Xylyltrioxorhenium – the first arylrhenium(VII) oxide applicable as an olefin epoxidation catalyst†
Stefan
Huber
a,
Mirza
Cokoja
a,
Markus
Drees
a,
János
Mínk
b and
Fritz E.
Kühn
*a
aChair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Str. 1, D-85747, Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13081
bChemical Research Center, Hungarian Academy of Sciences, Pusztaszeri út 59-67, H-1025 Budapest, Hungary
First published on 24th July 2012
Abstract
The catalytic activity of (2,6-dimethylphenyl)trioxorhenium (XTO) for the epoxidation of cyclooctene is presented. Solutions of hydrogen peroxide and tert-butyl hydrogen peroxide (TBHP) in THF or CH2Cl2 were applied as oxidizing agent–solvent mixtures. Cyclooctene oxide yields are higher with TBHP than with H2O2. Application of non-coordinating solvents as reaction media increases both yields and reaction rates. The possible active species is analyzed by 17O NMR and Raman spectroscopy.
Introduction
In 1974 Wilkinson et al. reported the synthesis of [(CH3)4ReO], the first high oxidation state alkyl oxo rhenium compound.1 Its derivative (CH3)ReO3 methyltrioxorhenium(VII) (MTO) was the first organorhenium(VII) oxide to be described.2 Only after the synthesis of [(pentamethylcyclopentadienyl)trioxorhenium](VII), reported independently by two different research groups in 19843 and the large scale synthesis and catalytic application of MTO in 1989,4 however, the compound class R–ReO3(VII) was systematically examined.5 Within a few years a plethora of compounds were obtained.5,6 Nevertheless, it turned out that all these derivatives, with the famous exception of MTO, appeared to be either not catalytically active or of very limited stability.5,7Little attention has been paid to the aryl trioxorhenium compounds of the type [Ar–ReO3] with respect to catalytic applicability.8 Aryl ligands are available for the full range of organic aromatic substitution chemistry.8b,c However, the synthesis of [Ar–ReO3] is quite intricate since both the starting materials and the products are at least in some cases highly sensitive. Furthermore, most compounds of this class decompose slowly when exposed to sunlight. Phenyltrioxorhenium, which is obtained from Re2O7 and ZnPh2, is quite sensitive to moisture and only stable at low temperatures (<−30 °C).8a,b Therefore, it is not a good choice for catalytic applications. Its dimethyl derivative xylyltrioxorhenium (XTO, 1), however, is stable in solution at room temperature and thus is a far better target molecule for catalytic examinations. In 1994, catalytic activity of XTO in the epoxidation of olefins with aqueous H2O2 has been mentioned.7a Since the compound is quite sensitive to moisture, the observed poor catalytic activity is not surprising. In this work, water-free oxidants (H2O2 in THF solution, TBHP in n-decane) are applied in order to avoid the water-sensitivity of XTO. The active species have also been investigated by 17O NMR and Raman spectroscopy.
Experimental section
General
All preparations and manipulations were carried out using standard Schlenk techniques or in an MBraun glove box under a strictly oxygen- and water-free argon atmosphere. All reaction vessels were protected from light. THF was of HPLC grade and distilled over Na–benzophenone under argon before use. Dichloromethane was dried according to literature methods.9 Yellow Re2O7,7a dixylyl zinc10 and 17O-labeled MTO11 were synthesized according to the literature. A 3.3 mol L−1 water free solution of H2O2 in THF was prepared by dissolving aqueous H2O2 in THF followed by drying over MgSO4.7a TBHP-D1 was obtained by mixing TBHP in n-decane with 10 equiv. of D2O, stirring overnight and subsequent drying over molecular sieves. CDCl3 was degassed and stored over molecular sieves prior to use. A 5.5 M solution of TBHP in n-decane was obtained from Aldrich. MnO2, electrolytically precipitated, was obtained from Acros. NMR spectra were obtained using a Bruker Avance DPX-400 or Jeol JNM-GC-400 spectrometer. Mass spectra were recorded on a Finnigan MAT 311 A and a MAT 90 spectrometer. Raman spectra were recorded on a Bio-Rad FTS60A Dedicated FT-Raman spectrometer. Mid-IR spectra were measured on a Varian FT-660 FTIR spectrometer using a micro ATR attachment. Microanalyses and mass spectroscopy were performed at the TUM (Garching) laboratories.Improved preparation of 1
Dixylyl zinc (0.14 g, 0.50 mmol) was dissolved in 10 mL THF and dropwise added to a solution of Re2O7 (0.48 g, 1.00 mmol) in 25 mL THF, which was previously cooled to −78 °C. The solution was slowly warmed to −5 °C and stirred for 2 h. The solvent was removed under vacuum and the residue was extracted with 6 × 10 mL of cold (0 °C) n-hexane. After removing the solvent under vacuum, yellow crystal needles of 1 (0.14 g, 0.41 mmol, 41% based on rhenium) were obtained, which could be further purified by recrystallization in cold n-hexane. The product was characterized by NMR and IR spectroscopy. The obtained values are in good accordance with previously published data.8bCatalytic reactions
cis-Cyclooctene (0.341 g, 3.09 mmol), oxidant (6.19 mmol, TBHP in decane, or H2O2 35% in water, or H2O2 in THF) and 1 (1 mol%, 0.031 mmol) as catalyst were applied. The reactions were carried out in 4 mL CH2Cl2. The oxidant was added into a thermostated reaction vessel and stirred for 24 h. The course of the reaction was monitored by quantitative GC analysis; samples were taken every 5 min up to 30 min, then every hour for 3 h and a final sample was taken after 24 h. For the destruction of the oxidant, a catalytic amount of electrolytically precipitated MnO2 was added. After the evolution of gas ceased, the resulting slurry was filtered and dried over MgSO4. The external standards indane and p-xylene, dissolved in iso-propanol, were added to the filtrate and the resulting mixture was injected into a calibrated VarianWS gas chromatograph. The conversion of cyclooctene and the formation of cyclooctene oxide were calculated from a calibration curve recorded prior to the catalytic reactions. The turnover frequencies were determined by the initial-rate method using the conversion of the first five minutes.17O NMR-experiments
17O-Labeled MTO (0.015 g, 0.059 mmol) and XTO (0.020 g, 0.059 mmol) were dissolved in 0.4 mL of dry CDCl3 and stirred for 10 min. Thereafter a 5.5 M solution of TBHP in n-decane was added (1–400 equiv.). The solution was stirred for another 10 min at −20 °C. 1H and 17O NMR spectra were recorded at −20 °C.Raman experiments
XTO (10 mg, 0.029 mmol) was dissolved in 0.2 mL decane. A 5.5 mol (1–10 equiv.) solution of TBHP or TBHP-D1 was added and after short mixing, spectra were recorded at room temperature.Computational details
All calculations have been performed with GAUSSIAN-03 using the density functional/Hartree–Fock hybrid model Becke3LYP12 and the split valence triple-ζ (TZ) basis set 6-311+G**.13 Re atoms have to be treated with an effective core potential, the Hay–Wadt LANL2DZ basis set was chosen for this metal.14 All optimizations were obtained without using constraint coordinates. Solvent effects were simulated with the PCM model15 as implemented in Gaussian 03. The solvent of choice was dichloromethane with a dielectric constant of ε = 2.02 and a relevant molecular radius of 2.27 Å.Results and discussion
Synthesis and catalytic properties of XTO
The reaction of Re2O7 with dixylyl zinc yields compound 1 in maximum 50% yield based on rhenium, since zinc perrhenate is formed as a stoichiometric byproduct accounting for the other 50% of the applied rhenium (see Scheme 1).8 The reaction is very sensitive to the quality of the dirhenium heptoxide used. Only highest quality grade, lemon-yellow Re2O7, produced by oxidation of rhenium powder under oxygen gas and direct sublimation, should be applied.7a Even tiny impurities decrease the yield significantly. By lowering reaction temperatures and protecting the reaction vessels from light to prevent decomposition, yields of up to 40–45% based on rhenium can be obtained.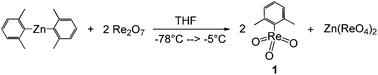 | ||
| Scheme 1 Synthesis of xylyltrioxorhenium (1). | ||
Previously, it had been stated that σ-phenyltrioxorhenium and σ-mesityltrioxorhenium show no catalytic activity when using aqueous H2O2 as an epoxidizing agent.7a However, no examinations applying non-aqueous H2O2 or TBHP were carried out. Fig. 1 summarizes the catalytic activity of 1 in the presence of various oxidants and solvents.
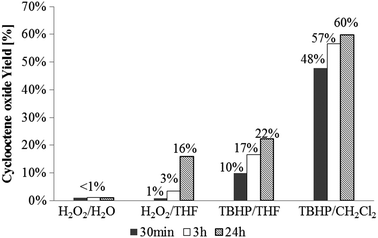 | ||
Fig. 1 Yields of cyclooctene epoxidation with compound 1 as catalyst precursor with different oxidants (H2O2, TBHP) in various solvents (H2O, THF, CH2Cl2). All reactions were performed at room temperature with a catalyst/substrate/oxidant ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100:200. :100:200. | ||
In general, oxidation reactions carried out using aqueous H2O2 as an oxidant show no significant activity. Using THF as solvent instead of water increases the activity, but the overall activity remains low, most likely due to the formation of water as a byproduct. Compound 1 reacts with water to yield perrhenate, which is not active in olefin epoxidation in polar or non-polar solvents. Changing the oxidant to TBHP in THF strongly increases the catalytic activity. Changing the solvent from coordinating THF to non-coordinating CH2Cl2 leads to overall epoxide yields of >60% and notably higher initial activities. In all cases no byproducts were detected by GC; especially no ring opening of the epoxide was observed. Thus, yields are equivalent to conversion. The reactions are, however, limited by catalyst decomposition. Similar experiments using the urea–hydrogen-peroxide adduct (UHP) as an oxidant in CH2Cl2 do not show any measurable activity, most likely due to the poor solubility of UHP in CH2Cl2. The activity of the system XTO/TBHP in olefin epoxidation is noteworthy. To the best of our knowledge, molecular organyl rhenium oxide complexes could so far only be activated by H2O2, whereas TBHP leads either to no reaction or to decomposition to perrhenate.5,16,17 TBHP, on the other hand, is known to be an efficient oxidant for some molecular Mo catalysts.18 The catalytic system XTO/TBHP apparently follows a somewhat different reactivity pattern than MTO/H2O2. Fig. 2 shows the dependence of the product yields on the reaction temperature. With increasing reaction temperature, both TOFs and yields increase. A temperature optimum can be found around 35 °C. Further increase in the reaction temperature (55 °C, CHCl3 as solvent) does not lead to a further increase in catalytic activity.
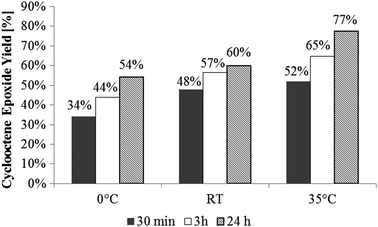 | ||
| Fig. 2 Yields of the cyclooctene epoxidation with catalyst 1 in CH2Cl2 with the oxidant TBHP at various temperatures with a catalyst/substrate/oxidant ratio of 1:100:200. | ||
Adding a base such as tert-butylpyridine, which is used in MTO catalysis to increase catalyst performance and to inhibit epoxide ring opening,19 almost completely inhibits the catalytic activity of XTO. In both MTO and XTO the α-carbon of the catalyst is shifted by 5 ppm in 13C NMR upon coordination of tert-butylpyridine. Therefore, the base seems to be equally strongly coordinated in both cases, but the sterically more demanding coordination sphere of XTO inhibits activation of the catalyst–base adduct. Table 1 shows the turnover frequencies (TOFs) obtained in the performed experiments. TOFs were determined by the initial rate method, using the conversion in the first 5 minutes, since no activation period could be observed. Comparison of the obtained results shows the same trends in temperature and solvent dependence given by comparing the yields. In the case of using TBHP as oxidant, using non-coordinating CH2Cl2 instead of coordinating THF leads to an increase in the initial activity by one order of magnitude. It can be assumed that THF coordinates to the Re center reducing its Lewis acidity, i.e. the catalytic activity.
NMR spectroscopic studies
In order to elucidate the mechanism of the epoxidation of cyclooctene catalyzed by 1, 17O-NMR studies were performed in order to shed light on the reaction of XTO with TBHP and the nature of the product. The oxygen atoms are suitable probes for answering the question, whether Re-oxo-peroxo species are formed or other reactions take place, analogously to the case of MTO/H2O2. There are several methods known to enrich the 17O-content of 1 to obtain detectable signals. Due to the high sensitivity to water it is not recommended to directly use 17O-enriched water for the labeling of 1, as is the case of MTO. Other methods for enriching compound 1 with 17O include the direct synthesis of 17O-labeled Re2O7 by oxidizing rhenium metal powder with 17O enriched oxygen gas,9 and carrying out the synthesis of XTO according to Scheme 1. Another possibility involves a multistep reaction, where Re2O7 is hydrolyzed in 17O-labeled water and precipitated with AgNO3. The resulting silver perrhenate would be reacted with (CH3)3SiCl and THF to yield [Re2O7(THF)2].11 The third and most convenient method, hence used in this work, draws on the ability of water-stable methyltrioxorhenium (MTO) to exchange its oxo ligands with (17O-enriched) water as well as with other rhenium-oxo compounds.7b It is therefore straightforward to label the easily available MTO and, in a second step, to transfer 17O-ligands to XTO. This method has also the advantage that the 17O-NMR spectra can be calibrated on the resonance of MTO. In all experiments 17O-labeled MTO is mixed with XTO in a 1:1 ratio in CDCl3. After 10 min, a solution of TBHP in decane is added and the 17O NMR spectrum is recorded at −20 °C.
The oxygen resonance originating from Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of 1 appears at 820 ppm (Fig. 3, bottom spectrum).22 Upon addition of TBHP, the signal of 1 at 820 ppm is slightly shifted to a broader signal at 819 ppm and a new signal at 726 ppm appears (Fig. 3, middle). The half width of the peak at 819 ppm is ca. 50 Hz, which is significantly broader than that of the peaks at 726 ppm (ca. 35 Hz) and 820 ppm (ca. 30 Hz), which indicates that this line is broadened by hydrogen exchange between terminal oxo ligands and hydroxo ligands. A weak signal at 65 ppm of tert-butanol (reported at 60 ppm20) is visible.
O of 1 appears at 820 ppm (Fig. 3, bottom spectrum).22 Upon addition of TBHP, the signal of 1 at 820 ppm is slightly shifted to a broader signal at 819 ppm and a new signal at 726 ppm appears (Fig. 3, middle). The half width of the peak at 819 ppm is ca. 50 Hz, which is significantly broader than that of the peaks at 726 ppm (ca. 35 Hz) and 820 ppm (ca. 30 Hz), which indicates that this line is broadened by hydrogen exchange between terminal oxo ligands and hydroxo ligands. A weak signal at 65 ppm of tert-butanol (reported at 60 ppm20) is visible.
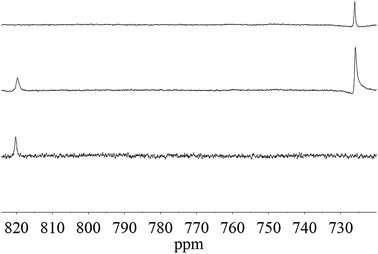 | ||
| Fig. 3 17O NMR spectra of the reaction of 1 with 17O-labeled MTO and various amounts of TBHP in decane (bottom: before addition of TBHP; middle: 200 equiv. TBHP; top: 400 equiv. TBHP). | ||
Upon increasing the concentration of TBHP the resonance at 819 ppm disappears and the resonance at 726 ppm becomes stronger. The signal for the monoperoxo complex of MTO, which is dominant at low H2O2 concentrations, is reported to appear at 763 ppm.20
Scheme 2 shows two possible complexes that may be the actual active species. Complex 2 is a linear TBHP–XTO complex which is proposed in analogy to the active species often discussed in [MoO2] catalysis with TBHP.19 Species 3 is proposed in analogy to the monoperoxo species of MTO, which is long known to be one of the catalytically active species of MTO.20
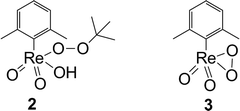 | ||
| Scheme 2 Possible active species of XTO (R = xylyl). σ-type-coordinated TBHP 2, monoperoxo complex 3. | ||
In order to assign this observation, the 17O NMR shifts of compounds 2 and 3, relative to MTO at 829 ppm, were calculated by DFT methods. Due to the fact that only ReO atoms can be exchanged with 17O by MTO, not all of the predicted peaks are visible in the real NMR experiments. A 17O shift of 721 ppm (and 707 ppm, which cannot be observed) for the ReO in the monoperoxo complex of XTO 3 was calculated. On the other hand a shift of 819 ppm (and 693 ppm, which cannot be observed) is predicted for the ReO moiety in the linear σ-type-coordinated XTO–TBHP complex 2. This shift can only be observed at lower concentrations of TBHP. Hence a mono-peroxo species like 3 seems to be the major species under catalytic conditions, where low catalyst amounts meet high oxidant concentrations.
Upon adding 400 equiv. TBHP, two broad peaks around 745 and 765 ppm emerge, indicating that MTO can be transformed to CH3-Re-peroxo complexes by TBHP, but in significantly smaller yields than for 1 (Fig. 3).21 Furthermore, MTO decomposes in the presence of TBHP to perrhenate and several other products, including tert-butyl methyl ether, methanol, methane and ethene.17
Raman spectroscopic studies
To further shed light on the active species, Raman spectra were measured. In order to identify characteristic OH bands, all experiments were performed with TBHP (tbutyl-OOH) as well as with TBHP-D1 (tbutyl-OOD). Other than 17O NMR, Raman spectroscopy is fast enough to resolve the difference between the oxo and hydroxo ligands of 2. In the first experiments, one equivalent of TBHP/TBHP-D1 was added to a concentrated solution of 1 in decane. A pale red solution formed, which was very unstable and formed a black residue within few minutes at room temperature. Table 2 shows the characteristic bands of 2 and their corresponding vibrational modes.| Raman band [cm−1] | Mode |
|---|---|
| 917 m (917 m) | ν A (ReO2) |
| 883 vs (884 vs) | ν S (ReO2) |
| 998 m (757 m) | δ(ReOH(D)) |
| 995 s (744 s) | |
| 556 m (520 m) | Re–OH(D) stretch |
| 538 w (510 w) |
Upon increasing the TBHP amount to 10 equiv., a yellow solution formed, which is stable at room temperature for several hours. Raman experiments show that the Re–OH bands (998, 995, 556 and 538 cm−1), previously observed in the initial spectrum, almost fully disappear and the characteristic bands of a Re-peroxo moiety, found at 510, 451 and 977 cm−1, appear. The Re(O)2 vibrational bands at 917 and 885 cm−1 are still visible, indicating the formation of a mono-peroxo complex. Table 3 summarizes the most important bands and their vibrational modes of 3 in solution.
| Raman band [cm−1] | Mode |
|---|---|
| 977 m | (O–O) stretch |
| 917 m | ν A (ReO2) |
| 885 vs | ν S (ReO2) |
| 510 m | ν S (Re(O–O)) |
| 451 m | ν A (Re(O–O)) |
The Raman experiments prove and extend the results of NMR experiments. At low concentrations, the unstable red compound 2 is dominant. It is responsible for the quick decomposition of the catalyst. At higher concentrations of TBHP the stable, yellow mono-peroxo species 3 is formed, which, in analogy to MTO, should also be the catalytically active species (Scheme 3).
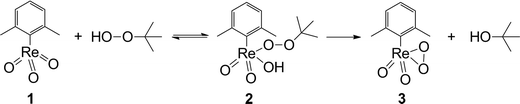 | ||
| Scheme 3 Possible activation pathway of 1 in the presence of TBHP. | ||
Decomposition of the catalyst
When treated with TBHP, XTO appears to form Re-peroxo species as is the case with MTO and H2O2. However, unlike MTO, XTO exhibits catalytic activity in olefin epoxidation with TBHP as oxidant. Finally it also decomposes to perrhenate, however, significantly slower than MTO.In 1999, Brittingham and Espenson suggested a radical pathway for the decay of the TBHP adduct of MTO, based on product quantifications and kinetic measurements.22 Product examinations in our case of the final reaction mixture by NMR also show the presence of perrhenate and bisxylyl, a typical radical decomposition product. Radical decomposition of XTO is further amplified by thermal and light induced cleavage of the aryl–rhenium bond. Most probably due to steric hindrance, decomposition of XTO in the presence of the bulky TBHP is significantly slower than decomposition of MTO, therefore catalytic activity can be observed before decomposition.
Periana et al. have discussed a non-radical decomposition mechanism of phenyltrioxorhenium23a and methyltrioxorhenium23b,c when using H2O2 in some detail using DFT methods. A key step in this mechanism is the insertion of oxygen into the rhenium alkyl(aryl)-bond. This mechanism might be responsible for the instant decomposition of the catalyst in aqueous H2O2 solution and the fast decomposition of the catalyst in H2O2 in THF. However, the main decomposition products of this pathway, 2,6-dimethylphenol and 2,6-dimethylphenol tert-butyl ether, could not be observed in 1H NMR experiments after treating XTO with TBHP. Anyway, the whole decomposition mechanism is not yet well understood. Accordingly, a detailed experimental and theoretical study of the decomposition pathways of XTO when using TBHP is currently being performed in our laboratories.
Conclusions
In the presence of TBHP as an oxidizing agent xylyltrioxorhenium shows significant epoxidation activity. XTO is the first literature-known Re(VII) catalyst showing catalytic activity in the presence of TBHP, while its congener MTO rapidly decomposes in the same environment. Using water-free reaction systems increases strongly the activity. Compound 1 is also an active catalyst in the presence of non-aqueous H2O2, but clearly shows the best results with TBHP due to catalyst decomposition to perrhenate in the presence of water. Application of non-coordinating solvents also increases catalytic rates, because coordinating solvents like THF inhibit the active site of the catalyst. NMR and Raman experiments indicate that the active species and hence the mechanism are the same as in the epoxidation with MTO. Differences in activity and oxidant tolerance seem to be controlled by the decomposition pathways.Acknowledgements
SH is grateful to the TUM Graduate School for financial support.References
- K. Mertis, J. F. Gibson and G. Wilkinson, J. Chem. Soc., Chem. Commun., 1974, 93a RSC.
- R. I. Beattie and P. J. Jones, Inorg. Chem., 1979, 18, 2318–2319 CrossRef.
- (a) W. A. Herrmann, R. Serrano and H. Bock, Angew. Chem., Int. Ed. Engl., 1984, 23, 383–385 CrossRef; (b) A. H. Klahn-Olivia and D. Sutton, Organometallics, 1984, 3, 1313–1314 CrossRef.
- W. A. Hermann, J. G. Kuchler, J. K. Felixberger, E. Herdtweck and W. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 394–396 CrossRef.
- (a) C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef; (b) W. A. Herrmann and F. E. Kühn, Acc. Chem. Res., 1997, 30, 169–180 CrossRef CAS; (c) J. H. Espenson, Chem. Commun., 1999, 479–488 RSC; (d) F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef.
- (a) W. A. Herrmann, F. E. Kühn, R. W. Fischer, W. R. Thiel and C. C. Romão, Inorg. Chem., 1992, 31, 4431–4432 CrossRef CAS; (b) W. A. Herrmann, F. E. Kühn, C. C. Romão, H. Tran Huy, M. Wang, R. W. Fischer, P. Kiprof and W. Scherer, Chem. Ber., 1993, 126, 45–50 CrossRef CAS; (c) F. E. Kühn, W. A. Herrmann, R. Hahn, M. Elison, J. Blümel and E. Herdtweck, Organometallics, 1994, 13, 1601–1606 CrossRef; (d) W. A. Herrmann, F. E. Kühn, C. C. Romão and H. Tran Huy, J. Organomet. Chem., 1994, 481, 227–234 CrossRef CAS; (e) W. A. Herrmann, F. E. Kühn and C. C. Romão, J. Organomet. Chem., 1995, 489, C56–C59 CrossRef CAS; (f) W. A. Herrmann, F. E. Kühn, M. U. Rauch, J. D. G. Correia and G. Artus, Inorg. Chem., 1995, 34, 2914–2920 CrossRef CAS; (g) W. A. Herrmann, F. E. Kühn and C. C. Romão, J. Organomet. Chem., 1995, 495, 209–213 CrossRef CAS.
- (a) W. A. Herrmann, R. W. Fischer, M. U. Rauch and W. Scherer, J. Mol. Catal., 1994, 86, 243–266 CrossRef CAS; (b) W. A. Herrmann, F. E. Kühn, M. U. Rauch, J. D. G. Correia and G. Artus, Inorg. Chem., 1995, 14, 2914–2920 CrossRef; (c) M. Högerl and F. E. Kühn, Z. Anorg. Allg. Chem., 2008, 634, 1444–1447 CrossRef.
- (a) W. A. Herrmann, M. Ladwig, P. Kiprof and J. Riede, J. Organomet. Chem., 1989, 371, C13–C17 CrossRef CAS; (b) C. de Méric de Bellefon, W. A. Herrmann, P. Kiprof and C. R. Whitaker, Organometallics, 1992, 11, 1072–1081 CrossRef; (c) M. H. P. Rietveld, L. Nagelholt, D. M. Grove, N. Veldman, A. L. Spek, M. U. Rauch, W. A. Herrmann and G. v. Koten, J. Organomet. Chem., 1997, 530, 150–167 CrossRef; (d) S. M. Bischof, M.-J. Cheng, R. J. Nielsen, T. B. Gunnoe, W. A. Goddard and R. A. Periana, Organometallics, 2011, 30, 2079–2082 CrossRef CAS; (e) S. Huber, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 1353–1357 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Butterworth-Heinemann, Amsterdam, 2003 Search PubMed.
- H. K. Hofstee, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1978, 144, 255–261 CrossRef CAS.
- W. A. Herrmann, F. E. Kühn and P. W. Roesky, J. Organomet. Chem., 1995, 485, 243–251 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (b) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- K. A. Brittingham and J. H. Espenson, Inorg. Chem., 1999, 38, 744–750 CrossRef CAS.
- (a) N. Grover and F. E. Kühn, Curr. Org. Chem., 2012, 16, 16–32 CrossRef CAS; (b) A. Raith, P. Altmann, M. Cokoja, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2010, 254, 608–634 CrossRef CAS; (c) F. E. Kühn, A. M. Santos and M. Abrantes, Chem. Rev., 2006, 106, 2455–2475 CrossRef; (d) M. Vasconcellos-Dias, C. D. Nunes, P. D. Vaz, P. Ferreira, P. Brandão, V. Félix and M. J. Calhorda, J. Catal., 2008, 256, 301 CrossRef CAS; (e) W. R. Thiel and T. Priermeier, Angew. Chem., Int. Ed. Engl., 1995, 34, 1737–1738 CrossRef CAS; (f) W. R. Thiel, J. Mol. Catal. A: Chem., 1997, 117, 449–454 CrossRef CAS.
- (a) J. Rudolph, K. L. Reddy, J. P. Chihang and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 6189–6190 CrossRef CAS; (b) C. Copéret, H. Adolfsson and K. B. Sharpless, Chem. Commun., 1997, 1565–1566 RSC; (c) W. A. Herrmann, H. Ding, R. M. Kratzer, F. E. Kühn, J. J. Haider and R. W. Fischer, J. Organomet. Chem., 1997, 549, 319–322 CrossRef CAS; (d) W. A. Herrmann, R. M. Kratzer, H. Ding, W. R. Thiel and H. Glas, J. Organomet. Chem., 1998, 555, 293–295 CrossRef CAS.
- D. G. Lonnon and J. M. Hook, Anal. Chem., 2003, 75, 4659–4666 CrossRef CAS.
- F. E. Kühn, A. M. Santos, P. W. Roesky, E. Herdtweck, W. Scherer, P. Gisdakis, I. V. Yudanov, C. Di Valentin and N. Rösch, Chem.–Eur. J., 1999, 5, 3603–3615 CrossRef.
- K. A. Brittingham and J. H. Espenson, Inorg. Chem., 1999, 38, 744–750 CrossRef CAS.
- (a) M.-J. Cheng, S. M. Bischof, R. J. Nielsen, W. A. Goddard III, T. B. Gunnoe and R. A. Periana, Dalton Trans., 2012, 41, 3758–3763 RSC; (b) B. L. Conley, S. K. Ganesh, J. M. Gonzales, W. J. Tenn III, K. J. H. Young, J. Oxgaard, W. A. Goddard III and R. A. Periana, J. Am. Chem. Soc., 2006, 128, 9018–9019 CrossRef CAS; (c) J. M. Gonzales, R. Distasio Jr., R. A. Periana, W. A. Goddard III and J. Oxgaard, J. Am. Chem. Soc., 2007, 129, 15794–15804 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20371e |
| This journal is © The Royal Society of Chemistry 2013 |