The role of negatively charged Au states in aerobic oxidation of alcohols over hydrotalcite supported AuPd nanoclusters†
Shun
Nishimura
a,
Yusuke
Yakita
a,
Madoka
Katayama
b,
Koichi
Higashimine
b and
Kohki
Ebitani
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: ebitani@jaist.ac.jp; Fax: +81-761-51-1149; Tel: +81-761-51-1610
bCenter for Nano Materials and Technology, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan
First published on 22nd May 2012
Abstract
The PVP-protected bimetallic gold–palladium nanoclusters (AuxPdy-PVP NCs) were prepared on the solid base hydrotalcite (HT) with various Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd (x:y) molar ratios. Transmission electron microscopy showed narrow particle size distributions of AuxPdy-PVP NCs with a mean diameter in the range of 2.6–3.0 nm regardless of Pd content. Aerobic oxidations of 1-phenylethanol over the AuxPdy-PVP/HT catalysts showed that their catalytic activities were significantly affected by the Pd content. Correlations between charge transfer between Au and Pd and catalytic activity of the AuxPdy-PVP/HT catalysts were investigated with X-ray photoelectron spectroscopy (XPS), X-ray absorption near-edge structure (XANES), Michaelis–Menten kinetic studies for alcohol oxidation, and other analytical techniques. The peaks of Au 4f in the XPS spectra were shifted to the lower energy side with increase of Pd content, indicating the electron transfer from Pd to Au atoms according to Pauling's electronegativity protocol. The electron densities in the Au 5d orbital in the AuxPdy-PVP/HT catalysts estimated by the Au L3-XANES spectra correlated well with their catalytic activities. Moreover, the kinetic studies also proposed that the electron rich Au 5d states, resulting from the intermetallic electron transfer from Pd atoms, strongly contributed to the rate-determining step in the alcohol oxidation. It was concluded that the electronic negativity of the Au 5d states controlled by the Pd content accelerated the rate-determining step in alcohol oxidation through highly active radical-like intermediates.
:Pd (x:y) molar ratios. Transmission electron microscopy showed narrow particle size distributions of AuxPdy-PVP NCs with a mean diameter in the range of 2.6–3.0 nm regardless of Pd content. Aerobic oxidations of 1-phenylethanol over the AuxPdy-PVP/HT catalysts showed that their catalytic activities were significantly affected by the Pd content. Correlations between charge transfer between Au and Pd and catalytic activity of the AuxPdy-PVP/HT catalysts were investigated with X-ray photoelectron spectroscopy (XPS), X-ray absorption near-edge structure (XANES), Michaelis–Menten kinetic studies for alcohol oxidation, and other analytical techniques. The peaks of Au 4f in the XPS spectra were shifted to the lower energy side with increase of Pd content, indicating the electron transfer from Pd to Au atoms according to Pauling's electronegativity protocol. The electron densities in the Au 5d orbital in the AuxPdy-PVP/HT catalysts estimated by the Au L3-XANES spectra correlated well with their catalytic activities. Moreover, the kinetic studies also proposed that the electron rich Au 5d states, resulting from the intermetallic electron transfer from Pd atoms, strongly contributed to the rate-determining step in the alcohol oxidation. It was concluded that the electronic negativity of the Au 5d states controlled by the Pd content accelerated the rate-determining step in alcohol oxidation through highly active radical-like intermediates.
Introduction
Bimetallic nanoparticles (NPs) have attracted great interest and potential in advanced materials science because they achieved unique performances different from those of monometallic NPs.1 Since the catalytic activities of nano-sized Au particles were reported by Haruta et al. in 1980s,2,3 breakthroughs have been made in the development of a series of Au and incorporated second element NPs as catalysts such as Au clusters, AuPd, AuPt, AuAg and so on.4–8 However, the contribution of these advanced catalysts to the reaction kinetics over the bimetallic catalyst is still questionable. In general, (i) each element promotes different elementary reaction steps (bifunction effect), (ii) the electron transfer among two elements improves reactivity (ligand effect), and (iii) the specific group of surface atoms developed by geometric growth (ensemble effect), were considered as driving forces for significant performances of heterometallic assemblies.9–11 The difficulties in the synthesis of uniform bimetallic NPs with various mixing ratios and/or morphologies become a barrier to discuss and compare their performances in the reaction mechanism.From the viewpoint of energy and environmental issues, aerobic oxidation of alcohols for the synthesis of fine chemicals over a highly active heterogeneous catalyst has been investigated.12–18 It leads to environmentally-friendly synthesis routes compared to the stoichiometric oxidations over transition metal complexes. Additionally, the traditional process produces a large amount of undesirable salts and needs energy for separation of the product from the reaction mixture. Therefore, the notable protocols for the active sites synthesis on a nanoscale have a great impact on development of highly functionalized heterogeneous catalysts. In this regard, supported polymer capped highly dispersed bimetallic NP catalysts, e.g. Au@Pd-PVA/TiO2,19 AuPt-PVA/MgO,20 and AuPd-PVA/C,21,22 have been extensively studied for alcohols oxidation into carbonyl compounds.
Herein, we examined the aerobic oxidation over the PVP-protected AuPd bimetallic (AuxPdy-PVP) NCs deposited onto the hydrotalcite (HT) catalysts with similar size distributions of AuxPdy-PVP NCs. The AuPd NPs are well-known as some of the most attractive active sites for the catalyst for various reactions such as not only alcohol oxidation,19–24 but also hydrogenation of 1,3-cyclooctadiene,25,26 the direct synthesis of H2O2,27–29 acetoxylation of ethylene,30 and oxygen reduction in electrodes.31,32 The HT has been known as an effective support for various reactions such as deoxygenation, chemoselective reduction, and oxidation reactions because it exhibits tunable surface basic sites, achieving high catalytic performance through proton abstraction and uniform deposition of active metal on the surface.33–39 It is supposed that the combination of AuPd NCs and HT becomes a significant heterogeneous catalyst for the alcohol oxidation. In addition, both the careful synthesis of bimetallic NCs and study of their reaction mechanism may give the clarification of their novelty in the reaction.
The effect of doping heteroatoms to Au nanoclusters (NCs) for catalytic activities has been widely focused by several researchers.40–42 Notably, Toshima et al. reported that the degree of electron transfer in the bimetallic core–shell NCs is proportional to the visible-light-induced hydrogen generation from water in the EDTA/[Ru(bpy)3]2+/MV2+/metal NC system. They clarified that the bimetallic NC can accept electrons more easily than the monometallic one, which accelerated the rate-determining step and production of methyl viologen cation radicals.40 They also suggested that differences in the ionic potential between Pd and Pt could provide an uneven distribution of electrons, and the formed positive Pd shell in the Pt@Pd-PVP NCs favored the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the diene substrate and provided good catalytic activity.41 It has been said that the intermetallic electron transfer in the bimetallic NCs seems to play an important role in the acceleration of the rate-determining step.9,43–45
C double bond of the diene substrate and provided good catalytic activity.41 It has been said that the intermetallic electron transfer in the bimetallic NCs seems to play an important role in the acceleration of the rate-determining step.9,43–45
In this study, we succeeded in the synthesis of extremely active Au60Pd40-PVP/HT possessing the highest turnover number (=395700) for aerobic oxidation of 1-phenylethanol (250 mmol). Thereafter, transmission electron microscopy, X-ray photoelectron spectroscopy, X-ray absorption spectroscopy, Michaelis–Menten kinetic studies for alcohol oxidation and other analytical techniques were employed in order to realize the novelty of the synthesized AuxPdy-PVP/HTs. By taking advantage of morphological and electronic analysis of AuPd-PVP NPs with kinetic studies in aerobic oxidation of alcohols with and without radical scavengers, it was also revealed that the remarkable activity of the AuxPdy-PVP/HT catalysts was strongly contributed by the electron rich Au 5d states, since the formed negative Au species induces the active radical-like peroxo-species formation which accelerated the rate-determining step of the alcohol oxidation.
Experimental
Chemicals and materials
Hydrogen tetrachloroaurate tetrahydrate (HAuCl4·4H2O), palladium chloride (PdCl2), potassium chloride (KCl), ethylene glycol (EG), sodium carbonate decahydrate (Na2CO3·10H2O) and benzyl alcohol were supplied by Wako Pure Chemical Ind., Ltd. Co. PVP (M.W. = 58000) and MgAl HT (Mg/Al = 5) were purchased from Acros Organics Co. and Tomita Pharmaceutical Co., Ltd., respectively. 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO) and 1-phenylethanol were provided by Tokyo Chemical Ind. Co. Ltd. 2,6-Di-tert-butyl-p-cresol, naphthalene and toluene were purchased from Kanto Chem. Co. Ltd.
Synthesis of the AuPd-PVP/HT catalyst
The AuxPdy-PVP NCs with various Pd content were prepared by the polyol reduction method according to the previous report46 with some modifications. Briefly, an aqueous solution (50 ml) of PdCl2 (y mmol) including KCl (0.1 g) and HAuCl4·4H2O (x mmol) were mixed with PVP (0.58 g) and EG (50 ml), then the obtained mixture was refluxed for 2 h. Thereafter, HT (1.0 g) was added to the formed colloidal dispersion to stabilize the formed AuxPdy-PVP NCs onto the surface of HT with stirring. The obtained precipitates were filtered, washed and dried in a vacuum overnight. The Pd content was varied in the range of 0 to 100, and the total amount of both metals in the mixed solution (x + y) was kept as 0.1 mmol; i.e. the prepared AuxPdy-PVP/HT catalysts contain 0.1 mmol metal per gram in stoichiometry.Aerobic oxidation of alcohols
All reactants and solvents were purified before use. Oxidation reactions were carried out in the glass tube attached to a reflux condenser. In a general procedure, 2 mmol of alcohol in 5 ml of toluene and the AuxPdy-PVP/HT catalyst were added to the glass tube, and purged with O2 flow before reaction under stirring (500 rpm). Subsequently, the mixture was stirred at desired temperature for a given time under O2 flow (20 ml min−1) at atmospheric pressure. After the reaction, resultant solution was filtered off with a Millex-LG 0.20 μm syringe. The products were analyzed by GC equipped with a DB-FFAP (30 m length, 0.25 mm id) or a DB-1 (30 m length, 0.25 mm i.d.) column with an FID detector using the internal standard curve method. The naphthalene was used as an internal standard to determine the conversion and yield.Characterizations
Transmission electron microscopy (TEM) data were acquired using a Hitachi H-7650 operating at 100 kV. Energy-dispersive X-ray (EDS) and the scanning TEM-high angle annular dark field (STEM-HAADF) analyses were performed with a JEOL JEM-ARM200F operating at 200 kV. Ultraviolet and visible (UV-vis) spectra were measured by a Perkin-Elmer Lambda35 spectrometer at room temperature with a light path length of 1 cm. X-ray photoelectron spectroscopy (XPS) was measured on a Shimadzu Kratos AXIS-ULTRA DLD spectrometer using an Al target at 15 kV and 10 mA. The binding energies were calibrated with the C 1s level (284.8 eV) as the internal standard reference. Induced couple plasma spectroscopy (ICP) was recorded with a Shimadzu ICPS-7000 Ver.2. The contents of Pd and Au on the catalyst were estimated by the standard curve method. X-ray absorption near edge structure (XANES) measurement was performed at the BL01B1 in SPring-8 of Japan Synchrotron Radiation Research Institute (JASRI). The Au L3-edge XANES spectra were recorded at room temperature using a Si(111) monochromator.Results and discussion
Aerobic oxidations of 1-phenylethanol to acetophenone were carried out with AuxPdy-PVP/HTs with various Pd content. The results are summarized in Table 1. Size distributions of the AuxPdy-PVP/HTs were also listed in Table 1, and these values were similar among AuxPdy-PVP/HTs except for Au100-PVP/HT (aggregation) (see Fig. S1, ESI†). Au100-PVP/HT showed no activity (entry 1). The bimetallic AuxPdy-PVP/HTs exhibited different activities, and Au60Pd40-PVP/HT achieved the most significant activity among AuxPdy-PVP/HTs (entries 2–5). The excellent yields with Au60Pd40-PVP/HT were also obtained even at 300 K (>99% yield, entry 3h) and under air conditions (>99 yield, entry 3i). It is likely that the Pd content in the AuxPdy-PVP/HTs has a strong influence on catalytic activity for aerobic oxidation of 1-phenylethanol. We also tested their reusability in aerobic oxidation of 1-phenylethanol, and found that they could be reused without significant loss of activity and selectivity after washing with acetone followed by a 10 wt% Na2CO3 aqueous solution and drying in a vacuum (Fig. S2, ESI†). The reaction over the Pd100-PVP/HT catalyst was rarely proceeded regardless of the small particle size (2.6 nm) whereas the bare Au NPs stabilized on the HT (Au100/HT) catalyst with a similar size distribution (2.6 nm) and metal amount (0.075 Au mmol g−1) showed little activity (entry 7). It is supposed that the Au atoms in AuxPdy-PVP NPs mainly act as an active site for the alcohol oxidation. Furthermore, scope for Au60Pd40-PVP/HT in aerobic oxidation of various alcohols to the corresponding aldehydes or ketones under mild reaction conditions was also examined. Though the presence of substituents such as p-H3CO– and p-Cl– on the aromatic ring affects the yield of the product (vide infra), the Au60Pd40-PVP/HT catalyst allows adaptive oxidation for various alcohol substrates (Table S1, ESI†).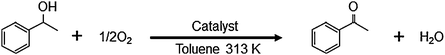
| Entry | Catalyst | Conv.b (%) | Yieldb (%) | Particle size (nm)c | Metal amount (mmol g−1)d | |
|---|---|---|---|---|---|---|
| Au | Pd | |||||
| a Reaction conditions: 1-phenylethanol (2 mmol), catalyst (0.2 g; Au + Pd = 0.02 mmol), mole ratio of alcohol/(Au + Pd) = 100 , toluene (5 ml), 313 K, 1 h, O2 flow (20 ml min−1). b Analyzed by GC using an internal standard technique. c Determined by TEM measurement about 500 NPs for each sample. d Estimated with ICP analysis. e Reduced by KBH4. f 1-phenylethanol (4 mmol). g 300 K, 3 h. h Air purge, 12 h. | ||||||
| 1 | Au100-PVP/HT | 2 | 0 | Agglomerate | 0.075 | 0 |
| 2 | Au80Pd20-PVP/HT | 100, 92f | 99, 92f | 3.1 | 0.115 | 0.034 |
| 3 | Au60Pd40-PVP/HT | 100, 100f, 100g, 100h | >99, >99f, >99g, >99h | 2.6 | 0.054 | 0.042 |
| 4 | Au40Pd60-PVP/HT | 58 | 57 | 2.6 | 0.052 | 0.098 |
| 5 | Au20Pd80-PVP/HT | 19 | 19 | 2.6 | 0.023 | 0.135 |
| 6 | Pd100-PVP/HT | 2 | 0 | 2.6 | 0 | 0.154 |
| 7 | Au100/HTe | 18 | 18 | 2.6 | 0.075 | 0 |
The turnover number (TON) and turnover frequency (TOF) of the oxidation of 1-phenylethanol (250 mmol) into acetophenone were up to 395700 and 69100 h−1, respectively, at 423 K for 24 h in the absence of solvents with 35% yield and 95% selectivity.47,48 These values are comparable to the previous reports (detailed information is listed in Table S2, ESI†); i.e. Au/HT (93% yield, TON = 200000, TOF = 8300 h−1),36 Au/CeO2 (TON = 250000 (after three recycles), TOF = 12500 h−1),12,13 PdHAP (37.8% yield, TON = 236000, TOF = 9800 h−1),49 and Au@Pd/TiO2 (TOF = 269000 h−1).19 These results clearly show that the present Au60Pd40-PVP/HT catalyst is an excellent catalyst for oxidation of alcohols. Investigation of different supports with Au60Pd40-PVP NCs also provided the novelty of the combination of AuPd bimetallic NCs and HT as a catalyst for alcohol oxidation reaction (Table S3, ESI†).
To clarify the reaction pathway over the Au60Pd40-PVP/HT, investigations using radical scavengers were performed. From the results shown in Fig. 1, it was found that TEMPO moderately influenced the oxidation rate of 1-phenylethanol. It has been reported that the alcohol oxidation over the monometallic metal supported catalysts proceeds by the metal-alcohoxide formation mechanism, which is not affected by the radical scavengers.16–18,37,49 Interestingly, our finding is different from such previous studies over monometallic catalysts (vide infra). Additionally, if the dominant reaction mechanism involves the formation of carbon-centered free radical intermediates over the AuxPdy-PVP/HT catalyst, the yield of acetophenone should be unchanged by such a small amount of the TEMPO radical scavenger (0.0032 mmol). The 2,6-di-tert-butyl-p-cresol (0.0454 mmol) also influenced the oxidation rate of 1-phenylethanol (67% yield after 1 h reaction). These results implied that another radical intermediate was seemed to be formed during the reaction (vide infra).
 | ||
| Fig. 1 Time-course of aerobic oxidation of 1-phenyl ethanol in the absence (closed circle) or presence (open circle) of TEMPO. Reaction conditions: 1-phenylethanol (2 mmol), Au60Pd40-PVP/HT catalyst (0.2 g), TEMPO (0 or 0.5 mg), toluene 5 ml, 313 K, O2 flow (20 ml min−1). | ||
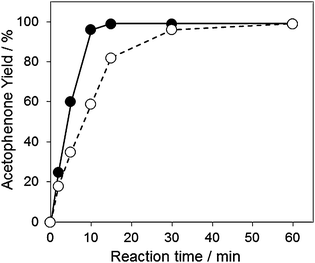
From comparison of the reaction rate between primary and secondary alcohols, the oxidation of 1-phenylethanol (TOF = 755 h−1) was faster than that of benzyl alcohol (TOF = 552 h−1), individually (Fig. S3, ESI†). In contrast, benzyl alcohol (TOF = 315 h−1) was oxidized faster than 1-phenylethanol (TOF = 69 h−1) using an equimolar mixture of alcohols (intermolecular competitive oxidations) (Fig. S4, ESI†). These observations were also reported previously.14,17 It is said that the faster oxidation of primary alcohol than secondary alcohol in competitive oxidation presumably supported the formation of metal-alcoholate intermediate species through the ligand exchange.50–52 To determine the presence of a carbocationic character in the transition state of the reaction, the logarithm of the rate constants, log(kX/kH), against substituent constant (σ) reported by Hammet53 was plotted for the aerobic oxidation of p-substituted benzyl alcohols. As shown in Fig. 2, there is a reasonable linearity between log(kX/kH) and the σ parameter in the following order of the reactivity; p-CH3O > p-CH3 > p-H > p-Cl > p-NO2. The Hammet ρ value was −0.240 (R2 = 0.96). The negative value indicates that a carbocationic character on the benzylic carbon is an intermediate species in the transition state of the alcohol oxidation over the AuxPdy-PVP/HT catalysts. Consequently, it is likely that the oxidation of alcohols over the AuxPdy-PVP/HT catalysts progressed via the alkoxide intermediate.
 | ||
| Fig. 2 Hammet plots for oxidation of benzyl alcohol and p-substituted benzyl alcohols. Reaction conditions: alcohol (0.5 mmol), toluene (5 ml), Au60Pd40-PVP/HT catalyst (10 mg), 313 K, 5 min, O2 flow (20 ml min−1). | ||
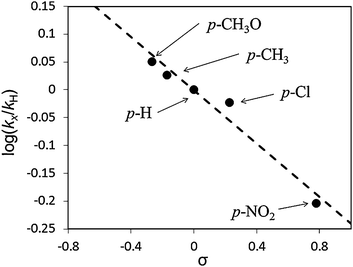
In order to understand the superiority of the Au60Pd40-PVP/HTs for alcohols oxidation, their morphologies and electronic states were also investigated. Fig. 3 shows the STEM-HAADF image and STEM-EDS line analysis of the Au60Pd40-PVP NCs. Since the STEM-HAADF analysis technique offers enhanced contrast proportional to Z2, the heavier Au atoms (atomic number; Z = 79) give rise to a brighter image than the lighter Pd atoms (Z = 46). However, the STEM-HAADF image of Au60Pd40-PVP NCs looks uniform contrast (Fig. 3(A)). As the result of quantitative analysis of the EDS profile, the Au60Pd40-PVP NCs were likely composed of homogeneously mixed 60.2% Au and 39.8% Pd atoms (Fig. 3(B)). These values agreed well with the stoichiometric ratio in the Au60Pd40 NC. Moreover, other components of AuxPdy-PVP NCs show similar images in the STEM-HAADF and the STEM-EDS line analyses (Fig. S5, ESI†), and the latter proposed that their Au/Pd ratios also suit the stoichiometric ratio of Au/Pd for each. The detailed morphologies of AuxPdy-PVP NCs could not be resolved due to the extremely small size of the NCs and the difficulty in obtaining electron beam diffraction. The UV-vis spectra of the AuxPdy-PVP NCs dispersed solutions show no specific surface plasmon (SPR) absorption of the Au NPs around 520–580 nm54 whereas the Au100-PVP NPs show the SPR feature at 527 nm (Fig. S6, ESI†). As mentioned above, the unimodels and narrow size distributions among AuxPdy-PVP NCs with various Pd content were observed (Fig. S1, ESI†). If the alloy is not formed, the size related to the isolated Au NPs will also be obtained. Therefore, distribution would lead to a broad or bimodal one due to different growth rates for the two metal NPs and the SPR phenomena. According to these results, it was suggested that the compositions of the AuxPdy-PVP NCs were different from that of Au or Pd mother clusters and the mixtures of isolated Au and Pd NCs, which indicates that each particle contains both Au and Pd elements in the form of alloy phase. It seems to be possible to form the alloy via the spontaneous alloying mechanism even at the low temperature.55–57
 | ||
| Fig. 3 (A) STEM-HAADF image and (B) STEM-EDS line analysis of the Au60Pd40-PVP NCs. The blue and green lines are corresponding to the presence of Au and Pd elements, respectively. | ||
The XPS spectra of AuxPdy-PVP NCs were measured in order to discuss the charge transfer between two metals. The peaks around Au 4f components are shown in Fig. 4(A). The humped peaks were attributed to Au 4f7/2 (at around 83 eV) and 4f5/2 (at around 87 eV). All AuxPdy-PVP NCs exhibited negative shifts in the both Au 4f binding energies compared to that of pure Au foil (84.0 and 88.0 eV).58 Importantly, increasing the Pd content induced more shifts in binding energy to the lower energy side (Fig. 4(B)). Thus, it is supposed that the charge transfer from Pd to Au atoms was facilitated by increase in Pd content of AuxPdy-PVP NCs. The largest negative shift in Au 4f7/2 was −1.96 eV in the case of Au20Pd80-PVP NCs. The negative shifts of binding energy in the Au 4f7/2 peak were also reported in the crown-jewel-structured Au12/Pd147-PVP NCs (−1.5 eV),9 Au90-Ag10-PVP alloy NCs (−1.4 eV),43 Au1@Pt4-Rh20-PVP NCs (−0.15 eV),44 and Au90Pt5Ag5-PVP NCs (−1.4 eV).45 The large number of negative shift in the Au 4f peak supposed that the AuxPdy-PVP NCs have a lot of agglutinations between Au and Pd atoms, which increased the opportunity for electron transfer from Pd to Au atoms. While, the XPS peaks in Pd 3d were hard to analyze because of weak intensities and energy overlapping at 335.0 eV corresponding to Pd 3d5/2 and Au 4d5/2 (Fig. S7, ESI†).58 It was reported that the Pd NCs with a diameter less than 2.0 nm have an amorphous structure whereas those with a diameter more than 2.5 nm have crystalline structures.59 It is indicated that the phase transfer of Pd NCs was found between 2.5 and 4.0 nm. In our case, the average size of AuxPdy-PVP NCs was below 2.7 nm. Therefore, the Pd atoms in the AuxPdy cluster supposedly have the amorphous structure of Pd aggregates and/or the less ordered AuPd structure, which makes the Pd related peaks broaden. From XPS analysis, the electron transfer from Pd to Au was elucidated, however, the correlation between the electron transfer and their catalysis still remains unclear.
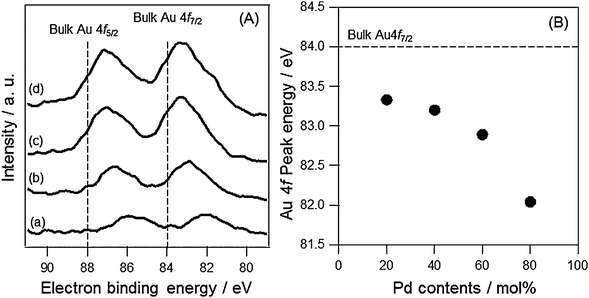 | ||
| Fig. 4 (A) XPS spectra of (a) Au20Pd80-PVP, (b) Au40Pd60-PVP, (c) Au60Pd40-PVP and (d) Au80Pd20-PVP NCs around Au 4f components, and (B) plots of the peak positions of Au 4f7/2 as a function of Pd content. | ||
To confirm the presence of the electronegative Au atoms in AuxPdy-PVP NCs, Au L3-edge XANES spectra were also measured for AuPd-PVP/HTs, as shown in Fig. 5(A). The white-line (WL) feature in the L3-edge spectrum is related to the transition of 2p electron to unoccupied 5d electron states. Though the unperturbed Au atom possesses no holes in the 5d orbital (electron configuration [Xe]6s14f145d10), the Au bulk L3-edge XANES spectra exhibit the WL feature due to the s–p–d hybridization which leads to the small amount of electron transformation from 5d to s–p states.60–62 The WL features around 11.925 eV in the AuxPdy-PVP/HTs indicated lower intensities than Au foil (Fig. 5(A)). Therefore, it was supposed that the AuxPdy-PVP/HTs contained more 5d electrons than Au foil. To further elucidate the differences in the 5d electron densities among AuxPdy-PVP/HTs, the areas of WL features in AuxPdy-PVP/HTs were calculated. Because the width in the WL feature depends on the lifetime of core holes, the dipole-transition matrix element, and the distribution of the density of states of the unoccupied d band at the Fermi level of the element,60 the range from 10 eV below the X-ray absorption edge (Eo) to 13 eV above Eo was applied for calculation.63,64 Here, μ3 = 107.7 cm2 g−1 and ρ = 19.32 g cm−3 are the X-ray absorption cross section at L3-edge jump and the density of Au, respectively. The areas as a function of Pd content were plotted with the yield of acetophenone in Fig. 5(B). The area of Au L3-edge XANES spectrum was attributed to the amount of 5d holes in Au atoms. In other words, the area decreased with increasing the 5d electrons. Therefore, it was clarified that Au60Pd40-PVP/HT possessed the largest number of the 5d electron-rich Au atoms among AuxPdy-PVP/HTs. Relationships between the areas and yield of acetophenone as a function of Pd content implied that the electron density of 5d states in Au atoms strongly contributed to the catalytic activity for aerobic oxidation (Fig. 5(B)). One other important feature is that the increase of intensities with increasing Pd content at around 11.935 keV was also observed in the Au L3-edge XANES spectra (Fig. 5(A)). It seemed to be evidence of the homogeneously-mixed AuPd alloy formation because these features were relatively sensitive to the interatomic redistribution of charge when Au and Pd atoms come closer together or further apart in AuPd NPs.65–68
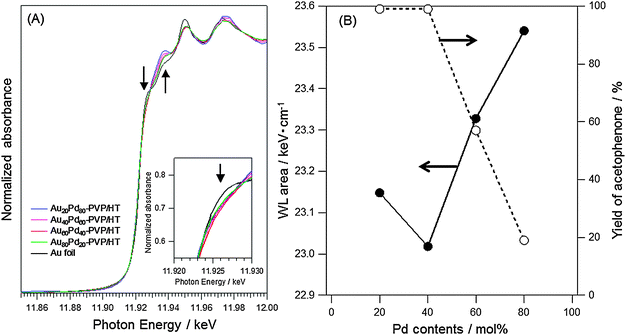 | ||
| Fig. 5 (A) The changes in Au L3-edge XANES spectra and (B) correlations between the area in the range of −10 eV < Eo < 13 eV among AuxPdy-PVP/HT catalysts with various Pd content. | ||
We proposed the catalytic cycle for alcohol oxidation over the AuxPdy-PVP/HT catalysts in Fig. 6, which proceeds via alkoxide intermediates and radical-like peroxo-species. Firstly, an O2 molecule adsorbed onto the negatively charged Au site on AuxPdy NCs, then the adsorbed molecular O2 is dissociated through the electron donation from Au 5d to the antibonding 2π* orbital of O2 (AuO2− peroxo- and/or AuO22− superoxo-species formation). Activating the adsorbed molecules (e.g. CO and O2) on anionic and/or neutral Au clusters was also suggested in the literature based on the experiment and theoretical studies.69–73 This proposed step agrees well with Au-PVP NC catalysts for aerobic oxidation of alcohol.74 Secondary, an alcohol molecule is dissociatively adsorbed on Au and/or Pd affording [metal-alkoxide]− (oxidative addition). The HT basicity promotes this process owing to abstraction of proton from alcohol involving [H–HT]+ formation. Third, the H atom on the α-carbon of the adsorbed alkoxide is transformed into the Au-peroxo- (and/or superoxo-) species (β-hydrogen elimination), then the corresponding carbonyl compound and Au-hydroperoxide species were formed.75 Finally, another O2 attacked and removed the hydroperoxide species from the Au surface with the formation of H2O, thereby, the catalytic cycle was completed.76 The AuxPdy NCs seem to have key factors in this step since the β-hydrogen elimination is considered as the rate-determining step in the alcohol oxidation via alkoxide formation in the previous reports.14,18
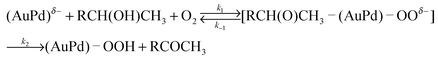 | (1) |
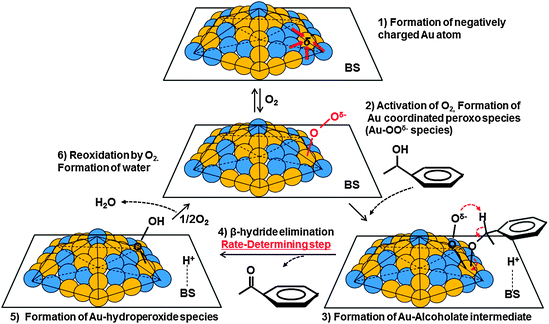 | ||
| Fig. 6 Proposed reaction mechanism of alcohol oxidation over AuxPdy-PVP/HT catalysts. | ||
According to the these reaction mechanisms, both the [RCH(O)CH3–(AuPd)–OOδ−] type of Michaelis complex and Michaels–Menten-type kinetics [eqn (1)] were assumed in the 1-phenylethanol oxidation over AuxPdy-PVP/HT catalysts. Because the Lineweaver–Burk plots in the oxidation of 1-phenyl ethanol over the Au60Pd40-PVP/HT catalyst showed good linear correlation between the inverse of initial rate (1/Ro) and the inverse of substrate concentration (1/substrate) (Fig. 7), the oxidation of 1-phenylethanol follows the Michaels–Menten-type kinetics. This is because the primary alcohol oxidation over the bare Au supported catalyst also follows Michaels–Menten-type kinetics [eqn (1)]. The alcohol oxidation over the bare Au100/HT catalyst also agrees well with the mechanism via the similar [RCH2O–Au] intermediate.14,37
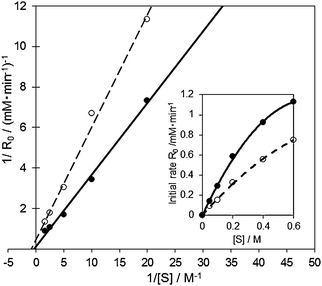 | ||
| Fig. 7 Lineweaver–Burk plot and Michaelis–Menten kinetics in the aerobic oxidation of 1-phenyl ethanol over the Au60Pd40-PVP/HT (solid line) and bare Au100/HT (dashed line) catalysts. Reaction conditions: 1-phenylethanol (0.25–3 mmol), catalyst (10 mg), toluene 5 mL, 313 K, 5 min, O2 flow (20 ml min−1). | ||
In the case of the Au60Pd40-PVP/HT catalyst, KM and k2 were calculated to be 3228 mM and 0.152 mM s−1, respectively, whereas over the bare Au100/HT catalyst, they were 1112 mM and 0.033 mM s−1. It was indicated that the rate of β-hydrogen elimination was drastically facilitated in the bimetallic AuPd-PVP/HT catalyst than that in the monometallic Au100/HT catalyst. To further investigate the reaction mechanism, the results of Lineweaver–Burk plot and Michaelis–Menten kinetics in the aerobic oxidation of 1-phenylethanol over the Au60Pd40-PVP/HT catalyst were compared in the presence and absence of TEMPO as a radical scavenger (Fig. 8). In the presence of TEMPO, both KM and k2 were decreased to 475 mM and 0.023 mM s−1, respectively, whereas the slopes in the Lineweaver–Burk plot (Km/Vmax) were similar irrespective of TEMPO. Therefore, TEMPO affected the reaction pathway by the uncompetitive inhibition mechanism; i.e. TEMPO is not combined with the active site but intermediate species. Since TEMPO cannot combine with the alkoxide intermediate,17,49 the prospective radical-like peroxo- species were protected by TEMPO, which makes the reaction slower. These results suggested that the remarkable activity of AuxPdy-PVP/HT catalysts for aerobic oxidation of alcohols might be originated from the negatively charged Au (5d states) generating the peroxo- and/or superoxo- species which strongly enhances the reaction rate in the β-hydrogen elimination of the alkoxide intermediates.
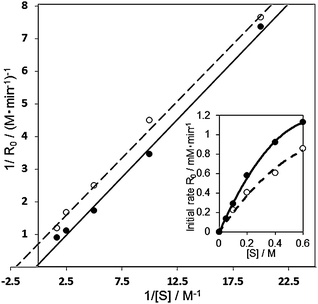 | ||
| Fig. 8 Lineweaver–Burk plot and Michaelis–Menten kinetics in the aerobic oxidation of 1-phenyl ethanol over the Au60Pd40-PVP/HT catalyst in the presence (solid line) and absence (dashed line) of the TEMPO radical scavenger. Reaction conditions: 1-phenylethanol (0.25–3 mmol), catalyst (10 mg), TEMPO (0.0032 mmol), toluene 5 ml, 313 K, 5 min, O2 flow (20 ml min−1). | ||
Conclusions
The effect of intermetallic charge transfer in bimetallic nanoparticles was examined by using AuxPdy-PVP NCs supported onto HT catalysts (AuxPdy-PVP/HT) with narrow particle size distributions. The Pd content in AuxPdy-PVP/HTs strongly influenced the activities for the aerobic oxidation of alcohols. The XPS analysis suggested that the electron transfer from Pd to Au atoms was facilitated by increase in Pd content of AuxPdy-PVP NCs. The close correlations between the electron density in the Au 5d state and reaction activity was obtained by the Au L3-edge XANES analysis of the AuxPdy-PVP/HTs. Moreover, the Michaels–Menten-type kinetic studies clearly indicated that the electron rich Au 5d states strongly accelerated the rate-determining step in the alcohol oxidation. These findings pointed out that design of electron transfer using heterometallic catalysts may form a guiding principle for further endeavors in the field of bimetallic nanocatalysis, and atomic-scale design of nanoparticles with remarkable catalytic activities.Abbreviations
| PVP | poly(N-vinyl-2-pyrrolidone) |
| PVA | poly(vinyl alcohol) |
| X@Y | X-core Y-shell (X, Y; element) |
Notes and references
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- A. Stephen, K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef CAS.
- G. J. Hutchings, Gold Bull., 1996, 29, 123–130 CrossRef CAS.
- M. Haruta, Nature, 2005, 437, 1098–1099 CrossRef CAS.
- T. Ishida and M. Haruta, Angew. Chem., Int. Ed., 2007, 46, 7154–7156 CrossRef CAS.
- P. Schwerdtfeger, Angew. Chem., Int. Ed., 2003, 42, 1892–1895 CrossRef CAS.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef.
- D. Xu, X. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS.
- T. Ebashi, Y. Ishida, Y. Nakagawa, S. Ito, T. Kubota and K. Tomishige, J. Phys. Chem. C, 2010, 114, 6518–6528 CAS.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. Garcia, Tetrahedron, 2006, 62, 6666–6672 CrossRef CAS.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS.
- K. Ebitani, K. Motokura, T. Mizugaki and K. Kaneda, Angew. Chem., Int. Ed., 2005, 44, 3423–3426 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353–4361 CrossRef CAS.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Tayor and G. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, S. Meenakshisub-daram, J. M. Anthonykutty, G. Brett, A. F. Carley, S. H. Taylor, D. W. Knight and G. J. Hutchings, Green Chem., 2009, 11, 1209–1216 RSC.
- A. Villa, C. Campione and L. Prati, Catal. Lett., 2007, 115, 133–136 CrossRef CAS.
- Y. Shi, H. Yang, X. Zhao, T. Cao, J. Chen, W. Zhu, Y. Yu and Z. Hou, Catal. Commun., 2012, 18, 142–146 CrossRef CAS.
- K. Kaizuka, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2010, 132, 15096–15098 CrossRef CAS.
- N. Toshima, M. Haruta, Y. Yamazaki and K. Asakura, J. Phys. Chem., 1992, 96, 9927–9933 CrossRef CAS.
- H. Liu, G. Mao and S. Meng, J. Mol. Catal., 1992, 74, 275–284 CrossRef CAS.
- J. K. Edwards, B. Solsona, N. E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kielyc and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiey and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917–1923 RSC.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS.
- J. H. Shim, J. Kim, C. Lee and Y. Lee, Chem. Mater., 2011, 23, 4694–4700 CrossRef CAS.
- M. Nie, P. K. Shen and Z. Wei, J. Power Sources, 2007, 167, 69–73 CrossRef CAS.
- T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545–5548 CrossRef CAS.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2012, 51, 136–139 CrossRef CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2009, 11, 793–797 RSC.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890–1896 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS.
- A. Tsuji, K. T. V. Rao, S. Nishimura, A. Takagaki and K. Ebitani, ChemSusChem, 2011, 4, 542–548 CrossRef CAS.
- N. Toshima, Pure Appl. Chem., 2000, 72, 317–325 CrossRef CAS.
- N. Toshima, M. Harada, T. Yonezawa, K. Kushihashi and K. Asakura, J. Phys. Chem., 1991, 95, 7448–7453 CrossRef CAS.
- H. Hakkine, S. Abbet, A. Sanchez, U. Heiz and U. Landman, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef.
- N. K. Chaki, H. Tsunoyama, Y. Negishi, H. Sakurai and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4885–4888 CAS.
- N. Toshima, R. Ito, T. Matsushita and Y. Shiraishi, Catal. Today, 2007, 122, 239–244 CrossRef CAS.
- H. Zhang, M. Okumura and N. Toshima, J. Phys. Chem. C, 2011, 115, 14883–14891 CAS.
- D. Ferrer, A. Torres-Castro, X. Gao, S. Sepulveda-Guzman, U. Ortiz-Mendez and M. Jose-Yacaman, Nano Lett., 2007, 7, 1701–1705 CrossRef CAS.
- The TON and TOF were estimated to be >682300 and 119260 h−1, respectively, assuming that only Au atoms could act as active sites in 0.2 g of the Au60Pd40-PVP/HT catalyst.
- Under the standard conditions for comparing the catalytic activities among heterogeneous catalysts (40 mmol scale of 1-phenylethanol), the TON was up to 207000 at 423 K for 24 h (>99% yield and selectivity).
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS.
- H. Tomioka, K. Takai, K. Oshima and H. Nozaki, Tetrahedron Lett., 1981, 22, 1605–1608 CrossRef CAS.
- S. Kanemoto, S. Matsubara, K. Takai, K. Oshima, K. Utimoto and H. Nozaki, Bull. Chem. Soc. Jpn., 1988, 61, 3607–3612 CrossRef CAS.
- A. Hanyu, E. Takezawa, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 1998, 39, 5557–5560 CrossRef CAS.
- L. P. Hammet, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- H. Yoshida, H. Mori, M. Komatsu and K. Takeda, Appl. Phys. Lett., 1992, 61, 2173–2174 CrossRef.
- H. Yoshida and H. Mori, Phys. Rev. Lett., 1992, 69, 3747–3750 CrossRef.
- Y. Shimizu, K. S. Ikeda and S. Sawada, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075412 CrossRef.
- J. F. Moulder, W. F. Stickel, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, ed. J. Chastain, Perkin-Elmer Co., Minnesota, 1992 Search PubMed.
- J. S. Bradley, E. W. Hill, S. Behal, C. Klein, B. Chaudret and A. Duteil, Chem. Mater., 1992, 4, 1234–1239 CrossRef CAS.
- T. K. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 1888–1902 CrossRef CAS.
- L. F. Mattheiss and R. E. Dietz, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 1663–1676 CrossRef CAS.
- V. V. Nemoshkalenko, V. N. Antonov, W. John, H. Wonn and P. Ziesche, Phys. Status Solidi B, 1982, 111, 11–52 CrossRef.
- A. N. Mansour, J. W. Cook and D. E. Sayers, J. Phys. Chem., 1984, 88, 2330–2334 CrossRef CAS.
- S. Nishimura, A. T. N. Dao, D. Mott, K. Ebitani and S. Maenosno, J. Phys. Chem. C, 2012, 116, 4511–4516 CAS.
- M. O. Pedersen, S. Helveg, A. Ruban, I. Stensgaard, E. Laegsgaard, J. K. Norskov and F. Besenbacher, Surf. Sci., 1999, 426, 395–409 CrossRef CAS.
- S. N. Reifsnyder and H. H. Lamb, J. Phys. Chem. B, 1999, 103, 321–329 CrossRef CAS.
- P. Dash, T. Bond, C. Fowler, W. Hou, N. Coombs and R. W. J. Scott, J. Phys. Chem. C, 2009, 113, 12719–12730 CAS.
- T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425–436 CrossRef CAS.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS.
- L. D. Socaciu, J. Hagen, T. M. Bernhardt, L. Woste, U. Hwiz, H. Hakkinen and U. Landman, J. Am. Chem. Soc., 2003, 125, 10437–10445 CrossRef CAS.
- D. Stolcic, M. Fischer, G. Gantefor, Y. D. Kim, Q. Sun and P. Jena, J. Am. Chem. Soc., 2003, 125, 2848–2849 CrossRef CAS.
- B. Yoon, H. Hakkinen and U. Landman, J. Phys. Chem. A, 2003, 107, 4066–4071 CrossRef CAS.
- B. E. Salisury, W. T. Wallace and R. L. Whetten, Chem. Phys., 2000, 262, 131–141 CrossRef.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tuskuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS.
- The electron transfer from the alkoxides to Au and/or Pd occurred during this step. The formation of Au-hydroperoxide species via β-hydride elimination through short life-timed hydride was also considerable though the presence of peroxo-species on the active sites might limit its formation.
- The H2O2 formation in the reaction medium could not be detected by an oxidation–reduction titration.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis, XPS, STEM-EDS, and results of alcohol oxidations. See DOI: 10.1039/c2cy20244a |
| This journal is © The Royal Society of Chemistry 2013 |