 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge-transfer excitation: unconventional yet practical means for controlling stereoselectivity in asymmetric photoreactions
Tadashi Mori
* and
Yoshihisa Inoue
Department of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka, 565-0871, Japan. E-mail: tmori@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7923; Tel: +81-6-6879-7921
First published on 5th July 2013
Abstract
In chiral donor–acceptor (D–A) systems, irradiation wavelength plays vital roles in determining the photochemical consequences. Selective excitation of a D–A complex at the charge-transfer (C-T) band affords an excited C-T complex (ECT), while the local-band excitation of D or A may lead to the formation of a conventional exciplex (EX) upon subsequent interaction with the D–A partner. These two excited species, generated from the same D–A pair, may be categorized formally as excited complexes or exciplexes, but should be distinguished, provided that they significantly differ in structure and reactivity. Indeed, ECT and EX exhibit distinctly different temperature-dependent photophysical and photochemical behaviours, which are assignable to the differences in relative stability, conformational flexibility and/or solvation properties. Fine-tuning excitation wavelength further enabled us to discriminate stereoisomeric intramolecular C-T complexes through preferential excitation, as C-T complexes are generally composed of an ensemble of various geometries. Besides temperature and solvent polarity, the excitation wavelength was shown to be employed as an unconventional yet practical tool for critically controlling the chemo-, regio- and stereoselectivities in molecular and supramolecular photochemistry.
 Tadashi Mori | Tadashi Mori was born in Kanazawa, Japan, in 1970. He obtained his PhD from Kyoto University in 1997 under the supervision of Prof. Hitomi Suzuki, working on the mechanism of aromatic nitration with combined ozone and nitrogen oxides. He then worked as a postdoctoral associate and a JSPS research fellow for his first stay abroad in Prof. Jay K. Kochi's group at Houston University, where he set about studies on the donor–acceptor or charge-transfer chemistry. In 1998, he joined Prof. Yoshihisa Inoue's group at Osaka University, where he started to combine the C-T chemistry with Yoshi's cutting-edge research on supramolecular asymmetric photochemistry. During these periods, he was a visiting professor at Georgetown University in 2003, working with Prof. Richard G. Weiss, and an Alexander von Humboldt Fellow at Westfälische Wilhelms-Universität Münster in 2005–2006, working with Prof. Stefan Grimme. Currently, as Associate Professor in Osaka, he focuses his research interest on the combined experimental and theoretical studies on the chiroptical properties of ground- and excited-state molecules and (supra)molecular complexes. |
 Yoshihisa Inoue | Yoshihisa Inoue was born in Nara, Japan, in 1949. He received his PhD from Osaka University in 1977 under the supervision of Profs Hiroshi Sakurai and Setsuo Takamuku, working on the vacuum UV photochemistry of simple alkenes. After a short period of stay at the Institute of Scientific and Industrial Research, Osaka University, as a JSPS fellow, he joined Prof. Tadao Hakushi's group as Assistant Professor in 1978 and started the work on supramolecular chemistry, and moved to Prof. Akira Tai's group in 1985 and worked on chiral photochemistry, both at Himeji Institute of Technology. In 1978–1979, he spent a year at Columbia University as a research associate to Prof. Nicholas J. Turro, working on the singlet oxygenation of strained alkenes. In 1994, he returned to Osaka University as a full professor at the Department of Applied Chemistry. He served as Director of ERATO Photochirogenesis and ICORP Entropy Control projects supported by JST in 1995–2008. He is currently interested in chiral photochemistry and supramolecular chemistry. |
1. Introduction
An aromatic electron donor (D) and acceptor (A) often form a weakly interacting complex in the ground state1–3 with accompanying new absorption at longer wavelengths assignable to the C-T band.Compared with the other weak interactions, such as hydrogen bonding,4 the C-T interaction is less robust and less directional. Hence, its use for controlling chemical reaction is often discouraged. It is also true however that a pre-equilibrium complex of C-T character, or more forthrightly a C-T complex, plays crucial roles in such (thermal) bimolecular reactions as electrophilic aromatic substitution and addition reactions.5,6 Accordingly, the properties of C-T complexes, as well as the structure–reactivity relationship, have been extensively investigated over the past three decades. In contrast, the importance of (ground-state) C-T interaction is not significantly appreciated or taken into account in general in the excited-state chemistry (including the photochemical reaction of D and A), although there has been a substantial dispute about its significance (vide infra). Photoirradiation of a C-T complex at its C-T band leads to the formation of an excited C-T complex (ECT), which may undergo electron transfer, addition, cleavage, rearrangement and/or other reactions, or revert to the ground state. Upon direct or local-band excitation of one component of a D–A pair at its local band, the excited D or A generated diffuses to encounter with the ground-state counterpart (A or D), affording a conventional exciplex (EX),7 which may also undergo some net reaction(s). By definition, these two excited-state species should be categorized as exciplexes in a broader sense, as they are undoubtedly excited-state complexes. We will however employ a more precise definition in this review, whenever it is feasible. Thus, ECT refers to an excited-state species or an ensemble formed upon selective excitation of a ground-state complex at the C-T band, while EX denotes a conventional excited-state complex formed through the diffusion of an excited D or A to encounter with a ground-state partner. Throughout the discussion to follow, we will not specifically discern the two situations that arise from the difference in the ensemble or in structure, since they lead to the distinctly different photochemical behaviours in any event. More precise discrimination remains to be elucidated in the future study.
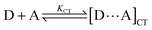
In the last decade, we have shown that the C-T interaction plays decisive roles in a variety of asymmetric photoreactions. Despite the low effective concentration (as a consequence of the generally small association constants), the selective C-T excitation was readily achieved by carefully choosing the irradiation wavelength. Remarkably, the ECT species thus generated exhibits the photochirogenic and temperature-dependence behaviours significantly different from those displayed by the EX species. In this review, we will emphasize with ample examples that such a simple, readily attainable, change as excitation wavelength can dramatically alter the relative contribution of the ground- and excited-state conformer ensembles of a C-T complex, allowing us to favourably modulate the photochemical consequences. As for the longstanding debate on the identity of ECT, we will unambiguously demonstrate that ECT differs from EX both in structure and reactivity.
2. Ground-state charge-transfer interaction
Before getting into the details of the photochemistry of the C-T complex, we briefly summarize the nature of the C-T complex in the ground state. C-T complexes do not involve covalent or any other strong interactions, but are formed upon association of D and A molecules and hence equilibrated with free D/A. Consequently, they are, in general, extremely sensitive to the alteration in temperature, concentration, and solvent. According to Nobel laureate Robert Mulliken, the ground and excited states of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 C-T complex formed between D and A can be described as a resonance of a pair of wavefunctions as follows:8
:1 C-T complex formed between D and A can be described as a resonance of a pair of wavefunctions as follows:8
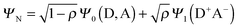 | (1) |
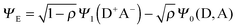 | (2) |
3. Excitation of the C-T complex
Selective excitation at the C-T band
Even when the ground-state C-T interaction is not apparent, the excited-state donor or acceptor (D* or A*), generated upon local-band excitation of D or A, may have stronger interaction with the ground-state partner to form a conventional exciplex (EX) in a diffusive manner within the lifetime of the excited state (typically 1–100 ns). It is to be noted that due to the possible spectral overlap with the local bands of D and/or A, the C-T band would also be excited to a certain extent upon direct excitation of a local-band.An apparently similar, but conceptually different, excited-state complex may also be generated by selectively exciting the ground-state C-T complex. Thus, the irradiation at the C-T band leads to the formation of singlet, and triplet (after intersystem crossing), excited state(s) of the C-T complex (ECT), which subsequently undergo the intra-complex electron transfer to give a geminate radical ion pair (RIP). ECT and RIP may decay through back electron transfer and/or undergo some chemical processes, but generally dissociate to yield free radical ions, which eventually lead to net chemical reaction.
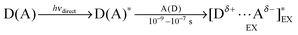
The photophysical processes of such reactive ECTs have been studied by time-resolved (picosecond) spectroscopies for a number of C-T systems.18,19 Upon selective photoexcitation at the C-T band, ECT immediately (≤0.1 ns) undergoes intra-complex electron transfer to give a singlet RIP. A recent femtosecond stimulated Raman spectroscopic study on the photoinduced charge-separation dynamics of a C-T complex of pyromellitic dianhydride and hexamethylbenezene showed that the structural relaxation is facilitated by solvation dynamics during the charge separation process and that the ECT already possesses the significant charge-separated character and spontaneously relaxes.20
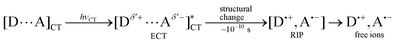
Conformational variety of ECT
The conformational variety of ECT, as well as the multidimensionality of the reaction coordinate, have become widely recognized recently, which may cause additional complexity in understanding the excited-state behaviour of the C-T complex. For instance, the transient absorption-stimulated emission spectroscopic studies on the relaxation dynamics of excited donor–acceptor auramine dyad revealed the existence of two distinct excited-state conformers which differ in geometry and dynamics.21 Another investigation on an excited intramolecular D–A system underlined two distinct C-T excited states, in addition to the locally excited state.22 Besides the intramolecular C-T systems (spectroscopically) distinguishable excited-state complexes have also been detected in the coupled electron–proton transfer process of hydrogen-bonded acid–base systems, for which the femtosecond transient absorption and coherent Raman spectroscopies confirmed the presence of two distinct excited states with different dynamic signatures.23 Furthermore, the vibrational quantum state was also utilized recently in controlling the excited-state behaviour and the product distribution upon oxidation of acetylene under the atmospheric conditions.24Spectroscopic discrimination of ECT from EX
Under the (thermally) fully equilibrated conditions, a common excited-state complex (or ensemble) is reached from ECT and EX through rapid relaxation. However, these species can exist as separate species if not freely interconverted within the lifetimes of these excited states due to the energy barrier or potential surface crossing (Fig. 1).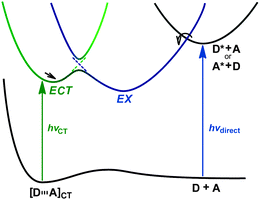 | ||
| Fig. 1 Potential energy profiles for the formation and possible equilibrium of conventional exciplex (EX) and excited C-T complex (ECT), generated through direct (local-band) excitation of donor (D) or acceptor (A) and selective C-T band excitations, respectively. | ||
Accordingly, the similarities and dissimilarities of ECT and EX have been intensively discussed for a number of D–A pairs. However, the results of the foregoing studies are rather controversial or opposed, probably because the photophysical and photochemical behaviours observed are highly system-dependent. Fluorescence spectra in solution are generally broad and structureless, and thus the difference in fluorescence should be difficult to detect or very small between ECT and EX, if exists. Nevertheless, several investigations disclosed that ECT and EX display appreciably different fluorescence spectra. For instance, the EX emission (λEXmax = 530 nm) was observed upon fluorescence quenching of 1,2,4,5-tetracyanobenzene by 2-phenylpropene. Interestingly, additional weak fluorescence, assignable to ECT with a geometry different from EX, was observed at shorter wavelengths (λECTmax = 480 nm) upon selective C-T excitation.25 For the 9,10-dicyanoanthracene–naphthalene pair, diverse fluorescence from ECT was observed at 490 nm in non-polar solvent at low temperatures, which is somewhat higher in energy than the EX fluorescence observed at 540 nm, and the lifetimes of these species showed different temperature-dependence behaviour.26,27 At higher temperatures, the difference in lifetime becomes diminished due to the faster solvent reorientation. Isomeric forms of the excited-state complexes have also been identified for anthracene–N,N-dimethylaniline and perylene–N-methylaniline pairs, but not for the anthracene–aniline or perylene–N,N-dimethylaniline pair for reasons not specified.28
Distinction between ECT and EX by product analysis
Photocycloaddition behaviour of (E)-stilbene (ST) with dimethyl fumarate has been compared at different excitation wavelengths.29 Excitation of the weak ground-state C-T complex resulted in the same chemical reactions as those obtained upon local-band excitation of (E)-ST. Fluorescence from the complex was also studied but indistinguishable from that of EX formed via the encounter of excited ST with dimethyl fumarate. As such, ECT was concluded to be indistinguishable from EX for this D–A pair. Contradictory observations have been reported for other D–A pairs. For instance, photoirradiation of acenaphthylene in the presence of tetracyanoethene led to the formation of a cycloadduct through the singlet excited state of the donor. However, the C-T excitation of the ground-state complex led to a complete recovery of the starting materials even upon prolonged irradiation in various solvents.30–32 It has been reported also that the adducts' yields were considerably dependent on the excitation wavelength in the photocyclization of acenaphthylene or indene derivatives with a variety of acceptors.33,34 These observations may be explained in terms of the geometry difference between EX and ECT. Such wavelength-dependent reactivities reported in the literature are indicative that ECT is an excited-state complex species different from EX in structure and reactivity, but how and to what extent they differ is an arduous question remained to be answered.4. Asymmetric photoreactions of the intermolecular C-T complex upon local-band versus C-T excitations
Diastereoselective photocyclization of (E)- or (Z)-stilbene with chiral fumarate
As discussed above, ECT and EX resemble each other and are difficult to differentiate. Oftentimes, the excitation mode (i.e., local-band versus C-T) does not appreciably affect the net photochemical outcomes (vide supra). Since the difference in free energy is relatively small in general for a pair of diastereomeric transition states leading to the end products, the relative rate constant, or diastereoselectivity, is likely to critically respond to the change in nature and geometry of the excited species. Hence, employing asymmetric photoreaction is expedient for experimentally differentiating ECT from EX by observing the change in the stereochemical outcome. We employed a chiral version of the well-established photocycloaddition of (E)-ST to alkyl fumarate,29 where (R)-1-methylpropyl was chosen as the simplest chiral alkyl group.35,36 In this system, the intervention of ECT versus EX was unambiguously sorted out by analysing the diastereoselectivities of the cyclobutanes obtained upon excitation at different wavelengths, as detailed below.Photoirradiation of a mixture of (E)-ST and bis((R)-1-methylpropyl) fumarate ((R)-F) afforded a mixture of cycloadducts (Scheme 1), irrespective of the excitation mode (local-band or C-T). Thus, besides the geometrical isomers of both substrates and a small amount (∼1%) of stilbene dimer, two stereoisomeric cyclobutanes, μ- and δ-truxinates (μ- and δ-CB), were obtained in fair to good yields (typically 10–60%), along with a considerable amount (40–60%) of butadiene products most likely derived from the oxetanes produced by the Paternó–Büchi reaction. Despite the substantial efforts, the diastereoselectivities of these oxetanes were not able to be determined, due to the instability (ready rearrangement to the butadienes) under the GC conditions employed. We therefore concentrated on the analyses of the product ratio (μ-CB/δ-CB) as well as the diastereoselectivity. Although the photoreaction of (Z)-ST with dimethyl fumarate was reported to proceed much slower than that of (E)-ST, we noticed that (Z)-ST reacts with (R)-F in nearly comparable efficiencies, and more interestingly the diastereoselectivity of oxetane was inverted. As a consequence of concurrent E–Z photoisomerization of stilbene, the observed diastereoselectivities were gradually decreased with increasing irradiation time, irrespective of the geometry of the starting material, (E)- or (Z)-ST. Therefore, comparisons were made by using the values extrapolated to zero irradiation time, to facilitate the direct assessment of selectivities caused upon distinct excitation.
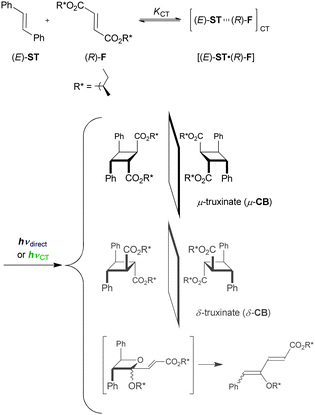 | ||
| Scheme 1 Photoreaction of (E)-stilbene ((E)-ST) with bis((R)-1-methylpropyl) fumarate ((R)-F). Note that diastereomeric pairs are illustrated with a mirror plane to emphasize the enantiomeric structures of the cyclobutane ring. | ||
Table 1 compares the product ratios and diastereomeric excesses (de's) obtained in the photocycloaddition of (E)- or (Z)-ST to (R)-F in toluene at various temperatures.35,36 Under all the reaction conditions employed, the same products were formed, but in varying ratios and de's. Upon cycloaddition to (R)-F, (E)-ST gave more μ-truxinate than δ-truxinate at all the examined temperatures, while (Z)-ST afforded more δ-truxinate. A similar switching phenomenon was observed for the de values of the oxetanes derived from (E)- and (Z)-ST, as indicated by the sign inversion of de. Crucially, both the product ratio and diastereoselectivity were highly dependent on the excitation mode, clearly demonstrating that ECT differs from EX in structure and reactivity. Temperature plays another important role in determining the (dia)stereoselectivities. Thus, the C-T excitation of the [(E)-ST·(R)-F] complex yields the cyclodimers in consistently high de's of ≥95% independent of the temperature, whereas the de significantly decreased at lower temperatures upon local-band excitation. The contrast in photobehaviour became even more apparent at higher temperatures upon local-band versus C-T excitation in the photoreaction of (Z)-ST with (R)-F. Intriguingly, this contrasting behaviour was strongly solvent-dependent, being observed in ether and toluene but not in methylcyclohexane, which implies the positive role of solvation in the excited-state relaxation dynamics of the ECT and EX species. Thus, in the local-band excitation, the product's de became more positive or less negative (favouring the formation of the same specific diastereomer) by raising the temperature, irrespective of the geometry of stilbene used. In contrast, the de value was kept high upon C-T excitation throughout the temperature range employed. Such contrasting temperature-dependence behaviours may be explained in terms of the differences in relative energy, equilibrium, and/or the cyclization rate of the corresponding diastereomeric transition states or precursor complexes. However, as the photoreaction of stilbene with fumarate gives rise to other products such as oxetane, the competition with the additional reaction channel(s) would also be taken into account in more detailed mechanistic elucidations.
| Donor | Temp. (°C) | Local-band excitation (313 nm) | C-T excitation (365 nm) | ||
|---|---|---|---|---|---|
| μ-CB/δ-CB | % Deb | μ-CB/δ-CB | % Deb | ||
| a [(E)-ST]0 = [(Z)-ST]0 = 0.1 M; [(R)-F]0 = 1.0 M in toluene.b De values obtained by extrapolation to zero irradiation time; see the text for reason. Note that the positive–negative signs for de refer to the preferred formation of the first/second-eluted diastereomers in GC analysis. | |||||
| (E)-ST | +50 | 1.0 | +97 | 1.0 | +98 |
| +25 | 1.3 | +91 | 1.4 | +95 | |
| 0 | 1.4 | +75 | 1.6 | +97 | |
| −25 | 1.8 | +59 | 2.1 | +96 | |
| −50 | 2.1 | +34 | 2.6 | +99 | |
| (Z)-ST | +50 | 0.39 | −32 | 0.03 | −94 |
| +25 | 0.26 | −59 | 0.03 | −94 | |
| 0 | 0.24 | −73 | 0.03 | −96 | |
| −25 | 0.17 | −84 | 0.03 | −96 | |
| −50 | 0.17 | −90 | 0.02 | −97 | |
The C-T complexation was substantially enhanced by applying hydrostatic pressure to give the cyclodimers at faster rates and in better yield upon excitation. Nevertheless, the diastereoselectivity did not show any appreciable changes,37 indicating that the molecular and activation volumes of the diastereomeric C-T complexes and transition states are equally reduced or compensated with each other.
Theoretical investigations of the ground- and excited-state structures of a diastereomeric complex pair of (E)-stilbene with chiral fumarate
Theoretical calculations provide us with valuable insights into the nature of the C-T complex in its ground and excited states. Thus, the geometries of the diastereomeric C-T complexes between (E)-ST and (R)-F in the ground state were theoretically optimized by the dispersion-corrected DFT method at the DFT-D3-B-LYP/TZVP level,38 which is known to properly incorporate the non-bonded interactions. Fig. 2 shows the most stable diastereomeric re,si- and si,re-stacked pair of the C-T complex, in which the reacting double bonds are stacked face-to-face and the fumarate's double bond is s-cis to both of the carbonyls. For simplicity purpose, the isomeric s-cis, s-trans- and s-trans, s-trans-rotamers will not be separately examined in the following discussion, but note that they are only slightly higher in energy (by 0.1–0.2 or 0.3–0.4 kcal mol−1) than the s-cis, s-cis-rotamer and should also contribute significantly as an ensemble to the overall diastereoselectivity. Compared with the parallel conformer, the other conformers with crossed double bonds were found to be considerably less stable (by >5 kcal mol−1) and hence ignored. Note however that the cross conformer, possessing the correct geometry, is a potential precursor to δ-truxinate, if the photocycloaddition is concerted. The most stable si,re-[ST·F] complex, in which the ST's si- and F's re-faces are confronting, differs in alkyl orientation from the diastereomeric re,si-[ST·F], but these two share essentially the same geometry around the reacting double bonds. The energy difference in gas phase was evaluated as 0.43 kcal mol−1 by the most accurate SCS-MP2/TZVPP calculations,39 indicating that the si,re- and re,si-complexes are populated in ca. 2:1 ratio (equivalent to 35% de) at 25 °C. The fact that a much higher diastereoselectivity of 95% de was achieved upon C-T excitation (Table 1) reveals that the product's de is not an immediate function of the ground-state diastereomer equilibrium but is also governed by the excitation efficiency and the excited-state dynamics upon excitation, relaxation, deactivation, intersystem crossing and cycloaddition. The ST and F molecules are in close contact at average spacings of 3.50 and 3.45 Å for the si,re- and re,si-complexes, respectively, while the facing double bonds are not exactly overlaid but slid by ∼1.5 Å. The spacing is smaller than the sum of the van der Waals radii, confirming the strong C-T interaction in the ground state. The slid-out benzene ring of ST is tilted by 10 and 20° in the si,re- and re,si-complexes, respectively, avoiding the steric clash with the facing alky group of F, while the benzene ring on the opposite side remains cofacial to retain the π overlap.
![Structures of the diastereomeric ground-state C-T complexes, re,si- and si,re-[(E)-ST·(R)-F] (left), optimized at the DFT-D3-B-LYP/TZVP level and of diastereomeric exciplexes, re,si- and si,re-[(E)-ST·(R)-F]* (right), optimized at the TD-DFT-BH-LYP/TZVP level. The most stable si,re-C-T complex are shown at the bottom.](/image/article/2013/CS/c3cs60117j/c3cs60117j-f2.gif) | ||
| Fig. 2 Structures of the diastereomeric ground-state C-T complexes, re,si- and si,re-[(E)-ST·(R)-F] (left), optimized at the DFT-D3-B-LYP/TZVP level and of diastereomeric exciplexes, re,si- and si,re-[(E)-ST·(R)-F]* (right), optimized at the TD-DFT-BH-LYP/TZVP level. The most stable si,re-C-T complex are shown at the bottom. | ||
As anticipated, the calculated degree of C-T was fairly small, merely amounting to 2.0 and 2.5% for the si,re- and re,si-complexes, respectively. On the whole, the ground-state C-T complex avoids the complete π overlap due to the electrostatic and Pauli repulsion and is less symmetrical. In contrast, the excited-state structure becomes almost symmetrical to maximize the π overlap. The structures of a diastereomeric exciplex pair of (E)-ST with (R)-F were theoretically predicted at the TD-DFT-BH-LYP/TZVP level (Fig. 2).40,41 Interestingly, the centre-to-centre distance between the double bonds of ST and F is significantly reduced to 3.14 and 3.08 Å in the excited-state si,re- and re,si-complexes, which are balanced by adjusting the ST conformation to avoid the steric congestions at the peripheries. Analogous bent structures have been reported for the excimers with relatively large π-systems.42 As a result of the steric repulsion, the inter-periphery distance is much larger for the exciplex than for the C-T complex. This explains at least in part why the C-T, rather than local-band, excitation is more effective in manipulating the stereochemical course of the subsequent photocycloaddition. In this context, it is crucial that the diastereotopic face preference predicted for the exciplex becomes opposite to that for the ground-state C-T complex, favouring the re,si-complex in a 53:47 ratio (ΔE = −0.07 kcal mol−1).
Such structural differences between the ground- and excited-state complexes have already been recognized in other D–A systems. For example, the X-ray crystallographic study of the [hexamethylbenzene·1,2,4,5-tetracycnobenzene] complex revealed that the centres of the two benzene rings are not completely overlapped but slid by ∼0.3 Å, while the theoretical calculations for the exciplex of the same D–A pair predict good overlap of the π-MOs.43–45
Diastereoselective Paternó–Büchi reaction of 1,1-diphenylethene with chiral p-cyanobenzoates
Paternó–Büchi reaction46,47 proceeds through the [2+2] photocycloaddition of excited carbonyl compound to ground-state alkene. Typically, the excited ketone triplet adds to an alkene to give an oxetane as the final product through a triplet 1,4-biradical intermediate. It has been reported that the C-T band excitation of the [ST·chloranil] complex affords a mixture of stereoisomeric oxetanes in a molar ratio identical to that obtained upon local-band excitation of chloranil.48 This suggests that differentiating ECT from EX becomes more arduous in the triplet manifold, due to a better chance to relax or equilibrate during the generally much longer lifetime of the triplet state. The [2+2] photocycloaddition of p-cyanobenzoate to 1,1-diphenylethene (DE) leads to the formation of an oxetane, which is however known to proceed rather exceptionally through a singlet exciplex.49 This allowed us to expand the scope of the wavelength-control concept to the diastereoselective Paternó–Büchi reaction of chiral alkyl p-cyanobenzoates (Scheme 2). Indeed, the chiral version of the Paternó–Büchi reaction turned out to be valuable in differentiating ECT from EX. Unlike the ST-F system, the Paternó–Büchi reaction was clean, yielding a pair of diastereomeric oxetanes as the sole product, absolute configuration of which was unambiguously determined by X-ray crystal structural analysis.50,51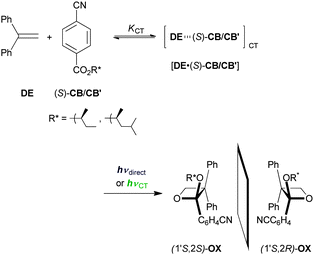 | ||
| Scheme 2 Diastereoselective Paternó–Büchi reaction of 1,1-diphenylethene (DE) with (S)-1-methylpropyl or (S)-1,3-dimethylbutyl p-cyanobenzoate ((S)-CB or (S)-CB′). | ||
Table 2 compares the de's obtained upon local-band versus C-T excitation in the photocycloaddition of DE to (S)-1-methylpropyl or (S)-1,3-dimethylbutyl p-cyanobenzoate ((S)-CB or (S)-CB′) in various solvents at different temperatures. In non-polar methylcyclohexane, the local-band excitation of benzoate gave the oxetane in good de's of 34–77% for (S)-CB and 68–74% for (S)-CB′, while the C-T excitation led to much lower 3–26% de for (S)-CB and 18–44% de for (S)-CB′. By increasing the solvent polarity, the diastereoselectivity was significantly decreased and the chiral sense of the product was inverted in polar solvent(s). Nevertheless, the local-band and C-T excitations consistently afforded different de values, exhibiting distinct temperature-dependence behaviours. These results unambiguously corroborate the discrete nature of ECT and EX, also for this D–A pair. Remarkably, the absolute de value was almost always higher for local-band rather than C-T excitation, a result in sharp contrast to the ST-F system, where the C-T excitation gave better de's.
| Solvent | Temp. (°C) | % Deb | |
|---|---|---|---|
| Local-band excitation (290 nm) | C-T excitation (330 nm) | ||
| a [DE]0 = 1.0 M, [(S)-CB]0 or [(S)-CB′]0 = 0.2 M.b Diastereomeric excess upon local-band (290 nm, 3 h) or C-T excitation (330 nm, 5−10 h) in the photoreaction of DE with (S)-CB, unless noted otherwise; the positive/negative de values indicate the preferential formation of (1′S,2R)- and (1′S,2S)-oxetane, respectively.c Values in the parentheses for the reaction of DE with more hindered (S)-CB′. | |||
| Methylcyclohexane | +50 | 77 (74)c | 3 (18)c |
| −50 | 34 (68)c | 26 (44)c | |
| Toluene | +50 | 74 | −11 |
| −50 | 37 | 21 | |
| Tetrahydrofuran | +50 | 57 | −12 |
| −50 | 32 | 12 | |
| Acetonitrile | +50 | −13 | −3 |
| −40 | −11 | −11 | |
The structures of the diastereomeric C-T complexes of DE with (S)-CB were simulated by the theoretical calculations at the DFT-D3-B-LYP/TZVP level.38 For the sake of simplicity, only the most stable Tg+ conformation of the chiral group is shown for the diastereomeric C-T complexes in Fig. 3, although an ensemble of all possible conformers was considered in the calculation.50,51 The calculated results revealed that the ground-state C-T complex is stabilized mostly by the π–π interaction between the benzene rings of DE and (S)-CB. Thus, the rather modest diastereoselectivity observed upon C-T excitation seems reasonable, since the chiral auxiliary is not only located at a remote position from the centre of interaction but also avoids effective contacts with the DE's another phenyl group. The preference for the (1′S,2R)-isomer upon C-T excitation at lower temperatures, where the entropic contribution is reduced, was nicely reproduced by the diastereomer ratio predicted by the theoretical calculation. The degrees of C-T calculated for the ground-state si- and re-[DE·(S)-CB] complexes (Fig. 3, left) were again as small as 0.3 and 1.0%, respectively. The structures of the diastereomeric si- and re-[DE·(S)-CB]* exciplexes (Fig. 3, right) were optimized at the TD-DFT-BH-LYP/TZVP level. At a glance, the exciplex structures, especially the re-exciplex, look only marginally different from those of the corresponding C-T complexes, but careful examinations of the non-bonded interaction distances revealed significant differences, providing some insights into the nature of the C-T complex and the exciplex. Thus, the facing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds were separated by 5.5 and 3.6 Å in the si- and re-C-T complexes, respectively, but by almost equal to 3.8 Å in the si- and re-exciplex. Quite interestingly, the facing benzene rings were nevertheless kept parallel at ≈3.7 Å in both C-T complexes. The first excited singlet state of (S)-CB calculated at the RI-CC2/TZVPP level revealed that the main contributor (MO #52 → #55), possessing significant n–π* character, amounts to nearly 65% (Fig. 4). Thus, the carbonyl group would actively contribute to the excited-state behaviour. This implies that the exciplex formation is more sensitive to the stereochemistry of the ester group and the chiral information is more effectively transferred in EX than in ECT, if both do not equilibrate in the excited state.
C and CO bonds were separated by 5.5 and 3.6 Å in the si- and re-C-T complexes, respectively, but by almost equal to 3.8 Å in the si- and re-exciplex. Quite interestingly, the facing benzene rings were nevertheless kept parallel at ≈3.7 Å in both C-T complexes. The first excited singlet state of (S)-CB calculated at the RI-CC2/TZVPP level revealed that the main contributor (MO #52 → #55), possessing significant n–π* character, amounts to nearly 65% (Fig. 4). Thus, the carbonyl group would actively contribute to the excited-state behaviour. This implies that the exciplex formation is more sensitive to the stereochemistry of the ester group and the chiral information is more effectively transferred in EX than in ECT, if both do not equilibrate in the excited state.
![Structures of the diastereomeric ground-state C-T complexes, si- and re-[DE·(S)-CB] (left), optimized at the DFT-D3-B-LYP/TZVP level and of the diastereomeric exciplexes, si- and re-[DE·(S)-CB]* (right), optimized at the TD-DFT-BH-LYP/TZVP level. Note that only the Tg+ conformation of the chiral group is shown, but the other conformers also contribute as ensemble.](/image/article/2013/CS/c3cs60117j/c3cs60117j-f3.gif) | ||
| Fig. 3 Structures of the diastereomeric ground-state C-T complexes, si- and re-[DE·(S)-CB] (left), optimized at the DFT-D3-B-LYP/TZVP level and of the diastereomeric exciplexes, si- and re-[DE·(S)-CB]* (right), optimized at the TD-DFT-BH-LYP/TZVP level. Note that only the Tg+ conformation of the chiral group is shown, but the other conformers also contribute as ensemble. | ||
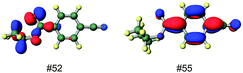 | ||
| Fig. 4 Optimized structure of (S)-CB at the DFT-D3-B-LYP/TZVP level and the relevant molecular orbitals associated with the first excited state. | ||
Temperature dependence of diastereoselectivity and activation parameters for ECT and EX
It is now clear that simply changing excitation wavelength can critically manipulate the net photochemical outcome of D–A pairs, especially the (dia)stereoselectivity of chiral photoreaction. The distinctly different photobehaviours observed upon local-band and C-T excitations indicate that ECT differs in structure and reactivity from the conventional EX. Then the subsequent question may follow: how and to what extent do the two species differ? The formation of EX is a dynamic process that involves structural relaxation of an encounter complex with accompanying solvent reorganization, while ECT is thought to retain some structural information of the ground-state C-T complex due to the π–π stacking interaction between D and A, and hence the solvent reorganization is anticipated to be less extensive. It is likely therefore that EX, rather than ECT, is more susceptible to such environmental variants as temperature and solvent. In order to gain further insights into the nature of ECT and EX, we examined the temperature-dependence behaviour of the product's de values obtained in the above-mentioned diastereoselective photocycloaddition reactions, which were then subjected to the Eyring analysis to determine the differential activation parameters.52,53 Thus, the logarithm of the diastereomer ratio was plotted against the reciprocal temperature to afford good straight lines of different slopes and intercepts (Fig. 5), indicating that a single but discrete diastereodifferentiation mechanism is operative in each system, but the activation parameters depend on the system and the excitation mode. This again confirms that the difference between ECT and EX is not an incidental phenomenon, but a common feature specific to the excited state behaviour of D–A system. Note that the plot was somewhat scattered for the C-T excitation of the (E)-ST–(R)-F pair, due to the high de's of ≥90%.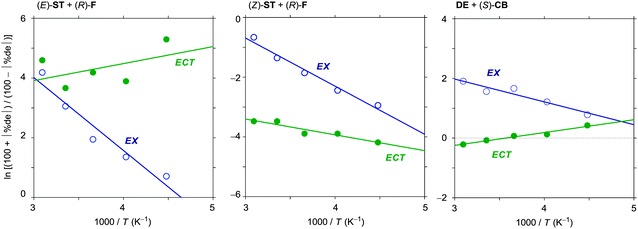 | ||
| Fig. 5 Eyring plots of the relative rate constant for the formation of the diastereomeric pairs in the photocycloaddition of (E)-ST to (R)-F (left), of (Z)-ST to (R)-F (middle), and of DE to (S)-CB (right) via EX generated upon local-band excitation and ECT generated upon C-T excitation in toluene. | ||
The Eyring treatment (eqn (3)) of the relative rate constant for competitive diastereomeric processes leads to the following equation (eqn (4)). From the slope and intercept of the regression line, the differential enthalpy (ΔΔH‡) and entropy (ΔΔS‡) of activation can be calculated.54 Accordingly, the differential activation parameters for each D–A system were evaluated for the local-band and C-T excitation (Table 3).
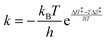 | (3) |
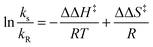 | (4) |
| C-T system | Solvent | Local-band excitation | C-T excitation | ||
|---|---|---|---|---|---|
| ΔΔH‡ | ΔΔS‡ | ΔΔH‡ | ΔΔS‡ | ||
| a Calculated from the slope and intercept of the regression lines obtained by plotting the logarithm of the diastereomer ratio against the reciprocal temperature for each C-T system, as exemplified in Fig. 5. | |||||
| (E)-ST + (R)-F | Toluene | +20 | +94 | ≈−4 | ≈+20 |
| (Z)-ST + (R)-F | Toluene | +13 | +34 | +6.9 | −4.2 |
| DE + (S)-CB | Methylcyclohexane | +6.4 | +39 | −3.3 | −10 |
| Toluene | +6.3 | +35 | −3.6 | −13 | |
| Tetrahydrofuran | +3.9 | +23 | −2.8 | −11 | |
| Acetonitrile | −0.2 | −2.7 | +1.0 | +2.4 | |
| DE + (S)-CB′ | Methylcyclohexane | +1.6 | +21 | −3.8 | −9 |
All of the C-T systems examined share a common feature that the absolute activation entropy |ΔΔS‡| is consistently and significantly larger for local-band rather than C-T excitation. This indicates that the diastereotopic face selectivity upon photocycloaddition is more critically controlled by the entropic factors in EX formed via local-band excitation. This seems reasonable because EX is a species formed dynamically in the excited state with significant desolvation and/or solvent reorganization, while the C-T complex which is originally solvated in the ground state suffers only modest solvent reorganization after excitation. It is also reasonable that the absolute activation parameters become smaller to reduce the product's de in polar solvents, irrespective of the excitation mode. Thus, both ECT and EX are considered to be more polarized than the relevant ground-state species and therefore better to be solvated in polar solvents (but not to the same extent), as schematically illustrated in Fig. 6.
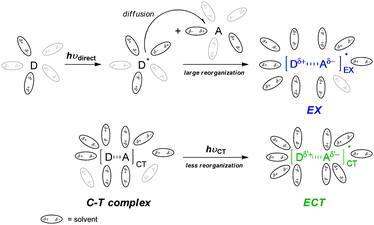 | ||
| Fig. 6 Schematic drawings of the desolvation and/or solvent reorganization behaviour upon local-band and C-T excitation of a D–A system. | ||
The enthalpic and entropic changes often compensate each other. Indeed, the ΔΔH‡ and ΔΔS‡ values obtained for the photocycloaddition of DE to (S)-CB in various solvents show the compensatory enthalpy–entropy relationship to afford distinct linear plots, passing through the origin, both for ECT and EX.55,56 These observations reinforce our claim that a single diastereodifferentiating mechanism is operative in each excitation mode and ECT and EX differ in structure and reactivity. In particular, the higher isodiastereodifferentiating temperature for EX (177 K) than for ECT (275 K) could be taken as experimental evidence in support of the greater flexibility of EX.
5. Asymmetric photoreaction of an intramolecular donor–acceptor system
Diastereoselective photocyclization of (S)-1,1-dicyano-2-methyl-4-phenyl-1-pentene
In order to explore the general validity of the wavelength control and also to further elucidate the mechanistic details, we now discuss the photochemical behaviours of intramolecular C-T systems upon local-band versus C-T excitation. In the intermolecular C-T systems discussed above, the intervening ECT and EX species are complicated conformer mixtures of different spacing, orientation and solvation state, each of which contributes to the overall photochemical outcomes. This situation more or less hinders further mechanistic elucidations. In contrast, the conformational variation is much reduced in intramolecular C-T systems with a tethered D–A pair, which allows us to more closely analyse the conformational behaviours in the ground and excited states, with the aid of spectroscopic and theoretical tools.4-Aryl-1,1-dicyano-1-butene derivatives are known to undergo competitive cyclization/rearrangement reactions upon irradiation.57 This class of compounds shows a C-T band at longer wavelengths, indicating strong intramolecular C-T interactions in its gauche conformation. Accordingly, the excitation mode can be switched by shifting the irradiation wavelength. A chiral D–A compound, (S)-1,1-dicyano-2-methyl-4-phenyl-1-pentene ((S)-DP), was employed for elucidating the mechanistic details of the photocyclization to a diastereomeric pair of indan derivative upon local-band and C-T excitation (Scheme 3).58,59
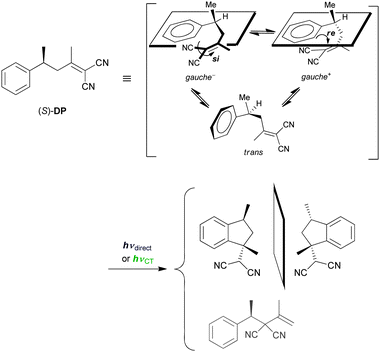 | ||
| Scheme 3 Diastereodifferentiating photocyclization and photorearrangement of (S)-1,1-dicyano-2-methyl-4-phenyl-1-pentene ((S)-DP). | ||
The effects of temperature and solvent on the cyclization/rearrangement ratio and the de of the cyclization product were examined in the photoreaction of (S)-DP, and the results are summarized in Table 4. Upon local-band excitation (254 nm), the formation of the (1R,3S)-product was favoured in dichloromethane but the opposing isomer, i.e., the (1S,3S)-product, designated by a negative sign for de in Table 4, was favoured in polar acetonitrile. The former isomer was always favoured upon C-T excitation over the entire temperature range examined, irrespective of the solvent polarity. At a higher concentration (20 mM), the wavelength effect became less extensive, due to the participation of intermolecular (self) quenching. The temperature and solvent effects are mostly ascribed to the shift of the ground-state conformer equilibrium, but the excited-state conformer equilibrium becomes competitive at higher temperatures. Whatever the reason, it is more crucial that the stereochemical outcome was dramatically manipulated by the mode of excitation also in this intramolecular C-T system. This implies that the C-T excitation leads to an excited-state conformer ensemble which is different from the one formed upon local-band excitation. Such a difference in the excited-state conformer ensemble is likely to be a source of the distinct nature of ECT and EX in the intermolecular C-T system.
| Solvent | Conc. (mM) | Temp. (°C) | % Deb (cyclization/rearrangement ratio)c | ||
|---|---|---|---|---|---|
| Local-band excitation (254 nm) | C-T excitation | ||||
| 280 nm | 300 nm | ||||
| a Local-band (254 nm, 1 h) and C-T excitation (280 or 330 nm, 2 h) under Ar.b Diastereomeric excess of the cyclization product; the +/− signs for de indicate the preferential formation of (1R,3S)- and (1S,3S)-isomer, respectively.c Cyclization/rearrangement ratio shown in the parentheses.d De value not determined due to the low yield of cyclization product. | |||||
| Methylcyclohexane | 0.1 | +20 | d (<0.01) | d (<0.01) | d (<0.01) |
| Dichloromethane | 0.1 | +20 | +20 (0.01) | +82 (0.02) | +90 (0.6) |
| Acetonitrile | 0.1 | +20 | −24 (0.1) | +28 (0.3) | +57 (0.9) |
| 0 | −17 (0.1) | +66 (0.7) | +63 (1.9) | ||
| −20 | −16 (0.3) | +52 (0.4) | +75 (2.3) | ||
| −40 | −14 (0.4) | +45 (0.5) | +51 (2.6) | ||
| 20 | +25 | +28 (0.2) | +65 (0.3) | +62 (1.0) | |
Wavelength effects upon C-T band excitation at different wavelengths
Another intriguing observation in the above system is that the diastereoselectivity was susceptive to the excitation wavelength even within the C-T band. As can be seen from Table 4, the de value became much higher when irradiated at the edge of the C-T band at 300 nm, rather than the middle of the C-T band at 280 nm.58,59The effects of excitation wavelength on the dynamics of C-T complexes have recently been investigated extensively by ultrafast time-resolved spectroscopy.60 The study disclosed that the wavelength effect can be categorized into three distinct types. The difference in photophysical behavior upon low- and high-energy C-T band excitation was ascribed to the different degree of borrowing local excitation in the overlapped C-T band and also to the different complex geometry.61 For instance, a C-T complex of anthracene dimer with tetracyanoethene shows two well-resolved C-T bands, individual excitations of which lead to clean cycloreversion to anthracene at significantly different quantum efficiencies. This wavelength-dependent cycloreversion upon C-T excitation at different wavelengths was explained by the unequal cycloreversion rates for the two distinct excited states of the C-T complex, where the excitation at the high-energy C-T band leads to the formation of an excited-state anthracene cation in the geminate ion pair, accelerating the cycloreversion.62 In addition, the relative contribution of the hot and relaxed (or thermally equilibrated) states also varies with irradiation wavelength.
In the present system, such further wavelength effect was noticed upon excitation at the red edge of the C-T band. Thus, the different product ratios and de values (Table 4) obtained upon C-T excitations at 280 and 300 nm may be ascribed to the different spectral profiles of the two diastereomeric conformers in the ground-state, leading to the unequal excitation efficiencies. Accordingly, even a small change in excitation wavelength within the C-T band allows us to manipulate the relative contribution of the excited-state ensemble components and hence the stereochemical outcomes. Similar observation has been reported recently in the wavelength-controlled supramolecular photocyclodimerization of 2-anthracenecarboxylic acid mediated by native and modified γ-cyclodextrins.63
6. Spectroscopic detection of the excited C-T complex
As mentioned above, the spectroscopic discrimination of ECT from EX has been controversial or at least inconclusive. Nevertheless, the obvious dissimilarities in photochemical behavior observed upon local-band versus C-T excitation of several D–A systems (vide supra) prompted us to further explore spectroscopic investigations. We reexamined the fluorescence behaviour of the representative inter- and intramolecular C-T complexes; i.e., the [(E)-ST·(R)-F] complex and 1,1-dicyano-2-methyl-4-p-anisyl-1-butene (unfortunately, the other substrates, (Z)-ST, (S)-CB and (S)-DE, are not appreciably fluorescent).Fluorescence spectra of a mixture of (E)-ST and (R)-F were measured at different excitation wavelengths (Fig. 7, left). Upon excitation at 350 nm, two fluorescence peaks were observed at ca. 400 and 420 nm, tentatively assignable to ECT and EX, respectively. The ECT fluorescence is slightly higher in energy than the EX fluorescence in consistent with the energy diagram (Fig. 1) depicted in consideration of the nature of the two excited species. Fluorescence lifetimes (τ) of these species (observed at the wavelength specific to each species) were in the same range, indicating that the two excited species resemble each other or the lifetime is relatively insensitive to the small conformational difference. The observed τ value of EX was 0.8–1.2 ns in methylcyclohexane and 1.3–1.4 ns in toluene, while that of ECT was only slightly longer or indistinguishable 1.1–1.3 ns in methylcyclohexane and 1.3–1.5 ns in toluene.
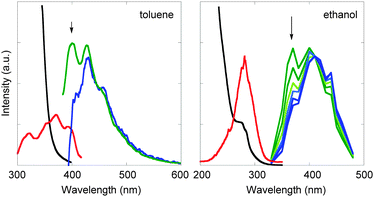 | ||
| Fig. 7 Steady-state and time-resolved fluorescence spectral investigations of ECT and EX. Left: fluorescence from ECT (excited at 350 nm, green) and EX (excited at 330 nm, blue) for a mixture of (E)-ST with (R)-F in toluene. Right: time-resolved fluorescence spectra (normalized at 410 nm) at 5–30 ns delay (from top to bottom) of 1,1-dicyano-2-methyl-4-p-anisyl-1-butene in ethanol glass (77 K). Red and black lines denote the excitation spectra monitored at the ECT fluorescence (indicated by arrows) and the absorption spectra, respectively. | ||
Two excited species were also detected in the lifetime measurement of 1,1-dicyano-2-methyl-4-p-anisyl-1-butene, an achiral analog of DP. Thus, the short-lived component (τ = 2.8–3.9 ns measured at 370 nm) appeared slightly higher in energy than the long-lived component (τ = 7.6–12 ns measured at 400 nm) in an ethanol matrix (Fig. 7, right). All of these observations are consistent with the photochemical results, both jointly revealing that ECT differs from EX in photophysical and photochemical behaviours.
7. Summary and outlook
By selectively exciting the C-T band of a ground-state complex, unconventional excited species ECT, which distinctly differs from EX, is formed. EX is usually formed upon encounter of an excited D or A with a ground-state A or D and hence dynamic in nature, while ECT more or less retains the original structure of the C-T complex pre-organized in the ground state mostly through orbital or π–π interactions. The two excited-state complex species could eventually equilibrate with each other or relax to a common geometry, but nevertheless have been proven to significantly differ in structure and reactivity as described above. Such differences may be better explained by considering an ensemble of excited-state conformers with different reactivities. Consequently, the distinction of ECT from EX has been a difficult controversial issue in the literature. By closely examining the diastereodifferentiating photoreactions that are expected to differ in activation parameter, we have sorted out the elusive ECT from the conventional EX for the representative inter- and intramolecular C-T systems. Thus, the diastereoselectivity was critically manipulated by alternating excitation wavelength to reveal the distinctly different nature of ECT versus EX, which were more specifically characterized by analyzing the activation parameters determined by the temperature-dependence studies. The Eyring plots obtained over a limited temperature range always gave good straight lines, indicating operation of a single diastereodifferentiation mechanism. Crucially, the obtained slopes and intercepts significantly differ for the photoreactions via ECT and EX, indicating the discrete reactivity specific to each species. The differential entropy of activation was consistently larger for EX than for ECT, reflecting the flexible nature of the former species, for which larger solvent reorganization in the excited state is necessary because no stabilization is expected in the ground state through the C-T interaction that is available to the latter. In this connection, it is important to examine the photochemical behaviour of ECT and EX at different temperatures, since the stereoselectivities obtained upon local-band and C-T excitation could incidentally become close at a given temperature. The theoretical calculations provided a wealth of insights into the structures and properties of C-T complexes, ECT and EX. The temperature-dependence behaviour may be more precisely understood or rationalized by theory if the entropic factors are appropriately treated in the calculation. Incorporating the solvent effect may also be encouraged in calculation, as solvation plays a vital role in bimolecular photochemical reactions, although such advanced theoretical issues remain to be elucidated in a future study.In addition to the conventional variants such as temperature and solvent polarity, we can exploit the excitation wavelength as an extra tool for critically controlling the photochemical consequences, in particular the stereoselectivity in chiral (supramolecular) photochemistry. Such an unconventional, yet practical, means should be widely examined in other donor–acceptor systems not only for better controlling the stereochemical outcomes but also for expanding the regime of photochemistry.
Acknowledgements
This review is dedicated to Profs Jay Kazuo Kochi (1927–2008, age 81)64,65 and Nicholas John Turro (1938–2012, age 74),66,67 both were not only the distinguished physical chemists but also outstanding mentors. This work was generously supported by Grant-in-Aid for Scientific Research (No. 23350018, 24655029, and 21245011) from JSPS, the Mitsubishi Chemical Corporation Fund, the Sumitomo Foundation, the Shorai Foundation for Science and Technology, the Kurata Memorial Hitachi Science and Technology Foundation, and the Tokuyama Science Foundation. We thank Profs Stefan Grimme at Universität Bonn, Andreas Dreuw at Universität Heidelberg, and Cornelia Bohne at University of Victoria for fruitful discussion on the theoretical treatment on the excited state and time-resolved fluorescence experiment, respectively. We also wish to express sincere gratitude to all of the students who carried out experimental work on C-T photochemistry over the years, especially to Dr Hideaki Saito, Mr. Kazuyuki Matsumura, and Ms. Emi Saito-Nishiuchi, who contributed to each of the three C-T systems.Notes and references
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken and W. B. Berson, Molecular Complexes, Wiley, New York, 1969 Search PubMed.
- S. P. McGlynn, Chem. Rev., 1958, 58, 1113–1156 CrossRef CAS , and references therein.
- Hydrogen bonding plays crucial roles also in the excited state and has been recently emphasized in the photophysical and photochemical dynamics. G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS , and references therein.
- S. V. Rosokha and J. K. Kochi, J. Org. Chem., 2002, 67, 1727–1737 CrossRef CAS.
- L. Lenoir, Angew. Chem., Int. Ed., 2003, 42, 854–857 CrossRef , and references therein.
- N. Mataga, H. Chosrowjan and S. Taniguchi, J. Photochem. Photobiol., C, 2005, 6, 37–79 CrossRef CAS , and references cited therein.
- For theoretical description of CT complex, see: C. J. Bender, Chem. Soc. Rev., 1986, 15, 475–502 RSC.
- W. D. McKim, J. Ray and B. R. Arnold, J. Mol. Struct., 2013, 1033, 131–136 CrossRef CAS , and references therein.
- J. E. Frey, T. Aiello, S.-L. Fu and H. Hutson, J. Org. Chem., 1996, 61, 295–300 CrossRef CAS.
- J. E. Frey, T. Aiello, D. N. Beaman, H. Hutson, S. R. Lang and J. J. Puckett, J. Org. Chem., 1995, 60, 2891–2901 CrossRef CAS.
- J. E. Frey, T. Aiello, D. N. Beaman, D. Combs, S.-L. Fu and J. J. Puckett, J. Org. Chem., 1994, 59, 1817–1830 CrossRef CAS.
- J. E. Frey, A. M. Andrews, S. D. Combs, S. P. Edens, J. J. Puckett, R. E. Seagle and L. A. Torreano, J. Org. Chem., 1992, 57, 6460–6466 CrossRef CAS.
- J. E. Frey, A. M. Andrews, D. G. Ankoviac, D. N. Beaman, L. E. Du Pont, T. E. Elsner, S. R. Lang, M. A. O. Zwart, R. E. Seagle and L. A. Torreano, J. Org. Chem., 1990, 55, 606–624 CrossRef CAS.
- For examples: D. Jérome, Chem. Rev., 2004, 104, 5565–5592 CrossRef.
- H. Peng and Z. Liu, Coord. Chem. Rev., 2010, 254, 1151–1168 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS , and references therein.
- J. M. Masnovi, J. K. Kochi, E. F. Hilinski and P. M. Rentzepis, J. Am. Chem. Soc., 1986, 108, 1126–1135 CrossRef CAS.
- E. F. Hilinski, J. M. Masnovi, C. Amatore, J. K. Kochi and P. M. Rentzepis, J. Am. Chem. Soc., 1983, 105, 6167–6168 CrossRef CAS.
- T. Fujisawa, M. Creelman and R. A. Mathies, J. Phys. Chem. B, 2012, 116, 10453–10460 CrossRef CAS.
- C. Singh, B. Modak, J. A. Mondal and D. K. Palit, J. Phys. Chem. A, 2011, 115, 8183–8196 CrossRef CAS.
- P. B. Coto, L. Serrano-Andrés, T. Gustavsson, T. Fujiwara and E. C. Lim, Phys. Chem. Chem. Phys., 2011, 13, 15182–15188 RSC.
- B. C. Westlake, M. K. Brennaman, J. J. Concepcion, J. J. Paul, S. E. Bettis, S. D. Hampton, S. A. Miller, N. V. Lebedeva, M. D. E. Forbes, A. M. Moran, T. J. Meyer and J. M. Papanikolas, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8554–8558 CrossRef CAS.
- D. R. Glowacki, J. Lockhart, M. A. Blitz, S. J. Klippenstein, M. J. Pilling, S. H. Robertson and P. W. Seakins, Science, 2012, 337, 1066–1069 CrossRef CAS.
- M. Irie, S. Tomimoto and K. Hayashi, J. Phys. Chem., 1972, 76, 1419–1424 CrossRef CAS.
- M. ltoh and T. Mimura, Chem. Phys. Lett., 1974, 24, 551–554 CrossRef.
- M. Itoh, T. Mimura, H. Usni and T. Okamoto, J. Am. Chem. Soc., 1973, 95, 4388–4392 CrossRef CAS.
- M. Castella, A. Tramer and F. Piuzzi, Chem. Phys. Lett., 1986, 129, 112–116 CrossRef CAS.
- B. S. Green, M. Rejto, D. E. Johnson, C. E. Hoyle, J. T. Simpson, P. E. Correa, T. I. Ho, F. McCoy and F. D. Lewis, J. Am. Chem. Soc., 1979, 101, 3325–3331 CrossRef CAS.
- N. Haga, H. Nakajima, H. Takayanagi and K. Tokumaru, J. Org. Chem., 1998, 63, 5372–5384 CrossRef CAS.
- N. Haga, H. Nakajima, H. Takayanagi and K. Tokumaru, Chem. Commun., 1997, 1171–1172 RSC.
- Y. Shirota, J. Nagata and H. Mikawa, Chem. Lett., 1972, 49–50 CrossRef CAS.
- N. Haga, H. Takayanagi and K. Tokumaru, Photochem. Photobiol. Sci., 2003, 2, 1215–1219 CAS.
- N. Haga, H. Takayanagi and K. Tokumaru, J. Chem. Soc., Perkin Trans. 2, 2002, 734–745 RSC.
- H. Saito, T. Mori, T. Wada and Y. Inoue, J. Am. Chem. Soc., 2004, 126, 1900–1906 CrossRef CAS.
- H. Saito, T. Mori, T. Wada and Y. Inoue, Org. Lett., 2006, 8, 1909–1912 CrossRef CAS.
- H. Saito, T. Mori, T. Wada and Y. Inoue, Chem. Commun., 2004, 1652–1653 RSC.
- S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916–926 CrossRef CAS , and reference cited therein.
- S. Grimme, J. Phys. Chem. A, 2005, 109, 3067–3077 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS.
- S. Grimme, Rev. Comput. Chem., 2004, 20, 153–218 CrossRef CAS.
- M. Kołaski, C. R. Arunkumar and K. S. Kim, J. Chem. Theory Comput., 2013, 9, 847–856 CrossRef.
- T. Kobayashi, K. Yoshihara and S. Nagakura, Bull. Chem. Soc. Jpn., 1971, 44, 2603–2610 CrossRef CAS.
- N. Niimura, Y. Ohashi and Y. Saito, Bull. Chem. Soc. Jpn., 1968, 41, 1815–1820 CrossRef CAS.
- See also: S. D. Bella, I. L. Fragalá, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 1993, 115, 682–686 CrossRef , and references therein.
- E. Paternó and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341–361 Search PubMed.
- G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327–4331 CrossRef.
- D. Sun, S. M. Hubig and J. K. Kochi, J. Org. Chem., 1999, 64, 2250–2258 CrossRef CAS.
- R. A. Neunteufel and D. R. Arnold, J. Am. Chem. Soc., 1973, 95, 4080–4081 CrossRef CAS.
- K. Matsumura, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2009, 131, 17076–17077 CrossRef CAS.
- K. Matsumura, T. Mori and Y. Inoue, J. Org. Chem., 2010, 75, 5461–5469 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- O. Exner, Nature, 1970, 227, 366–367 CrossRef CAS.
- See also: J. E. Leffler, J. Org. Chem., 1955, 20, 1202–1231 CrossRef CAS.
- R. C. Cookson, D. E. Sadler and K. Salisbury, J. Chem. Soc., Perkin Trans. 2, 1981, 774–782 RSC.
- E. Nishiuchi, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2012, 134, 8082–8085 CrossRef CAS.
- T. Ito, E. Nishiuchi, G. Fukuhara, Y. Inoue and T. Mori, Photochem. Photobiol. Sci., 2011, 10, 1405–1414 CAS.
- O. Nicolet, N. Banerji, S. Pagès and E. Vauthey, J. Phys. Chem. A, 2005, 109, 8236–8245 CrossRef CAS.
- D. Levy and B. R. Arnold, J. Phys. Chem. A, 2005, 109, 2113–2119 CrossRef CAS.
- J. M. Masnovi and J. K. Kochi, J. Am. Chem. Soc., 1985, 107, 6781–6788 CrossRef CAS.
- Q. Wang, C. Yang, C. Ke, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, Chem. Commun., 2011, 47, 6849–6851 RSC.
- S. J. Ainsworth, Chem. Eng. News, 2008, 86(35), 69 Search PubMed.
- R. Rathore, Angew. Chem., Int. Ed., 2008, 47, 9186 CrossRef CAS.
- S. J. Ainsworth, Chem. Eng. News, 2012, 90(50), 58 Search PubMed.
- V. Ramamurthy and J. Mattay, Angew. Chem., Int. Ed., 2013, 52, 1363–1364 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |