Glucose oxidation using Au-containing bimetallic and trimetallic nanoparticles
Haijun
Zhang
a and
Naoki
Toshima
*bc
aCollege of Materials & Metallurgy, Wuhan University of Science and Technology, Wuhan, Hubei Province 430081, China
bDepartment of Applied Chemistry, Tokyo University of Science Yamaguchi, SanyoOnoda-shi, Yamaguchi 756-0884, Japan. E-mail: toshima@rs.tus.ac.jp
cCREST, Japan Science and Technology Agency, Kawaguchi, Saitama 332-0012, Japan
First published on 6th July 2012
Abstract
Clean syntheses, based on the use of natural renewable reagents, in water solution under mild conditions, are highly desirable processes and often the catalytic step is a key factor for the successful application. Gold (Au) and Au-containing bimetallic and trimetallic nanoparticles (BNPs and TNPs) have been extensively investigated as the promising catalysts for the clean syntheses of gluconic acid, which is an important intermediate in the field of food industry and pharmaceutical applications, by glucose oxidation using atmospheric oxygen or hydrogen peroxide as an oxidant. With significant research efforts, a lot of new efficient catalytic systems, based on the peculiar properties of nanometric Au NPs, have been developed for aerobic glucose oxidation. In this minireview, we provide an overview of the recent development of Au-containing BNPs and TNPs for the improved catalytic performance toward glucose oxidation.
1. Introduction
The interest in the application of carbohydrates as chemical feedstock has considerably increased during the last decade.1–3 The renewable character of carbohydrates, as well as biodegradability of the materials derived from carbohydrates, have played a role in this respect. Gluconic acid is the main product resulting from the oxidation of glucose. The current annual worldwide production capacity for gluconic acid and its derivatives is estimated to be about 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t.4 Gluconic acid is useful as a food additive, an ingredient for concrete, and raw materials for medicines and biodegradable polymers.5,6
000 t.4 Gluconic acid is useful as a food additive, an ingredient for concrete, and raw materials for medicines and biodegradable polymers.5,6
Currently gluconic acid is industrially produced by fermentation, i.e., enzymatic oxidation of D-glucose. Besides this route, recent developments have shown the high potentiality of chemical, electrochemical and catalytic procedures for oxidizing glucose with oxygen or air. Chemical methods have the disadvantages of limited selectivity towards gluconic acid, environmental toxicity and biological hazards; whereas the rapid increase in the cost of electric power in recent years has made the electrolytic process noncompetitive.4,7 The fermentation process, however, has a few drawbacks in practical production,8 such as the difficulties in moulds and free enzymes separation, waste-water removal, low space–time yield, slow reaction rate,9 and dead microbes and accumulated excretory substances of microbes that should be disposed of.10
Owing to the problems of the chemical, electrochemical and fermentation methods for glucose oxidation, interest exists in alternative environmentally friendly technologies based on the use of atmospheric oxygen in water solution under mild conditions and with the aid of heterogeneous catalysis.11,12 The introduction of catalytic systems using oxygen from air is preferred for ‘green’ processing.13–15 The ideal “green” processes are carried out at atmospheric pressure and room temperature, in aqueous media or under solvent-free conditions, and using air as oxidant or molecular hydrogen as reductant. In recent years catalysis of glucose oxidation by metallic nanoparticles (NPs) has started to play a major role with respect to green chemistry.16,17 Many efforts have been made for developing new catalysts for glucose oxidation.18–21 Metal NPs, which exhibit different properties from their bulk materials and their isolated atoms, are known to be effective for applications in catalysis, electrochemistry, optics, surface-enhanced Raman scattering, and ultrasensitive chemical sensors.22–33 From the viewpoint of catalyst design,34–37 it is of great importance to develop metal catalysts arranged on the nanometer scale in order to provide superior functions to metal NPs with uniform size and narrow size distribution.
The advantages of using metal NPs as catalysts for glucose oxidation are the following: (1) the size and shape of metal NPs are easily controlled by the preparation conditions; the NPs with more uniform particle sizes can be obtained in the dispersed systems, especially under conditions of a high metal concentration, because the dispersed metal NPs can be easily concentrated by evaporating the solvent without changing the structures. (2) Metal NPs dispersed in solution can be used as catalysts in solution like homogeneous catalysts. Thus the temperature applied to the catalyst is below the boiling point of the solvent. Homogeneous catalysts (organometallic complex) which can in general be operated under mild reaction conditions contribute sufficient contact of active sites with reactants, exhibiting excellent performance in a wide range of reactions. (3) Metal NPs capped by organic polymers can be functionalized by modifying the organic polymers, and the high stability can be easily obtained due to good protection of active sites by a protective agent. (4) It is possible to prepare bimetallic nanoparticles (BNPs) and trimetallic nanoparticles (TNPs) with various compositions and structures. The catalytic activity and selectivity of these metal BNPs and TNPs can be controlled by varying the composition and structure. (5) Metal NPs dispersed in solution or immobilized on solid supports are usually more active and selective as catalysts under mild reaction conditions than the conventional industrial catalysts due to the so-called ‘quantum size effect’ and the high ratio of atoms located on the surface of NPs.
BNPs and TNPs have been used in various technologically important areas for many years due to their unique catalytic, electronic, and magnetic properties, which differ from their monometallic counterparts.38,39 BNPs and TNPs composed of 3d-transition metals and noble metals with specific structures will provide a great number of new candidates as catalysts for various chemical reactions, since the catalytic properties of BNPs and TNPs can be potentially tailored by both the electronic effect (ligand effect) and the geometrical effect (ensemble effect).40–42 In the case of the electronic effect the added metal can electronically affect the electron density of the metal at the catalytic site, which improves or deactivates the catalytic activity and selectivity. In this case, the atoms of the added metal should be adjacent to those of the catalytic metal. The reaction substrates interact only with catalytic metal atoms without direct interaction with added metal atoms. In the case of the geometrical effect, on the other hand, the substrate should interact not only with the catalytic metal atoms but also with the added metal atoms. Thus, both kinds of metal atoms can directly interact with a substrate and affect the activity and selectivity of the reaction of substrates. In recent years, great progress has been made and many examples have been reported on the preparation and catalysis of BNPs and TNPs with the great efforts of researchers all over the world. In this review, we briefly survey the research progress in the catalytic activity of Au NPs and Au-containing BNPs and TNPs for aerobic glucose oxidation. In the last section, we will briefly discuss the recent progress, expected improvement and future outlook in this research area.
2. Catalytic activity of monometallic nanoparticles (MNPs) for glucose oxidation
The first catalytic oxidation process of carbohydrates was discovered by von Gorup-Besanez in 1861 using a platinum catalyst.43 In the 1960s, the platinum catalyzed carbohydrate oxidation was studied more systematically.44 However, the platinum catalysts used in this work were neither very active nor selective. Later publications described the promotion of heterogeneous platinum and palladium catalysts with metals like Bi or Pb,45,46 which enhanced the selectivity and activity in glucose oxidation, selectivity up to 97% for sodium gluconate can be achieved. Unfortunately, these Pt and Pd catalysts showed no sufficient long-term stability and leaching of the second metal has been described.47–49 Another major progressive step in the area of the heterogeneous catalysis was the report in 1983 by Taramasso et al. on the synthesis of TS-1, a titanium silicalite with MFI (Mobil fifth Zeolite, ZSM-5) structure, which has found several applications in oxidation reaction under mild conditions with 30% aqueous hydrogen peroxide as the oxidant.50,51Gold (Au) has long been considered as an inactive catalyst,52 despite early examples using Au catalysts in hydrogenation reactions.53–55 Ever since the supported Au nanocluster was discovered by Haruta,56,57 Au has drawn increasing attention in the field of catalysis.58–62 Recently, gold has received growing interest as a catalyst for the selective oxidation of organic molecules using oxygen under mild conditions.63–65 The extension of gold catalysis to the selective oxidation of glucose led to promising results, thus suggesting a new route to gluconates in competition with enzymatic catalysis.61,66,67 An important feature of gold for glucose oxidation is that it is active both in the form of supported metal and in the form of nanometric colloidal particles (sol), and another advantage of gold with respect to biological systems is the possibility to work in a wide range of pH, from alkaline to acidic conditions.68
Au colloids were used for the oxidation of glucose and very high selectivity towards gluconic acid was found.69–71 U. Prüße's group synthesized Au colloids by reducing tetrachloroauric acid (HAuCl4) with sodium borohydride (NaBH4) in the presence of polymers as stabilizing agents in an aqueous solution.49 Their results showed that three polymers, i.e., poly(diallyl dimethyl ammonium chloride) (PDADMAC), chitosan and poly-2,2-trimethyl ammonium methylmethachloride (PTAMM), can generate colloids with substantial catalytic activity for glucose oxidation. The highest activity (157 mol glucose h−1 mol Au−1) and selectivity (99.8%) for glucose oxidation were obtained with the Au-PDADMAC colloid. The other two colloids display a lower catalytic reaction rate (70 and 50 mol glucose h−1 mol Au−1 for Au-chitosan, and Au-PTAMM, respectively) and thus, a lower selectivity, i.e., 99.2%, due to fructose formation by isomerisation.
Recently, Comotti et al. found that water-dispersed gold sol exhibits a surprising activity when used as ‘naked particles’, that is, in the absence of common protectors such as polyvinylalcohol (PVA) and poly(N-vinyl-2-pyrrolidone) (PVP).61 The “naked” Au NPs (∼3.6 nm) were reported to possess a high catalytic activity with an initial turnover frequency of 18043 mol glucose h−1 mol Au−1 (calculated with respect to the total gold), which is comparable to that of enzymatic systems.61 They also observed that, for the Au NPs ranging from 3 to 6 nm, there was a good correlation between activity and particle size, which indicated that only surface Au atoms were associated with the glucose oxidation reaction. However, these unprotected Au colloids were so active that they could maintain catalytic activity for only a few minutes, after which their performance quickly declined as a result of particle aggregation. In addition, the high catalytic activity of the “naked” Au colloidal particles may not be reproducible, and a lower TOF (turnover frequency) value of 4600 mol glucose h−1 mol Au−1 was obtained for the NPs in their another report.68
Very recently, our group prepared Au, Ag, Cu, Pt, Pd, and Rh NPs by rapid injection of NaBH4 into the corresponding metallic ion solutions with PVP as a protective agent, all the NPs were used as the colloidal dispersion catalysts for aerobic glucose oxidation at 60 °C. In the metal NPs protected by a polymer like PVP, a PVP molecule steadily binds to a metal NP at multiple sites on the surface of the NP in spite of the weak coordination by a single bond. The polymer-protected metal NPs can work as an active and stable catalyst because the polymer can still remain near the NP during the attack of the reaction substrates on the surface of the NPs (Scheme 1). The results (Table 1) showed that the catalytic activity of the Au NPs for aerobic glucose oxidation is the highest (4020 mol glucose h−1 mol Au−1) among all the MNPs.72
 | ||
| Scheme 1 Schematic illustration of polymer-protected NPs on the reaction substrate of aerobic glucose oxidation (large red circle: NPs; small dark yellow circle: D-glucose; small green circle: gluconic acid). | ||
| NPs | Preparation conditions | Activity/mol glucose h−1 mol metal−1 | Average size/nm | |
|---|---|---|---|---|
| R PVP | R NaBH4 | |||
| R PVP: PVP/metal molar ratio; RNaBH4: NaBH4/metal molar ratio. | ||||
| Au | 100 | 5 | 4020 | 1.4 ± 0.5 |
| Ag | 100 | 5 | 130 | 4.5 ± 2.2 |
| Cu | 100 | 10 | 1880 | 17 ± 9.2 |
| Pt | 100 | 5 | 1160 | 1.4 ± 0.4 |
| Pd | 100 | 5 | 590 | 2.7 ± 0.9 |
| Rh | 100 | 5 | 110 | 4.3 ± 2.9 |
Accordingly, Au NPs supported on various kinds of supports have been actively studied. Au clusters supported on polymers,73–75 activated carbon,76–78 and metal oxide particles79,80 have been reported as effective catalysts especially for aerobic oxidation. Prüβ and co-workers80,81 reported glucose oxidation using Au catalysts, mainly supported on metal oxides. They reported Au/Al2O3 catalysts with Au NPs as small as 2.8 nm (1 wt%) and 1.9 nm (0.3 wt%). Their Au/Al2O3 catalyst (0.3 wt%) has achieved MTY (metal time yield on the basis of the total number of metal atoms; mol glucose h−1 mol Au−1) of 22320 mol glucose h−1 mol Au−1 with selectivity of 100% to sodium gluconate. Glucose oxidation reactions using Au/C catalysts have been originally reported by Rossi et al.65 They reported a Au/C catalyst having better catalytic activity than Pt–Pd–Bi/C, Pd–Bi/C and Pt/C catalysts in terms of conversion and TOF at pH values of 9.5, 8 and 7. The Au/ZrO282 catalyst prepared by solid grinding was reported to possess a high catalytic activity, i.e., the TOF as high as 160000 mol glucose h−1 mol Au−1 for aerobic glucose oxidation.
MNPs, such as Cu, Ni, Fe, Pt and Au, were also studied for catalysis of electro-oxidation of glucose. At Cu, Ni and Fe electrodes, glucose oxidation reactions occur when positive potentials exceed ca. 0.6–0.7 V (vs. Ag/AgCl), which produce formic acid (a 12-electron oxidation product) as a main product.83–89 At Pt electrodes, reactions occur from −0.3 V, and glycolic acid (a 6-electron oxidation product) is obtained as the main product.90 When gold electrodes are used, oxidation reactions occur from ca. −0.6 V, and gluconolactone (a 2-electron oxidation product) is obtained with a current efficiency of 100% at an electrode potential of −0.3 V.91–93 The oxidation potential at gold is more negative in comparison to Cu, Ni, Fe, Pt electrodes, indicating that Au NPs are an attractive catalyst for electrocatalytic oxidation of glucose.90–94
In summary, Au NPs are superior to other metals for aerobic glucose oxidation among catalytic MNPs. The Au colloid as well as Au NPs supported catalysts have been widely investigated with a view to improving the activity and selectivity of the catalyst.
3. Catalytic activity of BNPs for glucose oxidation
BNPs have widespread applications in catalysis, electronics, and optics.95,96 One of the major reasons for the interest in BNPs is the fact that their chemical and physical properties may be tuned by varying the size, composition, and degree of mixing.Very recently, our group prepared Au–Ag, Au–Cu, Au–Pt, Au–Pd, and Au–Rh BNPs by rapid injection of NaBH4 into the corresponding metallic ion solutions with PVP as a protective agent, all the BNPs were used as the colloidal dispersion catalysts for aerobic glucose oxidation at 60 °C.72,97,98 The results (Table 2) showed that Au–Ag, Au–Pt, Au–Pd, and Au–Rh BNPs possess higher catalytic activities than Au MNPs among colloidal dispersion catalysts.72 The Au–Ag (9/1, in atomic ratio), Au–Pt (8/2) and Au–Pd (7/3) BNPs having an average particle size of less than 2.0 nm show a high mean catalytic activity of 14900, 14200, and 9230 mol glucose h−1 mol metal−1, respectively, in the range of 2 h for glucose oxidation.
| NPs | Preparation conditions | Activity/mol glucose h−1 mol metal−1 | Average size/nm | |
|---|---|---|---|---|
| R PVP | R NaBH4 | |||
| Au | 100 | 5 | 4020 | 1.4 ± 0.5 |
| Au–Ag (9/1) | 100 | 10 | 14900 |
1.6 ± 0.9 |
| Au–Cu (8/2) | 40 | 5 | 2150 | 2.7 ± 0.9 |
| Au–Pt (8/2) | 100 | 5 | 14200 |
1.6 ± 0.5 |
| Au–Pd (7/3) | 100 | 5 | 9230 | 1.9 ± 0.8 |
| Au–Rh (7/3) | 100 | 5 | 4250 | 2.2 ± 1.0 |
Among various structures of BNPs, the most interesting one could be a core–shell structure, in which atoms of one element form a core and those of the other element cover the core to form a shell. The surface element plays an important role in the functions of BNPs like catalytic and optical properties, but these properties can be tuned by addition of the second element which may be located on the surface or in the center of the particles adjacent to the surface element.41 Recently, our group successfully synthesized PVP-protected Agcore–Aushell BNPs of 1.4 nm in diameter by simultaneous reduction of the corresponding ions with rapid injection of NaBH4 (Scheme 2), and the catalyst can be kept stable for more than 2 months under ambient conditions.99
 | ||
| Scheme 2 Schematic representation of formation of core–shell structured Ag–Au BNPs by rapid injection of NaBH4. | ||
It is well known that if two metals are co-reduced at once and one of them is insignificantly more noble than the other, then the nobler metal is preferentially found at the core.100 However, our results show that the Agcore–Aushell NPs can be formed in the sample of Au90Ag10 BNPs by rapid injection of NaBH4, this result is promising. The mechanism for the formation of Agcore–Aushell nanostructures is still unclear and needs further investigation. The prepared Au90Ag10 BNPs were analyzed with HR-TEM, DF-STEM, and EDS (Fig. 1). The HR-TEM image (Fig. 1a), indicating an ordered fringe pattern, demonstrates that the prepared Au90Ag10 BNP is a single crystal. The elemental ratio of Au90Ag10 BNPs with rather large size was measured by EDS at various parts of a particle along a cross line (Fig. 1b). Since the size of an EDS electron beam is 1 nm in diameter, the composition of different parts of a nanoparticle can be examined independently. It is very interesting that the EDS results show the co-existence of Ag and Au in the center of the nanoparticle and the presence of only Au at the edge of the nanoparticle, which is suggestive of a core–shell structure with Ag as a core and Au as a shell for this particular particle of the prepared Au90Ag10 BNPs (Fig. 1c). The reason for the detection of small amounts of Ag on the edge may be due to the diffusion of the incident electron. The incident electron may diffuse into the central part of the particle and emit the characteristic X-ray of Ag.99
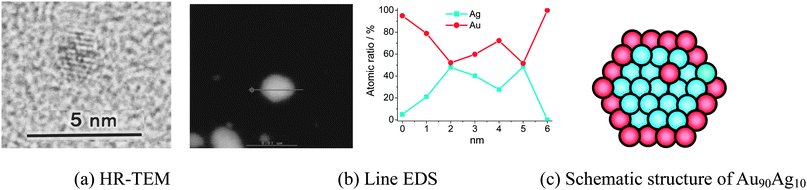 | ||
| Fig. 1 HR-TEM and DF-STEM images, and EDS of Au90Ag10 BNPs (final metal ion concentration = 0.20 mM, RPVP = 100). Adapted with permission from ref. 99. Copyright 2011, Elsevier. | ||
The Agcore–Aushell BNPs show a high and durable catalytic activity for the aerobic glucose oxidation. The Au–Ag BNPs were used as the catalyst for aerobic oxidation of glucose in water at 60 °C and pH 9.5. The catalytic activity (maximum activity) varies depending on the composition of the Au–Ag BNPs as shown in Fig. 2. The highest catalytic activity is achieved for Au80Ag20 BNPs. The TOF value is 16890 mol glucose h−1 mol metal−1, which is about 3 times higher than that of Au NPs (maximum TOF, 6230 mol glucose h−1 mol metal−1). It is interesting that the result is not caused by the difference in the surface area of the catalyst. In fact, when the activity is normalized to the total surface area (right Y axis in Fig. 2), the Au80Ag20 BNPs still have the highest catalytic activity. We think that the higher catalytic activity of the prepared BNPs than the usual Au NPs can be ascribed to: (1) the small average diameter, usually less than 2.0 nm, (2) good dispersion of the PVP-protected colloidal Au–Ag BNPs, and (3) the electronic charge transfer effect from adjacent Ag atoms and protecting PVP to Au active sites.
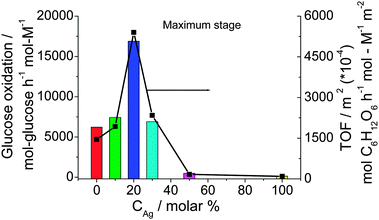 | ||
| Fig. 2 Catalytic activities of Au–Ag BNPs prepared at various feeding compositions (final metal ion concentration = 0.20 mM, RPVP = 100, RNaBH4 = 5) for glucose oxidation. | ||
Our group found that core–shell structured BNPs form spontaneously from the physical mixture of a dispersion of Ag NPs and that of another noble metal (Rh, Pd, or Pt) at room temperature.101–103 The experimental results showed that the initial step of such a spontaneous process is strongly exothermic. The strong exothermic interaction was considered to be the driving force for the formation of low entropy BNPs by the physical mixing of two kinds of MNPs. Based on the results of EF-TEM, preliminary EXAFS, and so on, we speculated the formation mechanism of core–shell-structured BNPs as shown in Scheme 3.101
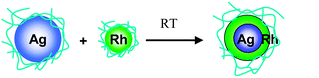 | ||
| Scheme 3 Schematic representation of formation of core–shell structured Ag–Rh BNPs by physical mixing of a dispersion of Ag and Rh NPs. | ||
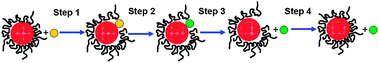
Core–shell-structured BNPs can be prepared by physical mixing of not only two kinds of MNPs, but also one kind of MNPs and another metal ion. Recently, we also found that core–shell-structured Au–Ag BNPs can be prepared by the addition of AgClO4 into aqueous dispersions of Au MNPs (Scheme 4).104 The Agcore–Aushell configuration was suggested by the data from energy dispersive X-ray spectra of prepared BNPs on a high resolution electron microscope (Fig. 3). STEM-EDS analysis (Fig. 3b) indicated that Ag contents in a nanoparticle were 19% (edge) and 39% (center), respectively. It suggests that the prepared BNP has a Agcore–Aushell structure. The EDS analysis of randomly chosen NPs suggested that most of the particles included Ag atoms. Based on the results of HR-TEM, STEM-EDS, UV-Vis and XPS, we speculated the formation mechanism of Agcore–Aushell BNPs as follows: Ag+ ions might be reduced by the natural light to metal atoms or tiny particles first, and then they could barge into the lattice of Au NPs, causing the aggregation of Au NPs, finally the mutual diffusion between Ag and Au atoms forms the Agcore–Aushell BNPs.
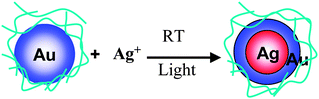 | ||
| Scheme 4 Schematic representation of formation of Agcore–Aushell BNPs prepared by physical mixing of Ag+ ions and Au NPs. | ||
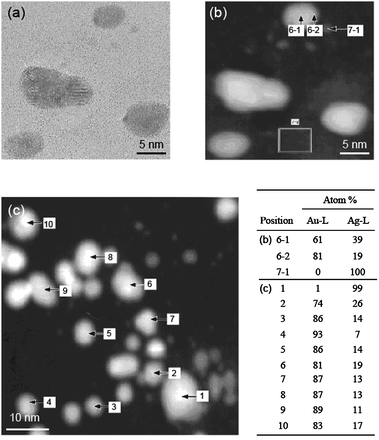 | ||
| Fig. 3 (a) HR-TEM and (b) small and (c) large scale STEM-EDS images of Ag–Au BNPs prepared by the addition of AgClO4 aqueous solution into PAANa-Au colloidal dispersion. Inset table represents the EDS analysis results of each position. Adapted with permission from ref. 104. Copyright 2010, American Chemical Society. | ||
Moreover, the catalytic activity of the Au–Ag BNPs prepared by physical mixing of Ag+ ions and Au NPs with various composition ratios for the glucose oxidation was investigated (Fig. 4). All Au–Ag NPs examined here had higher activity than that of the Au or Ag MNPs. The highest activity (4079 mol glucose h−1 mol M−1) among the NPs examined here was observed for the Au–Ag BNPs prepared with the metallic Au–Ag ratio of 1/0.25, which was 18 times higher than that of Au MNPs examined in the present experiments. It can be attributed to the electronic (ligand) effect between the core and shell atoms.41 The ionization potential of Au and Ag is 9.22 and 7.58 eV, respectively. Consequently, electronic charge could transfer from Ag in the core to Au in the shell (Scheme 5), leading to an increase in the electron density on the NP surface, which may activate the dissolved oxygen to attack the aldehyde group in the glucose.41
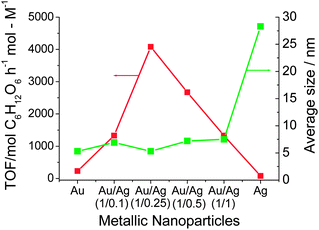 | ||
| Fig. 4 Catalytic activity and particle size of Au–Ag BNPs at various feed composition ratios and Au and Ag MNPs for glucose oxidation. | ||
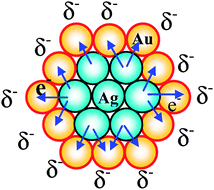 | ||
| Scheme 5 Schematic illustration of electronic charge transfer in the Agcore–Aushell BNPs. | ||
Glucose protected Au–Pt and Au–Pd BNP catalysts in the form of colloidal dispersions were tested in the aerobic oxidation of D-glucose to D-gluconic acid in water solution under mild conditions.68 The BNP colloidal particles appeared to be more stable towards agglomeration than Au MNPs, allowing higher conversions. After the 2.5 h test, Au–Pt and Au–Pd BNPs behave better than Au MNPs. The TOF values of Au–Pt (2/1) and Au–Pd (1/1) BNPs, and Au MNPs are 10500, 11600, and 4600 mol glucose h−1 mol M−1, respectively.
Taniguchi's group reported the electrocatalytic oxidation of glucose by carbon electrode-supported Au–Ag BNPs.92–94 It is interesting to note that the catalytic current at Au–Ag BNPs modified electrodes was observed from ca. −0.75 V, which represents a negative potential shift of ca. 0.1 V compared to that at Au MNPs modified electrodes. This result indicates that Au–Ag BNPs are effective catalysts for the electrocatalytic oxidation of glucose. Similar results were also observed in the case of the Au–Cu BNPs modified electrode.105 Electrochemical oxidation of glucose on Au–Pt nanocomposite electrodes was investigated with cyclic voltammetry.106 The Au–Pt nanocomposite electrodes display high electrocatalytic activity for the glucose oxidation in alkaline solution, showing a 0.30–0.35 V negative shift in peak potential as compared with bare Au electrodes, suggesting that Au–Pt nanocomposite electrodes display high electrocatalytic activity for the glucose oxidation.
In summary, BNPs usually show high catalytic activity for aerobic glucose oxidation than MNPs, the enhanced activity of BNP catalysts can be explained in terms of a geometrical and/or an electronic effect of the added second metal.
4. Catalytic activity of Crown Jewels-structured BNPs for glucose oxidation
Galvanic replacement reaction is an effective approach to fabricate core–shell BNPs with hollow interiors based on the differences in the reduction potentials of core and shell metal ions. Xia's group has pioneered a method to prepare hollow metal particles based on a galvanic replacement reaction carried out in aqueous solutions.107–110 Xia and coworkers also developed a lot of core–shell-structured BNPs using this strategy.111 The shell metal is mostly noble metals whereas core metal could be either noble or non-noble metals.112,113Mostly recently, we presented a novel concept of ‘Crown Jewel-structured catalysts’ for preparation of highly active Au catalysts having active Au atoms at the top (vertex or corner) positions of the metal nanoclusters (NCs).114–116 The Crown Jewel-structured Au–Pd (CJ-Au–Pd) catalysts were synthesized by a galvanic replacement reaction of the top atoms of the mother (Pd) NCs with Au atoms, since Au is known to be a catalytically active element for oxidation at a relatively low temperature. The schematic illustration of the preparation of the Crown Jewel-structured Au–Pd catalysts is shown in Scheme 6. The detailed compositions and preparation conditions of the Crown Jewel-structured Au–Pd catalysts are shown in Table 3.
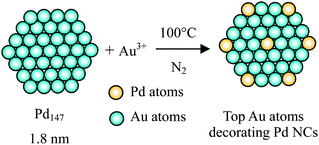 | ||
| Scheme 6 Schematic illustration of the deposition of top Au atoms on Pd mother clusters by a replacement reaction method. | ||
| Code | Feeding atomic ratio of Pd/Au3+ | Remarks |
|---|---|---|
| CJ-1 | 147/6 | Top Pd atoms are replaced by Au atoms |
| CJ-2 | 147/12 | |
| CJ-3 | 147/24 | Top and edge Pd atoms are replaced by Au atoms |
TEM images and size distribution histograms of the CJ-Au–Pd NCs, i.e., CJ-1, CJ-2, and CJ-3, with different Au contents show that all the NCs are spherical and well-isolated, and mainly distributed within the range from about 0.75 to 4 nm. The average sizes based on the TEM images are 1.8 ± 0.5 nm for CJ-1, 1.7 ± 0.5 nm for CJ-2, and 1.6 ± 0.5 nm for CJ-3. The structure of the Crown Jewel-structured Au–Pd NCs was also characterized by high-angle annular dark-field scanning transmission electron microscopy (HAADEF-STEM) accompanied by electron energy-loss spectroscopy (EELS), showing the presence of top Au atoms of the NCs (Scheme 7) and also suggesting the occurrence of the replacement reaction of Pd atoms with Au atoms at the top positions.
 | ||
| Scheme 7 Schematic illustration of Crown Jewel-structured Pd43Au12 NCs (yellow: Au atoms; blue: Pd atoms). | ||
What is of interest in this novel CJ structured catalyst is its unprecedented high activity. The catalytic activity was evaluated for glucose oxidation at 60 °C and at pH = 9.5 under O2 bubbling. The initial catalytic activity of the CJ-Au–Pd NCs for the glucose oxidation, normalized to Au mass, is much higher than that of Au, as shown in Fig. 5. The maximum catalytic activity (TOF) of the top Au atoms is about 194980 mol glucose h−1 mol Au−1. The specific activity is higher than that of Au (1.4 ± 0.5 nm in diameter, even if the activity was normalized to the surface atoms) and Pd mother clusters (1.8 ± 0.6 nm) by a factor of 20–30, and that of the Pd–Au alloy NCs by a factor of 8–10, although all of these NCs possess almost the same particle size (2.4 ± 0.8 nm for Pd–Au (8/2) alloy NCs and 1.9 ± 0.8 nm for Pd–Au (3/7) alloy NCs, prepared by rapid injection of NaBH4). This result demonstrates that the CJ-Au–Pd NCs possess a special structure different from that of the Pd and Au, and Pd–Au alloy NCs. Their activity is the highest reported to date for the glucose oxidation catalyzed by colloidal gold catalysts.
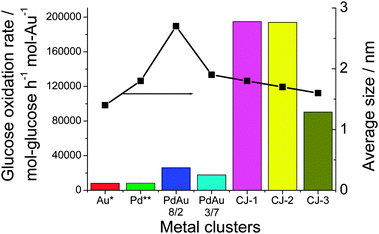 | ||
| Fig. 5 Comparison of the catalytic activities of CJ-Au–Pd, Au, Pd, and Pd–Au alloy NCs for aerobic glucose oxidation. (Au*, the activity was normalized to the number of surface Au atoms in NCs; Pd**, the activity was normalized to the number of surface Pd atoms.) | ||
It is interesting to note that CJ-3 NCs with increasing number of Au atoms show appreciably inferior catalytic performance compared to those of CJ-1 and CJ-2 NCs (Fig. 5). Based on the results of HAADF-STEM observation, all the Au atoms formed by replacement reaction can be assumed to predominantly locate at the top position on the surface layer of CJ-1 and CJ-2 NCs. In contrast, in the case of CJ-3 NCs containing a lot of Au atoms, part of the Au atoms formed by the replacement reaction could locate at the edge sites as well as the top sites. The low activity of CJ-3 NCs suggests that the presence of edge Au atoms will have a negative effect on the catalytic performance for glucose oxidation and that the top Au atoms are mainly responsible for the high activity. In other words, the activity of the Au atoms on the surface of Pd147 mother clusters should decrease in the following order: top Au atoms > edge Au atoms > face Au atoms (Scheme 8).
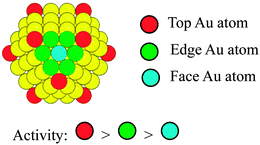 | ||
| Scheme 8 Schematic illustration of the activity of top, edge, and face Au atoms decorated on the Pd147 NCs (yellow spheres: Pd atoms). | ||
The high activities of the CJ-1 and CJ-2 NCs should be related to not only the unique geometrical structure but also the electronic properties of the top Au atoms. CJ-Au–Pd NCs protected by the low PVP content (RPVP = 5) were prepared and investigated by XPS to examine the electronic properties of the catalytically active CJ-Au–Pd NCs. The XPS results show that the electron apparent binding energy (BE) of Au 4f7/2 in the CJ NCs was about 1.5 eV lower than that of the bulk Au, and about 0.25 eV lower than that of the PVP-protected Au NCs with the mean diameter of 1.1 nm.117 The negative shift of the Au 4f BE suggests that a negative charge is deposited on the Au atoms of the CJ-Au–Pd NCs, and provides evidence that the Pd atoms donate electrons to the Au atoms. In addition, DFT calculations were also carried out to study the electronic states of the top Au atoms in the CJ-Au–Pd NCs. A more important result was that the negative charge density of the top atoms (Table 4) significantly increased when all of the top atoms of Pd55 clusters were replaced with Au atoms (−0.116). This result provides further evidence that the Pd atoms donate electrons to the Au atoms (Scheme 9).
| Site | Top | Edge | Face |
|---|---|---|---|
| Atom | Au | Pd | Pd |
| Mulliken charges | −0.116 | 0.056 | 0.030 |
 | ||
| Scheme 9 Schematic illustration of electronic charge transfer effects in CJ-1 NCs. | ||
Gold clusters have provided interesting catalysts so far. The origin of the catalytic activities of supported Au catalysts can be assigned to the perimeter interfaces between Au NCs and the support.118 However, the genesis of the catalytic activities of colloidal Au-based bimetallic NCs is unclear. The present catalytic activity results reveal that the Au atoms at the top positions having much higher catalytic activity than those at the other positions should be responsible for the catalytic activities of colloidal Au-based BNPs.
In summary, the Au atoms can be controllably assembled at the top position on the surface of the mother clusters, and then exhibit a high catalytic activity. We anticipate that the finding will initiate attempts for understanding the structure–activity relations of a catalyst on an atomic level. In addition, this concept of ‘Crown Jewel’ potentially provides a general design with a reactive metal embedded in less reactive materials.
5. Catalytic activity of TNPs for glucose oxidation
In contrast to MNPs and BNPs, tri- and multi-metallic NPs may possess an even greater degree of catalytic activity and selectivity because more variables are available for tuning. There are limited reports but rapidly growing interest in tri- and multimetallic NPs in the last two years.119–125 The introduction of a third/fourth metal into the NP catalyst is expected to produce a combination of effects such as reduction of the lattice distance, the addition of surface sites for the formation of a metal–oxygen bond and adsorption of OH−, and the modification of the d-band center. Thus TNPs and multimetallic NPs may possess an even higher catalytic activity and selectivity in comparison to MNPs and BNPs.Mostly recently, we have reported an effective method to synthesize Au–Pt–Ag alloy TNPs with an average diameter of 1.5 nm by reduction of the corresponding ions with rapid injection of NaBH4 (Scheme 10).125,126 The prepared alloy TNPs were characterized by UV-Vis, XRD, XPS, HR-TEM and EDS in HR-STEM. The elemental ratio of Au70Pt20Ag10 TNPs was measured by STEM-EDS at various parts of a particle on a cross line (Fig. 6). All the compositions of Au, Pt and Ag are similar to the feeding ratio in the nanoparticle, suggesting the formation of alloys in the TNPs. The catalytic activities (mean value during 2 h) of the Ag, Pt, Au, Au70Ag30, Au70Pt30, and Au70Pt20Ag10 NPs are shown in Fig. 7. The mean activity of Au70Pt20Ag10 TNPs during 2 h testing is about 200090 mol glucose h−1 mol metal−1, which is about 5 times higher than that of Au MNPs, although these two catalysts possess nearly the same particle size. In addition, the TOF value of the Au70Pt20Ag10 TNPs was about 3 and 1.6 times higher than that of Au70Ag30 and Au70Pt30 BNPs, respectively.
 | ||
| Scheme 10 Schematic representation of formation of Au–Pt–Ag TNPs prepared by rapid injection of NaBH4. | ||
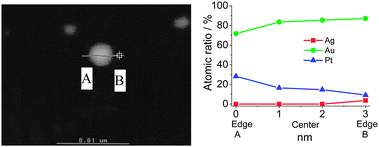 | ||
| Fig. 6 STEM images and line-EDS of Au70Pt20Ag10 TNPs prepared by rapid injection of NaBH4. Adapted with permission from ref. 126. Copyright 2011, Elsevier. | ||
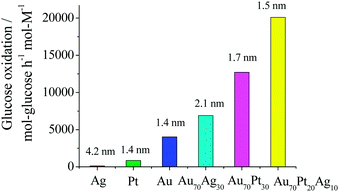 | ||
| Fig. 7 Comparison of catalytic activities of Ag, Pt, Au, Au70Ag30, Au70Pt30 and Au70Pt20Ag10 NPs prepared by rapid injection of NaBH4. RPVP was 100 and RNaBH4 was 5 for all catalysts. Numbers shown at the top of each bar indicate the average particles size of the NPs. | ||
In order to get more information on the effect of coexisting elements on the catalytic activity, the Au–Pt–Ag TNPs with various atomic ratios were used as the colloidal dispersion catalysts for aerobic glucose oxidation. The catalytic activity varies with the composition of the TNPs as shown in Fig. 8. The samples of Au70Pt20Ag10 TNPs show the highest mean catalytic activity during 2 h testing. It should be pointed out that the instantaneous maximum TOF of the TNPs is 23740 mol glucose h−1 mol metal−1. It is interesting that the high activity does not depend only on the surface area of the catalyst. In fact, even though the activity is normalized to the total surface area, the Au70Pt20Ag10 TNPs still have the highest catalytic activity (Fig. 8, right Y axis). This suggests that the ratio of Au–Pt–Ag = 70/20/10 is crucial for the preparation of Au–Pt–Ag TNPs with high activities.
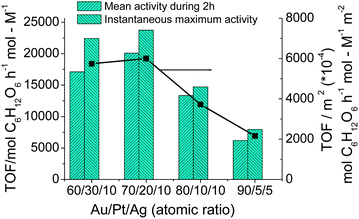 | ||
| Fig. 8 Catalytic activities and catalytic activities normalized to the surface area of Au–Pt–Ag TNPs prepared at various compositions (final metal ion concentration = 0.11 mM, RPVP = 100, RNaBH4 = 5, rapid injection of NaBH4) for glucose oxidation. | ||
The high catalytic activities of the Au–Pt–Ag TNPs can be ascribed to the following factors: (1) the small average size, about 1.5 nm in diameter, (2) stabilization of PVP for the colloidal dispersions of the TNPs, (3) long-term stability due to thermodynamically stable alloyed structure of the TNPs, and (4) the formed negatively charged Au atoms due to electron donation of Ag neighboring atoms and PVP acting as catalytically active sites for aerobic glucose oxidation. The presence of the negatively charged Au atoms was supported by XPS measurements and electron density calculation with density functional theory (DFT). The electron apparent binding energy (BE) of Au 4f7/2 in Au90Pt5Ag5 TNPs was about 1.4 eV lower than that of bulk Au (84.0 eV), and about 0.2 eV lower than that of the PVP-protected Au NPs (82.8 eV) with a mean diameter of 1.3 nm.127 The negative shift of the Au 4f BE suggests that a negative charge is deposited on Au atoms in the TNPs, and provides evidence that Ag atoms donate electrons to Au atoms. DFT calculations of a M55 model NP (Au37Pt12Ag6, the subscripts of Au, Pt and Ag stand for the number of atoms in the TNPs, similar compositions to that of Au70–Pt20–Ag10 TNPs) were examined for understanding the correlation between the electronic states and catalytic activity of the TNPs for aerobic glucose oxidation. A most important result was that Au atoms are indeed negatively charged (Mulliken charges: −0.015), while the Ag atoms have positive charges (Mulliken charges: 0.135) due to the electronic charge transfer from the Ag atom to Au and Pt atoms. We think that the generation of superoxo- or peroxo-like species resulted from electron transfer from anionic Au atoms to O2 plays a crucial role in the aerobic oxidation of glucose.
Electronic charge transfer effects between the various kinds of adjacent elements which were reported for the reasons of high catalytic activities of several cases of BNPs and TNPs,127–130 and this kind of consideration could be applied to the present Au–Pt–Ag TNP system. The possible electronic charge transfer effects in Au–Pt–Ag TNPs are illustrated in Scheme 11. Since Ag (ionization energy, 7.58 eV) is most electronegative among all elements in the TNPs (Pt ionization energy, 9.02 eV; Au ionization energy, 9.22 eV), Ag atoms would donate electrons to neighboring Au and Pt atoms due to the electronic charge transfer effect, and there are at least two modes of the charge transfer, i.e., from Ag to Au atoms and from Ag to Pt atoms. We infer that the synergistic effects of the two kinds of charge transfer modes might play an important role for the higher catalytic activity of the TNPs compared with that of the BNPs even though we have no strict evidence for it.
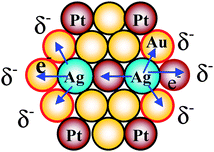 | ||
| Scheme 11 Schematic illustration of electronic charge transfer in the Au–Pt–Ag TNPs. | ||
In summary, PVP-protected Au–Pt–Ag TNPs containing 55 atoms with controlled composition exhibited higher catalytic activities for glucose oxidation than the Au NPs and Au-containing BNPs, although the TNPs have an alloy structure. The present results show that the electronic charge transfer effects of the adjacent elements are important to achieve high catalytic activities of the TNPs.
6. Concluding remarks
Catalysts based on metal NPs have recently received much attention due to their excellent properties. BNPs and TNPs with well-organized structure and composition usually show higher activity for glucose oxidation than MNPs, which can be mainly ascribed to the synergistic catalytic effect over heterometallic NPs based on the reports mentioned in the article as follows: (1) the electronic charge transfer effect among the different components along with geometric effects leads to appropriate modification of electronic structure, which benefits the catalytic activity enhancement. (2) Improved activity results from structural changes of nanocatalysts upon introducing another metal species.It should be pointed out that the present BNPs and TNPs are mostly based on precious metals such as Au, Pt, Pd, Rh, and Ag which have been proven to be efficient for aerobic glucose oxidation. Compared to BNP catalysts with noble metals, non-noble metal-based BNPs were much less studied. However, from the viewpoint of practical applications, the development of efficient, low-cost, and stable catalysts to further improve the kinetic properties under moderate conditions is therefore very important. A possible solution to this problem could be the development of catalysts based on the first row transition metals, such as Fe, Co, Ni, and Cu. Further efforts should be made to develop such effective and less expensive non-noble metallic BNP and TNP catalysts. In addition, BNPs and TNPs with a novel structure, like Crown Jewel, should receive more attention in the future. Our present catalytic activity results reveal that the Au atoms at the top positions have much higher catalytic activity than those at the other positions. This kind of the Crown Jewel-structured NCs are able to be prepared by using other pairs of metal elements, and will provide the most active catalyst for the reaction specific to the top metal.
References
- C. Baatz, N. Thielecke and U. Prüße, Appl. Catal., B, 2007, 70, 653–660 CrossRef CAS.
- N. Thielecke, M. Aytemir and U. Prüsse, Catal. Today, 2007, 121, 115–120 CrossRef CAS.
- Carbohydrates as Organic Raw Materials, ed. F. W. Lichtenthaler, VCH, Weinheim, 1991 Search PubMed.
- A. Mirescu, H. Berndt, A. Martin and U. Prüße, Appl. Catal., A, 2007, 317, 204–209 CrossRef CAS.
- J. Z. Liu, L. P. Weng, Q. L. Zhang, H. Xu and L. N. Ji, Biochem. Eng. J., 2003, 14, 137–141 CrossRef CAS.
- E. F. Bernstein, D. B. Brown, M. D. Schwartz, K. Kaidbey and S. M. Ksenzenko, Dermatol. Surg., 2004, 30, 189–196 CrossRef.
- Ullmann's Encyclopaedia of Industrial Chemistry, Gluconic Acid, Electronic Release, Wiley-VCH, 2002.
- H. Okatsu, N. Kinoshita, T. Akita, T. Ishida and M. Harut, Appl. Catal., A, 2009, 369, 8–14 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, J. Catal., 2004, 228, 282–287 CrossRef CAS.
- J. Bao, K. Furumoto, K. Fukunaga and K. Nakao, Biochem. Eng. J., 2001, 8, 91–102 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 2000, 57, 1 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. Hugh Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437(20), 1132–1135 CrossRef CAS.
- R. A. Sheldon, Stud. Surf. Sci. Catal., 1991, 66, 33–54 CrossRef.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- G. J. Hutchings, A golden future for green chemistry, Catal. Today, 2007, 122, 196–200 CrossRef CAS.
- T. Ishida and M. Haruta, Angew. Chem., Int. Ed., 2007, 46, 7154–7156 CrossRef CAS.
- J. Q. Tian, S. Liu, Y. L. Luo and X. P. Sun, Catal. Sci. Technol., 2012, 2(2), 432–436 CAS.
- Y. Onal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef CAS.
- C. Della Pina, E. Falletta, M. Rossi and A. Sacco, J. Catal., 2009, 263, 92–97 CrossRef CAS.
- D. L. An, A. H. Ye, W. P. Deng, Q. H. Zhang and Y. Wang, Chem.–Eur. J., 2012, 18(10), 2938–2947 CrossRef CAS.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354 CrossRef CAS.
- K. D. Hermnason, S. O. Lumsdon, J. P. Williams, E. W. Kaler and O. D. Velev, Science, 2001, 294, 1082 CrossRef.
- M. J. Feldstien, C. D. Keating, Y. H. Liau, M. J. Natan and N. F. Scherer, J. Am. Chem. Soc., 1997, 119, 6638 CrossRef.
- S. R. Emory and S. J. Nie, J. Phys. Chem. B, 1998, 102, 493 CrossRef CAS.
- T. Sun and K. Seff, Chem. Rev., 1994, 94, 857 CrossRef CAS.
- K. Aslan, S. N. Malyn and C. D. Geddes, J. Am. Chem. Soc., 2006, 128, 13372 CrossRef CAS.
- D. M. Dotzauer, J. H. Dai, L. Sun and M. L. Bruening, Nano Lett., 2006, 6, 2268 CrossRef CAS.
- F. J. Ibanez and F. P. Zamborini, Langmuir, 2006, 22, 9789 CrossRef CAS.
- S. Shanmukh, L. Jones, J. Driskell, Y. P. Zhao, R. Dluhy and R. A. Tripp, Nano Lett., 2006, 6, 2630 CrossRef CAS.
- L. Sun and L. Q. Mao, Talanta, 2006, 70, 68 CrossRef.
- J. Zhang and J. R. Lakowicz, J. Phys. Chem. B, 2006, 110, 2387 CrossRef CAS.
- N. F. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278 CrossRef CAS.
- H. Hirai and N. Toshima, in Tailored Metal Catalyst, ed. Y. Iwasawa, Reidel, Dordrecht, 1985, pp. 121–135 Search PubMed.
- H. Bonnemann, W. Brijoux, R. Brinkmann, E. Dinjus, T. Jouben and B. Korall, Angew. Chem., Int. Ed. Engl., 1991, 30, 1312 CrossRef.
- N. Toshima, Y. Shiraishi, T. Teranishi, M. Miyake, T. Tominaga, H. Watanabe, W. Brijoux, H. Bonnemann and G. Schmid, Appl. Organomet. Chem., 2001, 15, 178 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS.
- L. C. Cheng, J. H. Huang, H. M. Chen, T. C. Lai, K. Y. Yang, R. S. Liu, M. Hsiao, C. H. Chen, L. J. Her and D. P. Tsai, J. Mater. Chem., 2012, 22(5), 2244–2253 RSC.
- X. Y. Zhang, Q. Shen, C. He, C. Y. Ma, J. Cheng and Z. P. Hao, Catal. Sci. Technol., 2012, 2, 1059–1067 CAS.
- N. Toshima, Metal NPs used as catalysts. Encyclopedia of Nanoscience and Nanotechnology, Marcel Dekker, Madison Ave, New York, NY 10016, 2004, pp. 1869–1880 Search PubMed.
- N. Toshima, H. Yan and Y. Shiraishi, Recent Progress in Bimetallic NPs: Their Preparation, Structure and Functions, in Metal Nanoclusters in Catalysis and Materials Science: the Issue of Size-Control, ed. B. Corain, G. Schmid and N. Toshima, Elsevier, Amsterdam, 2007, pp. 49–75 Search PubMed.
- N. Toshima, Polymer-Assisted Composites of Trimetallic NPs with a Three-Layered Core-Shell Structure for catalyses, in Nanohybridization of Organic-Inorganic Materials, ed. A. Muramatsu and T. Miyashita, Springer, Heidelberg Dordecht, London, 2009, 55–79 Search PubMed.
- E. von Gorup-Besanez, Justus Liebigs Ann. Chem., 1861, 118, 257 CrossRef.
- K. Heyns and H. Paulsen, Adv. Carbohydr. Chem., 1962, 17, 169 CrossRef CAS.
- Kao. Corp. Process for producing gluconic acid, Ch. EP0142725 1985 Search PubMed.
- R. Freres, Process for the oxidation of aldoses, catalyst used in said process and products thus obtained, Ch. EP0233816 1987 Search PubMed.
- M. Wenkin, C. Renard, P. Ruiz, B. Delmon and M. Devillers, Studies in surface science and catalysis, in Heterogeneous Catalysis and Fine Chemicals IV, ed. H. U. Blaser, A. Baiker and R. Prins, Elsevier, Amsterdam, 1997, vol. 108, p. 391 Search PubMed.
- B. M. Despeyroux, K. Deller and E. Peldszus, in New Developments in Selective Oxidation, ed. G. Centi and F. Trifiro, Elsevier, Amsterdam, 1990, p. 159 Search PubMed.
- A. Mirescu and U. Prüße, Catal. Commun., 2006, 7, 11–17 CrossRef CAS.
- M. Taramasso, G. Perego and B. Notari, U.S. Patent 4.410.501 18.10.1983 Enichem.
- R. A. Sheldon, CHEMTECH, 1991, 21, 566 CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CAS.
- D. A. Buchanan and G. Webb, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 134 RSC.
- R. P. Chambers and M. Boudart, J. Catal., 1966, 5, 517 CrossRef CAS.
- P. A. Sermon, G. C. Bond and P. B. Wells, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 385 RSC.
- A. Haruta, Chem. Rec., 2003, 3, 75–87 CrossRef.
- M. Haruta, CATTECH, 2002, 6, 102–115 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS.
- M. Date and M. Haruta, J. Catal., 2001, 201, 221–224 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- M. Comotti, C. D. Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS.
- S. Salvatore, C. Carmelo, R. P. Maria, P. Giacomo and P. Alessandro, Appl. Catal., A, 2012, 417–418, 66–75 Search PubMed.
- L. Prati and M. Rossi, Stud. Surf. Sci. Catal., 1997, 110, 509 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242 CrossRef CAS.
- S. Biella, G. L. Castiglioni, C. Fumagalli, L. Prati and M. Rossi, Catal. Today, 2002, 72, 43 CrossRef CAS.
- M. Comotti, C. D. Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS.
- T. Tsukuda, H. Tsunoyama and H. Sakurai, Chem.–Asian J., 2011, 6, 736–748 CrossRef CAS.
- P. Beltrame, M. Comotti, C. D. Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS.
- M. Comotti, C. D. Pina, R. Matarrese and M. Rossi, Angew. Chem., 2004, 116, 5936–5939 CrossRef.
- N. Toshima and H. J. Zhang, 14th IUPAC International Symposium on MacroMolecular Complexes (MMC-14), Helsinki, Finland, 2011Aug 15 Search PubMed.
- H. Wu, Z. Liu, X. Wang, B. Zhao, J. Zhang and C. Li, J. Colloid Interface Sci., 2006, 302, 142–148 CrossRef CAS.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596 CAS.
- B. Corain, C. Burato, P. Centomo, S. Lora, W. M. Zaika and G. Schmid, J. Mol. Catal. A: Chem., 2005, 225, 189 CrossRef CAS.
- Z. J. Li, D. S. Huang, R. M. Xiao, W. Liu, C. Xu, Y. Jiang and L. D. Sun, Curr. Nanosci., 2012, 8(1), 26–28 CrossRef CAS.
- T. Ishida, K. Kuroda, N. Kinoshita, W. Minagawa and M. Haruta, J. Colloid Interface Sci., 2008, 323, 105 CrossRef CAS.
- E. G. Rodrigues, M. F. R. Pereira, X. W. Chen, J. J. Delgado and J. J. M. Órfão, J. Catal., 2011, 281(1), 119–127 CrossRef CAS.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134(3), 1560–1570 CrossRef CAS.
- C. Baatz and U. Prüße, J. Catal., 2007, 249, 34 CrossRef CAS.
- C. Baatz, N. Decker and U. Prüβ, J. Catal., 2008, 258, 165–169 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265 CrossRef CAS.
- Y. Zhang, F. G. Xu, Y. J. Sun, C. L. Guo, K. Cui, Y. Shi, Z. W. Wen and Z. Li, Chem.–Eur. J., 2010, 16(30), 9248–9256 CrossRef CAS.
- P. Parpot, N. Nunes and A. P. Bettencourt, J. Electroanal. Chem., 2006, 596, 65 CrossRef CAS.
- M. Z. Luo and R. P. Baldwin, J. Electroanal. Chem., 1995, 387, 87 CrossRef.
- N. Torto, T. Ruzgas and L. Gorton, J. Electroanal. Chem., 1999, 464, 252 CrossRef CAS.
- K. B. Male, S. Hrapovic, Y. Liu, D. Wang and J. H. T. Luong, Anal. Chim. Acta, 2004, 516, 35 CrossRef CAS.
- R. Ojani, J. B. Raoof and P. Salmany Afagh, J. Electroanal. Chem., 2004, 571, 1 CrossRef CAS.
- A. Salimi and M. Roushani, Electrochem. Commun., 2005, 7, 879 CrossRef CAS.
- K. B. Kokoh, J. M. Leger, B. Beden, H. Huser and C. Lamy, Electrochim. Acta, 1992, 37, 1909 CrossRef CAS.
- M. Tominaga, T. Shimazoe, M. Nagashima and I. Taniguchi, Electrochem. Commun., 2005, 7, 189 CrossRef CAS.
- M. Tominaga, T. Shimazoe, M. Nagashima, H. Kusuda, A. Kubo, Y. Kuwahara and I. Taniguchi, J. Electroanal. Chem., 2006, 590, 37–46 CrossRef CAS.
- M. Tominaga, M. Nagashima, K. Nishiyama and I. Taniguchi, Electrochem. Commun., 2007, 9, 1892 CrossRef CAS.
- M. Tominaga, T. Shimazoe, M. Nagashima and I. Taniguchi, J. Electroanal. Chem., 2008, 615, 51–61 CrossRef CAS.
- S. Hirasawa, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 1150–1152 CAS.
- S. Zafeiratos, S. Piccinin and D. Teschner, Catal. Sci. Technol., 2012, 2 10.1039/c2cy00487a.
- H. J. Zhang, N. Morita, K. Takasaki, M. Haba, S. Tokonami and N. Toshima, The 2010 International Chemical Congress of Pacific Basin Societies, Honolulu, Hawaii, USA, 2010, p. 225 Search PubMed.
- N. Toshima and H. J. Zhang, The 2010 International Chemical Congress of Pacific Basin Societies, Honolulu, Hawaii, USA, 2010, p. 167 Search PubMed.
- H. J. Zhang, J. Okuni and N. Toshima, J. Colloid Interface Sci., 2011, 354, 131–138 CrossRef CAS.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235 CrossRef CAS.
- N. Toshima, M. Kanemaru, Y. Shiraishi and Y. Koga, J. Phys. Chem. B, 2005, 109, 16326 CrossRef CAS.
- M. Kanemaru, Y. Shiraishi, Y. Koga and N. Toshima, J. Therm. Anal. Calorim., 2005, 81, 523 CrossRef CAS.
- K. Hirakawa and N. Toshima, Chem. Lett., 2003, 32, 78 CrossRef CAS.
- S. Tokonami, N. Morita, K. Takasaki and N. Toshima, J. Phys. Chem. C, 2010, 114, 10336–10341 CAS.
- M. Tominaga, Y. Taema and I. Taniguchi, J. Electroanal. Chem., 2008, 624(1–2), 1–8 CrossRef CAS.
- C. C. Jin and Z. D. Chen, Synth. Met., 2007, 157, 592–596 CrossRef CAS.
- Y. Xia, Anal. Chem., 2002, 74, 5297 CrossRef.
- Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641 CrossRef CAS.
- Y. Sun and Y. Xia, Adv. Mater., 2004, 16, 264 CrossRef CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892 CrossRef CAS.
- X. Lu, J. Chen, S. E. Skrabalak and Y. N. Xia, Proc. Inst. Mech. Eng., Part N: J. Nanoeng. Nanosyst., 2008, 221, 1–16 Search PubMed.
- W. R. Lee, M. G. Kim, J. R. Choi, J. I. Park, S. J. Ko, S. J. Oh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 16090–16097 CrossRef CAS.
- Y. Lu, Y. Zhao, L. Yu, L. Dong, C. Shi, M. J. Hu, Y. J. Xu, L. P. Wen and S. H. Yu, Adv. Mater., 2010, 22, 1407–1411 CrossRef CAS.
- H. J. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef.
- N. Toshima, H. Zhang, M. Okumura and M. Haruta, Japan-Australia Colloid and Interface Science Symposium, Cairns, Australia, 2011 Search PubMed.
- N. Toshima and H. Zhang, International Association of Colloid and Interface Scientists, Conference 2012 (IACIS 2012), Sendai, Japan, 2012 Search PubMed.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS.
- M. Haruta, Chem. Rec., 2003, 3, 75 CrossRef CAS.
- V. Mazumder, M. Chi, K. L. More and S. Sun, J. Am. Chem. Soc., 2010, 132, 7848–7849 CrossRef CAS.
- C. Wang, D. van der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. Sun, H. Daimon, G. Wang, J. Greeley, J. Pearson, A. P. Paulikas, G. Karapetrov, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2011, 11, 919–926 CrossRef CAS.
- P. P. Fang, S. Duan, X. D. Lin, J. R. Anema, J. F. Li, O. Buriez, Y. Ding, F. R. Fan, D. Y. Wu, B. Ren, Z. L. Wang, C. Amatore and Z. Q. Tian, Chem. Sci., 2011, 2, 531–539 RSC.
- H. Wang, R. Wang, H. Li, Q. Wang, J. Kang and Z. Lei, Int. J. Hydrogen Energy, 2011, 36, 839–848 CrossRef CAS.
- J. Zhao, K. Jarvis, P. Ferreira and A. Manthiram, J. Power Sources, 2011, 196, 4515–4523 CrossRef CAS.
- P. Gao, F. Li, F. K. Xiao, N. Zhao, W. Wei, L. S. Zhong and Y. H. Sun, Catal. Sci. Technol., 2012, 2, 1447–1454 CAS.
- H. J. Zhang, M. Okumura and N. Toshima, J. Phys. Chem. C, 2011, 115(30), 4883–14891 Search PubMed.
- H. J. Zhang, J. Okuni and N. Toshima, Appl. Catal., A, 2011, 400(1–2), 9–13 CrossRef CAS.
- N. K. Chaki, H. Tsunoyama, Y. Negishi, H. Sakurai and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4885–4888 CAS.
- N. Toshima, R. Ito, T. Matsushita and Y. Shiraishi, Catal. Today, 2007, 122, 239–244 CrossRef CAS.
- N. Toshima, T. Yonezawa and K. Kushihashi, J. Chem. Soc., Faraday Trans., 1993, 89, 2537–2543 RSC.
- N. Nishida, Y. Shiraishi, S. Kobayashi and N. Toshima, J. Phys. Chem. C, 2008, 51, 20284–20296 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |