Catalytic oxidation of rare sugars over gold catalysts
Bright T.
Kusema
and
Dmitry Yu.
Murzin
*
Laboratory of Industrial Chemistry and Reaction Engineering, Process Chemistry Centre, Department of Chemical Engineering, Åbo Akademi University, FI-20500 Åbo/Turku, Finland. E-mail: dmurzin@abo.fi; Fax: +358 2 215 4479; Tel: +358 2 215 4985
First published on 31st July 2012
Abstract
This minireview presents an overview on the recent developments for the heterogeneous catalytic aerobic oxidation of rare sugars over supported gold catalysts. The selective oxidation of several rare sugars; L-arabinose, D-galactose and D-lactose representing pentoses, hexoses and disaccharides, respectively, is reviewed. The critical evaluation of the aerobic oxidation of the different sugars presents a great opportunity to understand the fundamentals of the surprising reactivity and extraordinary properties of gold. This in turn can open up the possibilities for industrial exploitation of sustainable and green processes. Emphasis was put on catalysis, kinetics and mechanistic studies based on understanding of the fundamental reaction engineering.
 Bright T. Kusema | Dr. Bright T. Kusema studied and received his B.Sc. and M.Sc. degree in Chemical Engineering and Biotechnology at the D. I. Mendeleev University of Chemical Technology of Russia in Moscow (2001-2007). He obtained his PhD in Chemical Engineering at the Åbo Akademi Univesity in Åbo/Turku, Finland (2011) under the guidance of Prof. Dmitry Yu. Murzin and Prof. Tapio Salmi. He continues working as a post-doc researcher in the field of heterogeneous catalysis for the synthesis of fine chemicals. |
 Dmitry Yu. Murzin | Prof. Dmitry Yu. Murzin studied chemical technology at the Mendeleev University of Chemical Technology in Moscow (1980–1986). He obtained his PhD in 1989 and his DSc (1999) at Karpov Institute of Physical Chemistry, Moscow. He worked at the Universite Louis Pasteur, Strasbourg and Åbo Akademi University as a post-doc (1992–1994). Between 1995–2000 he was associated with BASF. Since 2000 he holds the Chair of Chemical Technology at Åbo Akademi University. He serves on several catalysis and chemical engineering editorial boards. He is co-author of a monograph, holds 3 patents and is a co-author of ca. 530 publications. |
1. Introduction
The concept of biorefineries is an integrated biomass processing entity producing multiple value-added products from a range of renewable feedstocks.1,2 Because of the on-going price increase of fossil resources, their uncertain availability, and particularly the environmental concerns, the feasibility of oil exploitation is predicted to decrease in the near future. Therefore, alternative solutions, which are able to mitigate environmental concerns and simultaneously reduce the dependence on fossil fuels should be promoted.3,4 In light of this, different alternative bio-compatible resources may play an important role in the production of fine and specialty chemicals. The biorefining concept has increasingly drawn much attention and is now widely accepted all over the world. The technologies behind this concept can provide a wide range of bio-based products, such as bio-chemicals, bio-materials and bio-fuels.5,6 An integrated biorefinery system should emphasize two aspects, namely; high value chemicals and material products; and self-fulfilling with regards to the energy consumption. The integration of green chemistry and green engineering into biorefineries, along with the use of low environmental impact technologies can be considered as a set of underlying principles to be applied.The sugar platform of the biorefinery system is based on (bio-) chemical conversion processes of biomass to sugar feedstocks. Woody biomass consists of lignocellulosic material, which is made up of three primary fractions: hemicellulose, cellulose, and lignin.7,8 Extraction of hemicelluloses and the rare and specialty sugars they contain can be easily achieved in water at moderate temperatures. The sugar feedstocks of mainly pentoses, hexoses and di-saccharides, such as L-arabinose, D-galactose and D-lactose, respectively, are considered in this work. These specialty or rare sugars are obtained from hemicelluloses, such as arabinogalactan, which account for 20–35% of lignocellulosic biomass.9,10D-Lactose is a disaccharide which consists of D-glucose and D-galactose moieties, and it is a by-product of the dairy industry.11,12 This makes them prime candidates for sustainable feedstocks for fine chemicals.
The important parameters for the (bio-) chemical processes of the sugar platform are temperature, pH, pressure, reaction time, substrate concentration, added reactants or catalysts. Catalysis and innovative process design will play an important role in the transformation of the sugar feedstocks into chemicals, materials and fuels. The immense importance of catalysis in the chemical industry is manifested by the fact that roughly 85–90% of all chemical products have undergone catalytic transformation during the course of production.13,14 The aldonic acids produced from L-arabinose, D-galactose and D-lactose as well as their derivatives have many applications in the food, pharmaceutical and cosmetic industries. Thus, the carbohydrates, a single class of natural products can be a major bio-feedstock and platform chemicals, from which it would be possible to develop both industrially and economically viable routes for the synthesis of fine chemicals.15
The application of heterogeneous catalysis for lignocellulosic biomass processing, which involves the transformation of carbohydrates to give products of greater value is a very promising field of research and in the past years the attention has intensified towards selective oxidation over noble-metal catalysts.16 Moreover, the major advantage of employing heterogeneous catalysts is the ease of separation and recycle, no waste or toxic material is generated and amenable to continuous processing. The development of an aerobic inorganic catalyst for the title reaction and related processes with an inexpensive oxidant, such as air or molecular oxygen and an aqueous solution, is a clean and elegant route employing mild reaction conditions, under which high activity and selectivity can be obtained. In addition, carbohydrate-based products have advantageous biodegradability and biocompatibility properties. Selective oxidation of carbohydrates over gold catalysts towards valuable sugar acids featured in this minireview is therefore a promising field of application.
2. Catalysis by gold
Gold has been known for centuries for its chemical inertness in the bulk form, because it neither reacts with air nor corrodes. It has, however, been discovered that Au has a hidden inner beauty for a scientist interested in catalysis, because it turns out that it becomes an incredibly reactive material on the nano-scale.17–19 Since the first major reports of the spectacular performance of gold nanoparticles in the aerobic oxidation reactions, a lot of work has been carried out to understand the fundamentals of this surprising reactivity.20–24 Supported Au nanoparticles exhibit an exceptionally low-temperature catalytic activity and high selectivity in the oxidation of carbohydrates, alcohols, hydrocarbons and CO.25–30 These studies demonstrated Au to be a suitable catalyst for the respective reactions, in contrast to previous reports on the poor catalytic activity of Au. Such levels of low-temperature activity and selectivity are not replicated by other metal catalysts used in oxidation reactions, such as palladium or platinum. Au also shows superior durability and stability because of its high resistance to deactivation caused by over-oxidation and poisoning of the active metal. Previous studies have elucidated that Au supported on alumina has a high activity in the oxidation of sugars under mild conditions.31,32In many cases, Au doped with other noble metals such as Pd and Pt shows an extra-high activity and a high resistance to deactivation due to synergetic effects. It has previously been demonstrated that bimetallic catalysts based on Au, Pd and Pt metals and supported on activated carbon are much more active in the liquid phase oxidation than monometallic ones.33,34 The properties of bimetallic Au–Pd catalysts depend strongly on the nature of the support used, catalyst preparation and treatments.35 Different materials, such as ceria, alumina, titania and activated carbon, were used as the supports for catalysts in selective oxidation of various alcohols and sugars. Among the reducible oxides, ceria is one of the most efficient supports, because of its capability to store oxygen and become reduced.36 Au nanoparticles supported on nanocrystalline cerium oxide were shown to be extremely effective in the oxidation of aldehydes to acids and also in the oxidation of allylic alcohols.37,38
It has been observed that the oxidation reactions exhibit structure sensitivity, i.e. the catalyst activity is influenced by the size of the gold nanoparticles.17,28 The Au nanoparticles can be prepared by using different methods and treatments that allow a wide range of particle diameters. It is well known that the preparation method is crucial in controlling the catalyst activity. Generally, an impregnation method tends to produce rather large particles (>10 nm) that may be undesirable for oxidation reactions, whereas deposition–precipitation and direct ion exchange methods result in small gold nanocrystals deposited on the surface of the support.39 The preparation methods can produce a wide range of materials, depending on the special needs as these methods involve the close control of a wide range of experimental variables which affect the nature of the Au species.40
There has been much debate concerning the nature of the active sites of Au catalysts. The active catalysts are typically found to comprise of small Au nanocrystallites of diameter 2–4 nm, supported on an oxide material. It has been proposed that Au atoms at the interface between the Au cluster and the oxide support material together with the availability of the defect sites at this interface are probably the important features of the active oxidation centers.41,42 Several research groups have proposed and reassessed the oxidation mechanism on the basis of the model that the substrate is activated by the adsorption on to Au0 on the surface of the Au nanoparticle, and that the O2 is activated by the Au atoms at the boundary between the support material and the Au nanocrystals. The Au atoms at the boundary have been proposed to be cationic in nature, possibly Au+ or Au3+.43–45
It is generally accepted that liquid-phase oxidation on noble metal catalysts takes place via an oxidative dehydrogenation mechanism which, in fact can be split into several steps according to Mallat and Baiker.46–49 It is believed that the first step is the dehydrogenation and oxidation of the hydrogen bound to the hemiacetal carbon atom which leads to the formation of the lactone. Hydrolysis of the lactone gives the aldonic acid. An alkaline pH is favorable for the saponification of the lactone.50 Alternative mechanisms such as the direct involvement of the oxidizing species in the rate determining step of dehydrogenation,51 and oxygen assisted dehydrogenation mechanisms,52 and the involvement of peroxidic intermediates have also been proposed.53 However, the exact reaction sequence through which the dehydrogenation mechanism occurs is still under dispute, due to the complex nature of the adsorbed species on the metal surface, the debated origin of the adsorbed oxygen species, and the roles played by the gold species.19,54 The understanding of the mechanism is far from complete as there is no consensus on the nature of the adsorbed intermediates and the reacting species. The nature of the adsorbed species mainly depends on the pH value of the reaction medium, the concentration of the oxidizing agent and the acidity of the catalyst.
3. Rare sugars – carbohydrates
Cellulose is the most abundant polysaccharide in Nature, whereas D-glucose is the most abundant monosaccharide. That is the reason why the oxidation of D-glucose is the process most intensively studied.55 On the other hand, hemicelluloses are the second most abundant polysaccharides in Nature, accounting for 20–35% of the lignocellulosic biomass.8 These macromolecular hetero-polysaccharides consist of a large number of different monomeric sugar units, pentoses and hexoses, such as L-arabinose, L-rhamnose, D-xylose, D-mannose, D-galactose and D-glucose linked to each other by glycosidic bonds.56 Arabinogalactans (AGs) are hemicelluloses, which appear in large quantities in larch species such as Larix sibirica. The structural basis of AG is a backbone of β-D-galactopyranose residues that are predominantly (1 → 3)-linked and most frequently branched with (1 → 6) β-D-galactopyranose, α-L-arabinofuranose and β-D-glucuronic acid side chains (Fig. 1). The average ratio of galactose, arabinose and glucuronic acid units in AG is about 5.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.08 and the molar mass is 20000–100000 g mol−1. The degree of polymerization of AG is about 100–300 and it is amorphous. Extraction of AG from larch wood chips and powder can be easily achieved in water at moderate temperatures on an industrial scale.9,10 The hemicelluloses are also released during the mechanical and chemical pulping processes and can be recovered on an industrial scale.57,58 It is, therefore, a sustainable feedstock for valuable chemicals. Hydrolysis of hemicelluloses to its monosaccharides in the presence of acidic homogeneous and heterogeneous catalysts in high yields without further degradation has been recently reported.59,60 The hydrolysis proceeds by the catalytic transformation of the monosaccharides into bio-based valuable products, in which the latter reactions can be selective oxidation to produce aldonic acids.
:1:0.08 and the molar mass is 20000–100000 g mol−1. The degree of polymerization of AG is about 100–300 and it is amorphous. Extraction of AG from larch wood chips and powder can be easily achieved in water at moderate temperatures on an industrial scale.9,10 The hemicelluloses are also released during the mechanical and chemical pulping processes and can be recovered on an industrial scale.57,58 It is, therefore, a sustainable feedstock for valuable chemicals. Hydrolysis of hemicelluloses to its monosaccharides in the presence of acidic homogeneous and heterogeneous catalysts in high yields without further degradation has been recently reported.59,60 The hydrolysis proceeds by the catalytic transformation of the monosaccharides into bio-based valuable products, in which the latter reactions can be selective oxidation to produce aldonic acids.
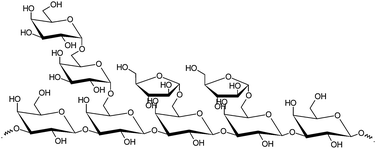 | ||
| Fig. 1 Structure of arabinogalactan. | ||
Lactose is a disaccharide consisting of D-glucose and D-galactose moieties joined by a β-1 → 4-glycosidic linkage (Fig. 2). Several million tons of lactose are produced annually as a by-product of the dairy industry, therefore it has a potential for the production of fine chemicals.11,12 Lactose can be hydrolyzed to the monosaccharides, D-glucose and D-galactose. Furthermore, isomerization in alkaline solution to lactulose, catalytic hydrogenation or oxidation to the corresponding polyhydric alcohol and acid, respectively, are industrially viable processes. The selective oxidation of a D-glucose moiety of D-lactose produces the corresponding aldonic acid, lactobionic acid.61
Sugars readily form cyclic hemiacetals, which in the aqueous solutions are in equilibrium with the open chain forms. The open-chain aldehydo-form could ring close and from this, different forms with furanose and pyranose rings are formed, which in addition can have either an α or a β anomeric conformation. The final proportions of these forms depend on the sugar molecule and type of solvent. In an aqueous solution, the cyclic aldehydo-form and pyranose ring are heavily favoured because of the relative thermodynamic stability.62 Therefore, it can be supposed that the predominant form of sugars under the mild reaction conditions is the hemiacetal pyranose. In the cyclic form, the hydroxyl group to be oxidized might be part of the hemiacetal, which is oxidatively dehydrogenated to the intermediate lactone with the subsequent transformation into the corresponding carboxylic acid.
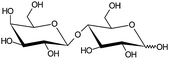 | ||
| Fig. 2 Structure of D-lactose. | ||
4. Selective oxidation of L-arabinose
Aqueous-phase selective oxidation of L-arabinose by molecular oxygen (Scheme 1) was studied in an isothermal shaking reactor under moderate conditions of 50–70 °C and pH values 6–9.63 The heterogeneous catalytic aerobic oxidation of the sugars was conducted in a semi-batch mode at constant pH values. Different Au catalysts on metal oxide and carbon supports were tested and compared to Pd catalysts. Au supported on metal oxides displayed high catalyst activities and selectivity to arabinonic acid with Au/Al2O3 proving to be the most suitable catalyst for the selective oxidation. Pd catalysts were characterized with low activity and selectivity due to the increase of the amount of by-products. A lower activity and selectivity of Pd and Pt catalysts in the oxidation of pentoses (arabinose, ribose, lyxose and xylose) were also reported in ref. 64–66. Au supported on metal oxide support such as titania was a very effective catalyst for the production of aldonic acids. In our previous works, the influence of the kinetic parameters such as pH, temperature, and oxygen flow rate was elucidated.63 Under an intrinsic kinetic regime, where only the true chemical kinetics plays a role and transport resistances are negligible, the amount of oxygen present in the liquid phase can greatly influence the catalytic activity. The kinetic experiments revealed that the oxidation rates increased with an increase in both temperature and oxygen flow rates. The selectivity over Au was high, 95–99% under these conditions and there were no degradation products. The catalyst over-oxidation is a major cause of deactivation under oxygen rich conditions for metals such as Pd and Pt. Higher temperatures may also lead to the sugar disproportionation leading to catalyst deactivation due to coking.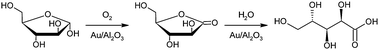 | ||
| Scheme 1 Selective oxidation of L-arabinose to arabinolactone and arabinonic acid by molecular oxygen over gold catalysts. | ||
Slightly alkaline conditions were favorable for high initial rates, catalyst activity and product selectivity, attaining a complete conversion and total selectivity towards arabinonic acid at both pH values 8 and 9 within 200 min. The acidic conditions retarded the sugar oxidation, due to the catalyst deactivation by the lactone and aldonic acid strongly adsorbed on the metal surface. At low pH values, product adsorption on the catalyst strongly inhibits the reaction (Fig. 3). A lower conversion of 23% and as well as a lower selectivity of 46% were achieved at pH value 6. Accordingly, the optimum conditions for the selective oxidation of arabinose were 60 °C, pH 8 and oxygen flow rate of 2.5 ml min−1, which are rather mild operation conditions achieving high selectivity >99% towards the desired product. Furthermore, under these conditions, the catalyst was demonstrated to be stable and it was reused at least 3 times with no loss of activity, Au particle sintering or leaching of the metal. In situ catalyst potential measurements during arabinose oxidation gave information about the extent of the oxygen accumulation on the metal surface from the reduced to the oxidized state and a correlation to activity was obtained (Fig. 4). The main driving forces for the catalyst potential changes are the rates of oxygen adsorption and oxygen consumption in the chemical reaction on the catalyst surface. The apparent activation energy Ea for the selective oxidation of arabinose was 23.8 kJ mol−1. This was close to the Ea found in ref. 28 for the oxidation of D-glucose over Au catalysts. However, it has been reported that different aldoses did not have the same reactivity because of their different configurations.64–66 The aldopentoses have equal rates of oxidation to the corresponding aldonic acids, whereas the rates of the consecutive oxidation to the aldaric acids follow the order xylo, arabino > lyxo, ribo.64 It is likely that a certain configuration of the substrate (axial OH 2 or 4) is more favorable with regard to the adsorption on the catalytic surface, as well as the reactivity, which may lead to higher catalytic activity.
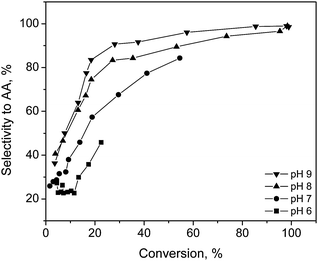 | ||
| Fig. 3 Selectivity to arabinonic acid as a function of L-arabinose conversion at different pH values over Au/Al2O3 catalysts at 60 °C and oxygen flow rate of 2.5 ml min−1. | ||
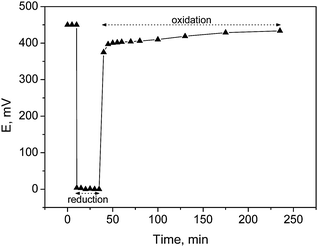 | ||
| Fig. 4 Catalyst potential for L-arabinose oxidation over Au/Al2O3 catalysts at 60 °C, pH 8 and oxygen flow rate 2.5 ml min−1. | ||
Selective oxidation of L-arabinose was further investigated over bimetallic Pd–Au catalysts supported on alumina and ceria.67 The catalysts were treated in hydrogen or oxygen at 300 °C, or by aqueous solution of formaldehyde at room temperature. Improvements in the catalyst activity, selectivity and stability can be achieved by using bi- and to some extent tri-metallic catalysts.33–35 Moreover, it has been pointed out that by alloying Au to Pt or Pd, the leaching of metals was avoided and the catalyst life was improved.68 The bimetallic Au–Pd catalysts demonstrated higher activity and selectivity in comparison with the monometallic Au and Pd catalysts. Thus, the presence of mixed Au–Pd species is a key factor for higher activity of these catalysts in oxidation of arabinose, similar to oxidation of different alcohols and other saccharides also described elsewhere in the literature.18 Activity and selectivity of catalysts were dependent on the catalyst treatment and the nature of the support used. The Au–Pd/CeO2 catalyst reduced by formaldehyde manifests the highest activity and selectivity demonstrated by a TOF value of 0.275 s−1 and 90% selectivity to arabinonic acid (Table 1).
| Support | Catalyst | |||
|---|---|---|---|---|
| Au | Au–Pd | |||
| TOF (s−1) | Selectivitya (%) | TOF (s−1) | Selectivitya (%) | |
| a Selectivity to arabinonic acid at 50% L-arabinose conversion. b FMD – formaldehyde. | ||||
| Al2O3–O2 | 0.093 | 80 | 0.149 | 81 |
| Al2O3–H2 | 0.094 | 86 | 0.122 | 85 |
| Al2O3–FMDb | 0.027 | 86 | 0.094 | 90 |
| CeO2–O2 | 0.092 | 82 | 0.171 | 75 |
| CeO2–H2 | 0.101 | 78 | 0.143 | 77 |
| CeO2–FMDb | 0.090 | 86 | 0.275 | 90 |
Because of the observed TOF values for bimetallic Au–Pd exceeding that of the monometallic Au catalysts, it can be inferred that the synergetic effect of the two metals contributed to the higher activity of the bimetallic catalysts. Mutual interactions of supported Au and Pd species depending on the support nature and catalyst treatment were observed. A more pronounced interaction between Au and Pd species was observed for Au–Pd/Al2O3 catalysts, while interactions of Au and Pd with reducible ceria coexist with the mutual interaction between these metals. Au metallic species seem to be responsible for the activation of arabinose while an easy redox transformation of Pd species can revoke oxygen activation. Thus, core–shell structures formed by Au species or Au–Pd alloy covered with thin PdO film manifest a synergetic effect on the selective arabinose oxidation by molecular oxygen.
5. Selective oxidation of D-galactose
The selective oxidation of a hexose, D-galactose to galactonic acid over gold catalysts was investigated under mild reaction conditions similar to the above-mentioned for a pentose, L-arabinose (Scheme 2). Au supported on alumina was demonstrated to be an effective catalyst for the oxidation of D-galactose by molecular oxygen at 60 °C and a slightly alkaline pH value (8–10).69 Under these conditions, D-galactose was completely oxidized with a total selectivity to the corresponding product, galactonic acid within 200 min. A comparison of different pH values showed significant increase in the catalyst activity as the pH value increases from 6 to 10 (Fig. 5). Slightly alkaline conditions were favorable for the oxidation of D-galactose, which was also the case for the L-arabinose oxidation. It also turned out that the variation in the pH value of the reaction medium did not only affect the catalyst activity, but influenced significantly the selectivity of the catalyst towards the main product, analogously to the case for L-arabinose oxidation. Inhibition of the catalytic activity was observed in the acidic medium. The intermediate lactone was present at low pH values, and this resulted in low conversion and selectivity to the main product. The selectivity during the oxidation of D-galactose towards galactonic acid was very high, ranging from 80–99% as a function of conversion when the oxidation was carried out under alkaline conditions (pH 8 and 10). However, under slightly acidic conditions (pH 6), only 44% D-galactose conversion was reached after 200 min with a maximum selectivity of 55% to galactonic acid. The only other product detected was galactonolactone. The concentration of the lactone increased with the decrease in pH value of the reaction medium. A correlation was drawn between the electrochemical potential shift of the catalyst, from the reduced state to the oxidized state, and the catalyst activity.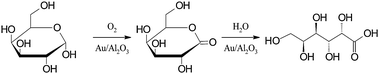 | ||
| Scheme 2 Selective oxidation of D-galactose to galactonolactone and galactonic acid by molecular oxygen over gold catalysts. | ||
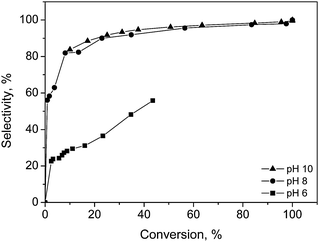 | ||
| Fig. 5 Selectivity to galactonic acid as a function of D-galactose conversion at different pH values over Au/Al2O3 catalysts at 60 °C and oxygen flow rate of 2.5 ml min−1. | ||
Comparative studies for the selective oxidation of hexoses such as D-glucose, D-galactose and D-mannose between Au and Pd and Pt derived catalysts also revealed the superiority of Au by a substantial margin.64,65 It was however revealed that Au displayed a higher activity in D-glucose compared to D-galactose or other glucose derivatives with a protective group such as acetyl or amine. The presence of these groups probably constricts the access of the catalyst to the carbon with the anomeric hydroxyl of the substrate.70,71 It appears that the structure of the sugars influences the activity of the Au catalyst. Nevertheless, Au catalysts in all the cases showed the highest catalytic activity and a permanently high selectivity towards the aldonic acid was found independent of the substrate investigated.
6. Selective oxidation of D-lactose
An extensive study on the kinetics of aerobic oxidation of D-lactose was carried out over different metals and supports in the aqueous phase.72–78 The catalyst electrochemical potential response was measured in situ and utilized in the reaction process characterization. D-Lactose is a disaccharide which contains D-galactose and D-glucose units. It was demonstrated that D-lactose oxidation is a consecutive reaction, forming primary and secondary products (Scheme 3).61,72 During the first stage of the reaction, D-lactose is oxidized at the hydroxyl group of the D-glucose hemiacetyl, to lactobionic acid which is further oxidized to 2-keto-lactobionic acid. Isomerization of lactose to lactulose is highly favoured in strong alkaline media, with pH values higher than 8. Analogous to the oxidation of pentoses and hexoses, strong alkaline medium also induces catalyst deactivation and a decrease of selectivity due to the formation of by-products.63,64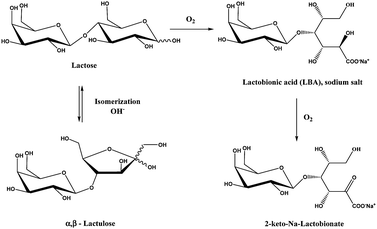 | ||
| Scheme 3 Reaction scheme for D-lactose oxidation by molecular oxygen. | ||
Among the supported metals tested, Au supported on alumina and ceria demonstrated high activity and selectivity to lactobionic acid. Pd catalysts resulted in the consecutive oxidation step to 2-keto-lactobionic acid. It was also found that Pd is more prone to deactivation due to over-oxidation and self-poisoning.73,74 Both the Au and Pd catalysts were, however, deactivated under conditions of high pH and high temperature. The influence of surface acidity on lactose oxidation over Pd supported on beta and MCM-22 zeolites was investigated.75 It was observed that Pd supported on MCM-22 zeolites were the most active catalysts. The acidity of the support as well as the method of Pd incorporation had a strong influence on the catalytic activity and selectivity. The surface acidity had a positive influence on the metal dispersion beneficial for the catalyst activity. However, the acidity has a negative effect on lactose oxidation reaction to lactobionic acid. Therefore, a compromise between the support porosity, acidity and metal loading is required to achieve both high activity and selectivity towards the desired product.
Various supported Au catalysts on metal oxides were utilized in the aerobic oxidation of D-lactose.74 Very high selectivity to D-lactobionic acid was achieved for all the Au catalysts which seems to be intrinsic characteristics of the Au catalysts. The catalyst activity was evidently dependent on the type and nature of support. Among the catalysts investigated, Au on alumina was the most active catalyst for the oxidation of D-lactose to the corresponding aldonic acid, analogous to pentoses and hexoses.63,64,69 The other Au catalysts were characterized with lower activity and higher rates of deactivation at prolonged reaction times. In some cases, the application of bimetallic catalysts and the addition of promoters were also demonstrated to increase the activity and selectivity of Pd and Pt based catalysts in the oxidation of D-lactose to lactobionic acid.76–78
A comparison on the aerobic oxidation of different disaccharides of various substrate configurations has also been reported.64,71 Maltose and cellobiose are composed of two units of D-glucose, whereas lactose and melibiose comprise D-galactose and D-glucose units. It was pointed out that the D-glucose presents a more preferential configuration than D-galactose, hence maltose and cellobiose possess a more beneficial configuration during the oxidation over Au catalysts. The presence of D-galactose in the structure of lactose and melibiose might induce a slower adsorption of the disaccharide on the catalyst surface, and implicitly, a lower catalytic activity. However, all the different disaccharides showed the same high selectivity towards the corresponding aldonic acids, similar to what was also observed for the oxidation of pentoses and hexoses over Au catalysts.63,69
Different reaction parameters, such as temperature and oxygen flow rates, were investigated and it was concluded that the moderate and optimized conditions of 60 °C, oxygen flow rate of 2.5 ml min−1 and pH 8 were sufficient to have a complete conversion of the sugars without further degradation of the products at prolonged reaction times. The above-mentioned reaction conditions were applied for L-arabinose, D-galactose and D-lactose oxidation. The same experimental conditions facilitate a direct comparison of the sugar structures, the monosaccharides, i.e. pentose in L-arabinose vs. hexose in D-galactose, and a disaccharide such as D-lactose, to the catalytic activity and selectivity. By using the same Au/Al2O3 and similar reaction conditions of 60 °C, pH 8 and oxygen flow rate of 2.5 ml min−1, high catalytic activity and a total selectivity with respect to the formed aldonic acids for all the sugars were found. The catalytic activity was 0.53, 0.34 and 0.67 mmol s−1 gAu−1 for L-arabinose, D-galactose and D-lactose, respectively.63,69,74 These values are in the same order of magnitude to other major works on selective catalytic oxidation of the corresponding sugars previously reported in the literature.
7. Structure sensitivity
The application of Au catalysts involves a possibility that the studied reactions exhibit structure sensitivity. It has been reported in the literature that the oxidation reactions demonstrate a structure sensitivity of the catalysts used, i.e. the catalyst activity is influenced by the size of the Au nanoparticles.17,18,28 Possible forms of dependence of the initial rate or the TOF on the particle size have been deduced, which may be (i) no dependence; (ii) negative/antipathetic dependence; (iii) positive/sympathetic; and (iv) rate/TOF passing through maximum.79–81 Particle size can be controlled by choosing the appropriate preparation method as well as the suitable support.39,40 In our previous studies for the selective oxidation of L-arabinose and D-lactose, gold catalysts on different metal oxide supports and carbon were investigated and it was concluded that alumina was the most suitable catalyst demonstrated by a combination of high activity and selectivity to the corresponding aldonic acid.63,74 For this reason, alumina was chosen as the support for further studies on the size effect. However, other inert supports such as carbon were also reported to show the size effect of gold nanoparticles. The gold catalysts, 2 wt% Au/Al2O3 were prepared by different methods and treatments allowing a wide range of particle diameters. The metal cluster size was varied by application of different preparation methods such as deposition–precipitation with urea and direct ion exchange, ammonia or water as the washing agents, varying calcination temperatures and concentration of the initial precursor solutions.82,83 The different particle sizes, ranging from 1–16 nm presented for the studied catalysts allowed a systematic evaluation of the influence of the average particle diameter on the catalytic activity.69,84The analysis of the structure sensitivity follows a classical approach based on the average values, obtained by using high resolution TEM. The calculations of the Au dispersions were based on the assumption that Au particles are cubooctahedral, even though particles around 1 nm have also icosahedral structures and above 1 nm decahedral crystals may appear. The influence of different Au particle sizes in the selective oxidation of L-arabinose and D-galactose on the catalyst activity is demonstrated in Fig. 6. A volcano-type relationship between the catalyst activity and the Au particle size demonstrated by the catalyst activity passing through maximum was deduced. The catalysts with an average particle diameter of around 2 nm were the most active in the L-arabinose and D-galactose oxidation. This result is explained as follows: carbohydrates are adsorbed on the gold atoms with low coordination numbers, i.e. corners, edges, steps, via the carbonyl group. The number of low-coordinated atoms has a maximum value around 2 nm. Below this size, there is efficient contact of Au nanoclusters with the support, whereas for the particles above 2 nm the number of these low-coordinated sites decreases. It can therefore be assumed that the catalyst with a diameter of 2.0 nm and 2.6 nm had the maximum number of these active sites for the adsorption of L-arabinose and D-galactose, respectively, and oxygen activation. However, the selectivity towards the aldonic acids of L-arabinose and D-galactose was not influenced by the different gold particle sizes. Similar observations were also revealed that there is an optimal metal particle size in a range of 3–10 nm giving the highest initial rates for the oxidation of D-glucose, D-lactose and rhamnose over supported metal catalysts.28,78,85,86 However, in all the cases, a deviation of the catalytic activity for particles larger than 6 nm has been detected, as well as an almost complete inactiveness for particles larger than 10 nm.
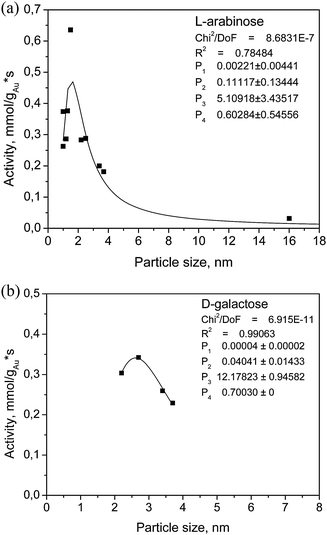 | ||
| Fig. 6 Dependence of catalyst activity on gold particle size of Au/Al2O3 in (a) L-arabinose and (b) D-galactose selective oxidation, respectively, at 60 °C, pH 8 and oxygen flow rate 2.5 ml min−1. | ||
A quantitative thermodynamic approach of the cluster size effect was used to describe the size-dependent Langmuir–Hinshelwood mechanism and the two-step catalytic cycle.80,81 The general treatment took the surface energy excess into account because of an intrinsic increase in the chemical potential with a decrease in size, as well as the changes in the chemical potential upon adsorption. An expression for the Langmuir–Hinshelwood mechanism was obtained from ref. 80 and 81 which could be modified and simplified leading to a four-parameter dependence for L-arabinose and D-galactose oxidation in the following way;
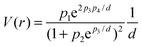 | (1) |
Comparison between the experimental and calculated data performed in ref. 69 and 84 demonstrated the applicability of the kinetic equations to treat the experimental data as a function of Au cluster size. Results of the calculations along with the values of parameters for L-arabinose and D-galactose are illustrated in Fig. 6, demonstrating that the reaction rates are passing through the maximum, confirming a good correspondence between the calculations and the model. The metal particle size is an important factor in the catalytic activity in sugar oxidation.
8. Reaction mechanism
Most of the studies conducted for the aerobic oxidation of alcohols and carbohydrates indicate that the reaction occurs via an oxidative dehydrogenation mechanism.45–54 It was demonstrated by accurate labeling experiments with 18O2 and H218O that oxygen atoms originating from hydroxide ions, instead of molecular oxygen, were in fact incorporated into the substrate during the oxidation reactions which are promoted by alkaline conditions at high pH values.87,88 This catalytic property is shared by Au, Pd and Pt supported nanoparticles. The reaction path was suggesting the involvement of both solution-mediated and metal-catalyzed elementary steps.89 In some cases, Au supported on titania was reported to show a “double-peak” catalytic activity in gas-phase selective oxidation of ethanol to acetaldehyde by molecular oxygen.90 The additional low-temperature peak probably corresponds to the specific participation of oxygen generated on the Au surface under mild reaction conditions. Other mechanistic elementary steps involving peroxidic intermediates have also been proposed to explain the detection of H2O2 as the reduction product of O2 during the oxidation reactions.53,91,92 In order to separate the effect of particle size, morphology and support on the mechanism of O2 activation, theoretical investigations on the adsorption and dissociation of molecular oxygen on different Au surfaces and nanoparticles have been carried out.19,89,93 The adsorption and dissociation of molecular O2 on extended gold surfaces, isolated gold nanoparticles of different size and shape, and small gold clusters supported on TiO2 have been investigated by means of DFT calculations. It has been found that O2 dissociation is very sensitive to the arrangement of the gold surface atoms. The most active sites for O2 dissociation are found at the metal–support interface and not at the gold particle. It is thus reasonable to assume that oxygen adsorbs on the edges, at the metal–support interface, and then migrates to the faces where the adsorption of the sugar occurs with subsequent reaction leading to the corresponding aldonic acid. In other works reported in the literature, the catalytic activity of unsupported colloidal gold nanoparticles (‘naked’ particles) has also been discovered.92,94 However, a short lifetime of the unsupported gold nanoparticles prevents them from being a practical catalyst. A stabilizing effect of the metal–support interactions on the original activity of the supported gold nanoparticles was demonstrated, which allows us to reach the total conversion in the oxidation of sugars.An oxidative dehydrogenation mechanism was proposed (Table 2) and a kinetic model taking into account the changes in the catalyst potential was developed for the selective oxidation of the sugars, L-arabinose and D-lactose.95,96 It is suggested that the sugars are activated by adsorption onto Au0 on the surface of the Au nanoparticles and that the dioxygen is activated by the atoms at the boundary between the support and the gold nanocrystals. The interface between the small Au particles and the support, together with the availability of defect sites at this interface are probably the important features of the active centers.41–45 This effect is thought to be highly significant for Au particles with diameter around 2 nm, which coincides with a dramatic enhancement in activity. The proposed oxidative dehydrogenation mechanism suggests that the reaction proceeds via the dehydrogenation of the adsorbed hydrogen atom bound to the hemiacetal carbon atom of sugars in the cyclic aldehydo-form, into the lactone, followed by the oxidation of the adsorbed hydrogen atom with dissociatively adsorbed oxygen. In a pure dehydrogenation mechanism, the rate determining step is considered to be the dehydrogenation of the hydroxyl group to the lactone species with its subsequent saponification favored by the alkaline pH into the acid product.
| Step | Reaction | N(1) | N(2) | N(3) | N(4) |
|---|---|---|---|---|---|
| N(1): 2A + O2 = 2B + 2H2O, N(2): B + H2O = C, N(3): O = O, N(4): 2H2O = 2OH− + 2H+. | |||||
| 1 | A + * ≡ A* | 0 | 0 | 0 | 0 |
| 2 | A + *′ ≡ A*′ | 2 | 0 | 0 | 0 |
| 3 | O2 + 2* ≡ 2O* | 1 | 0 | 0 | 0 |
| 4 | O* + *′ ≡ O*′ + * | 2 | 1 | 1 | 1 |
| 5 | A*′ + O*′ → B*′ + H2O + *′ | 2 | 0 | 0 | 0 |
| 6 | B*′ + OH−*′ → D*′ + *′ | 0 | 1 | 0 | 0 |
| 6a | D*′ + H+ → (fast) C + *′ | 0 | 1 | 0 | 0 |
| 7 | O*′ + H2O + 2e− → 2OH− + * | 0 | 0 | −1 | 1 |
| 8 | OH− + * → O* + H+ + 2e− | 0 | 1 | 0 | 2 |
| 9 | O*′ + H2O + 2e− → 2OH− + *′ | 0 | 1 | 1 | 1 |
| 10 | OH− + * ≡ OH−* | 0 | 0 | 0 | 0 |
| 11 | OH− + *′ ≡ OH−*′ | 0 | 1 | 0 | 0 |
| 12 | B*′ ≡ B + *′ | 2 | −1 | 0 | 0 |
A mechanistic kinetic model was used to describe the experimental data of L-arabinose and D-lactose oxidation over Au/Al2O3 taking into account the catalyst electrochemical changes.97–99 The model was based on the physico-chemical explanations of the observed phenomenon. The oxidative dehydrogenation mechanism was adopted and the following sequence of stages was suggested showing the selective oxidation pathway (Table 2).
It is assumed that the sugars, L-arabinose or D-lactose, can adsorb on both the faces and the edges of the Au clusters (steps 1 and 2, respectively). Oxygen is adsorbed on the edges with dissociation (step 3), thereafter migrating to the faces (step 4). The sugar oxidation involves dehydrogenation of the adsorbed hydroxyl of the hemiacetal to the lactone and oxygen reduction in step 5. The product, aldonic acid, is formed in steps 6 and 6a via the intermediate species, while steps 7 and 8 account for electron transfer, which involves oxygen adsorbed on the edges. Finally, as the oxidation rate was found to be dependent on pH, the adsorption of OH− on faces was also included in the model (step 11). Electron transfer involving adsorption of OH− on the edges was also included in the model (step 9). In addition, step 10 accounts for direct adsorption of OH− on the edges without electron transfer. Based on the suggested reaction mechanism and steps, kinetic equations were derived for the oxidation reaction rates along the first route N(1) and the second route N(2), respectively, in the following ways:
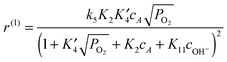 | (2) |
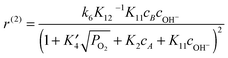 | (3) |
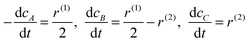 | (4) |
9. Conclusions
The application of catalysis gives new perspectives to the future exploitation of biomass derived feedstocks for the production of fine and specialty chemicals. The discovery of gold as a catalytic material, and its low temperature activity which is not replicated by other catalysts has only intensified the research in the field of aerobic oxidation. The selective oxidation of different carbohydrates, L-arabinose, D-galactose and D-lactose representing pentoses, hexoses and disaccharides, respectively, was studied with gold and gold–palladium catalysts. High selectivities were observed for the corresponding aldonic acids, the main products. The influence of kinetic parameters on the catalytic aerobic oxidation of sugars was elucidated. The aerobic oxidation of the sugars is pH dependent and slightly alkaline conditions (7–10) gave the best results. The dependence of the reaction rates on the gold particle size was investigated and the oxidation reactions were demonstrated to be structure sensitive. An oxidative dehydrogenation mechanism was proposed and a kinetic model taking into account the catalyst electrochemical potential was developed. The model was validated with the experimental observations.Acknowledgements
This work is part of the activities at Åbo Akademi University, Process Chemistry Centre (PCC) within the Finnish Centre of Excellence Program (2000–2011) appointed by the Academy of Finland. The financial support from the Academy of Finland is gratefully acknowledged.Notes and references
- B. Kamm, M. Kamm and P. Gruber, Biorefinery Systems-An Overview, in Biorefineries-Industrial Processes and Products. Status Quo and Future Directions, ed. B. Kamm, M. Kamm and P. Gruber, Wiley-VCH, Weinheim, 2006, vol. 1, pp. 3–40 Search PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS.
- A. Corma, S. Ibora and A. Velty, Chem. Rev., 2007, 107, 2411–2463 CrossRef CAS.
- D. Yu. Murzin and I. L. Simakova, Catal. Ind., 2011, 3, 218–249 CrossRef.
- P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Yu. Murzin, Catal. Rev. Sci. Eng., 2007, 49, 197–340 Search PubMed.
- E. Sjöstrom, Wood Chemistry-Fundamentals and Applications, San Diego, 2nd edn, 1993 Search PubMed.
- S. Willför, R. Sjöholm, C. Laine and B. Holmbom, Wood Sci. Technol., 2002, 36, 101–110 CrossRef.
- S. Willför and B. Holmbom, Wood Sci. Technol., 2004, 38, 173–179 CrossRef.
- P. Linko, Lactose and Lactitol, in Nutritive Sweeteners, ed. G. G. Birch and K. J. Parker, Applied Science Publishers, London and New Jersey, 1982, pp. 109–132 Search PubMed.
- T. A. Nickerson, Fundamentals of Dairy Industry Chemistry, ed. B. H. Webb, A. H. Johnson and J. A. Alford, Avi publishing Co., Wetport, 1974 Search PubMed.
- D. Murzin and T. Salmi, Catalytic Kinetics, Elsevier, Amsterdam, 2005 Search PubMed.
- F. W. Lichtenthaler, Ullmanns Encyclopedia of Industrial Chemistry, Carbohydrates, Electronic Release, Wiley-VCH, New York, 2002 Search PubMed.
- F. W. Lichtenthaler, The Key Sugars of Biomass Availability, Present Non-Food Uses and Potential Future Development Lines, in Biorefineries-Industrial Processes and Products, Status Quo and Future Directions, ed. B. Kamm, M. Kamm and P. Gruber, Wiley-VCH, Weinheim, 2006, vol. 2, pp. 3–59 Search PubMed.
- D. Yu. Murzin and T. Salmi, Catal. Lett., 2012, 142, 676–689 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Catalysis Science Series, ed. G. J. Hutchings, Imperial College Press, London, 2006, vol. 6 Search PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- G. C. Bond, Gold Bull., 1972, 5, 11–13 CrossRef CAS.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- C. H. Christensen, B. Jørgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651 CrossRef CAS.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS.
- A. Corma and M. E. Domine, Chem. Commun., 2005, 4042–4044 RSC.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS.
- N. Lopez, T. V. J. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Norskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- C. Baatz, N. Decker and U. Pruesse, J. Catal., 2008, 258, 165–169 CrossRef CAS.
- A. Mirescu and U. Prüße, Appl. Catal., B, 2007, 70, 644–652 CrossRef CAS.
- A. V. Tokarev, E. V. Murzina, J.-P. Mikkola, J. Kuusisto, L. M. Kustov and D. Yu. Murzin, Chem. Eng. J., 2007, 134, 153–161 CrossRef CAS.
- M. Comotti, C. Della Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS.
- N. Dimitratos, C. Messi, F. Porta, L. Prati and A. Villa, J. Mol. Catal. A: Chem., 2006, 256, 21–28 CrossRef CAS.
- D. I. Enache, D. W. Knight and G. J. Hutchings, Catal. Lett., 2005, 103, 43–52 CrossRef CAS.
- C. Campbell and C. Peden, Science, 2005, 309, 713–714 CrossRef CAS.
- A. Corma and M. E. Domine, Chem. Commun., 2005, 4042–4044 RSC.
- A. Abad, C. Almela, A. Corma and H. Garcia, Chem. Commun., 2006, 3178–3180 RSC.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105–114 CrossRef CAS.
- R. Zanella, L. Delannoy and C. Louis, Appl. Catal., A, 2005, 291, 62–72 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull. (Berlin, Ger.), 2000, 33, 41–52 CAS.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425–432 CrossRef CAS.
- M. Haruta, Gold Bull. (Berlin, Ger.), 2004, 37, 27–36 CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546–1548 CrossRef CAS.
- R. J. Davies, Science, 2003, 301, 926–927 CrossRef.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–284 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1995, 24, 143–150 CrossRef CAS.
- T. Mallat, C. Brönnimann and A. Baiker, J. Mol. Catal. A: Chem., 1997, 117, 425–438 CrossRef CAS.
- T. Mallat, C. Brönnimann and A. Baiker, Appl. Catal., A, 1997, 149, 247–284 CrossRef.
- V. R. Gangwal, J. van der Schaaf, B. F. M. Kuster and J. C. Schouten, J. Catal., 2004, 229, 389–403 CrossRef.
- C. Keresszegi, T. Brgi, T. Mallat and A. Baiker, J. Catal., 2002, 211, 244–251 CAS.
- J. Kluytmans, A. Markusse, B. Kuster, G. Marin and J. Schouten, Catal. Today, 2000, 57, 143–155 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Yu. Murzin, Chem. Rev., 2011, 111, 5638–5666 CrossRef.
- V. A. Babkin, Yu. A. Malkov, E. N. Medvedeva, N. N. Trofimova and N. V. Ivanova, Russ. Chem. J., 2011, 10–16 CAS.
- S. Willför, R. Sjöholm, C. Laine, M. Roslund, J. Hemming and B. Holmbom, Carbohydr. Polym., 2003, 52, 175–187 CrossRef.
- B. T. Kusema, C. Xu, P. Mäki-Arvela, S. Willför, B. Holmbom, T. Salmi and D. Yu. Murzin, Int. J. Chem. React. Eng., 2010, 8(A44), 1–16 Search PubMed.
- B. T. Kusema, G. Hilpmann, P. Mäki-Arvela, S. Willför, B. Holmbom, T. Salmi and D. Yu. Murzin, Catal. Lett., 2011, 141, 408–412 CrossRef CAS.
- J. Kuusisto, A. V. Tokarev, E. V. Murzina, M. U. Roslund, J.-P. Mikkola, D. Yu. Murzin and T. Salmi, Catal. Today, 2007, 121, 92–99 CrossRef CAS.
- P. M. Collins and R. J. Ferrier, Monosaccharides: Their Chemistry and Their Roles in Natural Products, John Wiley and Sons Ltd, England, 1995 Search PubMed.
- B. T. Kusema, B. C. Campo, P. Mäki-Arvela, T. Salmi and D. Yu. Murzin, Appl. Catal., A, 2010, 386, 101–108 CrossRef CAS.
- A. Mirescu and U. Prüße, Appl. Catal., B, 2007, 70, 644–652 CrossRef CAS.
- P. Vinke, D. de Witt, A. T. J. W. de Goede and H. van Bekkum, in Studies in Surface Science and Catalysis, ed. P. Ruiz and B. Delmon, 1992, vol. 72, pp. 1–20 Search PubMed.
- G. de Wit, J. J. de Vlieger, A. C. Kock-van Dalen, R. Heus, R. Laroy, A. J. van Hengstum, A. P. G. Kieboom and H. van Bekuum, Carbohydr. Res., 1981, 91, 125–138 CrossRef CAS.
- E. Smolentseva, B. T. Kusema, S. Beloshapkin, M. Estrada, E. Vargas, D. Yu. Murzin, F. Castillon, S. Fuentes and A. Simakov, Appl. Catal., A, 2011, 392, 69–79 CrossRef CAS.
- D. Liang, J. Gao, J. Wang, P. Chen and Z. Hou, Catal. Commun., 2009, 10, 1586–1590 CrossRef CAS.
- B. T. Kusema, B. C. Campo, O. A. Simakova, A.-R. Leino, K. Kordás, P. Mäki-Arvela, T. Salmi and D. Yu. Murzin, ChemCatChem, 2011, 3, 1789–1798 CrossRef CAS.
- U. Prüße, K. Heidkamp, N. Decker, M. Herrmann and K.-D. Vorlop, Chem. Ing. Tech., 2010, 82, 1231–1237 CrossRef.
- U. Prüße, K. Heidkamp, N. Decker, M. Herrmann and K.-D. Vorlop, Chem. Ing. Tech., 2010, 82, 1231–1237 CrossRef.
- A. V. Tokarev, E. V. Murzina, J. Kuusisto, J.-P. Mikkola, K. Eränen and D. Yu. Murzin, J. Mol. Catal. A: Chem., 2006, 255, 199–208 CrossRef CAS.
- A. V. Tokarev, E. V. Murzina, J.-P. Mikkola, J. Kuusisto, L. M. Kustov and D. Yu. Murzin, Chem. Eng. J., 2007, 134, 153–161 CrossRef CAS.
- E. V. Murzina, A. V. Tokarev, K. Kordás, H. Karhu, J.-P. Mikkola and D. Yu. Murzin, Catal. Today, 2008, 131, 385–392 CrossRef CAS.
- A. V. Tokarev, E. V. Murzina, P. K. Seelam, N. Kumar and D. Yu. Murzin, Microporous Mesoporous Mater., 2008, 113, 122–132 CrossRef CAS.
- A. V. Tokarev, E. V. Murzina, K. Eränen, H. Markus, A. J. Plomp, J. H. Bitter, P. Mäki-Arvela and D. Yu. Murzin, Res. Chem. Intermed., 2009, 35, 155–174 CrossRef CAS.
- P. Mäki-Arvela, E. V. Murzina, B. Campo, T. Heikkilä, A.-R. Leino, K. Kordas, D. Wolf, A. V. Tokarev and D. Yu. Murzin, Res. Chem. Intermed., 2010, 36, 423–442 CrossRef.
- P. Mäki-Arvela, A. V. Tokarev, E. V. Murzina, B. Campo, T. Heikkilä, J.-M. Brozinski, D. Wolf and D. Yu. Murzin, Phys. Chem. Chem. Phys., 2011, 13, 9268–9280 RSC.
- G. C. Bond, Metal-Catalysed Reactions of Hydrocarbons, Springer, New York, 2005 Search PubMed.
- D. Yu. Murzin, J. Mol. Catal. A: Chem., 2010, 315, 226–230 CrossRef CAS.
- D. Yu. Murzin, J. Catal., 2010, 276, 85–91 CrossRef CAS.
- S. Ivanova, V. Pitchon and C. Petit, J. Mol. Catal. A: Chem., 2006, 256, 278–283 CrossRef CAS.
- S. Ivanova, V. Pitchon, Y. Zimmermann and C. Petit, Appl. Catal., A, 2006, 298, 57–64 CrossRef CAS.
- O. A. Simakova, B. T. Kusema, B. C. Campo, A.-N. Leino, K. Kordas, V. Pitchon, P. Mäki-Arvela and D. Yu. Murzin, J. Phys. Chem. C, 2011, 115, 1036–1043 CAS.
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef.
- H. Okatsu, N. Kinoshita, T. Akita, T. Ishita and M. Haruta, Appl. Catal., A, 2009, 369, 8–14 CrossRef CAS.
- M. Ojeda and E. Iglesia, Chem. Commun., 2009, 352–354 RSC.
- J. Gong and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 16458–16459 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS.
- O. A. Simakova, V. I. Sobolev, K. Yu. Koltunov, B. Campo, A.-R. Leino, K. Kordás and D. Yu. Murzin, ChemCatChem, 2010, 2, 1535–1538 CrossRef CAS.
- C. Della Pina and E. Falletta, Catal. Sci. Technol., 2011, 1, 1564–1571 CAS.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538–8546 RSC.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS.
- B. T. Kusema, J.-P. Mikkola and D. Yu. Murzin, Catal. Sci. Technol., 2012, 2, 423–431 CAS.
- D. Yu. Murzin, E. V. Murzina, A. V. Tokarev and J.-P. Mikkola, Russ. J. Electrochem., 2009, 45, 1017–1026 CrossRef.
- G. Horanyi, Catal. Today, 1994, 19, 285–312 CrossRef CAS.
- M. I. Temkin, Adv. Catal., 1979, 28, 173–281 CrossRef CAS.
- K. D. Popovic, A. V. Trikovic and R. R. Adzic, J. Electroanal. Chem., 1992, 339, 227–245 CrossRef CAS.
- H. Haario, ModEst Users's Guide 6.1., ProfMath Oy, Helsinki, 2002 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |