Bulk gold (non-nanogold) catalysis of aerobic oxidations of amines, isocyanides, carbon monoxide, and carbene precursors
Received
24th May 2012
, Accepted 30th August 2012
First published on 7th September 2012

Robert J. Angelici
| Bob Angelici received his BS degree from St. Olaf College and his PhD with Prof. Fred Basolo from Northwestern University. After one year as an NSF Postdoctoral Fellow at the University of Münich with Prof. E. O. Fischer, he joined the chemistry faculty at Iowa State University in 1963. He and his research group have published 395 papers dealing primarily with transformations of organic ligands coordinated in transition metal complexes. In recent years, he has used principles of reactivity in organometallic chemistry to discover new reactions in heterogeneous catalysis. He is currently Distinguished Professor Emeritus at Iowa State University. |
1. Introduction
In recent years, gold has played a prominent role in catalyzing a broad range of reactions. Metal complexes with gold in the +1 or +3 oxidation state have been used as homogeneous catalysts for transformations of a variety of organic functional groups.1–6 There has also been an explosion of interest in using nanoparticles of gold metal as catalysts for a variety of reactions,7–10 including aerobic oxidations of H2, CO, alkanes, alkenes, alcohols, and amines.11–16 However, no Au-catalyzed reactions have yet been commercialized.17–19 Many of the studies of nanogold catalysts have been directed at understanding why nanosized gold particles, typically 5 nm or smaller, are such active catalysts for reactions that are poorly catalysed by much larger gold particles and especially by pure gold powder.20–24
Our studies of gold powder were initially directed at comparing reactivities of organic molecules adsorbed on Au powder with those of the same organic molecules coordinated to metals in organometallic complexes. This organometallic perspective of our early studies was summarized previously.25 Gold powder was chosen because it is not oxidized by O2 in air, and therefore we could expect that reactions on its surfaces would be promoted by Au in the zero oxidation rather than an ill-defined positive oxidation state that would be formed if the metal surface were to react with air to give a surface oxide. During the course of our studies of organic molecules on Au powder surfaces, we realized that some of these reactions were actually catalysed by the Au powder. Although we knew that gold powder did not catalyse at a significant rate reactions, such as CO + ½O2 → CO2, that were catalysed by nanogold particles supported on metal oxides, we found that Au powder did catalyse other reactions. Some of these reactions were of significant interest in the heterogeneous catalytic community because Au powder was previously identified as a poor catalyst.13 Our studies also suggested that larger (non-nano) gold particles on metal oxide supports could be used as stable catalysts in contrast to supported nanogold catalysts whose activities often decline with use due to agglomeration of the nanogold particles to form larger poorly-active gold particles. One reason for studying the catalytic properties of Au powder was to demonstrate that gold itself, rather than a combination of gold and a support, is capable of catalysing specific reactions. Moreover, mechanistic discussions of reactions on Au powder need not consider the involvement of a support.
The Au powder that we used in our research, which is described in this review, was prepared by the reduction of aqueous HAuCl4 with hydroquinone.26 After treating the initially-formed Au powder with H2O2/H2SO4, washing with water and methanol, and drying at 110 °C in air, it had a dull brown appearance.27 Electron microscopy (Fig. 1) of this powder showed that it contained spherical gold particles of approximately 500 nm size with spines jutting outward from the surface (Fig. 1a). As determined by EELS, the spines contained Au particles embedded in a carbon-containing matrix (Fig. 1b). The surfaces of the large spheres were also coated with a carbonaceous material, although HRTEM studies showed a lattice spacing that was too large for graphite; so, the nature of the carbon-containing material is not known. The catalytic activity of this dull brown gold powder in several different reactions that we studied was lower than that of the powder after it had been used in a reaction.
 |
| | Fig. 1 Electron micrographs of dull brown Au particles with sea urchin morphology. (a) Scanning electron micrograph (SEM) measured at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification; (b) transmission electron micrograph (TEM) measured at 64000× magnification, with the inset showing a HRTEM image focusing on the lattice fringes. The white arrow in (b) points to the area that is magnified in the high resolution image (inset).27 Reprinted with permission from the American Chemical Society. 000× magnification; (b) transmission electron micrograph (TEM) measured at 64000× magnification, with the inset showing a HRTEM image focusing on the lattice fringes. The white arrow in (b) points to the area that is magnified in the high resolution image (inset).27 Reprinted with permission from the American Chemical Society. | |
After one use of the dull brown gold powder catalyst in a reaction, it became a much more active catalyst. Among the reactions that were used to activate the gold powder were the gold-catalyzed coupling of ethyl diazoacetate (EDA) to give olefins,27 the reaction of EDA with amines and O2 to form enamines,28 and the oxidation of amines to imines using amine oxides.29 Typically these reactions were conducted in a stirred flask in a solvent at 60 °C. After an activating reaction, the dull brown color changed to a shiny gold. This shiny gold was significantly more catalytically active and was the form of gold that was typically used in our studies. This shiny gold powder consisted of particles that were much larger (∼50000 nm), and they no longer had a spherical shape (Fig. 2a). At higher SEM magnification (Fig. 2b), it is evident that the surfaces of the large particles exhibit ridges and have smaller particles attached to them. This clearly means there are many different types of gold sites on these catalytically-active gold powder surfaces. While this powder is an excellent catalyst for the reactions described in this review, it is not a practical catalyst because the vast majority of the gold atoms are in the interior of the particles and are therefore not accessible to the reactants. Moreover, the surface area of this powdered Au is very low (0.35 m2 g−1).30
 |
| | Fig. 2 Scanning electron micrograph of gold powder: (a) 500× magnification and (b) 50000× magnification. Black arrows point to small Au particles on the surface of the much larger Au particles.31 Reprinted with permission from Elsevier. | |
Although the gold powder studies demonstrate that large gold particles are active catalysts, they make inefficient use of the gold atoms in the particles. We therefore prepared a γ-Al2O3-supported Au catalyst by incipient wetness impregnation of HAuCl4 into γ-Al2O3 (158 m2 g−1).31 Calcining this 5% Au/Al2O3 catalyst at 700 °C for 68 h ensured that the gold particles on the alumina surface were larger than those typically found in nanogold–Al2O3 catalysts where the sizes are only a few nm in size.32,33
As seen in Fig. 3, STEM images of the 5% Au/Al2O3 catalyst show that the Au particles are mostly 50 nm or larger, but there are also particles in the 20–50 nm range.31 Although particles in the 4–20 nm range could have been detected by our STEM instrument, none was observed. This 5% Au/Al2O3 catalyst made much more efficient use of the Au atoms than Au powder as indicated by the result that 5 mg of Au in 5% Au/Al2O3 had a slightly higher catalytic activity than 1000 mg of Au in Au powder in the oxidation of amines to imines (discussed in more detail below).31 Thus, the large Au particles in our 5% Au/Al2O3 are excellent catalysts for certain reactions. In its supported form, Au metal therefore becomes a more practical catalyst. The focus of this review is not on practical gold catalysts but rather on expanding the range of reactions that bulk gold (non-nanogold) is capable of catalysing. Most of the discussion herein deals with gold powder as the catalyst.
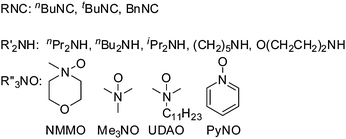
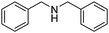
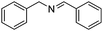
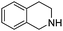
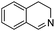
When secondary amines are stirred in a solvent under 1 atm of O2 in the presence of gold powder, oxidation of the amine to the imine is observed (eqn (1)).34 At 60 °C in MeCN solvent (Table 1), | | 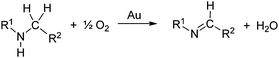 | (1) |
the yields of the imine products after 40 h are much higher for 1,2,3,4-tetrahydroisoquinoline (40%) and dibenzylamine (35%) than for the alkylamine di-i-propylamine (15%). Although the yields are much higher at 100 °C in toluene after 24 h, the same trend in amine reactivity is observed. The imine yield from N-benzylaniline however is much lower (entry 4) than from dibenzylamine (entry 1) perhaps because of a steric effect and the lower basicity of the nitrogen which would reduce its tendency to adsorb to the gold. Rather surprisingly, the addition of 0.4 mmol of pyridine to the 1,2,3,4-tetrahydroisoquinoline reaction (entry 2, Au powder, 100 °C) did not decrease the yield of the imine product,31 as might be expected if the pyridine were to compete with the amine for adsorption sites on the catalyst surface.
| Entry |
Substrate |
Product |
Yield (%) |
| 60 °C, Au powdera |
100 °C, Au powderb |
100 °C, Au/Al2O3c |
|
Reaction conditions: amine (0.2 mmol), O2 (∼1.0 L at ∼1 atm), gold powder (1.0 g) in 5 mL MeCN at 60 °C for 40 h.34
Reaction conditions: amine (0.2 mmol), O2 (∼1.0 L at ∼1 atm), gold powder (1.0 g) in 5 mL toluene at 100 °C for 24 h.34
Reaction conditions: amine (0.2 mmol), O2 (∼1.0 L at ∼1 atm), 5% Au/Al2O3 (100 mg) in 5 mL toluene at 100 °C for 24 h.31
|
| 1 |
|
|
35 |
64 |
97 |
| 2 |
|
|
40 |
87 |
89 |
| 3 |
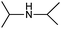
|
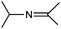
|
15 |
|
|
| 4 |
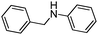
|
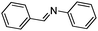
|
|
9 |
18 |
| 5 |

|
|
46 |
93 |
98 |
| 6 |

|
|
41 |
75 |
95 |
| 7 |
|
|
22 |
|
33 |
Although no mechanistic studies of these catalytic reactions have been reported, studies of the reaction of oxygen atoms on the surface of Au(111) with dimethylamine to form the MeN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 imine led Xu, Friend, and Madix to propose the mechanism in Scheme 1 for the reaction in eqn (1).35 Since O2 does not adsorb on Au(111), they introduced oxygen atoms onto the Au surface by reacting it with O3. There is abundant evidence to conclude that these O atoms are basic and deprotonate amines, as proposed in the first step of the mechanism. β-hydrogen transfer to the gold surface with release of H2O in the second step would give the imine product. Perhaps the O2 used in our gold powder-catalyzed reactions is cleaved to O atoms on low-coordinate gold sites on the irregular gold powder surfaces (Fig. 2b).
CH2 imine led Xu, Friend, and Madix to propose the mechanism in Scheme 1 for the reaction in eqn (1).35 Since O2 does not adsorb on Au(111), they introduced oxygen atoms onto the Au surface by reacting it with O3. There is abundant evidence to conclude that these O atoms are basic and deprotonate amines, as proposed in the first step of the mechanism. β-hydrogen transfer to the gold surface with release of H2O in the second step would give the imine product. Perhaps the O2 used in our gold powder-catalyzed reactions is cleaved to O atoms on low-coordinate gold sites on the irregular gold powder surfaces (Fig. 2b).
As no kinetic or other experimental studies have been reported that test the mechanism in Scheme 1, it is possible that some other mechanism is involved. A peroxide intermediate (rather than oxide) has been suggested for the mechanism of supported-gold-catalyzed oxidation (O2) of alcohols.12,36 The identification of H2O2 as a product of these oxidations is substantive evidence for a peroxide, rather than oxide, intermediate in these reactions. No attempts to identify H2O2 as a product of the oxidative-dehydrogenation of amines have been reported.
In order to determine the catalytic activity of gold supported on high surface area alumina (158 m2 g−1), some of these reactions were performed under the same conditions (100 °C, toluene, 1 atm O2) but using only 5 mg of Au on 5% Au/Al2O3 rather than 1000 mg of Au powder.31 The yields are comparable or better for the 5% Au/Al2O3 catalyst (Table 1, entries 1, 2, and 4), which means that gold in 5% Au/Al2O3 is approximately 200 times more active than the same mass of gold in gold powder. The higher activity of 5% Au/Al2O3 may be due to a variety of factors: the higher gold surface area, the smaller gold particles (∼50 nm) as compared with those in gold powder (∼50000 nm), effects of the alumina support, and other factors.
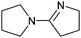
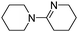
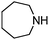
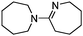
The aerobic oxidative-dehydrogenation of cyclic secondary amines is also catalysed by gold powder. Pyrrolidine ((CH2)4NH), piperidine ((CH2)5NH), and hexamethylene imine ((CH2)6NH) all give amidine products in which two amines are oxidized and coupled (Scheme 2, Table 1). In MeCN solvent at 60 °C with gold powder, yields of the products (Table 1) decrease as the ring size increases from the 5-membered amine (46%) to the 7-membered amine (22%) in a 40 h reaction period.
A proposed mechanism for the coupling of cyclic secondary amines is presented in Scheme 2.34 It involves initial oxidative-dehydrogenation (step 1) of the amine (1) to give the cyclic imine (2). This step is analogous to the oxidative-dehydrogenation reactions (eqn (1)) that non-cyclic secondary amines undergo (entries 1–4 in Table 1) under the same conditions. However, the less sterically-crowded cyclic imines react further to undergo attack by additional amine (1) to give 3. There is no GC/MS evidence for intermediate 2 or 3 in samples of the solution taken during the course of the reaction, which means that 2 and 3 must react faster than 1, and step 1 is the rate-determining step in the overall conversion of 1 to 4. To demonstrate that step 2 could occur under the conditions of the reaction, Δ1-pyrroline (2a) was prepared and reacted with 1a in the presence of gold powder under 1 atm of O2 (eqn (2)). This reaction gives product 4a
| | 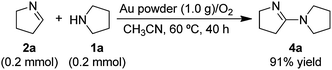 | (2) |
without any evidence for intermediate
3a. As step 2 in
Scheme 2 does not involve O
2, the reaction of
2a and
1a was also conducted under an argon atmosphere. However, there was no reaction, which suggests that the equilibrium in step 2 of
Scheme 2 lies to the left, which means that the oxidative-dehydrogenation of
3a to give
4a (step 3) provides the overall driving force for the conversion of
2a to
4a (
eqn (2)). It should also be noted that the reaction of
2a with
1a (
eqn (2)), which is nearly complete after only 20 h, is significantly faster than the overall conversion of
1a to
4a. Although it was not possible to study the Au powder-catalyzed oxidative-
dehydrogenation of compounds
3 because these compounds are unstable with respect to
1 and
2, a model compound with the same
aminal functionality as in
3 does undergo oxidative-
dehydrogenation under the conditions of the conversion of
1 to
4 to give the corresponding
amidine.
34 All of these studies support the mechanism for the oxidative-dehydrogenation of cyclic
amines outlined in
Scheme 2.
To test for the possibility that colloidal or soluble gold complexes in solution were the actual catalysts of reaction (1), the reaction of piperidine with O2 (Table 1, entry 6, at 60 °C in MeCN) was carried out for 6 h at which point the yield of the amidine product was only 9%. Then the solution was separated from the solid gold powder. Since the reaction did not continue to form the amidine product after the gold powder was removed, catalytic species must not have been present in the solution. Therefore, the solid gold powder must be the actual catalyst of the reaction. This same type of test was conducted on several other reactions discussed in this review. In all cases, it was found that the solid gold powder, rather than some soluble form of gold, was the actual catalyst of the reaction.
While the 5-, 6-, and 7-membered cyclic amines are converted to their amidine products (entries 5–7, Table 1) at 60 °C in MeCN using gold powder, their reactions are much faster at 100 °C in toluene. Using 5% Au/Al2O3, the amidine yields, also at 100 °C, are at least as high as those obtained using gold powder.
In addition to confirming the catalytic activity of gold powder in the aerobic oxidative-dehydrogenation of cyclic amines to amidines, Lambert and co-workers37 found that gold powder (1000 nm particles) and 1% Au/C (16 nm Au particles) had comparable catalytic activities, which indicated that the Au activity did not depend greatly on the size of the Au particles. On the other hand, Girrane, Corma and Garcia38 showed that the gold catalyst efficiency increased exponentially as the average gold particle size decreased. This conclusion was based on comparisons of TOF values for the aerobic oxidation of benzylamine at 100 °C using Au/TiO2 catalysts in which the Au particle sizes were 3.5, 5.5, and 25 nm.38 Using a series of polystyrene-based polymer-incarcerated gold nanoparticle catalysts, Kobayashi and co-workers39 found the aerobic oxidation of dibenzylamine to be faster for relatively large (>5 nm) Au particles than for small particles (1–3 nm). Considering the different gold particle sizes, the different reactions, and the different reaction conditions, it is not possible to draw any broad conclusions about the effect of particle size on the catalytic activity of gold in the oxidative-dehydrogenation of amines. Certainly in a variety of other reactions, such as the aerobic oxidation of CO, small nano-sized gold particles are essential for their high catalytic activity.7,8,13
An interesting extension of the oxidative-dehydrogenation-coupling of cyclic secondary amines (Scheme 2) is the finding40 that the amidine products undergo hydrolysis to give the corresponding lactams and original amines (eqn (3)). At 90 °C in
| |  | (3) |
toluene, the
amidine (20 mM),
H2O (200 μL) and 50 mg of Aerosil 200 (a fumed
silica) gave
lactam products after 24 h of reaction in yields that depend on the amine ring size:
n = 0 (35%),
n = 1 (53%),
n = 2 (70%). One reason for the non-quantitative yields is that some of the
lactam remains adsorbed on the Aerosil 200. Also, it is possible that some of the
lactam product polymerizes to the
polyamide or hydrolyzes to the
amino acid.
In order to explore the possibility that the cyclic amines can be converted to lactams in a one-pot reaction (eqn (4)), we added
| | 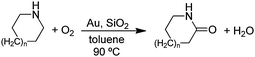 | (4) |
both the gold powder and Aerosil 200
catalysts to a
toluene solution of the cyclic
amine under 1 atm of O
2. Heating at 90 °C for 96 h gave the following yields of the
lactams:
n = 0 (35%),
n = 1 (51%),
n = 2 (11%). Turnover numbers (TON), based on the estimated number of surface gold atoms and assuming that all are active sites, for
n = 0, 1, and 2 are 12, 16, and 4, respectively. Although the TON values are not high, the conversion of cyclic
amines to
lactams using O
2 as the oxidant (
eqn (4)) offers the possibility that
nylon precursor
lactams can be produced in a more environmentally-friendly process.
In a related study, the aerobic oxidation of benzo-cyclic amines in the presence of excess NaOH and a Au:PVP catalyst gave lactams (eqn (5)).41 In this case, the size of the gold
| | 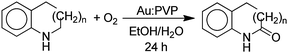 | (5) |
particles embedded in the poly(
N-vinyl-2-pyrrolidone) (PVP) was critical as nanogold particles larger than 10 nm were almost inert. For the cyclic
amine, where
n = 1, the yield of the
lactam was 91% at 27 °C. The larger cyclic
amine with
n = 2 required 90 °C in order to achieve a 96% yield, but the
amine with
n = 2 did not react even at 90 °C. This trend in decreasing reactivity with increasing ring size is similar to that for the simple cyclic
amines in
Scheme 2 and
Table 1. The reactions were proposed to occur by a mechanism in which the
amine was initially oxidized to the
imine which was hydrated to give an α-hydroxyamine intermediate which was oxidized to the
lactam product. The application of this method to the conversion of simple cyclic
amines to
lactams (
eqn (4)) was not reported.
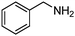
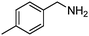
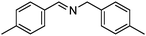
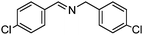
Under the same conditions (100 °C, toluene) used in the aerobic oxidative-dehydrogenation of secondary amines (Table 1), primary amines give the imine resulting from the oxidation and coupling of two amines (eqn (6)).31 As for the secondary amines,| | 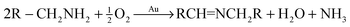 | (6) |
the aliphatic amine n-hexylamine (entry 1) gives a lower yield of the imine product than the benzylamines (Table 2, entries 2–4). Among the benzylamines, the highest yield is obtained with the electron-donating p-methyl-substituted derivative (entry 3). Perhaps this reactivity trend reflects the basicity of the amine group, which adsorbs more strongly to the Au surface when the amine is more basic.
| Entry |
Substrate |
Product |
Yield [%] |
| Au powder cat.a |
Au/Al2O3 cat.b |
|
Reaction conditions: amine (0.2 mmol), O2 (∼1.0 L at ∼1 atm), gold powder (1.0 g), in 5 mL toluene solvent for 24 h at 100 °C.31
Reaction conditions: amine (0.2 mmol), O2 (∼1.0 L at ∼1 atm), 5% Au/Al2O3 (100 mg), in 5 mL toluene solvent at 100 °C for 24 h.31
|
| 1 |

|
C6H13–NCHC5H11 |
5 |
29 |
| 2 |
|

|
56 |
92 |
| 3 |
|
|
61 |
96 |
| 4 |

|
|
7 |
59 |
As for the secondary amines (Table 1), the rates of reaction (eqn (6)) are faster for the 5% Au/Al2O3 catalyst with only 5 mg of Au than for the Au powder with 1000 mg of Au. This difference in activity is undoubtedly due at least in part to the higher Au surface area in the Al2O3-supported catalyst. When the 5% Au/Al2O3 was washed with CH2Cl2, EtOH, and hexanes and then re-used in the reaction of benzyl amine (entry 2), the yield of imine was only 71% as compared with 92% in its first use. It is not clear why the activity of the Au catalyst is reduced after use, but this is also observed in other reactions that are catalysed by gold powder where treatment of the used gold powder even with the strongly oxidizing H2O2/H2SO4 solution did not fully restore the Au powder to its original activity. Generally, it was necessary to convert the used Au back into HAuCl4 and then reduce it to Au powder, which then exhibited its original high activity.
Two reasonable mechanisms for the formation of imines from primary amines (eqn (6)) are proposed in Scheme 3. Both proceed by way of initial oxidative dehydrogenation of the amine to the imine intermediate RCHNH, presumably by the mechanism in Scheme 1. In path A, this intermediate is attacked by a second molecule of the primary amine to give an aminal which loses NH3 to give the coupled imine product RCHNCH2R. In path B, the initially formed imine reacts with H2O generated in the first step to give the aldehyde RCHO, which subsequently reacts with a second molecule of the amine to give the imine product. There is no conclusive experimental evidence to distinguish between paths A and B.
Although this review is focused on bulk gold (non-nanogold) catalysis, the aerobic oxidative-dehydrogenation of amines has been studied7 extensively using nanogold catalysts supported on ceria,42,43 polystyrene,44 alumina, titania,37 and carbon.45
In the course of our studies of the oxidation of amines with O2, we considered the possibility that O2 was a unique oxidant and that other oxygen sources may not be capable of providing oxygen atoms to the amine on the Au surface. However, we quickly discovered that amine oxides (R3N–O) are capable of oxidizing amines to the same products that were obtained with O2 under the same conditions (gold powder, 60 °C, CD3CN solvent).29 In fact, the reaction of dibenzylamine with N-methylmorpholine N-oxide (NMMO) to form the imine product (eqn (7)) was faster| | 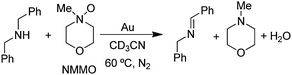 | (7) |
than the reaction with O2 (1 atm) (Fig. 4). The other aliphatic amine oxide Me3N–O was also an effective oxidant, but the reaction with pyridine N-oxide (PyNO) was much slower. Thus, it appears that the reactivities of the amine oxides decrease as their basicities decrease, as reflected by the pKas of their conjugate acids: NMMOH+ (4.75) > Me3NOH+ (4.56) > PyNOH+ (0.79). This trend suggests that the more basic amine oxides are better able to compete with the amine for adsorption sites on the gold surface. The fact that the rate of reaction (7) increased with increasing concentrations of NMMO is also consistent with a possible competitive adsorption step. However, it is still not known whether or not the amine oxide delivers oxygen atoms to the gold surface as is proposed in the O2 reactions (Scheme 1) or some other mechanism is involved.
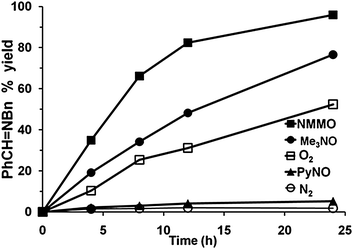 |
| | Fig. 4 Performance of different oxidants in the catalytic oxidative-dehydrogenation of Bn2NH to PhCHNBn. Conditions: Bn2NH (20 mM), oxidant: amine N-oxide (110 mM), O2 (∼1 L, ∼1 atm) or N2 (1 atm), Au (0.20 g), CD3CN (0.60 mL), 60 °C.29 Reprinted with permission from Springer Science and Business Media. | |
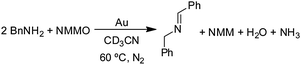
Other amines, 1,2,3,4-tetrahydroisoquinoline, pyrrolidine [(CH2)4NH] and piperidine [(CH2)5NH] also reacted with NMMO under the same conditions to give the same imine (eqn (1)) and amidine (Scheme 2) products that were obtained in their gold-catalyzed reactions with O2.29 The primary amine benzylamine also reacts with NMMO at 60 °C in CD3CN in the presence of 1.0 g of Au powder.29 As in the reaction of benzylamine with O2 (eqn (6), Table 2, entry 2), its reaction with NMMO gives N-benzylidenebenzylamine (eqn (8)) in 45% yield after 48 h of reaction.
| |  | (8) |
In all of the oxidative-dehydrogenation reactions of amines using amine oxides, additional products formed after the amine was consumed. These other products resulted from the Au powder-catalyzed decomposition of the amine oxide. When investigated in the absence of an amine, Me3N–O decomposed under the standard reaction conditions to give Me3N and HC(O)NMe2 (eqn (9)), which accounted for 78% of the
| | 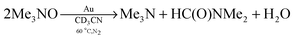 | (9) |
decomposed Me
3N–O. NMMO decomposed under the same conditions to give
N-methyl morpholine and
4-methyl-morpholine-2,3-dione (
eqn (10)). Approximately 56% of the
| | 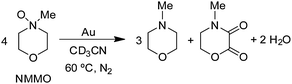 | (10) |
NMMO decomposed by the reaction in
eqn (10). In both of these reactions, other minor products were detected but not identified. Although the mechanism for gold powder-catalyzed
decomposition of amine oxides (
eqn (9) and (10)) to
amides by
oxidation of CH
2 groups is not known, the reaction of (CH
3)
2NH with oxygen atoms on Au(111) does give the
amide product HC(O)NHCH
3, in addition to other products (MeNCO, CO
2, and NO
2).
46 Perhaps
reactions (9) and (10) are initiated by transfer of the amine oxide (R
3N–O) oxygen atom to the Au surface, and it is these surface oxygen atoms that oxidize the CH
2 groups adjacent to the
nitrogen atom as proposed for the oxidation of (CH
3)
2NH to HC(O)NHCH
3.
46 It should be noted, however, that
reactions (9) and (10) do not occur when the more reactive secondary or
primary amine substrates are present, as these substrates are preferentially oxidized according to
eqn (7) and (8).
Besides O2 and amine oxides (R3N–O), NO2 is the only other oxidant that has been reported to convert secondary amines to imine or amidine products (Scheme 2) using a bulk gold metal catalyst.37 This study also showed that NO2 was a more effective oxidant of 1,2,3,4-tetrahydroisoquinoline to its imine (see Table 1, entry 2) than O2 using 500 mg of Au powder in toluene at 100 °C. Indeed this conversion gave a 76% yield of the imine when using 0.03 bar of NO2, but only a 23% yield even using 1.5 bar of O2. Also, the gold powder-catalyzed oxidation of pyrrolidine with NO2 gave the amidine (Scheme 2) in high yield (86%).
A variety of nanogold catalysts have been used to catalyse the aerobic oxidative-dehydrogenation of numerous alcohols to give aldehydes, ketones, and carboxylic acids under temperatures that range from 30 °C to 300 °C.7,11,12,15 The use of bulk gold catalysts for these reactions is much more limited. Guo, Repo and co-workers47 recently reported using gold foil (150 mg) to catalyse the reaction of benzyl alcohol with O2 (5 atm) at 90 °C in the presence of K2CO3 in toluene or heptane solvent. The major product was benzaldehyde, but the ester benzyl benzoate was a significant by-product (eqn (11)). As in gold powder-catalysis of | | 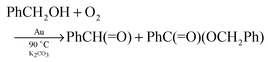 | (11) |
other reactions, these reactions are catalysed by the bulk gold, rather than colloidal or gold complex species that might be formed in solution. A variety of primary and secondary benzyl alcohols were successfully oxidized under these conditions; however, except for cinnamyl alcohol, other alcohols did not react.
Benzyl alcohol is oxidized slowly with O2 (1 atm) at 60 °C in CD3CN in the presence of Au powder to give only a 7% yield of benzaldehyde after 48 h.29 When the amine oxide NMMO (110 mM) was used as the oxidizing agent, a 52% yield of the aldehyde was obtained after 48 h under the same conditions (eqn (12)). When the temperature was increased to 85 °C, the yield of
| | 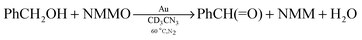 | (12) |
benzaldehyde was 63% after 48 h but it increased very little at longer reaction times. The results show that at 60 °C in CD
3CN, in the presence of Au powder, the oxidation of
benzyl alcohol is much more favourable with NMMO (110 mM) than with O
2 (1 atm).
4. Oxidative reactions of isocyanides (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N–R) with amines
N–R) with amines
4.1 Aerobic oxidative reactions of CN–R with amines
In our first studies of gold powder catalysis of reactions of n-BuNC, n-BuNH2, and O2, we discovered the formation of the carbodiimide (Bun–NCN–Bun) according to eqn (13).48| | 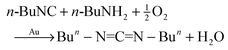 | (13) |
At 60 °C in hexane solvent, under an air atmosphere and stirring with 1.0 g of gold powder, n-BuNC (1.7 × 10−2 mmol) and n-BuNH2 (17 × 10−2 mmol) react over the course of 90 h to give a 73% yield of Bun–NCN–Bun. Since the n-BuNC had completely reacted, it meant that other products (27%) also formed. These products were not identified, but there were additional peaks at higher mass in the GC–MS, which might be attributed to oligomers of n-BuNC. If the reactions were conducted at higher concentrations of the isocyanide and amine, the percent yields decreased, presumably due to the formation of n-BuNC oligomers. These lower yields mean that we have not found conditions that make this reaction useful for the synthesis of larger amounts of carbodiimides. Mixed carbodiimides R–NCN–R′ were obtained when relatively low concentrations of a variety of isocyanides (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–R) were reacted with a variety of primary amines (R′NH2).48,49
N–R) were reacted with a variety of primary amines (R′NH2).48,49
In order to gain some understanding of the mechanism of this reaction, we first studied the adsorption of isocyanides on gold. From solution, e.g., 1,2-dichloroethane (DCE) or hexane, isocyanides readily adsorb on gold metal in all of its forms (films, powders, and nanoparticles).50 Typically, a ν(NC) frequency for an adsorbed isocyanide that is equal to or higher than that of the free isocyanide indicates that the isocyanide is adsorbed η1 through the carbon to one Au atom. In our studies, we observed for n-BuNC adsorbed on a gold film a ν(NC) value (2224 cm–1) that is 76 cm–1 higher than that (2148 cm–1) of free n-BuNC in DCE solution.48 Although the presence of only one ν(NC) band for the adsorbed isocyanide suggests that all isocyanide molecules are adsorbed on the same type of gold site, this cannot be the case because n-BuNC on gold films that are immersed in a pure solvent, such as DCE, desorb about 55% of the n-BuNC within one minute while the other 45% requires 4 h or longer. As the n-BuNC desorbs, the ν(NC) of the n-BuNC remaining on the gold increases slightly to 2232 cm–1. Thus, the adsorbed isocyanides may be classified into two groups: (1) those that are weakly adsorbed with ν(NC) = 2224 cm–1 and (2) those that are strongly adsorbed with a higher ν(NC) value (2232 cm–1). There are also two types of n-BuNC sites on powdered gold;51 one exhibits a ν(NC) value of 2225 cm–1 at saturation coverage, while the other shows a ν(NC) band at 2233 cm–1 for the strongly adsorbed n-BuNC which remains on the surface after the weakly adsorbed isocyanide is desorbed. While the ν(NC) values for the weakly and strongly adsorbed n-BuNC are the same on both gold film and gold powder, only 45% of n-BuNC is strongly adsorbed on gold film, whereas 70% is strongly adsorbed on gold powder. The much more irregular surface of gold powder would be expected to have more low-coordinate edge and corner gold atom sites than would exist on gold films with more high-coordinate gold atoms on (111) terrace sites.52 Therefore gold powder would be expected to bond more isocyanides strongly than gold film, as was observed.
In the presence of amine, as in reaction (13), we expected the adsorbed isocyanides to be attacked by the amine based on known reactions of isocyanide ligands in transition metal complexes. Isocyanide ligands whose ν(NC) values are more than 40 cm–1 higher than those of the free isocyanides have been observed to be attacked by amines (and other nucleophiles)53–55 to give diaminocarbene ligands (eqn (14)). Typically, the rate laws
| | 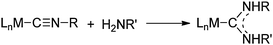 | (14) |
for these reactions are first order in the metal complex and
amine concentrations: Rate =
k[L
nM(CNR)][H
2NR′]. This rate law has been interpreted as supporting a mechanism that involves nucleophilic attack of the
amine on the
isocyanide carbon that is activated by a metal (M) in a positive oxidation state and/or by
ligands L that are weakly electron-donating. In some reactions, the rate law exhibits an
amine concentration dependence that is higher than first order. This is interpreted to indicate the involvement of a second
amine molecule, which promotes the rearrangement of the initial
isocyanide–
amine adduct to the diaminocarbene product.
53,55
As the ν(NC) value (2224 cm–1) for n-BuNC adsorbed on gold is much more than 40 cm–1 higher than that (2148 cm–1) of free n-BuNC, one expects n-BuNC adsorbed on gold metal to undergo attack by amines to give diaminocarbene groups on the surface, if one assumes that the 40 cm–1 guideline for isocyanides in metal complexes also applies to isocyanides adsorbed on metal surfaces. In fact, we observed48 that all of the n-BuNC (both weakly and strongly adsorbed) was removed within 20 s from a gold-plated slide containing adsorbed n-BuNC, when it was immersed into a hexane solution containing n-BuNH2. The reaction of the adsorbed n-BuNC is slower if the amine is less basic or more sterically bulky than n-BuNH2. There was no evidence in the diffuse reflectance infrared spectrum of the resulting gold film for diaminocarbene groups, which are expected to give a ν(N–CN) absorption near 1550 cm–1, which is characteristic of diaminocarbene ligands in metal complexes. This result suggested that the product of the reaction of the adsorbed isocyanide with the amine must have desorbed from the surface. By reacting n-BuNC adsorbed on 1.0 g of gold powder, which has a much larger surface area than the gold film, with n-BuNH2, it was possible to identify the product of the reaction as Bun–NCN–Bun together with a significant amount of n-BuNC, which presumably resulted from the rapid desorption of the weakly adsorbed n-BuNC. Thus, it appears that most of the reacting n-BuNC was the strongly adsorbed form, which would be expected to be more reactive toward nucleophiles because it has a higher ν(NC) value than the weakly adsorbed n-BuNC. If these solutions were allowed to stir for a longer time or were heated to 60 °C, the desorbed n-BuNC product was also converted to the carbodiimide product. This meant that the reaction of n-BuNC with n-BuNH2 to form Bun–NCN–Bun was catalyzed by gold metal. When the catalytic reaction of n-BuNC with n-BuNH2 in the presence of gold powder was conducted under an argon atmosphere, no carbodiimide product formed. This meant that O2 is a reactant and that H2O is the likely product. Therefore, the gold powder-catalyzed reaction of n-BuNC with n-BuNH2 and O2 is described by eqn (13).
Kinetic studies48 of the gold powder-catalyzed reaction (eqn (13)) of n-BuNC and n-BuNH2 at 60 °C in hexane show that the rate is independent of the O2 concentration/pressure, as indicated by the observation that the rate is the same in 1 atm of air or pure O2. However, the rate is first-order in the concentration of n-BuNH2. These results suggest a mechanism (Scheme 4) in which the isocyanide first adsorbs on the Au surface. As in metal complexes (eqn (14)), this makes the isocyanide susceptible to nucleophilic attack by the amine in step 1. This is the rate-determining step in the reaction. The resulting intermediate a either rearranges to a diaminocarbene group b (as occurs in metal complexes, eqn (14)) in step 3 or reacts with O2 to give H2O and the carbodiimide in step 2. Alternatively, the diaminocarbene intermediate b reacts with O2 to give the products. As the overall reaction rate does not depend on the O2 concentration, steps 2 and 4 must be fast as compared with step 1. Although step 1 is readily understandable in terms of a mechanism that is well-documented for reactions of isocyanide ligands with amines (eqn (14)), there is no comparable metal complex model for the conversion of a diaminocarbene ligand to a carbodiimide.
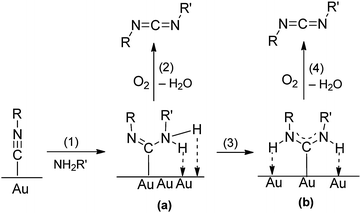 |
| | Scheme 4 Mechanism for the Au-catalyzed reaction of isocyanides (R–NC) with primary amines (H2NR′) and O2.48 Reprinted with permission from the American Chemical Society. | |
Steps 2 and 4 in Scheme 4 are unlikely to involve the generation of H2 from intermediate a or b, because H2 reacts with O2 to form H2O over gold powder only at temperatures above 130 °C.14,56 However, supported nanogold does catalyze the reaction of H2 and O2 to give H2O2 even below room temperature.57,58
Of course, carbodiimide products are only possible when the amine is a primary amine. With secondary amines, it is still possible for the amine to attack an adsorbed isocyanide, but the resulting intermediate a or b (Scheme 4) cannot react with O2 to give a carbodiimide product. However, isocyanides do react (eqn (15)) with secondary amines in the presence of gold powder, to
| | 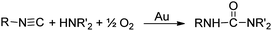 | (15) |
give
ureas.
59 The reactions were performed under the same conditions (60 °C,
hexane or MeCN
solvent, 1.0 g of gold powder) that were used with the gold-catalyzed reactions of
isocyanides with
primary amines (
eqn (13)). The reaction occurs under 1 atm of air or O
2, but the rate was slightly faster in the presence of pure O
2, although this dependence was significantly less than first order in the O
2 concentration/pressure. When the reaction was run under an argon atmosphere, only a small amount (∼1.5%) of the urea product formed, presumably due to the presence of adventitious O
2. This formation of
urea even under a very low pressure of O
2 means that the O
2 is highly reactive with intermediates on the gold surface. When
n-BuNC was reacted with different concentrations of HNPr
2n under 1 atm of O
2 in MeCN
solvent using 1.0 g of Au powder, the rate of the reaction increased as the
amine concentration increased, although it was somewhat less than first order in the
amine concentration. This
amine dependence suggests that a key step in the reaction is nucleophilic attack of the
secondary amine on the adsorbed
isocyanide. Then, one would expect the rate of the catalyzed reaction to depend also on the steric and electronic properties of the
amines. This is indeed observed as shown by the urea product yields (
Table 3) obtained in reactions of
n-BuNC with different
secondary amines under a standard set of conditions (60 °C, MeCN
solvent, 1 atm O
2, 1.0 g gold powder). The yields were determined after 24 h of reaction. As compared with the bulky HNPr
2n, the cyclic and therefore sterically-smaller
piperidine ((CH
2)
5NH) gives a higher yield (51%) of the corresponding urea. When
morpholine (O(CH
2CH
2)
2NH), which is less nucleophilic but sterically-similar to
piperidine, is reacted with
n-BuNC, the yield (22%) of the urea is significantly lower. The higher p
Ka of the protonated form of
piperidine (11.2) as compared with that for
morpholine (8.36) is an
indicator of the higher nucleophilicity of
piperidine. Therefore, both the steric and electronic properties of the
amine have predictable effects on the rates of reaction.
To test for the effect of the bulkiness of the isocyanide on the rate of reaction, 1,1,3,3-tetramethyl-n-butyl isocyanide (CH3)3CCH2C(CH3)2NC was reacted with HNPr2n and O2 under the standard conditions. As shown in the last entry in Table 3, this bulky isocyanide gives a significantly lower yield (19%) than n-BuNC (44%). The lower rate with the bulky isocyanide could reflect a lower isocyanide coverage on the Au surface60 or a reduced rate of amine attack on the sterically-bulky, adsorbed isocyanide.
Results of the above studies can be reasonably understood in terms of a mechanism (Scheme 5) that is related to that proposed for the gold-catalyzed reactions (eqn (13)) of isocyanides with primary amines (Scheme 4). The first step (1) in both of these mechanisms is amine attack on an adsorbed isocyanide to give an isocyanide–amine adduct a, which rearranges to the diaminocarbene intermediate b. The amine attack on adsorbed isocyanide is modeled by analogous reactions of amines with isocyanide ligands in metal complexes (eqn (14)). The rate of this attack in both mechanisms (Schemes 4 and 5) plays a major role in influencing the overall rate of catalysis. In reactions of n-BuNC with secondary amines, the rate of this first step (1) depends on the steric and electronic properties of the reacting amine, but the rate is somewhat less than first order in the amine concentration. The isocyanide–amine adduct (a) resulting from this attack presumably rearranges to the diaminocarbene intermediate b which reacts (step 3) with O2 to give the urea product. The observed small dependence of the rate of the gold-catalyzed reaction of n-BuNC with HNPr2n on the O2 concentration suggests that step 3 (Scheme 5) is faster than step 1 but not by a large amount. Details of step 3 are not known, and there are no examples of reactions of diaminocarbene transition metal complexes with O2 that lead to ureas. However, it is possible that the diaminocarbene groups on the gold surface add sufficient electron density to the gold metal that it adsorbs O2, which reacts with the diaminocarbene intermediate b to give the urea product. The notion that diaminocarbene groups would add electron density to the gold is supported by studies of diaminocarbene ligands in transition metal complexes,61–63 which show that these ligands are strongly electron-donating.
4.2 Amine oxide (R3N–O) reactions with CN–R and amines
As described in eqn (15), gold powder catalyzes the reaction of isocyanides (CN–R) and secondary amines 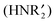 with O2 to give ureas
with O2 to give ureas 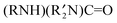 . Under the same conditions (1.0 g gold powder, CH3CN solvent, 60 °C), amine oxides (R3N–O) also react with isocyanides and secondary amines to give ureas (eqn (16)).64
. Under the same conditions (1.0 g gold powder, CH3CN solvent, 60 °C), amine oxides (R3N–O) also react with isocyanides and secondary amines to give ureas (eqn (16)).64| | 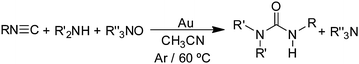 | (16) |
The rate of the reaction of nBuNC and nPr2NH with NMMO increases with the NMMO concentration up to 40 mM, above which there is no further increase in rate. The reaction rate actually decreases as the amine concentration increases. These concentration dependences are very different from those of reaction (15) in which the rate is almost first order in amine and nearly independent of O2 concentration. These differences suggest that the O2 (eqn (15)) and amine oxide (eqn (16)) reactions proceed by different mechanisms, as supported by further experiments discussed below.
With different amine oxides, the rates of reaction of nBuNC and nPr2NH are significantly faster with more basic, aliphatic amine oxides (NMMO, Me3NO, UDAO) than with pyridine N-oxide(PyNO) (Fig. 5). With different secondary amines (nPr2NH, nBu2NH, iPr2NH, piperidine morpholine), the initial rates of reaction with nBuNC and NMMO are all very similar. However, rates of the cyclic amine (piperidine and morpholine) reactions soon begin to decrease as their oxidative-dehydrogenation-coupling reactions (Scheme 2), become more important.
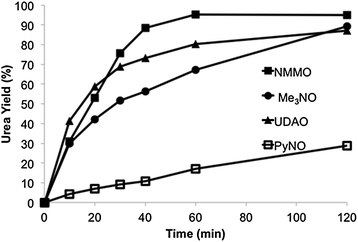 |
| | Fig. 5 Effect of various amine oxides in the reaction (eqn (16)) of 2.0 mM nBuNC, 10.0 mM nPr2NH, and 40.0 mM amine oxide in 5.0 mL of CH3CN using 1.00 g of Au at 60 °C.64 Reprinted with permission from Elsevier. | |
Primary amines (cyclohexylamine (CyNH2), n-hexylamine (nHexNH2) and aniline) also react with nBuNC and NMMO under the usual conditions (1.0 g gold powder, 60 °C in CH3CN solvent) to give the corresponding ureas (eqn (17)). The more
| | 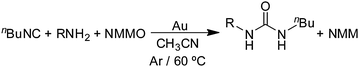 | (17) |
basic
amines (CyNH
2 and
nHexNH
2) react much faster than
aniline, but they also give the coupled
imine byproducts resulting from the oxidative-dehydrogenation of the
amine, as observed in the absence of
nBuNC (
eqn (8)).
Reaction (17) is especially interesting because the products of these reactions are
ureas and not the
carbodiimides that were observed when O
2 was used as the oxidizing agent (
eqn (13)). This is another major difference between amine oxides and O
2 in reactions of
isocyanides with
amines.
There are two reasonable mechanisms that might be considered for the reactions of isocyanides with amines and amine oxides to give ureas (Scheme 6).
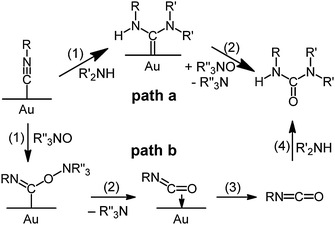 |
| | Scheme 6 Mechanisms for the reaction of adsorbed isocyanide with secondary amines and amine oxides to form urea products.64 Reprinted with permission from the American Chemical Society. | |
The first step in both mechanisms is the adsorption of the isocyanide on the Au surface. Numerous studies show that isocyanides adsorb on various gold metal surfaces (powder, film, and nanoparticles) from solutions48,50,51,60 or from the gas phase.50 The major difference between path a and b is that the amine attacks the carbon of the adsorbed isocyanide in path a, which is also the initial step in the mechanism for the reactions of O2 with isocyanides and amines (Schemes 4 and 5). In path b, it is the amine oxide that attacks the isocyanide, which leads to an isocyanate (OCN–R) that desorbs from the Au surface. This isocyanate then reacts with the amine (primary or secondary) to give the observed urea product.
If the amine oxide reactions (eqn (16) and (17)) were to proceed by path b, the reaction of an isocyanide with an amine oxide should give an isocyanate (R–NCO). In fact, nBuNC reacts with Me3N–O under the usual conditions to give a maximum yield (20%) of nBuNCO after 6 h (eqn (18)), but the nBuNCO is converted by reaction with adventitious water to give the urea (nBuNH)2CO which is formed in 92% yield after 24 h (eqn (19)). Consistent with the formation of an isocyanate
| | 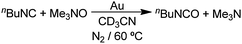 | (18) |
| | 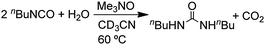 | (19) |
intermediate in
reactions (16) and (17) is the observation that the
nBuNCO reacts rapidly with
nBuNH
2 to give the urea product even in the absence of gold. Therefore, path
b is the most reasonable mechanism for reactions of
isocyanides,
amines (both secondary and primary), and amine oxides to form
ureas (
eqn (16) and (17)).
Since it is known that isocyanates also react with alcohols to form carbamates (RNCO + R′OH → (RHN)(R′O)CO), the gold-catalysed reaction of an isocyanide with an amine oxide and an alcohol should give a carbamate, according to the path b mechanism. Indeed, the reaction of nBuNC with Me3NO and nPrOH at 60 °C, gives a 92% yield of the carbamate product (eqn (20)).
| | 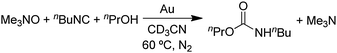 | (20) |
In order to understand why the amine oxide, rather than the
amine, attacks the adsorbed
isocyanide, it would be helpful to compare
amine oxide and
amine nucleophiles in their reactions with isocyanide
ligands in metal complexes. To my knowledge, there are no useful comparisons of these reactions with isocyanide
ligands; however, CO is pseudo-isoelectronic with C
N–R, and many of its complexes react with
amines (
eqn (21))
65 and amine oxides (
eqn (22)).
66,67 Both of these types of
| |  | (21) |
| |  | (22) |
reactions occur by nucleophilic attack of the
amine or
amine oxide on the CO
ligand. In general, the more positive the metal, the more likely both reactions will occur. On the other hand, a few complexes are known to react with the amine oxide but not the
amine. This is the situation with the M(CO)
6 (M = Cr, Mo, W) complexes, which react with Me
3N–O according to
eqn (22)68 but do not react with
amines according to
eqn (21). Likewise, the
cyclopentadienyl complex CpMn(CO)
3 reacts
66 with Me
3N–O but does not react with
amines.
69 Thus, it is not surprising that an
isocyanide adsorbed on a gold surface is attacked preferentially by an amine oxide (path
b) in the presence of an
amine, which leads to an
isocyanate intermediate enroute to the final urea product.
5. Aerobic oxidative reactions of CO with amines
Since carbon monoxide (CO) is isoelectronic with isocyanides (CN–R) and isocyanides react with primary amines and O2 to give carbodiimides (eqn (13)), it seemed possible that CO could react with H2NR and O2 to give isocyanates (OCN–R) according to eqn (23). Presumably the reaction would proceed by| | 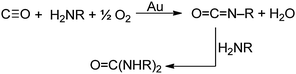 | (23) |
a mechanism similar to that proposed for isocyanides (Scheme 4). Such a mechanism would involve initial adsorption of CO on the gold surface, which would activate the CO to undergo attack by the amine as in Scheme 7. Although CO does not adsorb detectably to bulk gold at 1 atm and room temperature or above, it does adsorb at low temperatures (<150 K) and exhibits a ν(CO) value of ∼2100 cm–1. The significance of this value is that CO ligands in transition metal complexes with ν(CO) values higher than ∼2000 cm–1 react with amines to give carbamoyl complexes (eqn (21)).65,69 Therefore, we expected that CO adsorbed on Au would react with amines and give isocyanates according to eqn (23), although ureas would be the final product because the isocyanates would react further with the primary amine.
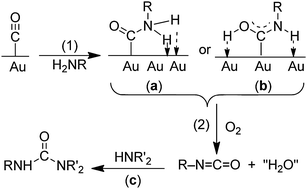 |
| | Scheme 7 Mechanisms for the gold-catalyzed reactions (eqn (23)) of CO with primary amines (H2NR) and O2.70 Reprinted with permission from the American Chemical Society. | |
When a MeCN solution of a primary amine was stirred for 24 h under an approximately 2.5:1 atmosphere of CO:O2 (∼1 atm total pressure) at 45 °C,70 ureas were formed (eqn (23), Table 4) in yields that decreased (nBuNH2 > sBuNH2 > tBuNH2) as the bulkiness of the amine increased. The yields also decreased as the basicity/nucleophilicity of the amine decreased (nBuNH2 > p-MeC6H4NH2 > PhNH2). These trends are consistent with a step in the mechanism that involves nucleophilic attack of the amine on an adsorbed CO. Other results that support the intermediacy of an isocyanate and the mechanism in Scheme 7 are the following: (1) Secondary amines (nPr2NH) do not react with CO/O2, which is expected because isocyanates (OCN–R) could not be formed without the loss of an –R group. (2) In a few reactions (entries 1, 4, 5, 6, 7 in Table 4), mostly those involving the weakly nucleophilic anilines, small amounts of the isocyanates were actually detected in the reaction solutions. (3) In the reaction of equimolar PhNH2 and nPr2NH with CO/O2 (entry 6), the mixed OC(NHPh)(NnPr2) was the only urea product. This result can easily be understood by realizing that only PhNH2, and not nPr2NH, could form an isocyanate (PhNCO), which would subsequently react with either PhNH2 or nPr2NH to give the urea product. As nPr2NH is more nucleophilic and reacts much faster than PhNH2 with PhNCO, the mixed urea OC(NHPh)(NnPr2) is the only observed product as expected in eqn (23) and Scheme 7. (4) The reaction of equimolar nBuNH2 and nPr2NH with CO/O2 (entry 7, Table 4) gave the mixed urea OC(NHnBu)(NnPr2) and OC(NHnBu)2 in a 39:5 ratio. That this ratio is nearly the same as that obtained from the reaction of nBuNCO with equimolar nBuNH2 and HNnPr2 also supports the intermediacy of the isocyanate.
| Entry |
Amine (mmol) |
Products |
| Urea (%) |
Isocyanate (%) |
|
At 45 °C in 5 mL MeCN solvent for 24 h with 1.00 g of Au powder; CO and O2 total pressure = ∼1 atm.
OC(NHPh)(NnPr2).
OC(NHnBu)(NnPr2).
OC(NHnBu)2.
|
| 1 |
n-BuNH2 (0.5) |
46 |
<1, trace |
| 2 |
s-BuNH2 (0.5) |
12 |
0 |
| 3 |
t-BuNH2 (0.5) |
2 |
0 |
| 4 |
PhNH2 (0.5) |
21 |
3 |
| 5 |
p-MeC6H4NH2 (0.5) |
24 |
3 |
| 6 |
PhNH2/HNPr2n (0.5/0.5) |
19b |
3, PhNCO |
| 7 |
n-BuNH2/HNPr2n (0.5/0.5) |
39c, 5d |
0, n-BuNCO |
In a related study, a nanogold catalyst consisting of gold particles (<10 nm) supported on an ion exchange styrene–divinylbenzene polymer (Merck Ion Exchange IV) catalysed the reaction of anilines with CO (40 atm) and O2 (∼10 atm) at 175 °C to give diarylureas.71 It is interesting that more strenuous temperature and pressure conditions were used than those in the gold powder-catalyzed reactions (Table 4) even though the catalyst consisted of nanoparticles. These results suggest that nanogold may not have an unusual catalytic activity, as compared with bulk gold, in reactions of the type in eqn (23).
6. Reactions of carbene precursors
6.1 Coupling of carbene precursors to give olefins
In the proposed mechanism of bulk gold-catalyzed reactions of isocyanides (CN–R) with secondary amines and O2 to form ureas (Scheme 5), a key step (3) is the reaction of a diaminocarbene intermediate with O2 to give the urea product. This possibility suggested that other carbene groups might react with O2 to give aldehyde or ketone products (eqn (24)). On the | | 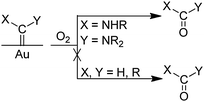 | (24) |
other hand, the reactivities of diaminocarbene ligands and their non-heteroatom analogs (:CR2) in transition metal complexes are very different. Diaminocarbene ligands are typically quite unreactive,72–74 whereas non-heteroatom carbenes are quite reactive.75–77 In binuclear metal complexes, where the carbene ligand can be either terminal or bridging (eqn (25)), the| |  | (25) |
diaminocarbene ligands form the terminal structure while the :CR2 carbenes favour the bridging structure.76–78 Therefore, it would not be surprising if diaminocarbene groups on metal surfaces were bonded to only one metal atom, while non-heteroatom carbenes bridge two or three metal atoms. Such differences in carbene bonding to metal surfaces would also lead one to expect the reactivities of :C(X)(Y) carbene groups on a metal surface to depend on the nature of X and Y. Although surface science studies show that CH2 precursors (CH2N2 or CH2I2) react with metal surfaces (Pt(111), Ru(001), Pd(100), Rh(111), or Cu(110)) to generate CH2 on these metal surfaces, the nature of the attachment of the CH2 group to one or more metal atoms has not been established.79,80
Non-heteroatom carbene groups (:CR2) are often incorporated into metal complexes by reacting an unsaturated metal complex LxM with a diazoalkane (R2CNN) to give the LxMCR2 complex with the elimination of N2.77,81,82 Several studies show that diazomethane reacts with gold surfaces to form polymethylene films.83 These reactions presumably occur by initial decomposition of the diazoalkane to N2 and surface-bound carbene (:CR2) groups, which couple to give the polymer. Colloidal gold catalyzes reactions of diazomethane and diazoethane to give polymethylene and polyethylidene, respectively, and small amounts of the olefins ethylene and 2-butene.84,85 Gold-coated silicon substrates, when reacted at 0 °C with solutions containing both diazomethane and ethyldiazoacetate (EDA, (EtO2C)HCN2), produce copolymer films that incorporate both the CH2 and (EtO2C)HC units. However, no polymeric film is produced from solutions of EDA alone.86
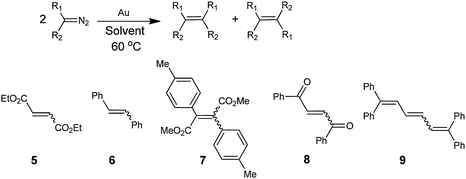
In our studies of solutions of EDA with gold powder at 60 °C, we find that the olefins diethyl maleate (cis-5) and diethyl fumarate (trans-5) are produced in a 52:48 ratio in a quantitative yield within 2 h (eqn (26)).27 The reaction proceeds in the same
| | 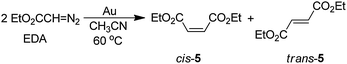 | (26) |
manner whether it is conducted in air or N
2, and there is no evidence for an
aldehyde product (EtO
2C)HC
O that might have resulted from the reaction of a surface (EtO
2C)HC:
carbene with O
2 (
eqn (24)). When the reaction was performed in the presence of 1 equiv. of
nBuNC,
pyridine, DABCO, or Ph
3P, no reaction occurred, presumably because these agents adsorbed on active gold sites.
The coupling of several other diazoalkanes to form the corresponding cis/trans mixtures of olefins was catalysed by gold powder under the same conditions (Table 5). A qualitative comparison of the rates of diazoalkane coupling shows that they decrease in the following order: PhHCN2 > EDA > (PhCO)HCN2 ∼ Me–MPDA ≫ (MeO2C)2CN2.
Table 5 Self-coupling of carbene precursors catalyzed by gold powdera
| Entry |
Carbene precursor |
Solvent |
Time (h) |
Yieldb (%) |
Product |
|
0.2 mmol of the diazo compound, 1.0 g of gold powder, 5 mL of solvent, 60 °C.
Yields were determined by 1H NMR with Ph3CH as the internal standard.
Low yield due to the formation of azine and some uncharacterized byproducts.27
|
| 1 |
EtO2CCHN2 (EDA) |
CH3CN |
2 |
100 |
5
|
| 2 |
PhCHN2 |
Toluene |
0.5 |
92 |
6
|
| 3 |
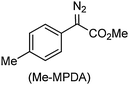
|
CH3CN |
24 |
76c |
7
|
| 4 |
PhCOCHN2 |
ClCH2CH2Cl |
24 |
86 |
8
|
| 5 |

|
CH3CN |
24 |
N.R. |
|
| 6 |
|
CH3CN |
4 |
82 |
9
|
Besides using diazoalkanes as precursors for the preparation of transition metal carbene complexes, 3,3-diphenylcyclopropene (DPCP) has been used as a reagent for the introduction of vinylcarbene ligands into transition metal complexes (eqn (27)).87 When DPCP is stirred with gold powder in MeCN
| | 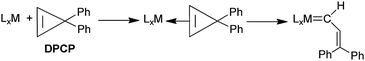 | (27) |
solvent at 60 °C for 4 h, an 82% yield of a 40/60
cis/
trans mixture of trienes is produced (
eqn (28)). This result suggests
| | 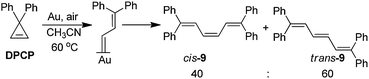 | (28) |
that DPCP is capable of rearranging to a vinylcarbene group on the gold surface as it does in the formation of metal vinylcarbene complexes (
eqn (27)).
When two carbene precursors (diazoalkanes and/or DPCP) were stirred with gold powder at 60 °C for 24 h, cross-coupled olefin products (eqn (29)) were obtained together with self-coupled
| | 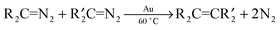 | (29) |
products
in the following precursor reactions: PhHC
N
2/DPCP, DPCP/EDA, DPCP/(PhCO)HC
N
2, EDA/(PhCO)HC
N
2, EDA/Me-MPDA, (PhCO)HC
N
2/Me-MPDA.
27 Only the following pairs of
carbene precursors did not give cross-coupled
olefin products: PhHC
N
2/EDA, PhHC
N
2/(PhCO)HC
N
2, DPCP/Me-MPDA. These observations can be reasonably understood by considering the relative rates of self-coupling (
Table 5) of each
carbene precursor which decrease in the following order: PhHC
N
2 ≫ DPCP > EDA > (PhCO)HC
N
2 > Me-MPDA. In reactions where one of the reactants,
e.g. PhHC
N
2, undergoes self-coupling rapidly and the other,
e.g. (PhCO)HC
N
2, is much slower, the rapidly-reacting PhHC
N
2 is completely self-coupled before any of the (PhCO)HC
N
2, which subsequently also undergoes self-coupling. In reactions where the self-coupling rates are similar,
e.g., EDA/(PhCO)HC
N
2, much of the product is the cross-coupled
olefin. The relative amounts of the cross-coupled products for all of the reactions can be broadly understood in terms of their self-coupling rates.
The overall trend in self-coupling rates (see above) is very similar to that observed in rates of transition metal complex-catalyzed self-coupling reactions of diazoalkanes.88,89 The generally accepted mechanism for these reactions involves initial formation of a metal carbene complex (MCR2) which is attacked at the carbon by another diazoalkane; this is followed by loss of N2 and formation of the olefin.75 In the gold powder-catalyzed reactions (eqn (26) and (28), Table 5), the diazoalkanes presumably form a carbene on the gold surface initially, although an attempt to identify such a species spectroscopically on the surface of a gold film that had been immersed in an Et2O solution of (Me3Si)HCN2 was unsuccessful,90 perhaps because of rapid coupling to give the (Me3Si)HCCH(SiMe3) olefin. The mechanism of the carbene coupling reaction on gold powder could involve initial formation of an adsorbed highly reactive carbene which undergoes attack by a diazoalkane in the solution phase, as proposed for the transition metal complex-catalyzed reactions. On the other hand, a carbene on the gold surface may bridge two (or more) gold atoms (eqn (25)) in which case the carbene coupling step may proceed by a mechanism (Scheme 8) that involves insertion of terminal carbene group into a gold–carbon bond of the bridging carbene group. Such a mechanism has been proposed for the reaction of dinuclear Ru and Os complexes with bridging CH2 carbene groups that react with diazoalkanes to give olefins.91,92
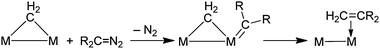 |
| | Scheme 8 Possible mechanism for carbene coupling. | |
6.2 Cyclopropanation of olefins with carbene precursors
Because gold powder catalyzes the self-coupling of carbene precursors to give olefins (eqn (26) and (28), Table 5), all other gold-catalyzed reactions of diazoalkanes and DPCP must compete with these self-coupling reactions. One of these reactions is gold-catalyzed cyclopropanation of olefins with carbene precursors (Table 6).27 In order for the cyclopropanation to be competitive with self-coupling, a 100 fold excess of the olefin was used; smaller excesses gave yields lower than those given in Table 6. Also, the diazoalkanes or DPCP were added dropwise over a period of 6 h to solutions of styrene containing gold powder, which produced an even higher styrene/carbene precursor ratio in the reaction solutions. The reaction of EDA with styrene at 80 °C in the presence of 1.0 g of gold for 24 h gave a 45% yield of the corresponding cyclopropane in a 25/75 cis/trans ratio (entry 1). Even without the gold catalyst, this reaction gave a small yield (10%) of the cyclopropane under the same conditions. The gold-catalyzed reaction of EDA with 4-methylstyrene gave a higher yield (54%) of the cyclopropane (entry 2) than with styrene (entry 1). Under the same conditions, 1-hexene, cyclohexene, and n-butyl vinyl ether did not react with EDA; only the self-coupled olefins were observed. The reaction of (PhCO)HCN2 with styrene gave none of the cyclopropane in ClCH2CH2Cl solvent. When styrene was used as the solvent, a 35% yield of the cyclopropane was produced (entry 3). The reaction of DPCP with styrene under the standard conditions (entry 4) gave a 19% yield of the monocyclopropane product, but the major product (39%) was a 32/68 cis/trans mixture of the triene (eqn (30)).| | 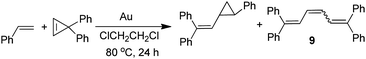 | (30) |
The mechanism of these gold-catalyzed cyclopropanation reactions presumably involves initial formation of a surface carbene species which reacts with the styrene. Given the limited number of carbene precursors and olefins that participate in these gold-catalyzed reactions, transition metal complexes are much more effective catalysts for olefin–diazoalkane cyclopropanation reactions.75,77,93,94
Table 6 Gold metal-catalyzed cyclopropanation of olefinsa
| Entry |
Alkene |
Carbene precursor |
Solvent |
Yield Ab (%) |
Yield Bb (%) |
|
0.1 mmol of the diazo compound, alkene/diazo = 100:1, 1.0 g of gold powder, 30 mL of solvent, 80 °C, 24 h under N2.
Yields were determined by 1H NMR with Ph3CH as the internal standard, based on the diazo compounds.27
|
| 1 |
Styrene |
EDA |
ClCH2CH2Cl |
45 |
10 |
| 2 |
4-Mestyrene |
EDA |
ClCH2CH2Cl |
54 |
5 |
| 3 |
Styrene |
PhCOCHN2 |
Styrene |
35 |
0 |
| 4 |
Styrene |
|
ClCH2CH2Cl |
19 |
39 |
6.3 Oxidative reactions of carbene precursors with amines and O2
Carbene ligands in metal complexes are known to be susceptible to attack by amines (eqn (31)).76,77 Such a reaction is proposed to| | 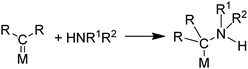 | (31) |
be a key step in the reaction of diazoalkanes with amines to give N–H insertion products (eqn (32)) when catalyzed by a variety of homogeneous transition metal complexes.75,95–97 Among these| | 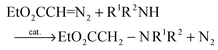 | (32) |
catalysts is an N-heterocyclic carbene complex of Au(I).98,99 Also relevant to the present discussion is the copper metal-catalyzed N–H insertion reaction (eqn (33)) of α-diazoacetophenone and aniline.100| |  | (33) |
If gold were to catalyze any reaction of diazoalkanes and amines, one would expect it to give N–H insertion products based on the reactions in eqn (32) and (33). Instead, it gives the unexpected enamine product, as illustrated by the reaction of EDA (eqn (34)).28 Studies of the reaction of 0.60 mmol| | 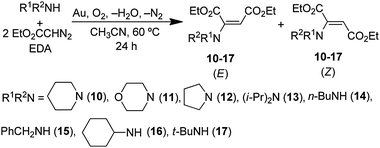 | (34) |
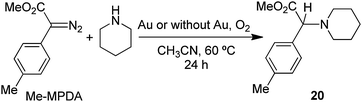
piperidine and 0.20 mmol EDA in 5 mL of CH3CN at 60 °C using 1.00 g of gold powder under 1.0 atm of O2 gave exclusively enamine (E)-10 in 94% yield after 24 h at 60 °C (eqn (35)).| | 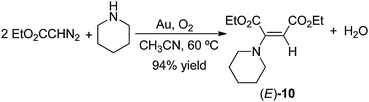 | (35) |
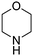
Products resulting from N–H insertion or dimerization of EDA were not detected. Oxygen is clearly a reactant because the reaction of EDA with piperidine under an argon atmosphere gives only 6% (E)-10, presumably due to the presence of adventitious O2; no other products were observed. When reaction (35) was conducted in toluene solvent, the (E)-10 yield was 82%, but it was accompanied by an approximately equal amount of the amidine (4b, Scheme 2) resulting from oxidative coupling of the piperidine.
The rate of reaction (35) was faster at higher piperidine concentrations, and yields of the enamine product (eqn (34)) depended on the nature of the amine used (Table 7). The cyclic secondary amines, piperidine (entry 1), morpholine (entry 2), and pyrrolidine (entry 3), and primary amines, n-BuNH2 (entry 5), cyclohexylamine (entry 7), and t-BuNH2 (entry 8) produced enamines in 78–94% yield. The enamine products from pyrrolidine (entry 3), diisopropylamine (entry 4), and benzylamine (entry 6) were accompanied by the competing formation of imines resulting from the gold catalyzed oxidative-dehydrogenation of these amines (eqn (1), Scheme 2). No reaction occurred between aniline or p-Me2NC6H4NH2 and EDA, even in the presence of added DABCO or Et3N base.
Table 7 Gold powder-catalyzed reactions of EDA with amines and O2 according to eqn (34)a
| Entry |
Amine |
Product |
Yieldb (%) |
E/Zc |
0.2 mmol of EDA and 0.6 mmol of amine in 5 mL of CH3CN with 1.00 g of Au powder at 60 °C under O2 (∼1 atm) for 24 h.
Yields were determined by 1H NMR with Ph3CH as the internal standard.
(E,Z) isomers were determined by 1H NMR, NOE and comparison of the data with authentic samples. (E/Z) ratio is the ratio of the olefinic protons of the (E)- and (Z)-isomers in 1H NMR spectra.
Imine 4b : (E)-12 = 0.1:1.
Imine (CH3)2CNCH(CH3)2: (E)-13 = 0.2:1.
Imine PhCHNCH2Ph: (Z)-15 = 0.25:1. : (E)-12 = 0.1:1.
Imine (CH3)2CNCH(CH3)2: (E)-13 = 0.2:1.
Imine PhCHNCH2Ph: (Z)-15 = 0.25:1.
|
| 1 |
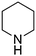
|
10
|
94 |
E
|
| 2 |
|
11
|
82 |
E
|
| 3 |

|
12
|
79 |
E
|
| 4 |
i
Pr2NH |
13
|
58 |
5:1e |
| 5 |
n
BuNH2 |
14
|
89 |
Z |
| 6 |
PhCH2NH2 |
15
|
78 |
1:3f |
| 7 |

|
16
|
86 |
1:12 |
| 8 |
t
BuNH2 |
17
|
85 |
1:8 |
Of particular interest are the relative amounts of the E and Z isomers produced from the different amines. Secondary amines give primarily E-isomers (entries 1–4), while primary amines favor Z-isomers (entries 5–8). The cyclic secondary amines produced exclusively E-isomers (entries 1–3), while primary amine n-BuNH2 yielded only the Z-isomer. In contrast, diisopropyl amine (entry 4, E/Z = 5:1), benzylamine (entry 6, E/Z = 1:3), and t-BuNH2 (entry 8, E/Z = 1:8) were less stereoselective.
As noted above, reactions of α-diazoacetophenone (PhCO)HCN2 with amines have been reported to yield N–H insertion products using Cu metal powder as the catalyst (eqn (33)).100 However, no N–H insertion occurred under conditions used in our gold metal catalyzed reactions. The reaction of (PhCO)HCN2 with piperidine provided only enamine (E)-18 in 90% yield after heating 29 h in CH3CN at 60 °C (eqn (36)); with the primary amine n-BuNH2, only the enamine (Z)-19 was obtained in 85% yield under the same conditions (eqn (37)). As in the reactions of EDA, the E isomer was obtained with the secondary amine, and the Z-isomer was the only product with the primary amine. Heating a mixture of (PhCO)HCN2 and aniline under the same conditions gave no N–H insertion product or enamine; however, it gave a 30% yield of the olefin (cis/trans = 1:3) (Table 5) resulting from the gold catalyzed dimerization of (PhCO)HCN2 and an oxazole product (35% yield) resulting from the reaction of the carbene intermediate with the MeCN solvent as reported previously.27
| | 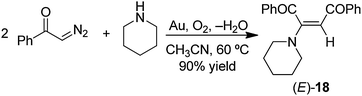 | (36) |
| | 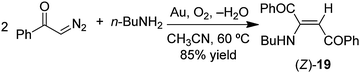 | (37) |
Instead of giving the
enamine product, phenyl diazomethane PhHC
N
2 reacts with
piperidine to give a 70% yield of
stilbene PhCH
CHPh (
cis/
trans 1
:
1) under the standard conditions; no
enamine or N–H insertion products were observed. The reaction of Me-MPDA with
piperidine provided a 50% yield of the N–H insertion product
20 and some other uncharacterized byproducts (
eqn (38)). But this reaction also gave a 66% yield of
20 even in the absence of the gold
catalyst under the same conditions.
| |  | (38) |
Although the formation of
enamines from gold powder-catalyzed reactions of
carbene precursors with
amines and O
2 was unexpected, one might consider possible mechanisms for this process (
eqn (34)–(37). Taking into account the known reactivities of diazoalkanes in the other gold powder-catalyzed reactions (Sections 6.1 and 6.2) and the reactivities of
carbene ligands in transition metal complexes, three mechanisms come to mind (
Scheme 9) for the reaction of EDA,
piperidine and O
2.
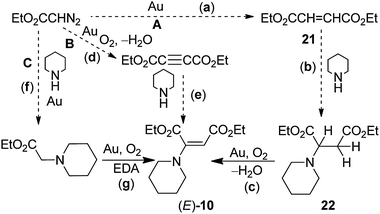 |
| | Scheme 9 Plausible mechanisms for gold powder-catalyzed reaction of EDA with piperidine and O2 (eqn (35)).28 Reprinted with permission from Springer Science and Business Media. | |
In pathway A, the first step (a) involves the coupling of carbene groups derived from EDA to give olefin 21, a reaction that is well-established in our previous work (eqn (26), Table 5).27 The second step (b) is a Michael addition of piperidine to 21 to give 22; we have shown that this reaction occurs in 100% yield in the absence of the gold catalyst under the conditions of reaction (35). The final step (c) in pathway A is the oxidative-dehydrogenation of 17 to give (E)-10. Unfortunately, this conversion did not occur under the conditions of the reaction, which suggests that A is not a likely pathway for reaction (35).
In pathway B, the oxidative-dehydrogenation of EDA in step (d) is followed by amine addition (e) to DEAD to give the product. Although the latter reaction (e) is well-known,101 step (d) does not occur as shown in the gold-catalyzed reaction of EDA in the absence of amine; this reaction gives the carbene coupled products (eqn (26)).27 Thus, pathway B is also unlikely. In pathway C, the first step (f) involves N–H insertion, which is plausible because of the known insertion reaction (eqn (38)) of Me-MPDA with piperidine. However, the conversion of this product (step g) to (E)-10 does not occur under the conditions of the reaction. Thus, none of the pathways, A, B, or C, is consistent with the experimental results.
Although it does not involve stable intermediates that can be studied independently, another possible mechanism is shown in Scheme 10. In the first step of this mechanism, a surface carbene is formed from EDA, as proposed in other gold-catalyzed reactions of EDA (Sections 6.1 and 6.2). As carbene ligands in transition metal complexes are susceptible to nucleophilic attack,76,77 we propose that this carbene 23 undergoes attack by piperidine to give 24, which couples to a surface carbene group to give 25. Oxidative-dehydrogenation of 25 in step (c) to give imine 26 is similar to the gold-catalyzed oxidative-dehydrogenation of amines to give imines (eqn (1).34 Product 10 is released from the surface in step (d). A key step in this mechanism is (a), nucleophilic attack of the amine on the surface carbene. If the amine is not sufficiently nucleophilic, step (a) is slower than carbene group coupling to form the EtO2CCHCHCO2Et products (eqn (26)). Thus, there is no reaction of EDA with the weakly nucleophilic anilines, PhNH2 and p-Me2NC6H4NH2. On the other hand, the more nucleophilic aliphatic amines (Table 7) give enamine products, presumably because their addition to the surface carbene is faster than the coupling of two carbene groups. The tendency of carbene groups to be attacked appears to depend on the presence of an electron-withdrawing group such as –CO2Et or –C(O)Ph as both EDA and (PhCO)HCN2 give enamine products with piperidine (eqn (34) and (36)). On the other hand, the reaction of PhHCN2, lacking an electron-withdrawing group, with piperidine gives none of the enamine but only stilbene (PhHCCHPh) resulting from coupling of carbene groups.
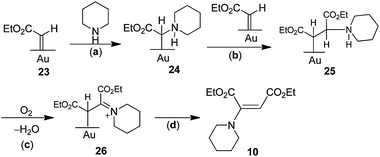 |
| | Scheme 10 Proposed mechanism for gold powder catalyzed reaction of EDA with piperidine and O2 (eqn (35)). | |
7. Conclusions
The investigations described in this review show that gold metal catalysts need not be nanosized in order to be active in a variety of reactions. Many of the reactions that have been discovered so far involve O2, although amine oxides (R3N–O) are capable of replacing O2 in some reported reactions. Thus, bulk gold is capable of providing pathways for delivering oxygen atoms from either O2 or R3N–O to amines and other substrates. Many of the reported bulk gold-catalyzed reactions are based on patterns of reactivity that were derived from known transformations of organic molecules coordinated in organometallic complexes, which suggests that other reactions of organic ligands in metal complexes can guide the discovery of new heterogeneous catalytic reactions.
Unlike nanogold catalysts, which tend to agglomerate into larger and less active forms under reaction conditions,102 bulk gold catalysts have the advantage that they are much less likely to suffer this type of deactivation. Although gold is an expensive metal, its practical applications will also depend on the catalyst lifetime and the value of the transformation that it catalyzes. It is therefore important to continue the exploration of reactions that are catalysed by bulk gold, especially when supported on materials with high surface areas.
Acknowledgements
I am grateful to Prof. L. Keith Woo, my collaborator on many of these investigations, and to the students and postdocs involved in the projects. Most of our work was supported by the U.S. Department of Energy through the Ames Laboratory under contract No. DE-AC02-07CH11358 with Iowa State University.
Notes and references
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS.
-
Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, John Wiley, 2012 Search PubMed.
- H. Huang, Y. Zhou and H. Liu, Beilstein J. Org. Chem., 2011, 7, 897 CrossRef CAS.
- A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS.
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS.
- R. Kang, H. Chen, S. Shaik and J. Yao, J. Chem. Theory Comput., 2011, 7, 4002 CrossRef CAS and references therein.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS.
-
S. A. C. Carabineiro and D. T. Thompson, in Gold: Science and Applications, ed. C. Corti and R. Holliday, CRC Press, Boca Raton, 2010, ch. 6, pp. 89–122 Search PubMed.
- B. Biannic and A. Aponick, Eur. J. Org. Chem., 2011, 6605 CrossRef CAS.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS.
- C. D. Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350 RSC.
- C. D. Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
-
G. Bond, C. Louis and D. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed.
- D. T. Thompson, Top. Catal., 2006, 38, 231 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- Z. Ma and S. Dai, Nano Res., 2011, 4, 3 CrossRef CAS and references therein.
- J. S. McPherson and D. T. Thompson, Top. Catal., 2009, 52, 743 CrossRef CAS.
- C. W. Corti, R. J. Holliday and D. T. Thompson, Top. Catal., 2007, 44, 331 CrossRef CAS.
- C. W. Corti, R. J. Holliday and D. T. Thompson, Appl. Catal., A, 2005, 291, 253 CrossRef CAS.
- A. Tost, D. Widmann and R. J. Behm, J. Catal., 2009, 266, 299 CrossRef CAS.
- J. C. Fierro-Gonalez and B. C. Gates, Chem. Soc. Rev., 2008, 37, 2127 RSC.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767 CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbaek, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Nørskov, Top. Catal., 2007, 44, 15 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Catal. Today, 2006, 111, 22 CrossRef CAS.
- R. J. Angelici, J. Organomet. Chem., 2008, 693, 847 CrossRef CAS.
- B. Block, Inorg. Synth., 1953, 4, 14 CrossRef CAS.
- Y. Zhou, B. G. Trewyn, R. J. Angelici and L. K. Woo, J. Am. Chem. Soc., 2009, 131, 11734 CrossRef CAS.
- Y. Zhou, R. J. Angelici and L. K. Woo, Catal. Lett., 2010, 137, 8 CrossRef CAS.
- E. R. Klobukowski, R. J. Angelici and L. K. Woo, Catal. Lett., 2012, 142, 161 CrossRef CAS.
- M. Robertson and R. J. Angelici, Langmuir, 1994, 10, 1488 CrossRef CAS.
- B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1 CrossRef CAS.
- A. C. Gluhoi, X. Tang, P. Marginean and B. E. Nieuwenhuys, Top. Catal., 2006, 39, 101 CrossRef CAS.
- Y.-F. Han, Z. Zhong, K. Ramesh, F. Chen and L. Chen, J. Phys. Chem. C, 2007, 111, 3163 CAS.
- B. Zhu and R. J. Angelici, Chem. Commun., 2007, 2157 RSC.
- B. Xu, C. M. Friend and R. J. Madix, Faraday Discuss., 2011, 152, 241 RSC.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74 CrossRef CAS.
- M. Turner, O. P. H. Vaughan and R. M. Lambert, Chem. Commun., 2008, 2316 RSC.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138 CrossRef CAS.
- H. Miyamura, M. Morita, T. Inasaki and S. Kobayashi, Bull. Chem. Soc. Jpn., 2011, 84, 588 CrossRef CAS.
- E. R. Klobukowski, M. L. Mueller, R. J. Angelici and L. K. Woo, ACS Catal., 2011, 1, 703 CrossRef CAS.
- W. Chavasiri and H. Sakurai, SYNLETT, 2011, 1121 Search PubMed.
- L. Aschwanden, T. Mallat, M. Maciejewski, F. Krumeich and A. Baiker, ChemCatChem, 2010, 2, 666 CrossRef CAS.
- L. Aschwanden, T. Mallat, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 309, 57 CrossRef CAS.
- Y. Perez, C. April, A. Corma and H. Garcia, Catal. Lett., 2010, 134, 204 CrossRef CAS.
- M.-H. So, Y. Liu, C.-M. Ho and C.-M. Che, Chem.–Asian J., 2009, 4, 1551 CrossRef CAS.
- B. Xu, L. Zhou, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2010, 49, 394 CrossRef CAS.
- H. Guo, A. Al-Hunaiti, M. Kemell, S. Rautiainen, M. Leskelä and T. Repo, ChemCatChem, 2011, 3, 1872 CrossRef CAS.
- M. Lazar and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 10613 CrossRef CAS.
-
B. Zhu and R. J. Angelici, unpublished results.
-
M. Lazar and R. J. Angelici, in Modern Surface Organometallic Chemistry, ed. J.-M. Basset, R. Psaro, D. Roberto and R. Ugo, Wiley-VCH, Weinheim, 2009, pp. 513–556 Search PubMed.
- A. C. Ontko and R. J. Angelici, Langmuir, 1998, 14, 3071 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- R. A. Michelin, A. J. L. Pombeiro and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2001, 218, 75 CrossRef CAS.
- J. Chatt, R. L. Richards and G. H. D. Royston, J. Chem. Soc., Dalton Trans., 1973, 1433 RSC.
-
B. Crociani, in Reactions of Coordinated Ligands, ed. P. Braterman, Plenum Press, New York, 1986, vol. 1, pp. 553–638 Search PubMed.
- A. F. Benton and J. C. Elgin, J. Am. Chem. Soc., 1927, 49, 2426 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanable, C. J. Kiely and G. J. Hutchings, J. Mater. Chem., 2005, 15, 4595 RSC.
- M. Lazar, B. Zhu and R. J. Angelici, J. Phys. Chem. C, 2007, 111, 4074 CAS.
- K.-C. Shih and R. J. Angelici, Langmuir, 1995, 11, 2539 CrossRef CAS.
- M.-T. Lee and C.-H. Lu, Organometallics, 2004, 23, 976 CrossRef CAS.
- R. A. Diggle, S. A. Macgregor and M. K. Whittlesey, Organometallics, 2004, 23, 1857 CrossRef CAS.
- G. Frenking, M. Solà and S. F. Vyboishchikov, J. Organomet. Chem., 2005, 690, 6178 CrossRef CAS.
- E. R. Klobukowski, R. J. Angelici and L. K. Woo, Organometallics, 2012, 31, 2785 CrossRef CAS.
- R. J. Angelici, Acc. Chem. Res., 1972, 5, 335 CrossRef CAS.
- M. O. Albers and N. J. Coville, Coord. Chem. Rev., 1984, 53, 227 CrossRef CAS.
- T. Y. Luh, Coord. Chem. Rev., 1984, 60, 255 CrossRef CAS.
- J. Shen, Y. Gao, Q. Shi and F. Basolo, J. Organomet. Chem., 1991, 401, 295 CrossRef.
- R. J. Angelici and L. J. Blacik, Inorg. Chem., 1972, 11, 1754 CrossRef CAS.
- B. Zhu and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 14460 CrossRef CAS.
- F. Shi and Y. Deng, J. Catal., 2002, 211, 548 CAS.
-
N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin/Heidelberg, 2007, vol. 21 Search PubMed.
-
Organometallic Oxidation Catalysis, T. Strassner, Springer, Berlin/Heidelberg, 2007, vol. 22, p. 125 Search PubMed.
-
N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, New York, 2007 Search PubMed.
-
Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, ed. M. P. Doyle, M. A. McKervey and T. Ye, Wiley-VCH, New York, 1998 and references therein Search PubMed.
- F. J. Brown, Prog. Inorg. Chem., 1980, 27, 1 CrossRef CAS.
-
K. H. Dötz, in Reactions of Coordinated Ligands, ed. P. Braterman, Plenum Press, New York, 1986, vol. 1, pp. 285–370 Search PubMed.
- D. I. Bezuidenhout, S. Lotz, D. C. Liles and B. van der Westhuizen, Coord. Chem. Rev., 2012, 256, 479 CrossRef CAS.
- B. E. Bent, Chem. Rev., 1996, 96, 1361 CrossRef CAS.
- F. Solymosi, Catal. Today, 1996, 28, 193 CrossRef CAS.
- M. Gandelman, B. Rybitchinski, N. Ashkenazi, R. M. Gauvin and D. Milstein, J. Am. Chem. Soc., 2001, 123, 5372 CrossRef CAS.
- J.-P. Djukic, D. A. Smith, V. G. Young and L. K. Woo, Organometallics, 1994, 13, 3020 CrossRef CAS.
- E. Ihara, Adv. Polym. Sci., 2010, 231, 191 CrossRef CAS.
- A. G. Nasini, L. Trossarelli and G. Saini, Makromol. Chem., 1961, 44–46, 530 Search PubMed.
- A. G. Nasini, G. Saini and L. Trossarelli, Pure Appl. Chem., 1962, 4, 255 CrossRef CAS and references therein.
- D. Bai and G. K. Jennings, J. Am. Chem. Soc., 2005, 127, 3048 CrossRef CAS.
- For a review of cyclopropenes, see M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS.
-
M. Regitz and G. Maas, Diazo Compounds: Properties and Synthesis, Academic Press, London, 1996 Search PubMed.
- T. Uchida and T. Katsuki, Synthesis, 2006, 1715 CAS.
- G. S. Tulevski, M. B. Myers, M. S. Hybertsen, M. L. Steigerwald and C. Nuckolls, Science, 2005, 309, 591 CrossRef CAS.
- R. G. Samant, T. W. Graham, B. D. Rowsell, R. McDonald and M. Cowie, Organometallics, 2008, 27, 3070 CrossRef CAS.
- M. Cowie, Can. J. Chem., 2005, 83, 1043 CrossRef CAS.
- D. Intrieri, A. Caselli and E. Gallo, Eur. J. Inorg. Chem., 2011, 5071 CrossRef CAS.
- C.-M. Che and J.-S. Huang, Coord. Chem. Rev., 2002, 231, 151 CrossRef CAS.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS.
- L. K. Baumann, H. M. Mbuvi, G. Du and L. K. Woo, Organometallics, 2007, 26, 3995 CrossRef CAS.
- I. Aviv and Z. Gross, Chem.–Eur. J., 2008, 14, 3995 CrossRef CAS.
- M. R. Fructos, T. R. Belderrain, P. de Fréémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284 CrossRef CAS.
- P. de Frémont, E. D. Stevens, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045 RSC.
- P. Yates, J. Am. Chem. Soc., 1952, 74, 5376 CrossRef CAS.
- D. Zewge, C. Chen, C. Deer, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2007, 72, 4276 CrossRef CAS.
- D. E. Starr, S. K. Shaikhutdinov and H.-J. Freund, Top. Catal., 2005, 36, 33 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
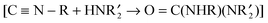 , (4) the reactions of CO with primary amines to give ureas [CO + H2N–R → O
, (4) the reactions of CO with primary amines to give ureas [CO + H2N–R → O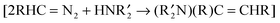 . In some of these reactions, amine oxides (e.g., Me3N–O) may be used in place of O2. Mechanisms of these and related reactions are examined. These studies show that bulk, non-nanogold is capable of catalysing a variety of different reactions.
. In some of these reactions, amine oxides (e.g., Me3N–O) may be used in place of O2. Mechanisms of these and related reactions are examined. These studies show that bulk, non-nanogold is capable of catalysing a variety of different reactions.
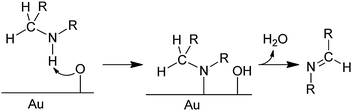
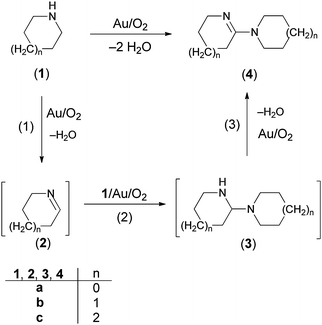
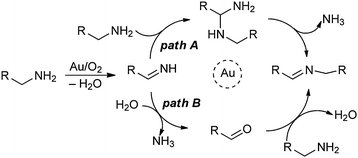

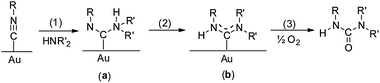
 and O2.59
and O2.59