Green oxidative synthesis of primary amides from primary alcohols or aldehydes catalyzed by a cryptomelane-type manganese oxide-based octahedral molecular sieve, OMS-2†
Received
23rd March 2012
, Accepted 1st May 2012
First published on 2nd May 2012
Abstract
In this study, a new green synthetic route to primary amides, that is, aerobic oxidative amidation of primary alcohols or aldehydes with ammonia, has been developed. In the presence of a cryptomelane-type manganese oxide-based octahedral molecular sieve (OMS-2), various kinds of structurally diverse primary alcohols or aldehydes including aromatic, olefinic, heteroaromatic, and aliphatic ones can be converted into the corresponding primary amides in moderate to high yields (20 examples from primary alcohols and 11 examples from aldehydes). Furthermore, gram-scale amidation is also effective, and the analytically pure primary amides can easily be isolated. The present catalysis by OMS-2 is truly heterogeneous in nature, and the retrieved OMS-2 catalyst can be reused several times (at least 12 times for the amidation of 2-pyridinemethanol). Though the formation rates of the corresponding primary amide are gradually decreased by repeating reuse experiments, OMS-2 can be regenerated by calcination. The present OMS-2-catalyzed amidation of primary alcohols is composed of four relay steps: (i) oxidative dehydrogenation of primary alcohols, (ii) dehydrative condensation of aldehydes with ammonia, (iii) oxidative dehydrogenation of aldimines, and (iv) hydration of nitriles to form the corresponding primary amides. All steps (i)–(iv) can be promoted by the presence of OMS-2.
1. Introduction
Amides are one of the most important compounds in modern chemistry as well as biology. They have widely been used as raw materials for engineering plastics, intensifiers of perfume, anti-block reagents, color pigments for inks, detergents, lubricants, and intermediates in peptide and protein synthesis.1 Even at present, aminolysis (or ammonolysis) of activated carboxylic acid derivatives with amines (or ammonia) is still being extensively utilized for synthesis of amides.2 However, this antiquated procedure requires stoichiometric reagents, e.g., thionyl chloride and carbodiimide, for pre-activation of carboxylic acids to acid chlorides or anhydrides, and at least equimolar amounts of by-products are formed during not only the aminolysis but also the pre-activation step.2 Thus, the development of green catalytic procedures for synthesis of amides from readily available and inexpensive starting materials is a very important subject.3
In 2007, Milstein and co-workers have reported dehydrogenative amidation of primary alcohols with amines catalyzed by a PNN pincer-type ruthenium complex.4 This amidation is composed of three relay steps: (i) dehydrogenation of primary alcohols, (ii) dehydrative condensation of aldehydes with amines to form hemiaminal intermediates, and (iii) dehydrogenation of the intermediates to the corresponding “secondary amides”.4–8 This amidation has several advantages: e.g., (i) readily available and inexpensive primary alcohols can be used as starting materials and (ii) only hydrogen (gas) is formed as a by-product. Since the Milstein's breakthrough work, this exceptionally green procedure has been emulated by using several precious metal-based complexes6,7 or heterogeneous catalysts.8
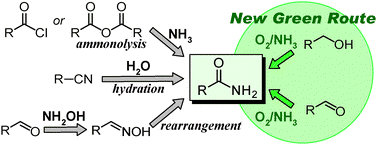
With regard to synthesis of “primary amides” (Fig. 1), hydration of nitriles9,10 and rearrangement of aldoximes11 are attractive procedures. Currently, benzonitrile (and its derivatives) and acrylonitrile are being industrially produced by ammoxidation. Thus, for synthesis of benzamide derivatives and acrylamide, hydration is a reliable procedure from both environmental and economical viewpoints. In contrast, other nitriles have generally been synthesized by costful non-green procedures,12 and hydration is not the best choice in these cases. Although the atom efficiency of rearrangement of aldoximes itself is theoretically 100%, the formation of hydrochloric acid or sulfuric acid is typically inevitable for preparation of aldoximes.
Amidation of alcohols with “ammonia” is attractive but a very difficult reaction. Until now, only a few systems for amidation of alcohols with ammonia have been reported.7,13,14 Togo and co-workers have reported metal-free amidation of primary alcohols with aqueous ammonia using I2 and hydrogen peroxide.13 We have also reported the step-by-step amidation of benzyl alcohol in the presence of a supported ruthenium hydroxide catalyst, Ru(OH)x/Al2O3.14 In the amidation with a rhodium diolefin amido complex, methylmethacrylate (3 equiv. with respect to alcohols) is required as a hydrogen acceptor, and easily-handled aqueous ammonia is not used.7a Although various secondary amides can be synthesized from alcohols and amines with a gold/DNA catalyst in the presence of LiOH·H2O (indispensable additive, 1.1 equiv. with respect to alcohols), the applicability of this system to synthesis of primary amides is only limited to benzamide, and the yield is low (50% yield).7b Subsequently, Kobayashi and co-workers reported a similar system using gold and gold-based nanoparticles.7c The applicability of Kobayashi's system is also limited to benzamide, and NaOH (1 equiv.) is required to promote the reaction. Thus, the development of widely usable, efficient procedures for aerobic oxidative amidation of primary alcohols using ammonia (without any additives) is still a challenging subject. In particular, the development of heterogeneously catalyzed amidation with readily available and inexpensive aqueous ammonia is highly desirable.
Very recently, we have reported that amidation of primary alcohols with “aqueous ammonia” is efficiently promoted by a cryptomelane-type manganese oxide-based octahedral molecular sieve (having the 2 × 2 hollandite structure with a one-dimensional pore, Fig. 2, KMn8O16, OMS-2‡15).16 Herein, we report the full details of the OMS-2-catalyzed aerobic oxidative amidation of primary alcohols or aldehydes with aqueous ammonia, e.g., the effects of catalysts, solvents, and nitrogen sources, the extension of the synthetic scope, the possible reaction path, and the role of OMS-2.
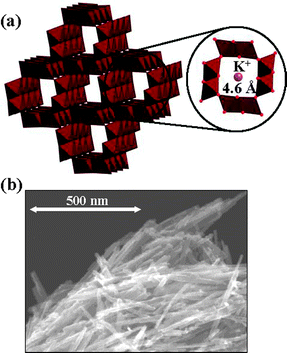 |
| | Fig. 2 (a) Structure and (b) SEM image of OMS-2. | |
2. Experimental
2.1 Instruments and reagents
GC analyses were performed on Shimadzu GC-2014 or GC-17A with a FID detector equipped with a TC-5, InertCap 5, or Rxi-5 Sil capillary column. GC mass spectra were recorded on Shimadzu GCMS-QP2010 at an ionization voltage of 70 eV. Liquid-state 1H and 13C NMR spectra were recorded on JEOL JNM-EX-270. 1H and 13C NMR spectra were measured at 270 and 67.8 MHz, respectively, with TMS as an internal standard (δ = 0 ppm). ICP-AES analyses were performed with Shimadzu ICPS-8100. BET surface areas were measured on micromeritics ASAP 2010 and calculated from the N2 adsorption isotherm with the BET equation. NH3-TPD profiles were measured on BEL Japan Multitask TPD with a quadrupole mass spectrometer. The sample was pretreated (evacuated at 150 °C for 1 h) and NH3 (2.7 kPa at 100 °C for 1 h) was adsorbed. After excess NH3 was evacuated, He (carrier gas) was allowed to flow into the cell (60 mL min−1). After the baseline was stabilized, the temperature was linearly increased to 800 °C at a rate of 10 °C min−1. The desorbed NH3 was quantified by a mass spectrometer with the fragment of m/z = 16. Commercially available CeO2 (Cat. No. 544841-25G, Aldrich) and CuO (Cat. No. 07503-30, Kanto Chemical) were used. Co3O417 and Ru(OH)x/Al2O314 were prepared according to the literature procedures. KMnO4 and MnSO4·H2O were obtained from Kanto Chemical and Aldrich (reagent grade), respectively, and used as received. Solvents and substrates were obtained from Kanto Chemical, TCI, Wako, or Aldrich (reagent grade), and purified prior to the use.18
2.2 Manganese-based oxides
OMS-2 was prepared as follows:15a an aqueous solution of KMnO4 (5.89 g, 100 mL) was added to an aqueous solution (30 mL) containing MnSO4·H2O (8.8 g) and concentrated HNO3 (3 mL). The resulting mixture was refluxed at 100 °C for 24 h. Then, the dark brown solid formed was filtered off, washed with a large amount of water (ca. 3 L), and dried under air at 120 °C, affording 8.2 g of OMS-2. Amorphous MnO2 was prepared as follows: an aqueous solution of KMnO4 (5.89 g, 100 mL) was added dropwise to an aqueous solution of MnSO4·H2O (8.8 g, 30 mL). The resulting mixture was stirred at room temperature for 10 min. Then, the dark brown solid formed was filtered off, washed with a large amount of deionized water (ca. 4 L), and dried under air at 150 °C for 12 h, affording 5.0 g of amorphous MnO2. Commercially available β-MnO2 (Cat. No. 133-09681, Wako) was used. Birnessite-type MnO2 was prepared according to the literature procedure.19 The properties and NH3-TPD profiles of manganese-based oxides used in this study (BET surface areas and amounts of acidic sites) are summarized in Table 1 and Fig. S1 (ESI†), respectively.
Table 1 Properties of manganese-based oxides used in this study
| Catalyst |
BET surface area (m2 g−1) |
Amount of acidic sitesa (mmol g−1) |
|
Determined by NH3-TPD measurements.
|
| OMS-2 |
138 |
1.23 |
| Amorphous MnO2 |
304 |
1.04 |
| Birnessite-type MnO2 |
36 |
0.33 |
| β-MnO2 |
54 |
0.36 |
2.3 Catalytic reactions
Oxidative amidation of primary alcohols was typically carried out as follows: OMS-2 (100 mg), 1,4-dioxane (2 mL), benzyl alcohol (1a, 0.5 mmol), and aqueous ammonia (28 wt%, 100 μL, ca. 2.6 equiv. with respect to 1a) were placed in a Teflon vessel with a magnetic stir bar. The Teflon vessel was attached inside an autoclave, and the reaction was carried out at 130 °C (bath temp.) in 3 atm of O2. The reaction rates were not affected by stirring rates from 500 to 1000 rpm. After the reaction was completed (3 h), the spent OMS-2 catalyst was separated by filtration (>95% catalyst recovery) and washed with ethanol and acetone. Then, the combined filtrate was analyzed by GC. The corresponding primary amide was isolated by evaporation of volatiles, followed by rinsing with n-hexane. The retrieved OMS-2 was washed with water, and then dried under air at 150 °C for 1 h prior to being used for the reuse experiment. Other transformations were carried out via a similar procedure. The products were confirmed by comparison of their GC retention times, GC mass spectra, and/or 1H and 13C NMR spectra with those of authentic data.
3. Results and discussion
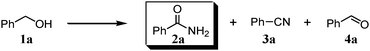
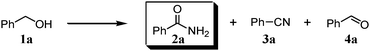
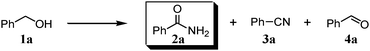
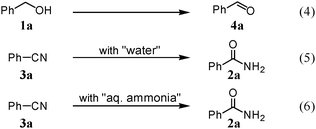
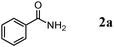
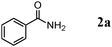
We expected that amidation of primary alcohols with ammonia can be realized through the dehydrogenation–hydration sequence. Thus, various kinds of active catalysts for both alcohol oxidation15b,20 and nitrile hydration10 were initially applied to the amidation of benzyl alcohol (1a) to benzamide (2a) in 1,4-dioxane using aqueous ammonia and O2 (Table 2). We found that manganese-based oxides gave the corresponding amide 2a, and that OMS-2 showed the best result. For example, when the amidation of 1a was carried out using aqueous ammonia (28 wt%, ca. 2.6 equiv. with respect to 1a) and O2 (3 atm) at 130 °C (bath temp.), 2a was obtained in 84% yield after 1 h with formation of benzonitrile (3a, 16% yield). Amorphous MnO2 prepared with KMnO4 and MnSO4·H2O (the same precursors as those for OMS-2) gave a moderate yield of 2a (40% yield). In the case of other manganese-based oxides such as birnessite-type MnO2 and β-MnO2, 2a was hardly produced (3–6% yields), and 3a and benzaldehyde (4a) were formed in moderate yields. Co3O4 and CeO2 did not give 2a, and the corresponding nitrile 3a was formed as a major product (18–57% yields). In the case of CuO, the reaction hardly proceeded. KMnO4 and MnSO4·H2O (precursors for OMS-2) were not effective for the amidation, suggesting that soluble manganese species are not active for the present amidation. Although a supported ruthenium hydroxide catalyst, Ru(OH)x/Al2O3, has been reported to be effective for the step-by-step amidation of 1a,14 Ru(OH)x/Al2O3 did not afford 2a under the conditions described in Table 2 because of the low ability to drive nitrile hydration. 1,4-Dioxane and N,N-dimethylformamide (DMF) were good solvents likely due to the high solubilities of 1a, ammonia, and water (Table 3). Toluene, dichloromethane, 1,2-dichloroethane, and water gave 3a as a major product with 10–20% yields of 2a (Table 3). Taking the solvent effect and the easiness of product isolation into account, we subsequently carried out amidation in 1,4-dioxane.
| Entry |
Catalyst |
Conv. (%) |
Yield (%) |
|
2a
|
3a
|
4a
|
|
Conditions: catalyst (100 mg), 1a (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv. with respect to 1a), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 1 h. Yields were determined by using GC using biphenyl as an internal standard.
Mn salt (1 equiv.).
Ru(OH)x/Al2O3 (100 mg, Ru: 5 mol%).
|
| 1 |
OMS-2 |
>99 |
84 |
16 |
<1 |
| 2 |
Amorphous MnO2 |
>99 |
40 |
59 |
<1 |
| 3 |
Birnessite-type MnO2 |
78 |
6 |
68 |
4 |
| 4 |
β-MnO2 |
38 |
3 |
15 |
3 |
| 5b |
KMnO4 |
43 |
<1 |
2 |
21 |
| 6b |
MnSO4·H2O |
3 |
<1 |
<1 |
3 |
| 7 |
Co3O4 |
68 |
<1 |
57 |
6 |
| 8 |
CeO2 |
21 |
<1 |
18 |
3 |
| 9 |
CuO |
3 |
<1 |
<1 |
3 |
| 10c |
Ru(OH)x/Al2O3 |
43 |
<1 |
18 |
22 |
| Entry |
Solvent |
Conv. (%) |
Yield (%) |
|
2a
|
3a
|
4a
|
|
Conditions: OMS-2 (100 mg), 1a (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv. with respect to 1a), solvent (2 mL), O2 (3 atm), 130 °C (bath temp.), 1 h. Yields were determined by using GC using biphenyl as an internal standard.
|
| 1 |
1,4-Dioxane |
>99 |
84 |
16 |
<1 |
| 2 |
DMF |
>99 |
85 |
15 |
<1 |
| 3 |
Toluene |
>99 |
20 |
74 |
<1 |
| 4 |
Dichloromethane |
98 |
14 |
79 |
2 |
| 5 |
1,2-Dichloroethane |
59 |
15 |
42 |
2 |
| 6 |
Water |
79 |
10 |
52 |
17 |
Next, the amidation of 1a was carried out with various ammonia surrogates (Table 4). Besides aqueous ammonia, (NH4)2CO3 (a mixture of ammonium bicarbonate and ammonium carbamate) and NH4HCO3 could act as effective nitrogen sources for the present amidation and gave 2a in 95% and 86% yields, respectively. It was confirmed by separate experiments that (NH4)2CO3 and NH4HCO3 thermally decomposed to ammonia under the same conditions described in Table 4. Urea also gave 2a in a moderate yield. In contrast, other ammonium salts such as HCOONH4, CH3COONH4, NH4HPO4, (NH4)2SO4, NH4HSO4, NH4NO3, and NH4Cl were not effective because these salts hardly decomposed to ammonia under the present conditions.
| Entry |
Nitrogen source |
Conv. (%) |
Yield (%) |
|
2a
|
3a
|
4a
|
|
Conditions: OMS-2 (100 mg), 1a (0.5 mmol), nitrogen source (2.6 equiv. with respect to 1a), water (65 μL), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 3 h. Yields were determined by using GC using biphenyl as an internal standard.
Benzoic acid was formed as a by-product (10% yield).
|
| 1 |
28% aq. ammonia |
>99 |
96 |
2 |
<1 |
| 2 |
Urea |
89 |
34 |
49 |
<1 |
| 3 |
(NH4)2CO3 |
>99 |
95 |
4 |
<1 |
| 4 |
NH4HCO3 |
97 |
86 |
7 |
<1 |
| 5 |
HCOONH4 |
34 |
17 |
10 |
1 |
| 6 |
CH3COONH4 |
29 |
1 |
22 |
1 |
| 7 |
(NH4)2HPO4 |
57 |
1 |
20 |
29 |
| 8 |
(NH4)2SO4 |
96 |
<1 |
5 |
84 |
| 9 |
NH4HSO4 |
45b |
1 |
1 |
28 |
| 10 |
NH4NO3 |
63 |
<1 |
1 |
57 |
| 11 |
NH4Cl |
26 |
<1 |
<1 |
24 |
3.2 Heterogeneous catalysis, catalyst reuse, and regeneration
In order to verify whether the observed catalysis is derived from solid OMS-2 or leached manganese species, the amidation of 1a to 2a was carried out under the conditions described in Table 2, and OMS-2 was removed from the reaction mixture by filtration at ca. 50% conversion of 1a. Then, the filtrate was again heated at 130 °C in 3 atm of O2. In this case, no further production of 2a and 3a was observed. It was confirmed by ICP-AES analysis that no manganese species were detected in the filtrate (below 0.001%). In addition, soluble manganese species such as KMnO4 and MnSO4·H2O were not effective for the amidation of 1a, as mentioned above (entries 5 and 6 in Table 2). All these results can rule out any contribution to the observed catalysis from manganese species that leached into the reaction solution, and the observed catalysis for the present amidation is truly heterogeneous.21
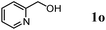
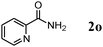
After the amidation was completed, OMS-2 was easily retrieved from the reaction mixture by simple filtration with >95% recovery. The retrieved OMS-2 catalyst could be reused for the amidation of 2-pyridinemethanol (1o); even for the twelfth reuse, 79% yield of 2o was still obtained (Fig. 3). Although the structure of OMS-2 was preserved even after the eleventh reuse experiment (Fig. 4), the formation rates of the corresponding primary amide 2o were gradually decreased by repeating reuse experiments (Fig. S2, ESI†). Fresh OMS-2 and OMS-2 after the twelfth reuse were characterized by means of N2 adsorption–desorption isotherms (Table 5 and Fig. S3, ESI†). The external and micropore surface areas of fresh OMS-2 were 117 m2 g−1 and 21 m2 g−1, respectively. After the twelfth reuse, the micropore was almost choked with carbonaceous materials (Table 5). This is not a serious problem because the present amidation does not proceed in the interior of OMS-2 (the pore diameter of OMS-2 is smaller than the kinetic diameters of substrates). Thus, the main reason for the deactivation is likely the deposition of carbonaceous materials on active site(s) on the external surface. By calcination at 300 °C for 1 h under an air atmosphere, the carbonaceous materials could completely be removed (burned out) (Table 5), and the structure of OMS-2 was also preserved (Fig. 4).
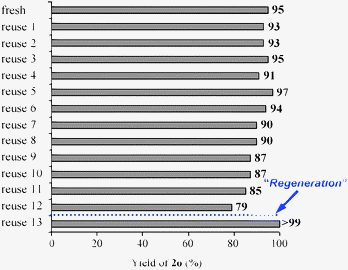 |
| | Fig. 3 Reuse experiments for the amidation of 2-pyridinemethanol (1o). The reaction was carried out with the retrieved OMS-2 catalyst under the conditions described in Table 2 for 1 h. | |
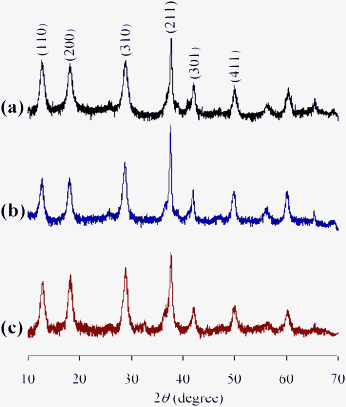 |
| | Fig. 4 XRD patterns of (a) as-prepared OMS-2, (b) OMS-2 retrieved after the eleventh reuse for the amidation of 2-pyridinemethanol (1o), and (c) OMS-2 regenerated after the twelfth reuse. The used OMS-2 was regenerated by calcination at 300 °C for 1 h under an air atmosphere. | |
Table 5 Characterization of fresh, used, and regenerated OMS-2
| OMS-2 |
Surface area/m2 g−1 |
Pore volume/cm3 g−1 |
Carbon content/wt% |
| External |
Micropore |
|
OMS-2 retrieved after the twelfth reuse for the amidation of 1o.
OMS-2 retrieved after the twelfth reuse was regenerated by calcination at 300 °C for 1 h under an air atmosphere.
|
| Fresh |
117 |
21 |
0.464 |
<0.01 |
| Useda |
102 |
<1 |
0.389 |
1.6 |
| Regeneratedb |
88 |
15 |
0.373 |
<0.01 |
When the amidation of 1o was carried out with the regenerated catalyst, the reaction rate and the final yield were almost the same as those with fresh OMS-2 (see the thirteenth reuse in Fig. 3).
3.3 Synthetic scope
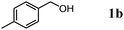
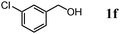
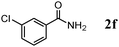
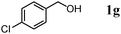
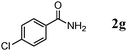
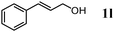
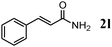
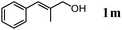
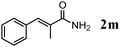
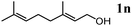
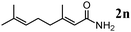
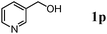
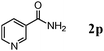
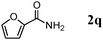
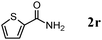
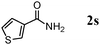
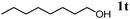
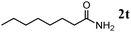
The scope of the present OMS-2-catalyzed amidation of primary alcohols was next examined. We have very recently reported the oxidative amidation of primary alcohols with aqueous ammonia.16 Herein, the scope is much extended to various kinds of structurally diverse primary alcohols, and ten new entries are added in Table 6.§22 The amidation of benzylic alcohols (1a–1j), which contain electron-donating as well as electron-withdrawing substituents at different positions, efficiently proceeded to afford the corresponding substituted benzamide derivatives in excellent yields (≥95% yields). The reaction rates for the amidation of ortho-, meta- and para-substituted benzyl alcohols (1c–1e) were almost the same, and the corresponding substituted benzamides were obtained in 93–98% yields. 1,2-Benzenedimethanol (1k) gave phthalimide (2k) as a major product (68% yield). In the amidation of aromatic allylic alcohols such as cinnamyl alcohol derivatives (1l and 1m), the corresponding aromatic unsaturated amides could be obtained in excellent yields (≥93% yields) without hydrogenation and hydration of the double bonds. In the case of the aliphatic allylic alcohol geraniol (1n), the corresponding aliphatic unsaturated amide was obtained in only 33% yield because of formation of unidentified by-products. Heteroaromatic alcohols such as pyridine (1o and 1p), furan (1q), and thiophene (1r and 1s) methanols gave the corresponding heteroaromatic amides in high yields (≥87% yields). Also, the aliphatic alcohol 1-octanol (1t) could be converted into the corresponding aliphatic amide in a moderate yield (65% yield).
| Entry |
Substrate |
Product |
Time (h) |
Yield (%) |
|
Conditions: OMS-2 (100 mg), primary alcohol (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv. with respect to substrate), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.). Yields were determined by using GC using naphthalene or biphenyl as an internal standard.
Phthalaldehyde was formed as a by-product (28% yield).
OMS-2 (200 mg).
|
| 1 |
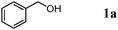
|
|
3 |
96 |
| 2 |
|
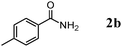
|
3 |
97 |
| 3 |
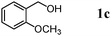
|
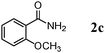
|
3 |
93 |
| 4 |
|
|
3 |
96 |
| 5 |
|
|
3 |
98 |
| 6 |
|
|
3 |
>99 |
| 7 |
|
|
3 |
97 |
| 8 |
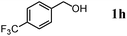
|
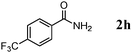
|
3 |
>99 |
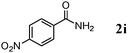
| 9 |
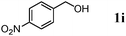
|
|
3 |
99 |
| 10 |
|
|
3 |
95 |
| 11 |
|
|
3 |
68b |
| 12 |
|
|
3 |
99 |
| 13 |
|
|
3 |
93 |
| 14c |
|
|
24 |
33 |
| 15 |
|
|
1 |
95 |
| 16 |
|
|
1 |
98 |
| 17 |
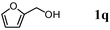
|
|
1 |
99 |
| 18 |
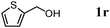
|
|
3 |
>99 |
| 19 |
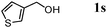
|
|
3 |
87 |
| 20c |
|
|
24 |
65 |
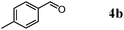
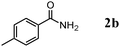
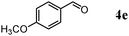
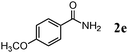
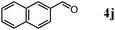
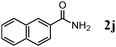
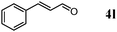
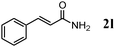
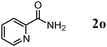
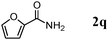
Besides primary alcohols, OMS-2 showed high catalytic activities and selectivities for amidation of various kinds of structurally diverse aldehydes including benzylic (4a, 4b, 4e, 4g, 4i, and 4j; ≥89% yields), olefinic (4l; 87% yield), heteroaromatic (4o, 4q, and 4r; ≥84% yields), and aliphatic (4t; 77% yield) ones to the corresponding primary amides in the presence of aqueous ammonia (Table 7).
| Entry |
Substrate |
Product |
Time (h) |
Yield (%) |
|
Conditions: OMS-2 (100 mg), aldehyde (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv. with respect to substrate), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.). Yields were determined by using GC using naphthalene or biphenyl as an internal standard.
OMS-2 (200 mg).
|
| 1 |
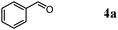
|
|
3 |
89 |
| 2 |
|
|
3 |
91 |
| 3 |
|
|
3 |
97 |
| 4 |
|
|
3 |
98 |
| 5 |
|
|
3 |
>99 |
| 6 |
|
|
3 |
>99 |
| 7 |
|
|
3 |
87 |
| 8 |
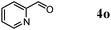
|
|
1 |
94 |
| 9 |
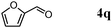
|
|
1 |
84 |
| 10 |
|
|
3 |
98 |
| 11b |
|
|
24 |
77 |
In order to show the practical usefulness of the present OMS-2-catalyzed amidation of primary alcohols to the corresponding primary amides, the gram-scale transformations of 1j (1.03 g) and 1p (1.09 g) were carried out (Scheme 1). These gram-scale transformations also efficiently proceeded without any decrease in the performance in comparison with the 0.5 mmol-scale transformations given in Table 6. After the amidation was completed, OMS-2 was separated by filtration and washed with ethanol and acetone. Evaporation of the combined filtrate gave crude mixtures (primary amides containing small amounts (1–2%) of remaining primary alcohols and nitriles). Primary amides are intrinsically insoluble, and the remaining primary alcohols and nitriles are soluble in n-hexane. Thus, the primary amide products could easily be purified by a simple rinse of crude mixtures with n-hexane, affording 1.06 g of 2j (95% isolated yield) and 1.09 g of 2p (89% isolated yield). The purities of isolated 2j and 2p were >99% (by GC and NMR analyses, no metal contamination).
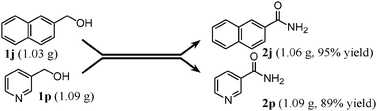 |
| | Scheme 1 Gram-scale amidation. Conditions for 1j: OMS-2 (1.3 g), 1j (1.03 g), 28% aq. ammonia (ca. 2.6 equiv.), 1,4-dioxane (26 mL), O2 (3 atm), 130 °C (bath temp.), 3 h. Conditions for 1p: OMS-2 (1 g), 1p (1.09 g), 28% aq. ammonia (ca. 2.6 equiv.), 1,4-dioxane (40 mL), O2 (3 atm), 130 °C, 2 h. | |
3.4 Possible reaction path and role of OMS-2
For the amidation of 1a, the addition of the radical scavenger 2,6-di-tert-butyl-p-cresol (5 mol% with respect to 1a) did not affect the reaction rate as well as the product selectivity (88% yield of 2a and 10% yield of 3a after 1 h, cf. entry 1 in Table 2), showing that free-radical intermediates are not involved in the present OMS-2-catalyzed amidation. When the transformation of 1a with methylamine (40% aq. solution) instead of aqueous ammonia was carried out, the corresponding secondary amide was not produced (reaction (1) in Scheme 2)¶, showing that the present amidation does not proceed through direct dehydrogenation of hemiaminal intermediates as has been reported for the precious metal-catalyzed Milstein's procedures.4,5 The rearrangement of p-methylbenzaldoxime hardly proceeded in the presence of OMS-2 (reaction (2) in Scheme 2), suggesting that the aldoxime path (oxidation of ammonia, followed by condensation with aldehydes) is negligible. In the case of the secondary alcohol 1-phenylethanol, only alcohol oxidation proceeded (reaction (3) in Scheme 2).
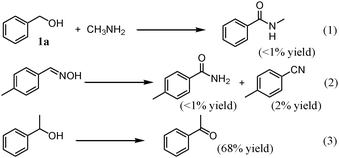 |
| | Scheme 2 Various transformations. Conditions for reaction (1): OMS-2 (100 mg), 1a (0.5 mmol), 40% aq. methylamine (ca. 2.6 equiv.), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temperature), 3 h. Conditions for reaction (2): OMS-2 (100 mg), aldoxime (0.5 mmol), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 3 h. Conditions for reaction (3): OMS-2 (100 mg), alcohol (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv.), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 1 h. | |
The reaction profile for the amidation of 1a to 2a showed that the corresponding nitrile 3a was initially produced as a major product, followed by formation of 2a (Fig. 5). During the transformation, the corresponding aldehyde 4a was also detected. Under the conditions described in Table 6, the absence of aqueous ammonia resulted in the quantitative conversion of 1a into 4a within 20 min. In addition, during the amidation of 1j to 2j, the corresponding aldimine (2-naphthalenemethanimine, 5j) could be detected by the GC mass analysis albeit in only a small amount (below 1% yield, Fig. 6). As mentioned above, OMS-2 showed high catalytic activities and selectivities for amidation of various kinds of structurally diverse aldehydes to the corresponding primary amides in the presence of aqueous ammonia (Table 7). In addition, the OMS-2-catalyzed hydration of various nitriles efficiently proceeded in the presence of aqueous ammonia, giving the corresponding primary amides in high yields (≥88% yields, Table S1, ESI†).
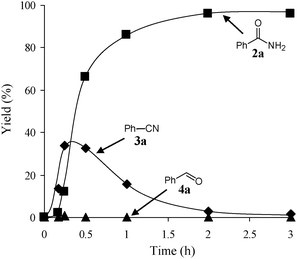 |
| | Fig. 5 Reaction profile for the amidation of benzyl alcohol (1a). The reaction was carried out under the conditions given in Table 6. | |
![Mass spectrum of an aldimine intermediate (5j) during the amidation of 2-naphthalenemethanol (1j). MS (EI): m/z (%): 156 (20) [M + H]+, 155 (100) [M+] (parent ion peak, M = C11H9N), 154 (96) [M – H]+, 153 (44), 128 (43) [C10H8]+, 127 (60) [C10H7]+, 126 (30), 77 (24) [C6H5]+, 76 (10), 75 (15), 74 (12), 63 (13), 51 (11).](/image/article/2013/CY/c2cy20178j/c2cy20178j-f6.gif) |
| | Fig. 6 Mass spectrum of an aldimine intermediate (5j) during the amidation of 2-naphthalenemethanol (1j). MS (EI): m/z (%): 156 (20) [M + H]+, 155 (100) [M+] (parent ion peak, M = C11H9N), 154 (96) [M – H]+, 153 (44), 128 (43) [C10H8]+, 127 (60) [C10H7]+, 126 (30), 77 (24) [C6H5]+, 76 (10), 75 (15), 74 (12), 63 (13), 51 (11). | |
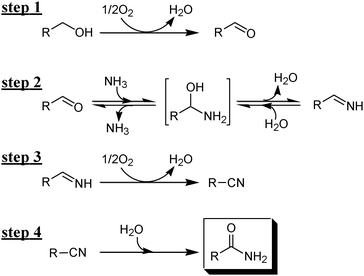
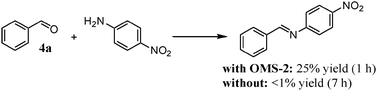
All the above-mentioned experimental evidence shows that the present OMS-2-catalyzed amidation possibly proceeds through the following four relay steps (Scheme 3): (i) aerobic oxidative dehydrogenation of primary alcohols to aldehydes (step 1), (ii) dehydrative condensation of the aldehydes and ammonia to aldimines via hemiaminal intermediates (step 2), (iii) aerobic oxidative dehydrogenation of the aldimines to nitriles (step 3), and (iv) successive hydration to the corresponding primary amides (step 4). The dehydrative condensation of 4a with less nucleophilic p-nitroaniline hardly proceeded in the absence of catalysts. In contrast, when the dehydrative condensation of 4a with p-nitroaniline was carried out in the presence of OMS-2, the corresponding N-alkylimine could be obtained in 25% yield (Scheme 4). OMS-2 possessed a large amount of acidic sites (1.23 mmol g−1) (Table 1). It is probable that OMS-2 can act as a solid acid catalyst to promote the condensation of aldehydes even with less nucleophilic amines. Therefore, all the above-mentioned relay steps 1–4 shown in Scheme 3 are likely promoted by the presence of OMS-2. As shown in Fig. 5, aldehyde 4a and an aldimine were hardly detected during the transformation (see also the profile for the amidation of 4a, Fig. S4, ESI†). In addition, 1a was rapidly oxidized in the absence of aqueous ammonia, and was quantitatively converted into 4a within 20 min under the conditions described in Fig. 5. Therefore, step 4 (nitrile hydration) is the slowest in the present amidation.
The catalytic activities of OMS-2 for the hydration of p-substituted benzonitrile derivatives increased in the order of p-methoxybenzonitrile (0.66) < p-methylbenzonitrile (0.93) < 3a (1.00) < p-chlorobenzonitrile (1.86) < p-nitrobenzonitrile (2.75), where the values in the parentheses are the relative rates, where the rate for 3a is taken as unity. The slope of the linear line in Fig. 7 gave the Hammett ρ value of +0.57. The positive value of the Hammett ρ can be interpreted as the formation of a negatively charged transition state. A very similar Hammett ρ value of +0.58 has been reported for the CeO2-catalyzed hydration of substituted 2-cyanopyridine derivatives, where the activation (polarization) of water on the surface of CeO2 followed by nucleophilic attack of OHδ− species to a nitrile carbon is proposed.10i In the same manner, the present OMS-2-catalyzed hydration would proceed through the above-mentioned reaction mechanism. In the hydration of 3a with OMS-2, the reaction rate with D2O (urea in D2O was used) was almost the same as that with H2O, and no kinetic isotope effect was observed (kH/kD = 1.1). Taking the above-mentioned results into account, the nucleophilic attack of OHδ− species to a nitrile carbon is likely included in the rate-limiting step for the present OMS-2-catalyzed nitrile hydration, i.e., overall amidation.
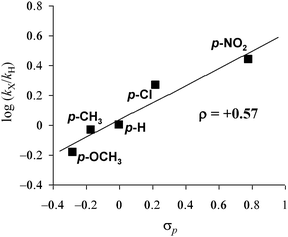 |
| | Fig. 7 Hammett plot for competitive hydration of benzonitrile (3a) and p-substituted benzonitrile derivatives. Conditions: OMS-2 (50 mg), 3a (0.25 mmol), p-substituted benzonitrile (0.25 mmol), 28% aq. ammonia (100 μL), 1,4-dioxane (2 mL), Ar (3 atm), 130 °C (bath temp.). | |
Next, dehydrogenation and hydration abilities of manganese-based oxides used in this study (OMS-2, amorphous MnO2, birnessite-type MnO2, and β-MnO2) were examined in more detail. For the oxidative dehydrogenation of 1a (reaction (4) in Table 8), the catalytic activity of OMS-2 was comparable to that of β-MnO2 but inferior to that of amorphous MnO2. The higher catalytic activity of amorphous MnO2 is likely due to the large BET surface area (Table 1). It has been reported that the catalytic activities of manganese-based oxides for aerobic alcohol oxidation increase with an increase in their specific surface areas and are almost independent of their physical properties.20c Thus, the results for reaction (4) in Table 8 are consistent with those of the previous report.20c Importantly, all manganese-based oxides could promote aerobic alcohol oxidation even in the presence of aqueous ammonia (reaction (3) in Scheme 2 and Table S2, ESI†).
| Entry |
Catalyst |
Yield (%) |
|
4a for (4) |
2a for (5) |
2a for (6) |
|
Conditions for reaction (4): catalyst (20 mg), 1a (0.5 mmol), 1,4-dioxane (1 mL), O2 (3 atm), 70 °C, 2 h. Conditions for reaction (5): catalyst (20 mg), 3a (0.5 mmol), water (65 μL), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 2 h. Conditions for reaction (6): catalyst (20 mg), 3a (0.5 mmol), 28% aq. ammonia (100 μL, ca. 2.6 equiv.), 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 2 h. Yields were determined by using GC using naphthalene or biphenyl as an internal standard.
|
| 1 |
OMS-2 |
14 |
20 |
63 |
| 2 |
Amorphous MnO2 |
53 |
74 |
16 |
| 3 |
Birnessite-type MnO2 |
8 |
4 |
2 |
| 4 |
β-MnO2 |
16 |
22 |
2 |
With regard to the hydration of 3a with water (in the absence of ammonia, reaction (5) in Table 8), the order of the activities was the same as that for reaction (4), and amorphous MnO2 showed the highest catalytic activity. Indeed, hydration of various kinds of nitriles with water efficiently proceeded in the presence of amorphous MnO2, giving the corresponding primary amides in moderate to excellent yields (≥54% yields, Table S3, ESI†). However, in the case of amorphous MnO2, birnessite-type MnO2, and β-MnO2, their catalytic activities were strongly suppressed by the presence of ammonia (entries 2–4 for reaction (6) in Table 8).|| Surprisingly, the yield of 2a for the OMS-2-catalyzed hydration with aqueous ammonia was much higher than that with just water (entry 1 for reactions (5) and (6) in Table 8). We confirmed in a separate experiment that the hydration did not proceed at all with aqueous ammonia alone. Thus, the OMS-2-catalyzed hydration is clearly promoted by the presence of ammonia (rather than suppressed as observed for other manganese-based oxides), which is one of the most important key points for realizing the present OMS-2-catalyzed aerobic amidation of primary alcohols with aqueous ammonia (see Tables 2, 6, and 7).
Finally, in order to clarify the active site(s) of OMS-2 for nitrile hydration (the most important key reaction), the hydration of 3a in the presence of 2,6-lutidine or pyridine was carried out. Pyridine can interact with both Brønsted and Lewis acid sites via protonation and coordination, respectively.23 In contrast, 2,6-lutidine can selectively interact with Brønsted acid sites and cannot interact with Lewis acid sites due to the steric hindrance.23
As shown in Scheme 5, the yield of 2a in the presence of 2,6-lutidine was almost the same as that in the absence.|| On the other hand, addition of pyridine significantly increased the yield of 2a from 20% to 51%. Therefore, coordination of nitrogen donors, i.e., pyridine and ammonia, to the Lewis acid (manganese) centers on the external surface of OMS-2 is very important in the present nitrile hydration. This is possibly explained as follows: the coordination of nitrogen donors to the Lewis acid centers would increase the electron density of neighboring oxygens, resulting in increased ability for activation (polarization) of water. Thus, nucleophilicity of OHδ− would be increased, resulting in promotion of nitrile hydration.
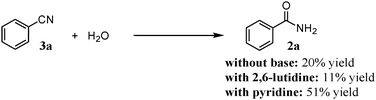 |
| | Scheme 5 Hydration of benzonitrile (3a) with or without bases. Conditions: OMS-2 (100 mg), 3a (0.5 mmol), water (65 μL), with (2.6 equiv. with respect to 3a) or without bases, 1,4-dioxane (2 mL), O2 (3 atm), 130 °C (bath temp.), 2 h. | |
4. Conclusions
We have successfully developed a new procedure for synthesis of primary amides via the OMS-2-catalyzed aerobic oxidative amidation of primary alcohols or aldehydes with aqueous ammonia. This system is applicable to various kinds of structurally diverse primary alcohols and aldehydes as starting materials. In addition, various nitriles could be used for synthesis of primary amides (nitrile hydration). The observed catalysis for amidation was truly heterogeneous, the product isolation was very simple, rather inexpensive manganese-based oxides (in comparison with precise metal-based catalysts) could be used, and the OMS-2 catalyst could be reused without an appreciable loss of its high performance. This convenient procedure with OMS-2 (heterogeneous catalyst) demonstrated herein will provide a new green route to primary amides, which can completely avoid utilization of (hazardous) stoichiometric reagents and formation of vast amounts of inorganic by-products, and will be one of the choices for green amide synthesis.
Acknowledgements
This work was supported in part by the Global COE Program (Chemistry Innovation through Cooperation of Science and Engineering), Japan Chemical Innovation Institute (JCII), and Grants-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology.
Notes and references
-
(a)
C. E. Mabermann, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, John Wiley & Sons, New York, 1991, vol. 1, pp. 251–266 Search PubMed;
(b)
D. Lipp, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, John Wiley & Sons, New York, 1991, vol. 1, pp. 266–287 Search PubMed;
(c)
R. Opsahl, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, John Wiley & Sons, New York, 1991, vol. 2, pp. 346–356 Search PubMed;
(d)
The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Material Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York, 2000 Search PubMed;
(e) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
-
(a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC;
(b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS.
-
(a) D. Milstein, Top. Catal., 2010, 53, 915 CrossRef CAS;
(b) R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef;
(c) C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20 RSC.
-
(a) L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672 CrossRef;
(b) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667 CrossRef CAS;
(c) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643 CrossRef CAS;
(d) J. H. Dam, G. Osztrovsky, L. U. Nordstrøm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820 CrossRef CAS;
(e) J. Zhang, M. Senthilkumar, S. C. Ghosh and S. H. Hong, Angew. Chem., Int. Ed., 2010, 49, 6391 CrossRef CAS;
(f) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159 CrossRef CAS.
-
(a) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef CAS;
(b) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917 CrossRef CAS;
(c) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550 CrossRef.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977 CrossRef CAS.
-
(a) T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287 CrossRef CAS;
(b) A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607 CrossRef CAS;
(c) R. S. Ramón, N. Marion and S. P. Nolan, Chem.–Eur. J., 2009, 15, 8695 CrossRef;
(d) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949 CrossRef CAS.
-
(a) P. Breuilles, R. Leclerc and D. Uguen, Tetrahedron Lett., 1994, 35, 1401 CrossRef CAS;
(b) C. G. Rao, Synth. Commun., 1982, 12, 177 CrossRef CAS;
(c) A. Solhy, A. Smahi, H. E. Badaoui, B. Elaabar, A. Amoukal, A. Tikad, S. Sebti and D. J. Macquarrie, Tetrahedron Lett., 2003, 44, 4031 CrossRef CAS;
(d) T. Mitsudome, Y. Mikami, H. Mori, S. Arita, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 3258 RSC;
(e) K. Mori, K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Commun., 2001, 461 RSC;
(f) K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576 CrossRef CAS;
(g) S. C. Roy, P. Dutta, L. N. Nandy, S. K. Roy, P. Samuel, S. M. Pillai, V. K. Kaushik and M. Ravindranathan, Appl. Catal., A, 2005, 290, 175 CrossRef CAS;
(h) H. Miura, K. Sugiyama, S. Kawasaki, T. Aoyama and T. Matsuda, Chem. Lett., 1982, 183 CrossRef CAS;
(i) M. Tamura, H. Wakasugi, K. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428 CrossRef CAS;
(j) Y. Gangarajula and B. Gopal, Chem. Lett., 2012, 41, 101 CrossRef CAS;
(k) K. Yamaguchi, Y. Wang, H. Kobayashi and N. Mizuno, Chem. Lett., 2012, 41, 574 CrossRef CAS.
-
(a) H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202 CrossRef CAS;
(b) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73 CrossRef CAS;
(c) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599 CrossRef CAS;
(d) H. Fujiwara, Y. Ogasawara, M. Kotani, K. Yamaguchi and N. Mizuno, Chem.–Asian J., 2008, 3, 1715 CrossRef CAS;
(e) M. Kim, J. Lee, H.-Y. Lee and S. Chang, Adv. Synth. Catal., 2009, 351, 1807 CrossRef CAS;
(f) D. Gnanamgari and R. H. Crabtree, Organometallics, 2009, 28, 922 CrossRef CAS;
(g) R. S. Ramón, J. Bosson, S. Diez-González, N. Marion and S. P. Nolan, J. Org. Chem., 2010, 75, 1197 CrossRef;
(h) M. A. Ali and T. Punniyamurthy, Adv. Synth. Catal., 2010, 352, 288 CrossRef CAS;
(i) C. L. Allen, C. Burel and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 2724 CrossRef CAS.
-
(a)
A. J. Fatiadi, Preparation and synthetic applications of cyano compounds, in Triple-Bonded Functional Groups, ed. S. Patai and Z. Rappaport, John Wiley & Sons, Ltd., Chichester, UK, vol. 2, 1983 Search PubMed;
(b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS;
(c) P. Magnus, D. A. Scott and M. R. Fielding, Tetrahedron Lett., 2001, 42, 4127 CrossRef CAS;
(d)
M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Inc., Hoboken, New Jersey, 6th edn, 2007 Search PubMed.
- R. Ohmura, M. Takahata and H. Togo, Tetrahedron Lett., 2010, 51, 4378 CrossRef CAS.
-
(a) T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286 CrossRef CAS;
(b) T. Oishi, K. Yamaguchi and N. Mizuno, Top. Catal., 2010, 53, 479 CrossRef CAS.
-
(a) R. N. DeGuzman, Y.-F. Shen, E. J. Neth, S. L. Suib, C.-L. O'Young, S. Levine and J. M. Newsam, Chem. Mater., 1994, 6, 815 CrossRef CAS;
(b) Y.-C. Son, V. D. Makwana, A. R. Howell and S. L. Suib, Angew. Chem., Int. Ed., 2001, 40, 4280 CrossRef CAS;
(c) S. L. Suib, J. Mater. Chem., 2008, 18, 1623 RSC;
(d) S. L. Suib, Acc. Chem. Res., 2008, 41, 479 CrossRef CAS;
(e) X. Yang, J. Han, Z. Dub, H. Yuan, F. Jin and Y. Wu, Catal. Commun., 2010, 11, 643 CrossRef CAS;
(f) T. Oishi, K. Yamaguchi and N. Mizuno, ACS Catal., 2011, 1, 1351 CrossRef CAS.
- K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544 CrossRef CAS.
- D. Pope, D. S. Walker and R. L. Moss, J. Catal., 1977, 47, 33 CrossRef CAS.
-
Purification of Laboratory Chemicals, ed. D. D. Perrin and W. L. F. Armarego, Pergamon Press, Oxford, U.K., 3rd edn, 1988 Search PubMed.
- O. Ghodbane, J.-L. Pascal and F. Favier, ACS Appl. Mater. Interfaces, 2009, 1, 1130 CAS.
-
(a) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef CAS;
(b) K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353 CrossRef CAS;
(c) F. Schurz, J. M. Bauchert, T. Merker, T. Schleid, H. Hasse and R. Gläser, Appl. Catal., A, 2009, 355, 42 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- Y. Wang, H. Kobayashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642 RSC.
-
(a) H. C. Brown and R. B. Johannesen, J. Am. Chem. Soc., 1953, 75, 16 CrossRef CAS;
(b) L. L. Murell and N. C. Dispenziere, Jr., J. Catal., 1989, 117, 275 CrossRef;
(c) A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S3. See DOI: 10.1039/c2cy20178j |
| ‡ OMS-2 has been recognized as a good catalyst or a support because of the following excellent properties: (i) large (external) surface area (ca. 100 m2 g−1), (ii) electron-conducting property, and (iii) dioxygen reduction ability.15 |
| § Very recently, we have also found that OMS-2 can act as an efficient heterogeneous catalyst for oxidative transformation of various primary amines to primary amides in the presence of aqueous ammonia.22 |
| ¶ In reaction (1) in Scheme 2, 2a, 3a, and 4a were formed in 23%, 29%, and 10% yields, respectively. During the transformation, methylamine was decomposed to ammonia and formaldehyde through the sequence of dehydrogenation and hydrolytic decomposition. |
| || In the case of other manganese-based oxides such as amorphous MnO2, birnessite-type MnO2, β-MnO2, the yields of 2a were much decreased with addition of 2,6-lutidine (decreased from 74% to 15% for amorphous MnO2, for example), suggesting that Brønsted acid sites are responsible for nitrile hydration. |
|
| This journal is © The Royal Society of Chemistry 2013 |