structure constituted by a 12MR linear channel system running parallel to [001] (Fig. 14e and f).276 ECS-14, in reality, is formed by layers terminating with [AlO4] tetrahedra, which share three O atoms with three different [CSiO3] tetrahedra while the fourth is coordinated with a Na ion lying in the interlayer region (Fig. 14e). Na ions are, in turn, tetrahedrally coordinated with four O atoms belonging to different [AlO4] groups. The Na–O distances, however, are shorter than expected supporting the hypothesis that a partial substitution of Na by Si may occur.276
![Crystal structures of ECS materials. (a) ECS-2 and (b) the cage defined by six phenylene rings.274 (c) ECS-3 and (d) projection of the inorganic layer showing the two independent 8MR.275 (e) The stacking of layers in ECS-14 and (f) the [001] projection showing the 12MR linear channels of the AFI framework type.276 [CSiO3] tetrahedra in yellow, [AlO4] tetrahedra in blue, C atoms in green and Na atoms in fuchsia.](/image/article/2013/CY/c2cy20510f/c2cy20510f-f14.gif) | ||
| Fig. 14 Crystal structures of ECS materials. (a) ECS-2 and (b) the cage defined by six phenylene rings.274 (c) ECS-3 and (d) projection of the inorganic layer showing the two independent 8MR.275 (e) The stacking of layers in ECS-14 and (f) the [001] projection showing the 12MR linear channels of the AFI framework type.276 [CSiO3] tetrahedra in yellow, [AlO4] tetrahedra in blue, C atoms in green and Na atoms in fuchsia. | ||
A significant development of this new and fascinating class of materials is easily predictable, the only limit being the availability of bis-silylated organic precursors, whose synthesis is in many cases very difficult and expensive. However, the huge amount of work performed on hybrid mesostructured materials (PMO) has demonstrated that they can be obtained with a large number of organic groups. This is really encouraging and it is our opinion that efforts in this direction will surely lead to the preparation of true hybrid organic–inorganic zeolite structures, of which the first clear example is constituted by ECS-14. In addition, it is necessary to solve another problem that limits somewhat the potential applications of these materials. The problem is, in fact, the very low Si/Al ratio in the solids that, similarly to what happens in zeolites, prevents the exchange in acid form and the applications of these materials in the field of acid catalysis.
Conclusions and outlook
We have discussed herein some recent developments in the field of zeolites, which form the most important class of microporous solids due to their practical importance in different technological areas. It is quite surprising to realize that, in spite of the 50+ years of research, there is still space for the innovation in this field and that makes zeolites special in the world of advanced materials. In several cases, in fact, results obtained at the laboratory level and considered breakthroughs in reality have little or no practical implications because of, e.g., the impossibility of scaling up the material in real devices, the costs, etc.The world of microporous solids is in reality formed by several different classes of materials. We have decided, however, to focus the attention on zeolites because the driving forces for the innovation arise not only from the needs of the chemical, petrochemical and refinery industries but even from the opportunities given by advanced technological areas, recently attracted by this class of materials. We have neglected other important classes of materials that constitute by themselves a well-defined branch of science (e.g. MOF) or that, after the initial enthusiasm, were practically abandoned (e.g. octahedral-pentahedral-tetrahedral, OPT, metallosilicates). Moreover, of the several areas of innovation in zeolites science, we have selected those that, in our opinion, could have the highest practical implications.
One of them is aimed at synthesising materials with unprecedented porous systems, particularly important if characterized by extra-large pores (14MR openings or more) or by multidimensional channel systems. Decisive, in this regard, are the deeper knowledge of the phenomena occurring during the zeolite nucleation and growth of the optimal characteristics of the organic additives (SDAs) as well as of the peculiar role of the heteroatoms in the stabilization of specific secondary building units (e.g. D4R with Ge), necessary for the formation of low-density frameworks. Furthermore, the development of systems for high-throughput experimentation allowing performing simultaneously even hundreds of synthesis runs has improved the screening capabilities and the possibility of discovering new materials, saving costly reagents and time.
The impressive results achieved in recent years make it possible other interesting developments in the synthesis of new materials but also raise questions about the perspectives of using new low-density framework topologies with extra-large and/or multidimensional pore systems. Although interesting results have been obtained in the catalytic tests performed with these new materials, the use of Ge and of complex commercially unavailable organic additives has a dramatic impact on the costs of the final materials, limiting their potential applications, at least in the petrochemical and refining processes that require high amounts of the catalyst. It should be considered, in fact, that the current catalysts are based on the use of zeolites synthesized in the purely inorganic system (e.g. USY employed in the formulation of the FCC catalyst) or in the presence of simple and relatively inexpensive SDAs (e.g. TEAOH for the zeolite beta or TPAOH for the zeolite ZSM-5). These new materials can compete with the zeolites currently in use only in the event of outstanding catalytic performances combined with a reduction of production costs obtainable with the elimination of Ge and the concurrent addition of Al in the synthesis recipe and with the use of cheaper SDAs.
We do not want to belittle the significance of the results achieved so far and consider the search for new framework topologies of secondary importance. Instead, we want to emphasize the problems that must be solved to promote these materials from scientific curiosity to the primary components of important transformation processes.
It is in this context that we place the other promising research topics. One of them, still not fully explored, is related to 2D zeolites. The discovery that some zeolites crystallize naturally (e.g. MWW) or upon choosing the appropriate SDA and condition (e.g. PREFER) in the form of layered precursors has opened new perspectives in the engineering of new materials. The layered precursor, in fact, can be transformed into the 3D ordered structure upon calcination or can be swollen in the presence of a surfactant to produce delaminated materials in which the layers form a sort of house of cards in which most of the layer surface is accessible. Moreover, the presence of silanol groups on the surface of the layers ensures anchoring points for the attachment of single molecules (e.g. dichlorodimethylsilane) or oligomers of various nature (e.g. silica or alumina). In the first case, one can produce 3D ordered structures in which the interlayer distance is expanded (hence the term Interlayer Expanded Zeolites, IEZ) with the formation of pores with dimensions greater than those due to the original zeolite. In the second case, we obtain structures with a hierarchical porous structure: the mesoporosity is generated by the pillars while the microporosity is a property of the zeolite layers. The family of zeolites 2D has recently expanded with the synthesis of ZSM-5 nanosheets, achieved by using a suitably designed SDA and with the first case of 3D → 2D conversion of a preformed zeolite with UTL topology, thus opening new opportunities in the synthesis of new materials.
Even the change of textural characteristics of known zeolites can improve their catalytic properties. The synthesis of hierarchical porous structures in which two levels of porosity (mesopores and micropores) coexist in the same particle (single crystals or agglomerates of nanocrystals) is one of the hottest topics in modern zeolite science. In fact, it is seen as a method of increasing the accessibility of the active sites located inside the crystals, avoiding the diffusion limitations imposed by the pore sizes. Similar to the early methods based on the demetallation of preformed zeolites, several other approaches have been proposed taking advantages of the knowledge accumulated in the synthesis of both crystalline microporous and amorphous mesostructured materials. Also in this area, the creativity of the researchers has been decisive in proposing new synthesis approaches that sometimes led to the preparation of materials of high potential interest. The attention so far has been directed toward the modification of known zeolite structures and this has demonstrated the correctness of the concept of hierarchical porous system. However, some aspects deserve to be studied in detail. In particular, we refer to the properties and the stability of crystalline nano-domains and to the characteristics of the active sites, especially in those materials whose synthesis requires the presence of a dual template system. Furthermore, in some cases, the true benefits associated with the presence of the mesoporous structure with respect to the “state of the art” catalyst are still not demonstrated. Only after this comparison it will be possible to assess whether these materials can compete with the best currently available catalysts.
Lastly, we focused the attention on a research topic, still largely unexplored, but we believe very attractive: the synthesis of hybrid organic–inorganic zeolites. The availability of crystalline silicates or aluminosilicates with organic groups in the framework could indeed open new possibilities not only in the fields of catalysis and separation but also in technologically advanced sectors (e.g. sensors, optical devices, nonlinear optics, …). When considering the zeolites as such, attempts to derivatise the crystalline structure with organic groups via grafting or co-condensation were unsuccessful or, at least, of limited applicability. On the other hand, the incorporation of an organic group in the zeolite framework suffers the strict limits imposed by the crystalline structure. The only option now demonstrated with reasonable certainty (although still questioned by some authors) is to replace part (25% maximum) of framework O atoms with methylene groups (–CH2–) by using a trialkoxysilylmethane as silica source. This kind of isomorphous substitution of the anions is not expected to be possible for other even slightly bigger organic groups (e.g. dimethylene, –CH2CH2–) because their incorporation in the framework would induce strain in the crystal structure and is predictably unfeasible from an energetic point of view. The bis-silylated molecule, on the other hand, can be used for binding preformed building blocks, such as, for example, the layers of 2D zeolite precursors (see the case of the Interlayer Expanded Zeolites, IEZ). The recent synthesis of the ECS class of materials has demonstrated that it is possible to obtain hybrid organic–inorganic crystalline porous aluminosilicates, with characteristics of the inorganic layers related to those of the zeolites. The research on this class of materials is still in the early stages and many issues are still open. Preliminary results seem to indicate that this system is quite versatile in the sense that, for each bis-silylated precursor, there are several possible crystalline phases with different organization of both the inorganic and the organic layers. However, the syntheses are rather complex, requiring an accurate control of the conditions to avoid hydrolysis of the Si–C bond and the consequent formation of conventional zeolites. The increase of the low Si/Al ratio (a limiting factor for possible applications in acid catalysis) is a problem still unsolved. Therefore, efforts are necessary to understand in more detail the decisive parameters in the synthesis of these materials.
Obviously, we have examined just some of the topics attracting the attention of the scientific community and which are, in our opinion, very promising. There are, however, other emerging research topics on microporous solids, which are much less explored, but which are expected to lead to interesting developments. One of these is represented by the chiral zeolites, a topic that only recently came to the attention of researchers. In fact the availability of chiral zeolite structures would expand the use of microporous solids for applications in enantioselective processes (catalysis, separations) of high interest, e.g., to the pharmaceutical, agrochemical and fragrance industries. Although the concept of chiral zeolites is now well assessed and verified experimentally, unfortunately there are some difficult challenges to face, the most important being that related to the preparation of pure enantiomeric forms of zeolites. The research in this field is still in its infancy but interesting developments are expected. For the moment, we refer the interested reader to some recent papers that illustrate the concept of chirality in microporous solids, the problems that exist today and the possible solutions.279–282
In conclusion, what reported here clearly demonstrates how research on microporous solids in general and zeolites in particular is still attracting the attention of many research groups both from the academia and from the industry. The involvement of research teams of major industrial groups reflects the importance of the topics discussed here. We believe that this is an incentive for the entire scientific community working in the different fields of the science and technology of zeolites and related microporous materials to increase efforts for producing materials with improved properties, providing new solutions to the growing demand for efficient technologies, with lower energy consumption and a lower environmental impact than those available today.
Notes and references
- A. F. Cronstedt, Kongl. Vetenskaps Acad. Handl. Stockh., 1756, 17, 120 Search PubMed.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663 CrossRef CAS.
- C. Colella, Curr. Phys. Chem., 2012, 2, 126 CAS.
- R. M. Barrer and P. J. Denny, J. Chem. Soc., 1961, 971 RSC.
- Database of zeolite structures: http://www.iza-structure.org/databases, as in October 2012.
- J. Rocha and Z. Lin, Rev. Mineral. Geochem., 2005, 57, 173 CrossRef CAS.
- C. K. King'ondu, N. Opembe, C.-H. Chen, K. Ngala, H. Huang, A. Iyer, H. F. Garcés and S. L. Suib, Adv. Funct. Mater., 2011, 21, 312 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- J. Coronas, Chem. Eng. J., 2010, 156, 236 CrossRef CAS.
- R. E. Morris, Top. Catal., 2010, 53, 1291 CrossRef CAS.
- J. Čejka, G. Centi, J. Perez-Pariente and W. J. Roth, Catal. Today, 2012, 179, 2 CrossRef.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS.
- F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608 RSC.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756 CrossRef CAS.
- S. L. Lawton and W. J. Rohrbaugh, Science, 1990, 247, 1319 CAS.
- J. P. Arhancet and M. E. Davis, Chem. Mater., 1991, 3, 567 CrossRef CAS.
- H. van Koningsveld, H. van Bekkum and J. C. Jansen, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 127 CrossRef.
- H. Gies and B. Marler, Zeolites, 1992, 12, 42 CrossRef CAS.
- H. Gies, B. Marler and U. Werthmann, in Molecular Sieves: Science and Technology–Vol. 1 Synthesis, ed. H. G. Karge and J. Weitkamp, Springer, Berlin, 1998, p. 35 Search PubMed.
- Y. Kubota, M. M. Helmkamp, S. I. Zones and M. E. Davis, Microporous Mater., 1996, 6, 213 CrossRef CAS.
- P. Wagner, Y. Nakagawa, G. S. Lee, M. E. Davis, S. Elomari, R. C. Medrud and S. I. Zones, J. Am. Chem. Soc., 2000, 122, 263 CrossRef CAS.
- S. I. Zones, Zeolites, 1989, 9, 458 CrossRef CAS.
- S. I. Zones, Y. Nakagawa, L. T. Yuen and T. V. Harris, J. Am. Chem. Soc., 1996, 118, 7558 CrossRef CAS.
- Y. Nakagawa, G. S. Lee, T. V. Harris, L. T. Yuen and S. I. Zones, Microporous Mesoporous Mater., 1998, 22, 69 CrossRef CAS.
- R. Millini, L. Carluccio, F. Frigerio, W. O. Parker Jr. and G. Bellussi, Microporous Mesoporous Mater., 1998, 24, 199 CrossRef CAS.
- S. Zanardi, G. Cruciani, L. C. Carluccio, G. Bellussi, C. Perego and R. Millini, Angew. Chem., Int. Ed., 2002, 41, 4109 CrossRef CAS.
- R. Millini, D. Berti, D. Ghisletti, W. O. Parker Jr., L. C. Carluccio and G. Bellussi, Stud. Surf. Sci. Catal., 2002, 142, 61 CrossRef.
- S. I. Zones, A. W. Burton, G. S. Lee and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 9066 CrossRef CAS.
- H. Koller, R. F. Lobo, S. L. Burkett and M. E. Davis, J. Phys. Chem., 1995, 99, 12588 CrossRef CAS.
- D. F. Shantz, J. Schmedt auf der Gunne, H. Koller and R. F. Lobo, J. Am. Chem. Soc., 2000, 122, 6659 CrossRef CAS.
- W. O. Parker Jr. and R. Millini, J. Am. Chem. Soc., 2006, 128, 1450 CrossRef.
- B. J. Campbell, G. Bellussi, L. Carluccio, G. Perego, A. K. Cheetham, D. E. Cox and R. Millini, Chem. Commun., 1998, 1725 RSC.
- C. M. Freeman, C. R. A. Catlow, J. M. Thomas and S. Brode, Chem. Phys. Lett., 1991, 186, 137 CrossRef CAS.
- D. W. Lewis, C. M. Freeman and C. R. A. Catlow, J. Phys. Chem., 1995, 99, 11194 CrossRef CAS.
- D. W. Lewis, D. J. Willock, C. R. A. Catlow, J. M. Thomas and G. J. Hutchings, Nature, 1996, 382, 604 CrossRef CAS.
- D. J. Willock, D. W. Lewis, C. R. A. Catlow, G. J. Hutchings and J. M. Thomas, J. Mol. Catal. A: Chem., 1997, 119, 415 CrossRef CAS.
- D. W. Lewis, G. Sankar, J. K. Wyles, J. M. Thomas, C. R. A. Catlow and D. J. Willock, Angew. Chem., Int. Ed. Engl., 1997, 36, 2675 CrossRef CAS.
- P. A. Barrett, R. H. Jones, J. M. Thomas, G. Sankar, I. J. Shannon and C. R. A. Catlow, Chem. Commun., 1996, 2001 RSC.
- M. M. J. Treacy, K. H Randall, S. Rao, J. A. Perry and D. J. Chadi, Z. Kristallogr., 1997, 212, 768 CrossRef CAS.
- M. M. J. Treacy, I. Rivin, E. Balkovsky, K. H. Randall and M. D. Foster, Microporous Mesoporous Mater., 2004, 74, 121 CrossRef CAS.
- M. D. Foster, A. Simperler, R. G. Bell, O. Delgado Friedrichs, F. A. Almeida Paz and J. Klinowski, Nat. Mater., 2004, 3, 234 CrossRef CAS.
- D. J. Earl and M. W. Deem, Ind. Eng. Chem. Res., 2006, 45, 5449 CrossRef CAS.
- J. M. Thomas and J. Klinowski, Angew. Chem., Int. Ed., 2007, 46, 7160 CrossRef CAS.
- S. M. Woodley and R. Catlow, Nat. Mater., 2008, 7, 937 CrossRef CAS.
- R. A. van Santen, J. Phys. Chem., 1984, 88, 5768 CrossRef CAS.
- M. A. Camblor, A. Corma and S. Valencia, Chem. Commun., 1996, 2365 RSC.
- M. J. Díaz-Cabañas, P. A. Barrett and M. A. Camblor, Chem. Commun., 1998, 1881 RSC.
- M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Top. Catal., 1999, 9, 59 CrossRef CAS.
- M. A. Camblor, P. A. Barrett, M. J. Díaz-Cabañas, L. A. Villaescusa, M. Puche, T. Boix, E. Pérez and H. Koller, Microporous Mesoporous Mater., 2001, 48, 11 CrossRef CAS.
- S. I. Zones, R. J. Darton, R. Morris and S. J. Hwang, J. Phys. Chem. B, 2005, 109, 652 CrossRef CAS.
- P. Caullet, J.-L. Paillaud, A. Simon-Masseron, M. Soulard and J. Patarin, C. R. Chim., 2005, 8, 245 CrossRef CAS.
- S. I. Zones, S.-J. Hwang, S. Elomari, I. Ogino, M. E. Davis and A. W. Burton, C. R. Chim., 2005, 8, 267 CrossRef CAS.
- G. Perego, R. Millini and G. Bellussi, in Molecular Sieves: Science and Technology–Vol. 1 Synthesis, ed. H. G. Karge and J. Weitkamp, Springer, Berlin, 1998, p. 187 Search PubMed.
- R. Millini, G. Perego and G. Bellussi, Top. Catal., 1999, 9, 13 CrossRef CAS.
- A. Burton, S. Elomari, C. Y. Chen, R. C. Medrud, I. Y. Chan, L. M. Bull, C. Kibby, T. V. Harris, S. I. Zones and E. S. Vittoratos, Chem.–Eur. J., 2003, 9, 5737 CrossRef CAS.
- D. L. Dorset and G. J. Kennedy, J. Phys. Chem. B, 2005, 109, 13891 CrossRef CAS.
- A. W. Burton, S. Elomari, I. Chan, A. Pradhan and C. Kibby, J. Phys. Chem. B, 2005, 109, 20266 CrossRef CAS.
- S. Elomari, A. W. Burton, K. Ong, A. R. Pradhan and I. Y. Chan, Chem. Mater., 2007, 19, 5485 CrossRef CAS.
- S. Elomari, A. Burton, R. C. Medrud and R. Grosse-Kunstleve, Microporous Mesoporous Mater., 2009, 118, 325 CrossRef CAS.
- D. Xie, L. B. McCusker and Ch. Baerlocher, J. Am. Chem. Soc., 2011, 133, 20604 CrossRef CAS.
- S. Merlino, Eur. J. Mineral., 1990, 2, 809 CAS.
- O. V. Petersen, G. Giester, F. Brandstätter and G. Niedermayr, Can. Mineral., 2002, 40, 173 CrossRef CAS.
- G. Giuseppetti, F. Mazzi, C. Tadini and E. Galli, N. Jb. Miner. Mh., 1991, 7, 307 Search PubMed.
- A. K. Cheetham, H. Fjellvåg, T. E. Gier, K. O. Kongshaug, K. P. Lillerud and G. D. Stucky, Stud. Surf. Sci. Catal., 2001, 135, 158 CrossRef.
- M. J. Annen, M. E. Davis, J. B. Higgins and J. L. Schlenker, J. Chem. Soc., Chem. Commun., 1991, 1175 RSC.
- C. C. Freyhardt, R. F. Lobo, S. Khodabandeh, J. E. Lewis, M. Tsapatsis, M. Yoshikawa, M. A. Camblor, M. Pan, M. M. Helmkamp, S. I. Zones and M. E. Davis, J. Am. Chem. Soc., 1996, 118, 7299 CrossRef CAS.
- L. B. McCusker, R. W. Grosse-Kunstleve, Ch. Baerlocher, M. Yoshikawa and M. E. Davis, Microporous Mater., 1996, 6, 295 CrossRef CAS.
- C. Röhrig and H. Gies, Angew. Chem., Int. Ed. Engl., 1995, 34, 63 CrossRef.
- H. Li and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 10569 CrossRef CAS.
- T. Conrandsson, M. S. Dadachov and X. D. Zou, Microporous Mesoporous Mater., 2000, 41, 183 CrossRef.
- Y. Mathieu, J.-L. Paillaud, P. Caullet and N. Bats, Microporous Mesoporous Mater., 2004, 75, 13 CrossRef CAS.
- L. A. Villaescusa, P. A. Barrett and M. A. Camblor, Angew. Chem., Int. Ed., 1999, 38, 1997 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas and V. Fornés, Angew. Chem., Int. Ed., 2000, 39, 2346 CrossRef CAS.
- T. Blasco, A. Corma, M. J. Díaz-Cabañas, F. Rey, J. A. Vidal-Moya and C. M. Zicovich-Wilson, J. Phys. Chem. B, 2002, 106, 2643 CrossRef.
- A. Corma, M. T. Navarro, F. Rey, J. Rius and S. Valencia, Angew. Chem., Int. Ed., 2001, 40, 2277 CrossRef CAS.
- O. V. Shvets, N. Kasian, A. Zukal, J. Pinkas and J. Čejka, Chem. Mater., 2010, 22, 3482 CrossRef CAS.
- D. E. Akporiaye, I. M. Dahl, A. Karlsson and R. Wendelbo, Angew. Chem., Int. Ed., 1998, 37, 609 CrossRef CAS.
- D. Akporiaye, I. Dahl, A. Karlsson, M. Plassen, R. Wendelbo, D. S. Bern, R. W. Broach, G. J. Lewis, M. Miller and J. Moscoso, Microporous Mesoporous Mater., 2001, 48, 367 CrossRef CAS.
- B. Jandeleit, D. J. Schaefer, T. S. Powers, H. W. Turner and W. H. Weinberg, Angew. Chem., Int. Ed., 1999, 38, 2494 CrossRef CAS.
- L. M. Knight and G. J. Lewis, Stud. Surf. Sci. Catal., 2004, 154, 171 CrossRef.
- Ø. B. Vistad, D. E. Akporiaye, K. Mejland, R. Wendelbo, A. Karlsson, M. Plassen and K. P. Lillerud, Stud. Surf. Sci. Catal., 2004, 154, 731 CrossRef.
- Y. Song, J. Yu, Y. Li, Y. Zhang, M. Zhang and R. Xu, Stud. Surf. Sci. Catal., 2004, 154, 1028 CrossRef.
- K. Choi, D. Gardner, N. Hilbrandt and T. Bein, Angew. Chem., Int. Ed., 1999, 36, 2891 CrossRef.
- J. Klein, C. W. Lehmann, H.-W. Schmidt and W. F. Maier, Angew. Chem., Int. Ed., 1998, 37, 3369 CrossRef CAS.
- J. M. Newsam, T. Bein, J. Klein, W. F. Maier and W. Stickert, Microporous Mesoporous Mater., 2001, 48, 355 CrossRef CAS.
- M. Moliner, J. M. Serra, A. Corma, E. Argente, S. Valero and V. Botti, Microporous Mesoporous Mater., 2005, 78, 73 CrossRef CAS.
- A. Cantin, A. Corma, M. J. Diaz-Cabanas, J. L. Jordá and M. Moliner, J. Am. Chem. Soc., 2006, 128, 4612 CrossRef.
- A. Corma, M. J. Díaz-Cabanas, M. Moliner and C. Martínez, J. Catal., 2006, 241, 312 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, C. Martínez and M. Moliner, Nature, 2006, 443, 842 CrossRef CAS.
- J. Jiang, J. L. Jorda, J. Yu, L. A. Baumes, E. Mugnaioli, M. J. Diaz-Cabanas, U. Kolb and A. Corma, Science, 2011, 333, 1131 CrossRef CAS.
- J. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 4986 CrossRef CAS.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120 CrossRef CAS.
- K. G. Strohmaier and D. E. W. Vaughan, J. Am. Chem. Soc., 2003, 125, 16035 CrossRef CAS.
- J.-L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, F. Rey, S. Nicolopoulus and K. Boulahya, Chem. Commun., 2004, 1356 RSC.
- J. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997 CrossRef CAS.
- M. Estermann, L. B. McCusker, Ch. Baerlocher, A. Merouche and H. Kessler, Nature, 1991, 352, 320 CrossRef CAS.
- M. G. Wu, M. W. Deem, S. A. Elomari, R. C. Medrud, S. I. Zones, T. Maesen, C. Kibby, C.-Y. Chen and I. Y. Chan, J. Phys. Chem. B, 2002, 106, 264 CrossRef CAS.
- G. S. Lee, Y. Nakagawa, S.-J. Hwang, M. E. Davis, P. Wagner, L. Beck and S. I. Zones, J. Am. Chem. Soc., 2002, 124, 7024 CrossRef CAS.
- R. Castañeda, A. Corma, V. Fornés, F. Rey and J. Rius, J. Am. Chem. Soc., 2003, 125, 7820 CrossRef.
- D. L. Dorset, S. C. Weston and S. S. Dhingra, J. Phys. Chem. B, 2006, 110, 2045 CrossRef CAS.
- D. L. Dorset, G. J. Kennedy, K. G. Strohmaier, M. J. Díaz-Cabañas, F. Rey and A. Corma, J. Am. Chem. Soc., 2006, 128, 8862 CrossRef CAS.
- D. L. Dorset, K. G. Strohmaier, C. E. Kliewer, A. Corma, M. J. Díaz-Cabañas, F. Rey and C. J. Gilmore, Chem. Mater., 2008, 20, 5325 CrossRef CAS.
- L. B. McCusker, Ch. Baerlocher, S. T. Wilson and R. W. Broach, J. Phys. Chem. C, 2009, 113, 9838 CAS.
- B. K. Marcus and B. M. Lok, US Patent 4.857,288, 1989 Search PubMed.
- M. Dodin, J.-L. Paillaud, Y. Lorgouilloux, P. Caullet, E. Elkaïm and N. Bats, J. Am. Chem. Soc., 2010, 132, 10221 CrossRef CAS.
- M. Moliner, J. González, M. T. Portilla, T. Willhammar, F. Rey, F. J. Llopis, X. Zou and A. Corma, J. Am. Chem. Soc., 2011, 133, 9497 CrossRef CAS.
- A. Moini, K. D. Schmitt, E. W. Valyoscik and R. F. Polomski, Zeolites, 1994, 14, 504 CrossRef CAS.
- S.-H. Lee, C.-H. Shin, D.-K. Yang, S.-D. Ahn, I.-S. Nam and S. B. Hong, Microporous Mesoporous Mater., 2004, 68, 97 CrossRef CAS.
- S.-H. Lee, C.-H. Shin, G. J. Choi, T.-J. Park, I.-S. Nam, B. Han and S. B. Hong, Microporous Mesoporous Mater., 2003, 60, 237 CrossRef CAS.
- A. Corma, M. Puche, F. Rey, G. Sankar and S. J. Teat, Angew. Chem., Int. Ed., 2003, 42, 1156 CrossRef CAS.
- M. J. Díaz-Cabañas, M. A. Camblor, Z. Liu, T. Ohsuna and O. Terasaki, J. Mater. Chem., 2002, 12, 249 RSC.
- Z. Liu, T. Ohsuna, O. Terasaki, M. A. Camblor, M. J. Díaz-Cabañas and K. Hiraga, J. Am. Chem. Soc., 2001, 123, 5370 CrossRef CAS.
- F. Gramm, Ch. Baerlocher, L. B. McCusker, S. J. Warrender, P. A. Wright, B. Han, S. B. Hong, Z. Liu, T. Ohsuna and O. Terasaki, Nature, 2006, 444, 79 CrossRef CAS.
- S. B. Hong, H.-K. Min, C.-H. Shin, P. A. Cox, S. J. Warrender and P. A. Wright, J. Am. Chem. Soc., 2007, 129, 10870 CrossRef CAS.
- Ch. Baerlocher, F. Gramm, L. Massüger, L. B. McCusker, Z. He, S. Hovmöller and X. Zou, Science, 2007, 315, 1113 CrossRef CAS.
- A. Corma, F. Rey, S. Valencia, J. L. Jordá and J. Rius, Nat. Mater., 2003, 2, 493 CrossRef CAS.
- Ch. Baerlocher, D. Xie, L. B. McCusker, S.-J. Hwang, I. Y. Chan, K. Ong, A. W. Burton and S. I. Zones, Nat. Mater., 2008, 7, 631 CrossRef CAS.
- A. Jackowski, S. I. Zones, S.-J. Hwang and A. W. Burton, J. Am. Chem. Soc., 2009, 131, 1092 CrossRef CAS.
- A. J. Blake, K. R. Franklin and B. M. Lowe, J. Chem. Soc., Dalton Trans., 1988, 2513 RSC.
- S. J. Andrews, M. Z. Papiz, R. McMeeking, A. J. Blake, B. M. Lowe, K. R. Franklin, J. R. Helliwell and M. M. Harding, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 73 CrossRef.
- B. Marler, M. A. Camblor and E. Gies, Microporous Mesoporous Mater., 2006, 90, 87 CrossRef CAS.
- R. Millini, G. Perego, W. O. Parker Jr., G. Bellussi and L. Carluccio, Microporous Mater., 1995, 4, 221 CrossRef CAS.
- S. L. Lawton, A. S. Fung, G. J. Kennedy, L. B. Alemany, C. D. Chang, G. H. Hatzikos, D. N. Lissy, M. K. Rubin, H.-K. C. Timken, S. Steuernagel and D. E. Woessner, J. Phys. Chem., 1996, 100, 3788 CrossRef CAS.
- W. J. Roth and J. Čejka, Catal. Sci. Technol., 2011, 1, 43 CAS.
- S. Zanardi, A. Alberti, G. Cruciani, A. Corma, V. Fornés and M. Brunelli, Angew. Chem., Int. Ed., 2004, 43, 4933 CrossRef CAS.
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892 CrossRef CAS.
- D. L. Dorset and G. J. Kennedy, J. Phys. Chem. B, 2004, 108, 15216 CrossRef CAS.
- B. Marler, N. Ströter and H. Gies, Microporous Mesoporous Mater., 2005, 83, 201 CrossRef CAS.
- Y. Wang, B. Marler, H. Gies and U. Müller, Chem. Mater., 2005, 17, 43 CrossRef CAS.
- P. S. Wheatley and R. E. Morris, J. Mater. Chem., 2006, 16, 1035 RSC.
- W. L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, J. Chem. Soc., Chem. Commun., 1995, 2187 RSC.
- W. L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259 CrossRef.
- U. Oberhagenmann, P. Bayat, B. Marler, H. Gies and J. Rius, Angew. Chem., Int. Ed. Engl., 1996, 35, 2869 CrossRef.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246 CrossRef CAS.
- W. J. Roth and D. L. Dorset, Microporous Mesoporous Mater., 2011, 142, 32 CrossRef CAS.
- W. J. Roth, C. T. Kresge, J. C. Vartuli, M. E. Leonowicz, A. S. Fung and S. B. McCullen, Stud. Surf. Sci. Catal., 1995, 94, 301 CrossRef CAS.
- Y. J. He, G. S. Nivarthy, F. Eder, K. Seshan and J. A. Lercher, Microporous Mesoporous Mater., 1998, 25, 207 CrossRef CAS.
- J.-O. Barth, A. Jentys, J. Kornatowski and J. A. Lercher, Chem. Mater., 2004, 16, 724 CrossRef CAS.
- J. Kornatowski, J.-O. Bath, K. Erdmann and M. Rozwadowski, Mesoporous Mater., 2006, 90, 251 CrossRef.
- W. J. Roth and C. T. Kresge, Microporous Mesoporous Mater., 2011, 144, 158 CrossRef CAS.
- P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 8178 CrossRef CAS.
- A. Corma, U. Díaz, T. García, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011 CrossRef CAS.
- J. Ruan, P. Wu, B. Slater, Z. Zhao, L. Wu and O. Terasaki, Chem. Mater., 2009, 21, 2904 CrossRef CAS.
- H. Gies, U. Müller, B. Yilmaz, T. Tatsumi, B. Xie, F.-S. Xiao, X. Bao, W. Zhang and D. De Vos, Chem. Mater., 2011, 23, 2545 CrossRef CAS.
- H. Gies, U. Müller, B. Yilmaz, M. Feyen, T. Tatsumi, H. Imai, H. Zhang, B. Xie, F.-S. Xiao, X. Bao, W. Zhang, T. De Baedemaeker and D. De Vos, Chem. Mater., 2012, 24, 1536 CrossRef CAS.
- A. Corma, V. Fornes, S. B. Pergher, Th. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353 CrossRef CAS.
- A. Corma, V. Fornés, J. M. Guil, S. B. Pergher, Th. L. M. Maesen and J. G. Buglass, Microporous Mesoporous Mater., 2000, 38, 301 CrossRef CAS.
- P. Wu, D. Nuntasri, J. Ruan, Y. Liu, M. He, W. Fan, O. Terasaki and T. Tatsumi, J. Phys. Chem. B, 2004, 108, 19126 CrossRef CAS.
- S. Maheshwari, E. Jordan, S. Kumar, F. S. Bates, R. L. Penn, D. F. Shantz and M. Tsapatsis, J. Am. Chem. Soc., 2008, 130, 1507 CrossRef CAS.
- I. Ogino, M. M. Nigra, S.-J. Hwang, J.-M. Ha, T. Rea, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2011, 133, 3288 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, Angew. Chem., Int. Ed., 2000, 39, 1499 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, J. Am. Chem. Soc., 2000, 122, 2804 CrossRef CAS.
- E. A. Eilertsen, I. Ogino, S.-J. Hwang, T. Rea, S. Yeh, S. I. Zones and A. Katz, Chem. Mater., 2011, 23, 5404 CrossRef CAS.
- A. Corma, V. Fornés and U. Díaz, Chem. Commun., 2001, 2642 RSC.
- T. Moteki, W. Chaikittisilp, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2008, 130, 15780 CrossRef CAS.
- Z. Li, B. Marler and H. Gies, Chem. Mater., 2008, 20, 1896 CrossRef CAS.
- L. Massüger, Ch. Baerlocher, L. B. McCusker and M. A. Zwijnenburg, Microporous Mesoporous Mater., 2007, 105, 75 CrossRef.
- T. Ikeda, Y. Akiyama, F. Izumi, Y. Kiyozumi, F. Mizukami and T. Kodaira, Chem. Mater., 2001, 13, 1286 CrossRef CAS.
- T. Ikeda, Y. Oumi, K. Honda, T. Sano, K. Momma and F. Izumi, Inorg. Chem., 2011, 50, 2294 CrossRef CAS.
- Y. Die, M. Torii, N. Tsunoji, M. Sadakane and T. Sano, Chem. Commun., 2012, 48, 7073 RSC.
- K. Na, C. Jo, J. Kim, W.-S. Ahn and R. Ryoo, ACS Catal., 2011, 1, 901 CrossRef CAS.
- J. Wang, L. Xu, K. Zhang, H. Peng, H. Wu, J. Jiang, Y. Liu and P. Wu, J. Catal., 2012, 288, 16 CrossRef CAS.
- K. Na, M. Choi, W. Park, Y. Sakamoto, O. Terasaki and R. Ryoo, J. Am. Chem. Soc., 2010, 132, 4169 CrossRef CAS.
- W. J. Roth, O. V. Shvets, M. Sharnzhy, P. Chlubná, M. Kubù, P. Nachtigall and J. Čejka, J. Am. Chem. Soc., 2011, 133, 6130 CrossRef CAS.
- A. Corma, C. Corell, J. Pérez-Pariente, J. M. Guil, R. Guil-López, S. Nicolopoulos, J. Gonzalez Calbet and M. Vallet-Regi, Zeolites, 1996, 16, 7 CrossRef CAS.
- C. Perego, S. Amarilli, R. Millini, G. Bellussi, G. Girotti and G. Terzoni, Microporous Mater., 1996, 6, 395 CrossRef CAS.
- A. Corma, V. Martínez-Soria and E. Schnoeveld, J. Catal., 2000, 192, 163 CrossRef CAS.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910 CAS.
- G. Sastre, C. R. A. Catlow and A. Corma, J. Phys. Chem. B, 1999, 103, 5187 CrossRef CAS.
- M. A. Climent, A. Corma and A. Velty, Appl. Catal., A, 2004, 263, 155 CrossRef CAS.
- J. Aguilar, S. B. C. Pergher, C. Detoni, A. Corma, F. V. Melo and E. Sastre, Catal. Today, 2008, 133–135, 667 CrossRef CAS.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494 CrossRef CAS.
- S. C. Larsen, J. Phys. Chem. C, 2007, 111, 18464 CAS.
- C. Madsen and C. J. H. Jacobsen, Chem. Commun., 1999, 673 RSC.
- I. Schmidt, C. Madsen and C. J. H. Jacobsen, Inorg. Chem., 2000, 39, 2279 CrossRef CAS.
- C. J. H. Jacobsen, C. Madsen, T. V. W. Janssens, H. J. Jakobsen and J. Skibsted, Microporous Mesoporous Mater., 2000, 39, 393 CrossRef CAS.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and A. Peral, Chem. Mater., 2006, 18, 2462 CrossRef CAS.
- D. P. Serrano, J. Aguado, G. Morales, J. M. Rodríguez, A. Peral, M. Thommes, J. D. Epping and B. F. Chmelka, Chem. Mater., 2009, 21, 641 CrossRef CAS.
- J. Aguado, D. P. Serrano and J. M. Rodríguez, Microporous Mesoporous Mater., 2008, 115, 504 CrossRef CAS.
- J. Aguado, D. P. Serrano, J. M. Escola and A. Peral, J. Anal. Appl. Pyrol., 2009, 85, 352 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, K. D. Schmidt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- F. Di Renzo, A. Galarneau, P. Trens and F. Fajulà, in Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, Wiley-VCH, Weinheim, 2002, ch. 5, p. 1311 Search PubMed.
- J.-W. Da, C.-M. Song, L. Qian, J.-M. Su and X.-Z. Xu, J. Porous Mater., 2008, 15, 189 CrossRef.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev. Sci. Eng., 2003, 45, 297 CAS.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896 CrossRef CAS.
- J. Pérez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530 RSC.
- R. Chal, C. Gérardin, M. Bulut and S. van Donk, ChemCatChem, 2011, 3, 67 CrossRef CAS.
- K. R. Kloetstra, H. van Bekkum and J. C. Jansen, Chem. Commun., 1997, 2281 RSC.
- M. J. Verhoef, P. J. Kooyman, J. C. Van der Waal, M. S. Rigutto, J. A. Peters and H. van Bekkum, Chem. Mater., 2001, 13, 683 CrossRef CAS.
- Y. Fang and H. Hu, J. Am. Chem. Soc., 2006, 128, 10636 CrossRef CAS.
- S. Habib, F. Launay, M.-A. Springuel-Huet, F. Guenneau, V. Semmer-Herlédan, N. N. Tušar, V. Kaučič and A. Gédéon, New J. Chem., 2006, 30, 1163 RSC.
- J. Wang, A. Vinu and M.-O. Coppens, J. Mater. Chem., 2007, 17, 4265 RSC.
- J. Čejka and S. Mintova, Catal. Rev., 2007, 49, 457 Search PubMed.
- D. Trong On and S. Kaliaguine, Angew. Chem., Int. Ed., 2002, 41, 1036 CrossRef.
- D. Trong On, A. Nossov, M.-A. Springuel-Huet, C. Schneider, J. L. Bretherton, C. A. Fyfe and S. Kaliaguine, J. Am. Chem. Soc., 2004, 126, 14324 CrossRef.
- D. Trong On and S. Kaliaguine, J. Am. Chem. Soc., 2003, 125, 618 CrossRef.
- K. R. Kloetstra, H. W. Zandbergen, J. C. Jansen and H. van Bekkum, Microporous Mater., 1996, 6, 287 CrossRef CAS.
- A. Karlsson, M. Stöcker and R. Schmidt, Microporous Mesoporous Mater., 1999, 27, 181 CrossRef CAS.
- L. Huang, W. Guo, P. Deng, Z. Xue and Q. Li, J. Phys. Chem. B, 2000, 104, 2817 CrossRef CAS.
- R. H. P. R. Poladi and C. C. Landry, J. Solid State Chem., 2002, 167, 363 CrossRef CAS.
- R. H. P. R. Poladi and C. C. Landry, Microporous Mesoporous Mater., 2002, 52, 11 CrossRef CAS.
- S. M. Solberg, D. Kumar and C. C. Landry, J. Phys. Chem. B, 2005, 109, 24331 CrossRef CAS.
- Z. Wang, L. Xu, J. Jiang, Y. Liu, M. He and P. Wu, Microporous Mesoporous Mater., 2012, 156, 106 CrossRef CAS.
- Y. Liu, W. Zhang and T. J. Pinnavaia, J. Am. Chem. Soc., 2000, 122, 8791 CrossRef CAS.
- Y. Liu, W. Zhang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2001, 40, 1255 CrossRef CAS.
- Z. Zhang, Y. Han, L. Zhu, R. Wang, Y. Yu, S. Qiu, D. Zhao and F.-S. Xiao, Angew. Chem., Int. Ed., 2001, 40, 1258 CrossRef CAS.
- Z. Zhang, Y. Han, F.-S. Xiao, S. Qiu, L. Zhu, R. Wang, Y. Yu, Z. Zhang, B. Zou, Y. Wang, H. Sun, D. Zhao and Y. Wei, J. Am. Chem. Soc., 2001, 123, 5014 CrossRef CAS.
- Y. Han, F.-S. Xiao, S. Wu, Y. Sun, X. Meng, D. Li, S. Lin, F. Deng and X. Ai, J. Phys. Chem. B, 2001, 105, 7963 CrossRef CAS.
- J. Liu, X. Zhang, Y. Han and F.-S. Xiao, Chem. Mater., 2002, 14, 2536 CrossRef CAS.
- Y. Han, N. Li, L. Zhao, D. Li, X. Xu, S. Wu, Y. Di, C. Li, Y. Zou, Y. Yu and F.-S. Xiao, J. Phys. Chem. B, 2003, 107, 7551 CrossRef CAS.
- Y. Han, X. Meng, H. Guan, Y. Yu, L. Zhao, X. Xu, X. Yang, S. Wu, N. Li and F.-S. Xiao, Microporous Mesoporous Mater., 2003, 57, 191 CrossRef CAS.
- Y. Xia and R. Mokaya, J. Mater. Chem., 2004, 14, 3427 RSC.
- Y. Liu and T. J. Pinnavaia, J. Mater. Chem., 2004, 14, 1099 RSC.
- M. Mrak, N. Novak Tušar, N. Zabukovec Logar, G. Mali, A. Kljajić, I. Arčon, F. Launay, A. Gedeon and V. Kaučič, Microporous Mesoporous Mater., 2006, 95, 76 CrossRef CAS.
- G. A. Eimer, I. Díaz, E. Sastre, S. G. Casuscelli, M. E. Crivello, E. R. Herrero and J. Perez-Pariente, Appl. Catal., A, 2008, 343, 77 CrossRef CAS.
- G. Coudurier, C. Naccache and J. C. Vedrine, J. Chem. Soc., Chem. Commun., 1982, 1413 Search PubMed.
- C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116 CrossRef CAS.
- I. Schmidt, A. Boisen, E. Gustavsson, K. Ståhl, S. Pehrson, S. Dahl, A. Carlsson and C. J. H. Jacobsen, Chem. Mater., 2001, 13, 4416 CrossRef CAS.
- A. Boisen, I. Schmidt, A. Carlsson, S. Dahl, S. Brorson and C. J. H. Jacobsen, Chem. Commun., 2003, 958 RSC.
- A. H. Janssen, I. Schmidt, C. J. H. Jacobsen, A. J. Koster and K. P. de Jong, Microporous Mesoporous Mater., 2003, 65, 59 CrossRef CAS.
- I. Schmidt, A. Krogh, K. Wienberg, A. Carlsson, M. Brorson and C. J. H. Jacobsen, Chem. Commun., 2000, 2157 RSC.
- D.-Y. Ok, N. Jiang, E. A. Prasetyanto, H. Jin and S.-E. Park, Microporous Mesoporous Mater., 2011, 141, 2 CrossRef CAS.
- M. Y. Kustova, P. Hasselriis and C. H. Christensen, Catal. Lett., 2004, 96, 205 CrossRef CAS.
- X. Wei and P. G. Smirniotis, Microporous Mesoporous Mater., 2006, 89, 170 CrossRef CAS.
- Z. Pavlačková, G. Košová, N. Zilková, A. Zukal and J. Čejka, Stud. Surf. Sci. Catal., 2006, 162, 905 CrossRef.
- K. Egeblad, M. Kustova, S. K. Klitgaard, K. Zhu and K. H. Christensen, Microporous Mesoporous Mater., 2007, 101, 214 CrossRef CAS.
- J.-B. Koo, N. Jiang, S. Saravanamurugan, M. Bejblová, Z. Musilová, J. Čejka and S.-E. Park, J. Catal., 2010, 276, 327 CrossRef CAS.
- C. H. Christensen, K. Johannsen, I. Schmidt and C. H. Christensen, J. Am. Chem. Soc., 2003, 125, 13370 CrossRef CAS.
- C. H. Christensen, K. Johannsen, E. Törnqvist, I. Schmidt, H. Topsøe and C. H. Christensen, Catal. Today, 2007, 128, 117 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- T.-W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601 CrossRef CAS.
- A. Sakthivel, S.-J. Huang, W.-H. Chen, Z.-H. Lan, K.-H. Chen, T.-W. Kim, R. Ryoo, A. S. T. Chiang and S.-B. Liu, Chem. Mater., 2004, 16, 3168 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 727 CrossRef CAS.
- W. Fan, M. A. Snyder, S. Kumar, P.-S. Lee, W. C. Yoo, A. V. McCormick, R. L. Penn, A. Stein and M. Tsapatsis, Nat. Mater., 2008, 7, 984 CrossRef CAS.
- H. Chen, J. Wydra, X. Zhang, P.-S. Lee, Z. Wang, W. Fan and M. Tsapatsis, J. Am. Chem. Soc., 2011, 133, 12390 CrossRef CAS.
- I. I. Ivanova, A. S. Kuznetsov, V. V. Yuschenko and E. E. Knyazeva, Pure Appl. Chem., 2004, 76, 1647 CrossRef CAS.
- I. I. Ivanova, A. S. Kuznetsov, O. A. Ponomareva, V. V. Yuschenko and E. E. Knyazeva, Stud. Surf. Sci. Catal., 2005, 158, 121 CrossRef.
- V. V. Ordomsky, V. Y. Murzin, Yu. V. Monakhova, Y. V. Zubavichus, E. E. Knyazeva, N. S. Nesterenko and I. I. Ivanova, Microporous Mesoporous Mater., 2007, 105, 101 CrossRef CAS.
- S. Wang, T. Dou, Y. Li, Y. Zhang, X. Li and Z. Yan, Catal. Commun., 2005, 6, 87 CrossRef CAS.
- S. Inagaki, M. Ogura, T. Inami, Y. Sasaki, E. Kikuchi and M. Matsukata, Microporous Mesoporous Mater., 2004, 74, 163 CrossRef CAS.
- J. Qi, T. Zhao, X. Xu, F. Li, G. Sun, C. Miao and H. Wang, Catal. Commun., 2009, 10, 1523 CrossRef CAS.
- J. Qi, T. Zhao, F. Li, G. Sun, X. Xu, C. Miao, H. Wang and X. Zhang, J. Porous Mater., 2010, 17, 177 CrossRef CAS.
- J. Y. Ying and J. Garcia-Martinez, US Patent 7,589,041, 2009, assigned to Massachusetts Institute of Technology Search PubMed.
- J. Garcia-Martinez, US Patent 7,807,132, 2010, assigned to Rive Technology, Inc Search PubMed.
- J. Garcia-Martinez and M. M. Johnson, US Patent Appl. 2009/0110631, 2009, assigned to Rive Technology, Inc Search PubMed.
- B. Speronello, J. Garcia-Martinez, A. Hansen and R. Hu, Refinery Operations, 2011, 4, 1 Search PubMed.
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118 RSC.
- A. Corma, M. Iglesias, C. del Pino and F. Sanchez, J. Chem. Soc., Chem. Commun., 1991, 1253 RSC.
- F. Sanchez, M. Iglesias, A. Corma and C. del Pino, J. Mol. Catal., 1991, 70, 369 CrossRef CAS.
- A. Carmona, A. Corma, M. Iglesias, A. San Jose and F. Sanchez, J. Org. Chem., 1995, 492, 11 CrossRef CAS.
- A. Cauvel, D. Brunel, F. Di Renzo, P. Moreau and F. Fajula, Stud. Surf. Sci. Catal., 1994, 94, 286 CrossRef.
- K. Maeda, Y. Kiyozumi and F. Mizukami, Angew. Chem., Int. Ed. Engl., 1994, 33, 2335 CrossRef.
- K. Maeda, J. Akimoto, Y. Kiyozumi and F. Mizukami, J. Chem. Soc., Chem. Commun., 1995, 1033 RSC.
- K. Maeda, J. Akimoto, Y. Kiyozumi and F. Mizukami, Angew. Chem., Int. Ed. Engl., 1995, 34, 1199 CrossRef CAS.
- K. Maeda, A. Sasaki, K. Watanabe, Y. Kiyozumi and F. Mizukami, Chem. Lett., 1997, 879 CrossRef CAS.
- W. Yan, E. W. Hagaman and S. Dai, Chem. Mater., 2004, 16, 5182 CrossRef CAS.
- C. W. Jones, K. Tsuji and M. E. Davis, Nature, 1998, 393, 52 CrossRef CAS.
- C. W. Jones, K. Tsuji and M. E. Davis, Microporous Mesoporous Mater., 1999, 29, 339 CrossRef.
- C. W. Jones, K. Tsuji and M. E. Davis, Microporous Mesoporous Mater., 1999, 33, 223 CrossRef CAS.
- C. W. Jones, K. Tsuji and M. E. Davis, Microporous Mesoporous Mater., 2001, 42, 21 CrossRef CAS.
- K. Yamamoto, Y. Sakata, Y. Nohara, Y. Takahashi and T. Tatsumi, Science, 2003, 300, 470 CrossRef CAS.
- K. Yamamoto, Y. Nohara, Y. Domon, Y. Takahashi, Y. Sakata, J. Plévert and T. Tatsumi, Chem. Mater., 2005, 17, 3913 CrossRef CAS.
- K. Yamamoto and T. Tatsumi, Chem. Mater., 2008, 20, 972 CrossRef CAS.
- U. Díaz, J. A. Vidal-Moya and A. Corma, Microporous Mesoporous Mater., 2006, 93, 180 CrossRef.
- B. L. Su, M. Roussel, K. Vause, X. Y. Yang, F. Gilles, L. Shi, E. Leonova, M. Edén and X. Zou, Microporous Mesoporous Mater., 2007, 105, 49 CrossRef CAS.
- R. Astala and S. M. Auerbach, J. Am. Chem. Soc., 2004, 126, 1843 CrossRef CAS.
- S. Inagaki, S. Guan, T. Ohsuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS.
- Y. Goto, T. Ohsuna, N. Mizoshita, T. Tani and S. Inagaki, Solid State Sci., 2011, 13, 729 CrossRef CAS.
- G. Bellussi, A. Carati, E. Di Paola, R. Millini, W. O. Parker Jr., C. Rizzo and S. Zanardi, Microporous Mesoporous Mater., 2008, 113, 252 CrossRef CAS.
- G. Bellussi, E. Montanari, E. Di Paola, R. Millini, A. Carati, C. Rizzo, W. O. Parker Jr., M. Gemmi, E. Mugnaioli, U. Kolb and S. Zanardi, Angew. Chem., Int. Ed., 2012, 51, 666 CrossRef CAS.
- G. Bellussi, R. Millini, E. Montanari, A. Carati, C. Rizzo, W. O. Parker Jr, G. Cruciani, A. de Angelis, L. Bonoldi and S. Zanardi, Chem. Commun., 2012, 48, 7356 RSC.
- J. M. Bennett, J. P. Cohen, E. M. Flanigen, J. J. Pluth and J. V. Smith, ACS Symp. Ser., 1983, 218, 109 CrossRef CAS.
- R. Bialek, W. M. Meier, M. Davis and M. J. Annen, Zeolites, 1991, 11, 438 CrossRef CAS.
- K. D. M. Harris and J. M. Thomas, ChemCatChem, 2009, 1, 223 CrossRef CAS.
- W. Lin and S. J. Lee, in Chirality at the Nanoscale Nanoparticles, Surfaces, Materials and more, ed. D. B. Amabilino, Wiley-VCH, Weinheim, 2009, p. 391 Search PubMed.
- C. Dryzun, Y. Mastai, A. Shvalb and D. Avnir, J. Mater. Chem., 2009, 19, 2062 RSC.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |