Nanoscale (111) faceted rock-salt metal oxides in catalysis
Christopher A.
Cadigan
,
April R.
Corpuz
,
Feng
Lin
,
Christopher M.
Caskey
,
Kenneth B. H.
Finch
,
Xue
Wang
and
Ryan M.
Richards
*
Colorado School of Mines Department of Chemistry and Geochemistry, 1500 Illinois St, Golden, CO 80401, USA. E-mail: rrichard@mines.edu; Fax: +1 (303) 273-3629; Tel: +1 (303) 273-3612
First published on 19th September 2012
Abstract
Facet-specific growth is highly desirable for catalysts, as conversion and selectivity can be altered with increasing amounts of particular active sites. The (100) surface of a rock-salt structure is comprised of alternating oxygen anions and metal cations, similar to the (110) facet. The (111) surface differs substantially in that an ideal (111) surface would consist solely of oxygen anions or metal cations, and cannot exist as-is. However, wet chemical syntheses of MgO(111) and NiO(111) have recently been reported; theory and experiments show the (111) surface is stabilized as a hydroxylated surface. These (111) faceted metal oxides exhibit catalytic properties that differ significantly from their (100) counterparts. Here, we discuss the theory and performance of the (111) rock-salt metal oxides in catalysis, as supports, and as adsorbents.
1. Introduction
Metal oxides have a diverse range of electronic and physical properties which make them useful for a variety of applications such as insulators in cables, semiconductors in diodes, different types of glasses and gels, and doping agents or catalysts to name a few. Development of new materials with improved conversions and/or selectivities is one of the main objectives in catalyst research. Metal oxides can take a variety of structures, morphologies, and metal to oxygen ratios, all of which can affect their catalytic properties. Specifically, in heterogeneous catalysis, materials might show different catalytic properties based on their primary exposed facet(s).One of the simplest structures of metal oxides is the rock-salt structure. Rock-salt metal oxides are face-centered cubic crystals with one metal ion surrounded by six nearest-neighbor oxygen ions and vice versa. The rock-salt metal oxides (MgO, CaO, NiO, CoO, MnO, SrO and EuO) are extensively used as catalysts and catalyst supports. Some examples are the catalytic effect of MgO(100) on the combustion of methane,1 the catalytic abilities of MgO(111) nanosheets and NiO(111) nanosheets,2,3 and the catalytic conversion of NO to NO2 on nickel oxide.4,5
Among the metal oxides mentioned above, MgO is the most widely studied and is reviewed by Spoto et al.6 The large number of studies on MgO is primarily due to its simplistic structure and high ionic character that make it a good model compound for computational and surface science studies. Experiments have been performed on both single crystals and thin films7–28 as well as powdered MgO.29–38 Investigations involving single crystal and/or thin film MgO samples typically require ultra-high vacuum conditions where more pure surface science information can be obtained that require the sample to be cleaved or grown in situ.
However, some of the more common methods of preparing MgO powders include: heating/combustion of solid magnesium in air,39–41 aerogel preparations that include precipitation from liquid solution and supercritically dried in an autoclave,29,31–33,35,36,42–54 chemical vapor deposition (CVD),55–63 solvothermal and hydrothermal treatments,34,37,64–67 and decomposition and topotactic dehydration of Mg(OH)2 under vacuum.40,68–73 Direct combustion of Mg ribbon in the presence of O2, and often combined with physical vapor deposition (PVD), can produce well-defined MgO cubes with (100) faces.39–41 These materials can be extremely defect-free and have high crystallographic quality. By simply varying oxygen partial pressures, MgO nanomaterials (orthogonally branched, nanocubes and straight nanowires) can also been obtained.74 Numerous other MgO nanomaterials including rods, belts, wires and fishbone fractal nanomaterials have also been synthesized utilizing the above synthesis techniques.74–82 Various NiO nanostructures can also be prepared via the same techniques as those mentioned above for MgO.83–92
Starting in the 1990s, Klabunde et al. at Kansas State University developed a method to producing metal oxides via a sol–gel methodology coupled with supercritical drying. This synthesis first converts a metal/metal salt into a methoxide and then into a hydroxide gel. Once hypercritically dried, a fine metal oxide (100) powder results with surface areas upwards of 500 m2 g−1.29,31–33,35,36,42–54
Complete understanding of the conditions for high performing metal oxide catalysts is not yet achieved. However, it is typically thought that free coordination sites on the surfaces of metal oxides such as steps, corners, and vacancies are important for catalysis.50,93–102 Lower coordination sites, or coordinatively unsaturated sites, play a crucial role in binding reactants or adsorbing molecules in order for reactions to proceed. Atoms on the surface of a material can have a coordination number that ranges between 3 and 5 due to the many surface defects on a crystal. Since site-specific chemistry is often difficult to measure under realistic catalytic conditions, coordination sites and their effectiveness are often measured indirectly. For example, results from methane adsorption on MgO(100), measured with infrared spectroscopy and analyzed using density functional theory (DFT), suggest that only three-coordinated O2− sites have the ability to bind methane. Meanwhile, the four- and five-coordinated O2− sites, along with the low coordinated Mg2+ sites, demonstrated a repulsive interaction with respect to binding methane.59 This clearly shows that the type of coordination site is important and can be highly specific for successful catalysis.
Edges and point defects are usually found to be more active than terraces (“flat” surface sites), as the former are more coordinately unsaturated.50,93–102 However, there are terraces with many active sites that are also thought to be very active.2,103–107 One example of this is a rock-salt (111) surface that can be classified as a Tasker “type 3” surface. A Tasker “type 3” surface is one with alternating layers of cations and anions. According to Tasker, such a surface, if bulk-terminated, would be unstable; the alternating layers of cations then anions would create a diverging electric field with an infinite surface energy.108 Since the surface energy of a material cannot be infinite, charge compensation must occur in order to alleviate the infinite surface dipole. While conceivably there are many possible modes of stabilization for a Tasker 3 surface, theory points to three primary scenarios to mitigate the surface dipole: surface reconstruction, electronic relaxation, and adsorption of species that stabilize the electric field.97,106,109 Stabilization can also change depending on the nanoscale properties of the material.110,111 Through some mode of stabilization, many different types of stabilized Tasker type 3 surfaces have been synthesized (e.g., single crystals,112–114 films,8,110,115,116 nanowires,106 nanocrystals2,3,103,106,107), and often show higher catalytic activity than the other surfaces of the same material.2,104,105,107
The majority of the literature to date, involving the catalytic activity of metal oxides with rock-salt structures, has been done on the (100) surface. This is due to the (100) materials being the most readily obtained and most thermodynamically stable form of most rock-salt metal oxides. For rock-salt type metal oxides, the (100) surface is a checkerboard of alternating metal cations and oxygen anions, as shown in Fig. 1. While most methods produce materials dominated by (100) surfaces, decomposition of metal hydroxides such as Mg(OH)2 and Ni(OH)2, can initially yield materials with hydroxylated (111) surfaces via topotactic dehydration.6,46,117 Following the development of techniques that allow for the deliberate preparation of materials with primarily (110) and (111) surfaces, came interest in potentially new catalytic abilities of these surfaces. An example of these surfaces for rock-salt type metal oxides is also shown in Fig. 1. In particular, the (111) surface is a Tasker type 3 surface; the (111) surface is comprised of alternating layers of reactive anions and cations. The alternating cation–anion layers cause the (111) surfaces to be very polar.97,108,118,119 Due to the polarity of the (111) surfaces, they have been theorized to be a “unique playground for catalysis” as well as an intriguing support material.110 In an effort to minimize surface (and structural) energy, the polar (111) surfaces are often hydrogen terminated, which makes (111) materials interesting for hydrogen storage or water splitting applications.118
 | ||
| Fig. 1 A prototypical rock-salt structured metal oxide, MgO, with the (100), (110), and (111) facets shown. Note that the (100) and (110) facets are composed of alternating cations and anions, while the (111) facet is made up of a single type of ion (either all cations or anions, depending on how the face is cut). Reproduced from ref. 125 by permission of The Royal Society of Chemistry. | ||
The present review is directed towards recent advances on the controlled synthesis and catalytic activity of MgO(111) and NiO(111) nanoparticle aerogels using wet chemical preparations.2,3,120–126 Although these two materials have nearly identical lattice spacings, MgO is a more insulating metal oxide, based on its experimental band gap of 7.8 eV,127 and is expected to have strong Lewis acid/base sites,50 while NiO, on the other hand, is a transition metal oxide and a p-type semiconductor with a wide band gap of 3.6–4.0 eV.128 Due to these differences between MgO and NiO as metal oxides, this system allows comparison of geometrically identical but electronically different surfaces. An additional consideration is that Ni, as a transition metal, has d-electrons and thus the capability to adopt oxidation states other than 2+.
2. Surface structure studies
If comparing (100), (110), and (111) facets of a rock-salt metal oxide in ideal dry conditions, the (100) facet has the lowest cleavage energy, and is the most thermodynamically stable surface.129 Subsequently, the (100) surface of alternating metal cations and oxygen anions along any line of nearest neighbors is typically reported in most papers dealing with the synthesis of rock-salt metal oxides. However, the facet stability is highly dependent on environmental conditions, and experiments have indicated that in wet conditions the (111) surface is more thermodynamically stable.130–132 Therefore, under proper conditions, the (111) surface, which consists of a plane of metal cations or oxygen anions, can be produced. Studies focused on a fundamental understanding of the rock-salt (111) surface have yielded increasingly more detailed analyses of the surface structures. Additionally, MgO(111) and NiO(111) are naturally formed in wet environments occurring as periclase and bunsenite respectively.There is some debate as to whether the surfaces of metal oxide (111) particles thermally microfacet to more stable (110) or (100) facets.133 Theoretical and experimental studies show that the treatment method of the metal oxides plays a crucial role and can alter the results. For example, in one study, acid etching was responsible for the faceting of the (111) surfaces to pyramids.134
In 1998, Plass et al. proposed that, above 1200 °C, reconstructions of the MgO(111) surface involving groups of oxygen trimers (ozone) and/or single oxygen atoms, occur periodically on the surface.135 The (√3 × √3)R30° (Rt3) reconstruction is comprised of equilateral oxygen trimers. The (2 × 2) and Rt3 reconstructions have both oxygen trimers and single oxygen atoms on the surface. At the time, shown by transmission electron diffraction, these reconstructions were the current standing models of the MgO(111) surface above 1200 °C.135
In 2003, ab initio studies using DFT on both MgO(111) and NiO(111) surfaces was the subject of investigation by Wander et al.129 The authors calculated that a hydroxylated (111) surface is energetically more stable than a clean (100) surface and more favorable than a dry octopolar reconstruction, which is a relatively low energy reconstruction of pyramids in a 2 × 2 array containing three oxygen atoms with a nickel atom on top or vice versa. The values of the calculated energies can be found in Table 1.129
In a work by Lazarov et al. in 2005, MgO(111) single crystals were studied with a range of ultra-high vacuum (UHV) techniques, including reflection high-energy electron diffraction (RHEED), low-energy electron diffraction (LEED), X-ray photoelectron diffraction (XPD), and Auger electron diffraction (AED).118 XPD/AED analysis favored the models with O- and OH-terminated surfaces instead of Mg-terminated surfaces. The experimental XPD curve of the MgO(111) single crystal was compared to curves of oxygen-, hydroxyl-, and magnesium-terminated surfaces based on theoretical methods – single scattering XPD in cluster geometry, multiple scattering XPD in cluster geometry, and multiple scattering XPD in slab geometry. The theoretical methods modeling oxygen- and hydroxyl-terminated surfaces yielded XPD curves similar to the experimental XPD curve of an MgO(111) single crystal; the theoretical Mg-terminated XPD curve was not similar to the experimental XPD curve. Experimental X-ray photoemission spectroscopy (XPS) data of the MgO(111) single crystal showed a surface-specific 2 eV shift towards higher binding energy when comparing the high-energy-resolution O 1s spectra at normal and grazing emission. Comparing this with theoretical XPS data of oxygen-, hydroxyl-, and magnesium-terminated surfaces generated by DFT shows a hydroxyl-terminated surface to be the most likely. DFT of the O 1s state for an oxygen-terminated surface predicted a 1 eV shift to lower energy, no shift for a magnesium-terminated surface, and a 1 eV shift to higher energy for a hydroxyl-terminated surface.118
More recently in 2009, Ciston et al. analyzed the Rt3 and 2 × 2 reconstructions on the MgO(111) surface by utilizing transmission high-energy electron diffraction (THEED), transmission electron microscopy (TEM), XPS, and DFT calculations.136 Direct methods analysis of THEED data revealed that possible Rt3 reconstruction structures include Rt3-Mg (a Mg-terminated surface with 2/3 of the bulk-like Mg atoms missing) and the Babinet Rt3-O structure where the two structures are indistinguishable using in-plane diffraction data. Additionally, the insertion of a hydrogen atom per unit cell to the second layer of a Rt3-Mg structure (Rt3-MgH) and to the termination layer of Rt3-O as a hydroxyl (Rt3-OH) results in two more possible structures. The THEED data for the 2 × 2 reconstruction is explained by a 2 × 2-α structure that maintains the underlying cation framework and has three surface sites that can be occupied with Mg, O or other species. XPS data from annealed samples showed significant chemisorbed hydroxyl groups for both Rt3 and 2 × 2 reconstructions and therefore the Rt3-Mg and Rt3-O are rejected as likely structures. DFT calculations yield an Rt3-OH surface structure that is only slightly lower in energy than the Rt3-MgH. For 2 × 2 reconstructions, DFT analysis demonstrated that the 2 × 2-α structures with lower energy were the ones that contained oxygen at each surface site with one uni-coordinated and one tetrahedral oxygen surface site terminated with hydrogen. While 2 × 2-octapolar structures are low in surface energy, they are not consistent with electron diffraction data. A kinetic model, driven by adsorption/desorption and dissociation of water at the surface, details a structural progression from a 1 × 1 H precursor to an Rt3-OH structure and further to a 2 × 2-α-OH structure. While previously reported 2 × 2-octapole surfaces are shown to be energetically favorable, the requirement of oxygen and magnesium exchange proves to be too slow kinetically to exist over a hydroxyl terminated Rt3 reconstruction.136 In addition to work on MgO(111), Ciston et al. investigated NiO(111), using the same methods, and concluded that the data supports significant coverage of hydroxyl groups and the surface structures are nearly identical as those on MgO(111).137
In 2011, Enterkin et al. used a chemical bonding approach to investigate polar oxide surfaces that included MgO(111) and NiO(111).138 The study concentrated on bond valence sum (BVS) calculations to find proper valence neutral surfaces by using ions and their bond distances, which give insight into structural chemistry and chemical bonding to predict what surface structures may form. BVS analysis (with geometrical considerations) on MgO(111) structures, and their resulting surface stabilities, were all consistent with the conclusions drawn by Ciston et al.136 Moreover, BVS analysis supported the structural evolution proposed by Ciston et al. and confirmed which sites in the 2 × 2-α structure preferentially desorb water molecules when transitioning to the Rt3-OH structure upon annealing/dehydration. BVS trends also correlate well with NiO(111) structures and DFT results presented by Ciston et al.137 with the only exception being hydroxylated NiO 2 × 2-α structures, in which case the trends are opposite. Overall, the BVS analysis supports earlier works by Ciston et al.136,137 with a chemical bonding perspective.
3. Preparation and characterization of (111) metal oxides
Wet chemical methods have been employed to produce metal oxides with a surface that is predominantly the (111) facet, including sol–gel chemistry coupled with templating agents that produce (111) materials in a modified aerogel process with supercritical drying. The resulting materials have been characterized with high-resolution transmission electron microscopy (HRTEM), nitrogen physisorption/BET analysis, X-ray diffraction (XRD), electron diffraction (ED), in situ diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS), and temperature programmed desorption (TPD).MgO(111)
The wet chemical synthesis of MgO(111) was first reported by Zhu et al.2 The MgO(111) nanosheets are synthesized by a modified aerogel method. Briefly, clean magnesium is dissolved in methanol under inert atmosphere to form a solution of Mg(OCH3)2. 4-Methoxybenzyl alcohol is then added as a templating agent by interacting with the hydroxyl group of the intermediate Mg(OH)–(OCH3) more strongly than methanol due to a higher acidity, to form a material with a predominantly (111) surface. In the absence of the 4-methoxybenzyl alcohol templating agent, the MgO(111) nanosheets were not observed. The addition of water induces hydrolysis and MgO nanosheets form. The resulting white sol–gel is then transferred to an autoclave reactor where it is purged with argon and then pressurized to 10 bar before heating to 265 °C. Upon heating, the pressure in the reactor increases to reach a pseudo-supercritical state where it is maintained. Pseudo-supercritical drying is performed by releasing the pressure while still hot, resulting in the dry white powder precursor, Mg(OH)(OCH3). Calcination in air at 500 °C removes all carbon species and MgO(111) nanosheets are obtained.The resulting materials were primarily characterized by HRTEM, nitrogen physisorption/BET analysis, X-ray diffraction, electron diffraction, in situ diffuse reflectance infrared Fourier transform spectroscopy, and temperature programmed desorption.2,126 HRTEM studies provided evidence of the (111) surfaces of MgO(111) by looking at nanosheets that were parallel to the optic axis of the TEM as seen in Fig. 2. This view allowed the measurement of the lattice spacings between 0.24 and 0.25 nm, which are in good agreement with the theoretical {111} lattice spacings of magnesium oxide. An MgO(111) nanosheet laying perpendicular to the optic axis of the TEM, displayed ED analysis lattice fringes of the 111 zone, signifying that the sheets lie on the (111) plane. MgO(111) nanosheets are 200–500 nm in diameter and 3–5 nm thick yielding a BET surface area of 198 m2 g−1 determined by nitrogen physisorption analysis.
 | ||
| Fig. 2 HRTEM images of two MgO(111) nanosheets viewed edge-on (parallel to the optic axis of the TEM). Insets are local Fourier transforms showing the lattice fringe measurements. (Reproduced with permission from ref. 2. Copyright 2006, Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
DRIFT spectra of MgO(111) nanosheets treated under high vacuum and heated to different temperatures are shown in Fig. 3. At room temperature, bands observed at 1642 cm−1 and 3300 cm−1 are due to the O–H bending and stretching of water molecules on the surface of the MgO(111). A clean surface is obtained after heating to 500 °C for 1 h. Bands observed between 3300 and 3800 cm−1 are due to surface hydroxyl groups with varying coordination to metal cations. There is a corresponding decrease in the stretching vibration frequency as the coordination number of the OH group increases. It was observed that OH stretching bands on commercial MgO with exposed (100) facets produced 3700 cm−1 stretching bands, corresponding to hydroxyl groups with a coordination number of 5. The 3723 cm−1 stretching bands observed for MgO(111) at 500 °C can reasonably be assigned to OH groups with a coordination number of 3. Shoulder bands at 3637 and 3538 cm−1 are produced by surface defects, and the band at 3391 cm−1 comes from surface O2− species. It is reasoned that OH groups on the surface of MgO(111) stabilize the polar (111) surface. This characterization finds both oxygen anions and hydroxyl groups on the (111) surface with oxygen defects.126
 | ||
| Fig. 3 DRIFT spectra of MgO nanosheets under high vacuum at (a) room temperature, (b) 100 °C, and (c) 500 °C. Reprinted with permission from ref. 126, Copyright 2007 American Chemical Society. | ||
TPD experiments were carried out with CO2 to measure the basicity of surface sites. Prior to adsorption of CO2, a blank TPD was performed up to 800 °C to verify that there was no CO2 desorption. The TPD profile seen in Fig. 4 shows several CO2 desorption peaks that correlate to CO2 desorbing from different basic surface sites of varying strengths. Peaks in the TPD profile suggest basic sites increasing in strength as the temperature increases, with the identification of the basic sites and their corresponding base strength order: hydroxyl groups < oxygen in Mg2+ and O2− pairs < low coordinated oxygen anions. This order coincides with the temperature ranges seen in Fig. 4 between 20 and 160 °C, 160 and 400 °C, and above 400 °C. Peaks that come off in the range between 20 and 160 °C can be assigned to CO2 interacting with weakly basic hydroxyl groups on the MgO(111) surface. Peaks in the second temperature range between 160 and 400 °C can be attributed to the association with mildly basic Mg2+ and O2− pairs. Finally, peaks appearing above 400 °C identify an absorbance with strongly basic isolated O2− sites. A high number of sites that produce the peak at 350 °C indicate a large quantity of medium basic sites from ionic pairs that is due to the alternating ionic layers of the MgO(111) structure. Overall, TPD results suggest that the MgO(111) surface is mostly covered by medium basic Mg2+ and O2− pairs, followed by surface hydroxyl groups.126
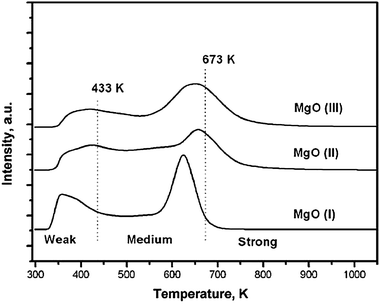 | ||
| Fig. 4 TPD of CO2 over different nanocrystalline MgO catalysts. Studied samples include: MgO(111) nanosheets (MgO(I)), conventionally prepared MgO (MgO(II)) and aerogel prepared MgO (MgO(III)). Reproduced from ref. 125 by permission of The Royal Society of Chemistry. | ||
NiO(111)
Hu et al. first reported the wet chemical synthesis of NiO(111).3 NiO(111) nanosheets are similarly prepared via a modified aerogel preparation. In a typical preparation, Ni(NO3)2·6H2O is dissolved in absolute methanol. Once dissolved, benzyl alcohol and urea are added to the solution to provide a structure-directing agent and pH neutralizer respectively. The solution is stirred before being transferred to an autoclave and purged with argon. Once purged, the reactor is pressurized with argon before being heated to 200 °C. The temperature is then raised to 265 °C and held before releasing the pressure. Pseudo-supercritical drying is achieved by releasing the pressure, which leaves behind a light green powder precursor. The final NiO(111) material is obtained by calcination at 500 °C in air.General characterization of the prepared NiO was done by TEM and BET. TEM images show that the nanosheets are up to 1 micrometer in diameter with a thickness of 1–5 nm. The sheets also show numerous hexagonal holes in the material that can be up to 100 nm across, as shown in Fig. 5. The surface area of the material is 88 m2 g−1 as determined by BET. To determine the main exposed facet of the NiO(111) surface, ED analysis revealed that the 111 plane is parallel to the main surface of the nanosheets. This result was accomplished by focusing the incident electron beam along the [111] zone axis and analyzing the resulting hexagonal diffraction pattern. ED analysis also revealed that the material is single crystalline. The (111) structure was further supported by XRD studies. By depositing an ethanolic solution of nickel oxide nanosheets on a silicon wafer, followed by evaporation of solvent, the nanosheets assemble themselves flat onto the silicon wafer, as it would be difficult for all of the nanosheets to be standing on edge (especially since the edges are rough). The results showed only the (111) diffraction peak. If other types of facets were present on the surface, they too would have displayed a peak on the XRD; this shows that it is the (111) facet that is the main surface of the nanosheets.3
 | ||
| Fig. 5 HRTEM image and ED pattern of single crystalline NiO(111) nanosheet with hexagonal holes, where angles between straight lines (AB, BC, and AC) are oriented at 120°. (Reproduced with permission from ref. 3. Copyright 2008, Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
For both the preparation of NiO(111) and MgO(111) a structure-directing agent is vital to produce a metal oxide with the primary surface being the (111) surface. While benzyl alcohol is necessary to obtain the (111) facet in the NiO(111) preparation and has been shown experimentally to be a good agent for tailoring certain metal oxides, the exact mechanism is not well understood. It is likely that the structure-directing agent either promotes crystal growth in the (111) plane, or somehow poisons growth in the direction of other planes. Although urea plays a role in the size of the NiO(111) nanosheets, it is mainly important in the synthesis method because it neutralizes the nitric acid byproduct that is formed from the reaction between nickel nitrate, methanol, and water. Too much acid hinders the formation of nickel oxide precursor.
4. Catalysis
On the basis of theory and the surface science studies, the catalytic properties of the (111) faceted metal oxides are of interest for a broad portfolio of processes. Initial studies have focused on methanol oxidation, condensations and transesterification as well as their role as catalyst supports.A. Methanol oxidation
Methanol is used as a common probe molecule for studying catalyst surfaces, and as such the adsorption of methanol on the NiO(111) aerogel was studied by using in situ DRIFTS. After exposing the NiO(111) surface to methanol vapor pressures of 1 and 0.005 torr, formate species were identified in the IR. After exposure to methanol at 70 °C for 5 minutes, a large amount of CO2 was measured on the NiO(111) surface, and this amount increased with time. From this information, the mechanism involves dissociation of methanol on the NiO(111) surface, formation of a formate intermediate species, and resulting in CO2 and H2 production, with the loss of a surface oxygen on the NiO(111) surface. Methanol adsorption was also studied on conventionally prepared NiO (CP-NiO), made by nickel acetate decomposition at 500 °C for 5 hours, producing NiO(100) with a surface area of 32 m2 g−1. After exposure of the CP-NiO surface to methanol vapor pressures of 1 and 0.005 torr, no dissociative adsorption or oxidation of methanol occurred. After exposure to methanol at 70 °C for 5 minutes, no CO2 was formed and a small amount of CO was formed. From these studies, it was concluded that the (111) facet of the NiO aerogel was responsible for the dissociation and oxidation of methanol.3 The proposed mechanism, as suggested by the DRIFTS data, can be found in Fig. 6. Details and spectra of the DRIFTS work can be found in ref. 3.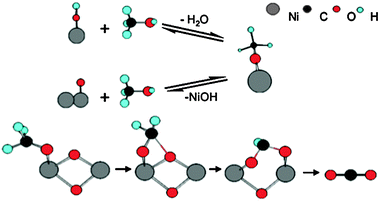 | ||
| Fig. 6 Proposed mechanism for methanol decomposition to H2 and CO2 over NiO(111). (Reproduced with permission from ref. 3. Copyright 2008, Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
In situ DRIFTS was also utilized to investigate the adsorption of methanol on the surface of MgO(111) nanosheets. Studies on conventional MgO(100) have shown that methanol does not adsorb on smooth crystalline surfaces of (100), but adsorbs when defects are present on the (100) surface. When methanol vapor was introduced to clean MgO(111), DRIFTS studies showed gas phase methanol as well as weakly adsorbed methanol on the MgO(111) surface. Methoxyl groups and formate species were also observed in the DRIFT spectra at room temperature. Methanol was removed by slowly lowering the pressure with vacuum which produced a decrease in the amount of methanol seen in the infrared, implying that adsorption of methanol on the MgO(111) surface is reversible. When MgO(111) was heated to 70 °C in the presence of methanol, within 25 minutes the majority of the methanol had been oxidized to CO2. Over the 25 minutes, the CO2 peak area gradually increased while the C–H peaks decreased. Comparing the room temperature spectra with the 70 °C spectra shows minor change in the O–H region, which suggests that adsorbed methanol species interact primarily with oxygen anions and defects (as opposed to hydroxyl groups). The same DRIFTS procedures were also carried out on commercial MgO(100), and it was found that only trace amounts of methanol were adsorbed on the MgO(100) surface with mostly reversible adsorption; a small amount of irreversible methanol adsorption occurred at, presumably, defect sites. When heated to 70 °C, methanol over MgO(100) forms carbonyl species that increase with time, but no formation of CO2 was detected. Additionally, methanol over MgO(111) at 70 °C shows that some carbonyl species are formed, but the peak intensity remains weak throughout the experiment, suggesting that the carbonyl species decompose into CO2.126
B. Lewis base condensation
The Claisen–Schmidt condensation is a carbon–carbon bond forming reaction of great value to the pharmaceutical and fine chemical industries.139,140 Additionally, the elimination of water at the end of the reaction increases the gravimetric energy content of reactant molecules making the Claisen–Schmidt or aldol condensations of potential value for alternative chemical fuels. The reaction can be catalyzed by free acids and bases, as well as by catalysts containing acid or base sites.141 This reaction is often studied in terms of the reaction between benzaldehyde and acetophenone, because water can only be eliminated from one position and the distribution of products is simple. Several mechanisms have been proposed for this reaction, and one commonly accepted involves the formation of acetophenonate ions which attack the carbonyl group of the benzaldehyde (see ref. 142 and references therein).Fourier transform infrared spectroscopy (FTIR) provides a good method of unambiguously characterising the adsorption and reaction of molecules on oxides. The adsorption of acetophenone on metal oxides has been studied by in situ FTIR, and the chemisorbed acetophenonate ions have been observed. However, the weakly adsorbed acetophenonate ions have never been reported.143,144 It is likely that most metal oxides are not active enough to produce observable concentrations of acetophenonate. In 2006, Zhu et al. compared the Claisen–Schmidt activity of MgO(111) nanosheets to several existing MgO systems, and the MgO(111) performed exceptionally well. This is noteworthy in that it points toward the heterogenization of the Claisen–Schmidt reaction, and toward the benefits accompanying heterogeneous processes including easier product recovery and catalyst recycling.2
Metal oxides have been used extensively as solid state Lewis bases, and a 2006 review by Corma and Iborra provides an excellent summary of research to that date.145 Basic sites on metal oxides consist of the oxygen anion or the oxygen atom associated with protons. From an argument of charge density, the expected strength of basic sites increases as anionic strength increases: H3O+ < H2O < OH− < O2−. It has been proposed that the O2− ion is the active site for some condensation reactions, and the OH− ion the active site for others. Additionally, the coordination number of the atom in the active site will have a profound effect on the activity and selectivity.145 Oxygen anions having 3, 4, 5, and 6 nearest neighbors are designated O2−(3c), O2−(4c), O2−(5c), and O2−(6c), respectively. The 2006 review by Corma and Iborra states that, “it becomes apparent that the population of basic sites and consequentially the activity and selectivity of the catalyst can also be changed by control of the size and shape of the MgO crystals through the preparation procedure, because this controls the relative number of atoms located at corners, terraces, or faces, i.e., O2−(3c), O2−(4c), and O2−(5c).”145 The unique geometry of the (111) surface has a high number of O2−(3c) sites146 and would therefore would be well suited to some reactions.
In 2004, Choudary et al. published a study which was in part devoted to the comparison of catalytic activities of various morphologies of MgO.139 The researchers compared commercial MgO (surface area 30 m2 g−1) with conventionally prepared MgO (surface area 250 m2 g−1) and an aerogel prepared MgO (surface area 590 m2 g−1).33,53 The conventionally prepared MgO is a nanocrystalline MgO catalyst from NanoScale Materials Inc, made by boiling commercial MgO with deionized water followed by heat treatment under vacuum. The resulting material exhibits hexagonally shaped disks with primarily (100) surfaces as hexagonal and rectangular faces, as well as some (110) rectangular surfaces.29,36,51,53 The aerogel prepared MgO is nanocrystalline from NanoScale Materials Inc. (also known as MgO Plus) with mainly square (100) faces; detailed methods on synthesizing aerogel prepared MgO can be found in ref. 29, 33, and 53. Choudary et al. found that the aerogel prepared MgO (NAP-MgO) was the most active, for the Claisen–Schmidt condensation of benzaldehyde and acetophenone (Scheme 1), followed by silylated NAP-MgO, conventionally prepared MgO (NA-MgO), silylated NA-MgO, and then commercial MgO.139 The aerogel prepared MgO(100)33,53 is the most similar to the aerogel prepared MgO(111) in terms of preparation techniques. MgO(111) (surface area 190 m2 g−1) was compared to the aerogel prepared MgO(100) found to have even higher catalytic activity for the Claisen–Schmidt condensation of benzaldehyde and acetophenone;2 this is shown in Fig. 7. The fact that MgO(111), with a lower surface area, outperformed the higher surface area aerogel prepared MgO(100) in this reaction, indicates that faceting is an essential factor in determining activity.
 | ||
| Scheme 1 Claisen–Schmidt condensation of benzaldehyde and acetophenone to benzalacetophenone. | ||
 | ||
| Fig. 7 Conversion of benzaldehyde with various MgO catalysts. Reactions were run in toluene at reflux (110 °C) with excess acetophenone. Figure adapted from ref. 2, with data for aerogel prepared and conventionally prepared MgO from ref. 139. | ||
C. Transesterification
Typical catalysts used for biodiesel transesterification are homogeneous base catalysts such as KOH and NaOH.147 While these catalysts are effective, there are issues that arise with biodiesel produced using these methods. Extraneous cations from these homogeneous catalysts can degrade engines and engine parts during the process of combustion in current diesel engines. To avoid these issues, great care must be taken to remove excess cations present in the fuel produced.147 Using solid (heterogeneous) base catalysts would circumvent the particular problem of cations introduced by the catalyst. There have been some recent studies that utilize solid basic oxides catalysts like CaO/MgO that have activity for transesterification, but they are susceptible to poisoning by free fatty-acids in the input stream as well as water and other acidic impurities.148Recent studies of basic oxides have shown that MgO mixed metal oxides have a very good affinity for converting triglycerides to fatty-acid methyl-esters (FAMEs), which are the major constituents in current diesel fuels.148 The reaction scheme is shown in Scheme 2. MgO(111) was shown to be an effective catalyst for transesterification of sunflower and rapeseed oil with methanol to biodiesel, better than its (110) and (100) counterparts.125Fig. 8 shows that MgO(I) (aerogel prepared MgO (111)) has a slightly higher reactivity and selectivity over the conventionally prepared MgO(110) and aerogel prepared MgO(100) [MgO(II) and MgO(III) respectively].29 Verziu et al., were also able to show that MgO(111) had activity over more cycles than the (100) and (110) catalysts.125
 | ||
| Scheme 2 Transesterification of triglycerides with methanol. | ||
 | ||
| Fig. 8 Transesterification of sunflower oil on nanoscale MgO catalysts in autoclave conditions (23 mL sunflower oil, 5 mL methanol, 300 mg MgO, 343 K, 2 h) over MgO(111) nanosheets (MgO(I)), conventionally prepared MgO (MgO(II)) and aerogel prepared MgO (MgO(III)). Reproduced from ref. 125 by permission of The Royal Society of Chemistry. | ||
D. MgO(111) as a support
Utilizing the (111) surface of MgO as a support for metal nanoparticles is of great interest because the surface containing all oxygen sites provides a very different environment than that of a typical (100) surface of alternating magnesium cation and oxygen anion sites. Analyzing catalytic activity and density functional theory (DFT) calculations of MgO(111) catalysts alongside MgO(100) catalysts may help identify the catalytically active site and help shed light on the role of the support for supported metal catalysts.MgO(111) as a support for gold nanoparticles has shown increased catalytic activity in the solvent-free benzyl alcohol aerobic oxidation when compared to gold nanoparticles deposited on a typical MgO aerogel. These catalytic results are supported by DFT calculations that explain that the high activity of Au/MgO(111), in the oxidation of benzyl alcohol, is attributed to the properties of the (111) support and/or the interface between the metal nanoparticles and the (111) support (Fig. 9).120
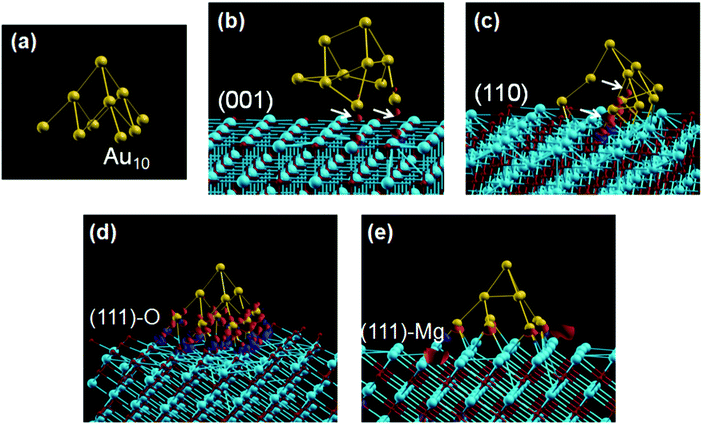 | ||
| Fig. 9 Relaxed atomic structure of (a) a Au10 cluster and (b–e) Au10/MgO, with electronic density transferred at the interface with various MgO substrates. The isosurfaces of the charge density difference are drawn at values of ±0.1 electrons Å−3, with the excess electron density shown in red, and deficiency shown in blue. Reproduced from ref. 120 by permission of the PCCP Owner Societies. | ||
It has been well established that gold catalyst supports have an effect on the oxidation of CO, and the activity is altered with varying preparation routes.149–151 Furthermore, reducible metal oxide supports tend to be more active than non-reducible metal oxides.152–158 In effort to study how the facet of an MgO support effects catalytic activity, gold nanoparticles were deposited on synthesized MgO(111) and commercial MgO (purchased from NanoScale Corporation and referred to as NanoActive MgO29,33,53). These materials were tested as catalysts in the oxidation of benzyl alcohol. Benzyl alcohol oxidation was chosen as it has been extensively used to establish gold catalyst activity, and is a model reaction for primary alcohol oxidation. In the study, at 120 °C, Au/MgO(111) showed a benzyl alcohol conversion nearly 5 times higher than Au/MgO (NanoActive), MgO(111), and MgO (NanoActive) while keeping the same selectivity to benzaldehyde.120
DFT calculations were utilized to provide an explanation for the observed catalytic results, by looking at how a model pyramidal Au10 cluster adsorbs onto different MgO surfaces.120 By first studying surface charges, results using experimental lattice constants show that the (100) and (110) surfaces of MgO have charges similar to those found in bulk MgO, while the (111) surface has surface ions with charges that are significantly different than that of bulk MgO. An O-terminated (111) surface is electron poor while an Mg-terminated (111) surface is electron rich (compared to bulk MgO values). This suggests that the (111) surface should be highly reactive. During chemisorption, oxygen atoms on an O-terminated (111) surface will gain electron density from adsorbates in order to try and achieve bulk MgO charge values. Similarly, magnesium atoms on an Mg-terminated (111) surface will donate electron density to adsorbates. This trend has been verified by a comparable study looking at Pd on MgO(111).159
Further DFT investigations show that the pyramidal Au10 cluster undergoes structural deformations when relaxed on the surfaces of MgO(100) and (110), while maintaining its overall structure when relaxed on the (111) surface as seen in Fig. 9. These large deformations of the Au10 cluster cause the particle to break away from the surfaces, leading to low surface-support interactions. However, for the (111) surface, the Au10 cluster experiences much smaller deformations and the average Au–O and Au–Mg distances (for O-terminated and Mg-terminated surfaces respectively) are much smaller. This leads to significantly stronger binding energies that also allow for better charge transfer between the support and Au10 cluster.120
While this DFT study shows that the cluster retains its pyramid structure and binds more strongly to the MgO(111) surface, it does not explain if the reaction is taking place at the interface, on the cluster, or on the surface of the support. However, from charge distribution analysis, electrons are donated from the gold cluster to the support, which creates electron rich oxygen atoms that may be the source of the increased catalytic activity.
5. Adsorbents
With the development of the textile industry, the treatment of wastewater has caused great attention in the world. Among various textile processes, dyeing procedures use large amounts of water and produce huge amounts of wastewater that contain toxic wastes such as suspended solids, auxiliary chemicals, and unreacted dyestuffs.160,161 According to a recent study, approximately 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 tons of different dyes are produced annually in the world, and more than 60% of the world dye production is consumed by textile industries.162 The wastewater from the textile industry causes tremendous problems due to the presence of hazardous wastes and toxic pollutants.123
000000 tons of different dyes are produced annually in the world, and more than 60% of the world dye production is consumed by textile industries.162 The wastewater from the textile industry causes tremendous problems due to the presence of hazardous wastes and toxic pollutants.123
Much research has gone into investigating the techniques for the treatment of wastewater, including different physical, chemical, thermal and biological processes.160 Researchers have also studied advanced oxidation processes like ozonation, photocatalytic oxidation,163–165 electrochemical oxidation,166etc. Various mechanisms have been used on decolorization of textile dyes.
(1) Microbial decolorization, by using bacteria, fungi, yeast and algae.167
(2) Advanced oxidation processes (AOPs), among which, heterogeneous photocatalytic processes using titanium dioxide nanomaterials was most effective due to the catalyst’s inert nature and photostability.168 Other AOPs involve ultrasound (US), ultraviolet (UV), hydroperoxide (H2O2) and ozone (O3).169
(3) Adsorption techniques, using low-cost adsorbents for the removal of dye from waste water. Activated carbon is one of the more common adsorbents, but it cannot be used widely due to its relatively high cost. Therefore, it is important to research alternative low cost adsorbents.170
(4) Combination of two or three of the techniques above.
With regard to adsorption technologies, high specific surface area (SSA) metal oxides have an advantage over activated carbon in that the adsorbed species can be combusted and the adsorbent reused. Regeneration of activated carbon remains challenging, thus the entire adsorbent/adsorbate system must be disposed.124 Hu et al. reported that MgO(111) nanosheets have adsorption properties for the dye pollutants from wastewater.123 The thickness of the MgO(111) nanosheets is 3 to 5 nm, with an average specific surface area of 198 m2 g−1. The authors studied the adsorption properties at different dye concentrations, solution pH, salt concentrations, and temperatures in a batch reactor using Congo red and reactive brilliant red X-3B as dye. The results showed that MgO(111) nanosheets exhibited much more favorable adsorption properties than other materials with high surface area, such as activated carbon (SSA = 1500 m2 g−1). The interaction of dye and MgO(111) can be explicated by the Langmuir model according to the isotherm evaluation. In addition, the adsorbent MgO(111) has the advantage in that it can be readily regenerated by a simple calcination process and reused without loss of activity.
Song et al. reported that NiO(111) nanosheets can also be used as efficient and recyclable adsorbents for dye pollutant removal from wastewater.124 Three typical textile dyes, reactive brilliant red X-3B, Congo red and fuchsin red were chosen as dyes to be investigated. The results revealed that the NiO(111) nanosheets exhibited much more favorable adsorptive properties than conventionally prepared nickel oxide powder obtained from thermal decomposition of nickel nitrate. The isotherm evaluations revealed that the Langmuir model demonstrated a better fit to experimental equilibrium data than the Freundlich model. Their studies infer that NiO(111) nanosheets possess desirable properties for use in adsorption and combustion applications.
These studies indicate that these surfaces possess selective adsorption properties that may be utilized in conjunction with catalytic processes in the future.
6. Future work
Theoretical studies indicate that the (111) facets of rock-salt structured metal oxides may be well suited for realizing some of the grand challenges in catalysis. The recent discoveries of wet chemical preparations for these materials have now allowed researchers to investigate their properties under working experimental conditions. While there have been some exciting results associated with the catalytic properties of (111) faceted MgO and NiO, there is much work to be done to gain a fundamental understanding of these materials and their potential. In particular, study of their stability in a variety of conditions would allow for their broader implementation as catalysts and/or catalyst supports. A full understanding of their formation mechanism may allow for design of systems with higher index facets or expanding the number of metal oxides with tailored surfaces. Further, probing the active sites and developing comprehensive structure activity profiles is a necessity for determining the breadth of possible applications.References
- M. Berg and S. Jaras, Appl. Catal., A, 1994, 114, 227 CrossRef CAS.
- K. K. Zhu, J. C. Hu, C. Kubel and R. Richards, Angew. Chem., Int. Ed., 2006, 45, 7277 CrossRef CAS.
- J. C. Hu, K. K. Zhu, L. F. Chen, H. J. Yang, Z. Li, A. Suchopar and R. Richards, Adv. Mater., 2008, 20, 267 CrossRef CAS.
- B. D. Zion and S. J. Sibener, J. Chem. Phys., 2007, 127, 154720 CrossRef CAS.
- M. Bender, O. Seiferth, A. F. Carley, A. Chambers, H. Freund and M. W. Roberts, Surf. Sci., 2002, 513, 221 CrossRef CAS.
- G. Spoto, E. N. Gribov, G. Ricchiardi, A. Damin, D. Scarano, S. Bordiga, C. Lamberti and A. Zecchina, Prog. Surf. Sci., 2004, 76, 71 CrossRef CAS.
- M. Sgroi, C. Pisani and M. Busso, Thin Solid Films, 2001, 400, 64 CrossRef CAS.
- M. Kiguchi, S. Entani, K. Saiki, T. Goto and A. Koma, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115402 CrossRef.
- D. Caceres, I. Colera, I. Vergara, R. Gonzalez and E. Roman, Vacuum, 2002, 67, 577 CrossRef CAS.
- J. M. Cho, K. H. Lee, C. I. Cheon, N. I. Cho and J. S. Kim, J. Eur. Ceram. Soc., 2010, 30, 481 CrossRef CAS.
- J. W. He, C. A. Estrada, J. S. Corneille, M. C. Wu and D. W. Goodman, Surf. Sci., 1992, 261, 164 CrossRef CAS.
- J. W. He, J. S. Corneille, C. A. Estrada, M. C. Wu and D. W. Goodman, J. Vac. Sci. Technol., A, 1992, 10, 2248 CAS.
- K. Kato, H. Omoto, A. Takamatsu and T. Tomioka, J. Cryst. Growth, 2011, 333, 59 CrossRef CAS.
- Z. N. Yu, W. Xue, D. X. Zheng and J. Sun, Plasma Sci. Technol., 2007, 9, 284 CrossRef CAS.
- A. M. Ferrari, S. Casassa and C. Pisani, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 155404 CrossRef.
- S. A. Chambers, Surf. Sci. Rep., 2000, 39, 105 CrossRef CAS.
- D. K. Fork, F. A. Ponce, J. C. Tramontana and T. H. Geballe, Appl. Phys. Lett., 1991, 58, 2294 CrossRef CAS.
- J. Wollschlager, D. Erdos and K. M. Schroder, Surf. Sci., 1998, 402, 272 CrossRef.
- J. Wollschlager, D. Erdos, H. Goldbach, R. Hopken and K. M. Schroder, Thin Solid Films, 2001, 400, 1 CrossRef CAS.
- X. Xu, W. S. Oh and D. W. Goodman, Langmuir, 1996, 12, 4877 CrossRef CAS.
- H. J. Freund, Surf. Sci., 2007, 601, 1438 CrossRef CAS.
- T. J. Zhu, L. Lu and X. B. Zhao, Mater. Sci. Eng., B, 2006, 129, 96 CrossRef CAS.
- Z. N. Yu, J. W. Seo, D. X. Zheng and J. Sun, Surf. Coat. Technol., 2003, 163, 398 CrossRef.
- S. Valeri, S. Altieri, A. di Bona, C. Giovanardi and T. S. Moia, Thin Solid Films, 2001, 400, 16 CrossRef CAS.
- S. Benedetti, P. Torelli, S. Valeri, H. M. Benia, N. Nilius and G. Renaud, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195411 CrossRef.
- D. Ochs, W. MausFriedrichs, M. Brause, J. Gunster, V. Kempter, V. Puchin, A. Shluger and L. Kantorovich, Surf. Sci., 1996, 365, 557 CrossRef CAS.
- M. C. Wu, J. S. Corneille, C. A. Estrada, J. W. He and D. W. Goodman, Chem. Phys. Lett., 1991, 182, 472 CrossRef CAS.
- S. Altieri, L. H. Tjeng and G. A. Sawatzky, Thin Solid Films, 2001, 400, 9 CrossRef CAS.
- R. Richards, W. F. Li, S. Decker, C. Davidson, O. Koper, V. Zaikovski, A. Volodin, T. Rieker and K. J. Klabunde, J. Am. Chem. Soc., 2000, 122, 4921 CrossRef CAS.
- B. Q. Xu, J. M. Wei, H. Y. Wang, K. Q. Sun and Q. M. Zhu, Catal. Today, 2001, 68, 217 CrossRef CAS.
- A. Khaleel, P. N. Kapoor and K. J. Klabunde, Nanostruct. Mater., 1999, 11, 459 CrossRef CAS.
- E. Lucas, S. Decker, A. Khaleel, A. Seitz, S. Fultz, A. Ponce, W. F. Li, C. Carnes and K. J. Klabunde, Chem.–Eur. J., 2001, 7, 2505 CrossRef CAS.
- K. J. Klabunde, J. Stark, O. Koper, C. Mohs, D. G. Park, S. Decker, Y. Jiang, I. Lagadic and D. J. Zhang, J. Phys. Chem., 1996, 100, 12142 CrossRef CAS.
- Y. Ding, G. T. Zhang, H. Wu, B. Hai, L. B. Wang and Y. T. Qian, Chem. Mater., 2001, 13, 435 CrossRef CAS.
- J. V. Stark, D. G. Park, I. Lagadic and K. J. Klabunde, Chem. Mater., 1996, 8, 1904 CrossRef CAS.
- H. Itoh, S. Utamapanya, J. V. Stark, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1993, 5, 71 CrossRef CAS.
- C. L. Yan and D. F. Xue, J. Phys. Chem. B, 2005, 109, 12358 CrossRef CAS.
- E. Ruckenstein and H. Y. Wang, Appl. Catal., A, 2000, 198, 33 CrossRef CAS.
- J. P. Coulomb, T. S. Sullivan and O. E. Vilches, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 4753 CrossRef CAS.
- L. Marchese, S. Coluccia, G. Martra and A. Zecchina, Surf. Sci., 1992, 269, 135 CrossRef.
- D. Scarano, S. Bertarione, F. Cesano, G. Spoto and A. Zecchina, Surf. Sci., 2004, 570, 155 CrossRef CAS.
- B. M. Choudary, R. S. Mulukutla and K. J. Klabunde, J. Am. Chem. Soc., 2003, 125, 2020 CrossRef CAS.
- P. Jeevanandam and K. J. Klabunde, Langmuir, 2002, 18, 5309–5313 CrossRef CAS.
- Y. X. Li and K. J. Klabunde, Langmuir, 1991, 7, 1388 CrossRef CAS.
- Y. X. Li, H. Li and K. J. Klabunde, Environ. Sci. Technol., 1994, 28, 1248 CrossRef CAS.
- M. S. Mel'gunov, V. B. Fenelonov, E. A. Mel'gunova, A. F. Bedilo and K. J. Klabunde, J. Phys. Chem. B, 2003, 107, 2427 CrossRef CAS.
- I. V. Mishakov, A. F. Bedilo, R. M. Richards, V. V. Chesnokov, A. M. Volodin, V. I. Zaikovskii, R. A. Buyanov and K. J. Klabunde, J. Catal., 2002, 206, 40 CrossRef CAS.
- S. Rajagopalan, O. Koper, S. Decker and K. J. Klabunde, Chem.–Eur. J., 2002, 8, 2602 CrossRef CAS.
- K. T. Ranjit and K. J. Klabunde, Chem. Mater., 2005, 17, 65 CrossRef CAS.
- R. M. Richards, A. M. Volodin, A. F. Bedilo and K. J. Klabunde, Phys. Chem. Chem. Phys., 2003, 5, 4299 RSC.
- J. V. Stark and K. J. Klabunde, Chem. Mater., 1996, 8, 1913 CrossRef CAS.
- N. J. Sun and K. J. Klabunde, J. Am. Chem. Soc., 1999, 121, 5587 CrossRef CAS.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175 CrossRef CAS.
- M. Atteya and K. J. Klabunde, Chem. Mater., 1991, 3, 182 CrossRef CAS.
- O. Diwald, M. Sterrer and E. Knozinger, Phys. Chem. Chem. Phys., 2002, 4, 2811 RSC.
- O. Diwald, M. Sterrer, E. Knozinger, P. V. Sushko and A. L. Shluger, J. Chem. Phys., 2002, 116, 1707 CrossRef CAS.
- E. Knozinger, K. H. Jacob, S. Singh and P. Hofmann, Surf. Sci., 1993, 290, 388 CrossRef.
- E. Knozinger, O. Diwald and M. Sterrer, J. Mol. Catal. A: Chem., 2000, 162, 83 CrossRef CAS.
- H. Knozinger, Science, 2000, 287, 1407 CrossRef CAS.
- S. Stankic, M. Muller, O. Diwald, M. Sterrer, E. Knozinger and J. Bernardi, Angew. Chem., Int. Ed., 2005, 44, 4917 CrossRef CAS.
- M. Sterrer, O. Diwald and E. Knozinger, J. Phys. Chem. B, 2000, 104, 3601 CrossRef CAS.
- M. Sterrer, O. Diwald, E. Knozinger, P. V. Sushko and A. L. Shluger, J. Phys. Chem. B, 2002, 106, 12478 CrossRef CAS.
- M. Sterrer, T. Berger, O. Diwald and E. Knozinger, J. Am. Chem. Soc., 2003, 125, 195 CrossRef CAS.
- F. M. Labajos, V. Rives and M. A. Ulibarri, J. Mater. Sci., 1992, 27, 1546 CrossRef CAS.
- M. M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808 CrossRef CAS.
- L. Yan, J. Zhuang, X. M. Sun, Z. X. Deng and Y. D. Li, Mater. Chem. Phys., 2002, 76, 119 CrossRef CAS.
- J. C. Yu, A. W. Xu, L. Z. Zhang, R. Q. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64 CrossRef CAS.
- E. Garrone, A. Zecchina and F. S. Stone, Philos. Mag. B, 1980, 42, 683 CAS.
- A. Zecchina, D. Scarano, S. Bordiga, G. Ricchiardi, G. Spoto and F. Geobaldo, Catal. Today, 1996, 27, 403 CrossRef CAS.
- A. Zecchina, D. Scarano, S. Bordiga, G. Spoto and C. Lamberti, Adv. Catal., 2001, 46, 265 CrossRef CAS.
- K. Itatani, K. Koizumi, F. S. Howell, A. Kishioka and M. Kinoshita, J. Mater. Sci., 1988, 23, 3405 CrossRef CAS.
- K. Itatani, K. Koizumi, F. S. Howell, A. Kishioka and M. Kinoshita, J. Mater. Sci., 1989, 24, 2603 CrossRef CAS.
- G. Martra, T. Cacciatori, L. Marchese, J. S. J. Hargreaves, I. M. Mellor, R. W. Joyner and S. Coluccia, Catal. Today, 2001, 70, 121 CrossRef CAS.
- Y. F. Hao, G. W. Meng, C. H. Ye, X. R. Zhang and L. D. Zhang, J. Phys. Chem. B, 2005, 109, 11204 CrossRef CAS.
- Y. Q. Zhu, W. K. Hsu, W. Z. Zhou, M. Terrones, H. W. Kroto and D. R. M. Walton, Chem. Phys. Lett., 2001, 347, 337 CrossRef CAS.
- Y. G. Yan, L. X. Zhou, J. Zhang, H. B. Zeng, Y. Zhang and L. D. Zhang, J. Phys. Chem. C, 2008, 112, 10412 CAS.
- L. Hu, Y. X. Li, J. P. Qu, Z. X. Huang, X. T. Huang, X. X. Ding, C. Tang and S. R. Qi, J. Nanosci. Nanotechnol., 2004, 4, 1071 CrossRef CAS.
- C. C. Tang, Y. Bando and T. Sato, J. Phys. Chem. B, 2002, 106, 7449 CrossRef CAS.
- Y. D. Yin, G. T. Zhang and Y. N. Xia, Adv. Funct. Mater., 2002, 12, 293 CrossRef CAS.
- N. Sutradhar, A. Sinhamahapatra, S. K. Pahari, P. Pal, H. C. Bajaj, I. Mukhopadhyay and A. B. Panda, J. Phys. Chem. C, 2011, 115, 12308 CAS.
- S. Kar and S. Chaudhuri, J. Nanosci. Nanotechnol., 2006, 6, 1447 CrossRef CAS.
- L. A. Ma, Z. X. Lin, J. Y. Lin, Y. A. Zhang, L. Q. Hu and T. L. Guo, Physica E (Amsterdam), 2009, 41, 1500 CrossRef CAS.
- J. Park, E. Kang, S. U. Son, H. M. Park, M. K. Lee, J. Kim, K. W. Kim, H. J. Noh, J. H. Park, C. J. Bae, J. G. Park and T. Hyeon, Adv. Mater., 2005, 17, 429 CrossRef CAS.
- M. Tadic, M. Panjan and D. Markovic, Mater. Lett., 2010, 64, 2129 CrossRef CAS.
- Y. Wang, Q. S. Zhu and H. G. Zhang, Chem. Commun., 2005, 5231 RSC.
- H. W. Yan, C. F. Blanford, B. T. Holland, M. Parent, W. H. Smyrl and A. Stein, Adv. Mater., 1999, 11, 1003 CrossRef CAS.
- X. Ni, Y. Zhang, D. Tian, H. Zheng and X. Wang, J. Cryst. Growth, 2007, 306, 418 CrossRef CAS.
- Y. Ren and L. A. Gao, J. Am. Ceram. Soc., 2010, 93, 3560 CrossRef CAS.
- S. Q. Shang, K. Y. Xue, D. R. Chen and X. L. Jiao, CrystEngComm, 2011, 13, 5094 RSC.
- X. Y. Wang, L. J. Wan, T. Yu, Y. Zhou, J. Guan, Z. T. Yu, Z. S. Li and Z. G. Zou, Mater. Chem. Phys., 2011, 126, 494 CrossRef CAS.
- S. L. Xiong, C. Z. Yuan, X. G. Zhang and Y. T. Qian, CrystEngComm, 2011, 13, 626 RSC.
- P. D. Yang and C. M. Lieber, J. Mater. Res., 1997, 12, 2981 CrossRef CAS.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713 CrossRef CAS.
- T. Kendelewicz, P. Liu, C. S. Doyle, G. E. Brown, E. J. Nelson and S. A. Chambers, Surf. Sci., 2000, 453, 32 CrossRef CAS.
- H. A. Al-Abadleh and V. H. Grassian, Surf. Sci. Rep., 2003, 52, 63 CrossRef CAS.
- P. V. Sushko, J. L. Gavartin and A. L. Shluger, J. Phys. Chem. B, 2002, 106, 2269 CrossRef CAS.
- C. Noguera, J. Phys.: Condens. Matter, 2000, 12, R367 CrossRef CAS.
- T. Zambelli, J. Wintterlin, J. Trost and G. Ertl, Science, 1996, 273, 1688 CrossRef CAS.
- J. K. Norskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275 CrossRef CAS.
- P. Gambardella, Z. Sljivancanin, B. Hammer, M. Blanc, K. Kuhnke and K. Kern, Phys. Rev. Lett., 2001, 87, 056103 CrossRef CAS.
- R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Laegsgaard, B. S. Clausen, J. K. Norskov and F. Besenbacher, Nat. Mater., 2005, 4, 160 CrossRef CAS.
- I. E. Wachs, L. E. Briand, J. M. Jehng, L. Burcham and X. T. Gao, Catal. Today, 2000, 57, 323 CrossRef CAS.
- S. W. Yang and L. Gao, J. Am. Chem. Soc., 2006, 128, 9330 CrossRef CAS.
- J. Tabatabaei, B. H. Sakakini and K. C. Waugh, Catal. Lett., 2006, 110, 77 CrossRef CAS.
- L. Chan and G. L. Griffin, Surf. Sci., 1985, 155, 400 CrossRef CAS.
- L. E. Greene, M. Law, D. H. Tan, M. Montano, J. Goldberger, G. Somorjai and P. D. Yang, Nano Lett., 2005, 5, 1231 CrossRef CAS.
- A. McLaren, T. Valdes-Solis, G. Q. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540 CrossRef CAS.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977 CrossRef CAS.
- M. Gajdardziska-Josifovska, R. Plass, M. A. Schofield, D. R. Giese and R. Sharma, J. Electron Microsc., 2002, 51, S13 CrossRef.
- R. Arita, Y. Tanida, S. Entani, M. Kiguchi, K. Saiki and H. Aoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 235423 CrossRef.
- J. Goniakowski and C. Noguera, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115413 CrossRef.
- V. K. Lazarov, Z. H. Cai, K. Yoshida, K. Zhang, M. Weinert, K. S. Ziemer and P. J. Hasnip, Phys. Rev. Lett., 2011, 107, 056101 CrossRef.
- A. Barbier and G. Renaud, Surf. Sci., 1997, 392, L15 CrossRef CAS.
- A. Ohtomo, K. Tamura, K. Saikusa, K. Takahashi, T. Makino, Y. Segawa, H. Koinuma and M. Kawasaki, Appl. Phys. Lett., 1999, 75, 2635 CrossRef CAS.
- E. D. L. Rienks, N. Nilius, H. P. Rust and H. J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 241404 CrossRef.
- C. Tusche, H. L. Meyerheim and J. Kirschner, Phys. Rev. Lett., 2007, 99, 026102 CrossRef CAS.
- H. J. Freund, Angew. Chem., Int. Ed. Engl., 1997, 36, 452 CrossRef.
- V. K. Lazarov, R. Plass, H. C. Poon, D. K. Saldin, M. Weinert, S. A. Chambers and M. Gajdardziska-Josifovska, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115434 CrossRef.
- J. Goniakowski, F. Finocchi and C. Noguera, Rep. Prog. Phys., 2008, 71, 55 CrossRef.
- Z. Li, C. V. Ciobanu, J. C. Hu, J. P. Palomares-Baez, J. L. Rodriguez-Lopez and R. Richards, Phys. Chem. Chem. Phys., 2011, 13, 2582 RSC.
- L. F. Chen, J. C. Hu, R. Richards, S. Prikhodko and S. Kodambaka, Nanoscale, 2010, 2, 1657 RSC.
- A. Corpuz and R. Richards, Nanoscale Mater. Chem.: Environ. Appl., 2010, 1045, 51 CrossRef CAS.
- J. C. Hu, Z. Song, L. F. Chen, H. J. Yang, J. L. Li and R. Richards, J. Chem. Eng. Data, 2010, 55, 3742 CrossRef CAS.
- Z. Song, L. F. Chen, J. C. Hu and R. Richards, Nanotechnology, 2009, 20, 275707 CrossRef.
- M. Verziu, B. Cojocaru, J. C. Hu, R. Richards, C. Ciuculescu, P. Filip and V. I. Parvulescu, Green Chem., 2008, 10, 373 RSC.
- J. C. Hu, K. Zhu, L. F. Chen, C. Kubel and R. Richards, J. Phys. Chem. C, 2007, 111, 12038 CAS.
- O. E. Taurian, M. Springborg and N. E. Christensen, Solid State Commun., 1985, 55, 351 CrossRef CAS.
- H. Sato, T. Minami, S. Takata and T. Yamada, Thin Solid Films, 1993, 236, 27 CrossRef CAS.
- A. Wander, I. J. Bush and N. M. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 233405 CrossRef.
- P. Geysermans, F. Finocchi, J. Goniakowski, R. Hacquart and J. Jupille, Phys. Chem. Chem. Phys., 2009, 11, 2228 RSC.
- R. Hacquart and J. Jupille, Chem. Phys. Lett., 2007, 439, 91 CrossRef CAS.
- K. Refson, R. A. Wogelius, D. G. Eraser, M. C. Payne, M. H. Lee and V. Milman, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 10823 CrossRef CAS.
- V. E. Henrich, Surf. Sci., 1976, 57, 385 CrossRef CAS.
- R. Plass, J. Feller and M. Gajdardziska-Josifovska, Surf. Sci., 1998, 414, 26 CrossRef CAS.
- R. Plass, K. Egan, C. Collazo-Davila, D. Grozea, E. Landree, L. D. Marks and M. Gajdardziska-Josifovska, Phys. Rev. Lett., 1998, 81, 4891 CrossRef CAS.
- J. Ciston, A. Subramanian and L. D. Marks, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 085421 CrossRef.
- J. Ciston, A. Subramanian, D. M. Kienzle and L. D. Marks, Surf. Sci., 2010, 604, 155 CrossRef CAS.
- J. A. Enterkin, A. E. Becerra-Toledo, K. R. Poeppelmeier and L. D. Marks, Surf. Sci., 2012, 606, 344 CrossRef CAS.
- B. M. Choudary, M. L. Kantam, K. V. S. Ranganath, K. Mahendar and B. Sreedhar, J. Am. Chem. Soc., 2004, 126, 3396 CrossRef CAS.
- L. T. Higham, U. P. Kreher, C. L. Raston, J. L. Scott and C. R. Strauss, Org. Lett., 2004, 6, 3257 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and J. Primo, J. Catal., 1995, 151, 60 CrossRef CAS.
- The Flavonoids: Advances in Research, ed. S. B. Harborne and T. J. Mabry, Chapman and Hall, New York, 1982 Search PubMed.
- J. Lichtenberger, S. C. Hargrove-Leak and M. D. Amiridis, J. Catal., 2006, 238, 165 CrossRef CAS.
- I. Ahmad, J. A. Anderson, T. J. Dines and C. H. Rochester, Spectrochim. Acta, Part A, 1998, 54, 319 CrossRef.
- A. Corma and S. Iborra, Adv. Catal., 2006, 49, 239 CrossRef CAS.
- K. Hermansson, M. Baudin, B. Ensing, M. Alfredsson and M. Wojcik, J. Chem. Phys., 1998, 109, 7515 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS.
- P. Patil, V. G. Gude, S. Pinappu and S. G. Deng, Chem. Eng. J., 2011, 168, 1296 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Catal. Today, 2006, 111, 22 CrossRef CAS.
- D. W. Goodman, Catal. Lett., 2005, 99, 1 CrossRef CAS.
- M. Valden and D. W. Goodman, Isr. J. Chem., 1998, 38, 285 CAS.
- M. Valden, S. Pak, X. Lai and D. W. Goodman, Catal. Lett., 1998, 56, 7 CrossRef CAS.
- B. K. Min, W. T. Wallace and D. W. Goodman, Surf. Sci., 2006, 600, L7 CrossRef CAS.
- J. Goniakowski and C. Noguera, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 16120 CrossRef CAS.
- M. M. Naim and Y. M. El Abd, Sep. Purif. Methods, 2002, 31, 171 CrossRef CAS.
- S. J. Doh, C. Kim, S. G. Lee, S. J. Lee and H. Kim, J. Hazard. Mater., 2008, 154, 118 CrossRef CAS.
- R. S. Blackburn, Environ. Sci. Technol., 2004, 38, 4905 CrossRef CAS.
- A. B. Prevot, C. Baiocchi, M. C. Brussino, E. Pramauro, P. Savarino, V. Augugliaro, G. Marci and L. Palmisano, Environ. Sci. Technol., 2001, 35, 971 CrossRef CAS.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 40, 271 CrossRef CAS.
- W. B. Zhang, T. C. An, M. M. Mao, H. M. Fu, G. Y. Sheng, M. C. Cui and G. Y. Li, Appl. Catal., A, 2003, 255, 221 CrossRef CAS.
- D. Rajkumar and J. G. Kim, J. Hazard. Mater., 2006, 136, 203 CrossRef CAS.
- K. Singh and S. Arora, Crit. Rev. Environ. Sci. Technol., 2011, 41, 807 CrossRef CAS.
- A. R. Khataee and M. B. Kasiri, J. Mol. Catal. A: Chem., 2010, 328, 8 CrossRef CAS.
- N. N. Mahamuni and Y. G. Adewuyi, Ultrason. Sonochem., 2010, 17, 990 CrossRef CAS.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, J. Hazard. Mater., 2010, 177, 70 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |