 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme immobilisation in biocatalysis: why, what and how†
Roger A.
Sheldon
*ab and
Sander
van Pelt
b
aDepartment of Biotechnology, Delft University of Technology, Julianalaan 136, Delft 2628BL, Netherlands. E-mail: r.a.sheldon@tudelft.nl; Fax: +31 (0)15 2781415; Tel: +31 (0)15 2782675
bCLEA Technologies B. V., Delftechpark 34, 2628 XH Delft, Netherlands. E-mail: s.vanpelt@clea.nl; r.sheldon@clea.nl; Tel: + 31 (0)15 7600300
First published on 27th March 2013
Abstract
In this tutorial review, an overview of the why, what and how of enzyme immobilisation for use in biocatalysis is presented. The importance of biocatalysis in the context of green and sustainable chemicals manufacture is discussed and the necessity for immobilisation of enzymes as a key enabling technology for practical and commercial viability is emphasised. The underlying reasons for immobilisation are the need to improve the stability and recyclability of the biocatalyst compared to the free enzyme. The lower risk of product contamination with enzyme residues and low or no allergenicity are further advantages of immobilised enzymes. Methods for immobilisation are divided into three categories: adsorption on a carrier (support), encapsulation in a carrier, and cross-linking (carrier-free). General considerations regarding immobilisation, regardless of the method used, are immobilisation yield, immobilisation efficiency, activity recovery, enzyme loading (wt% in the biocatalyst) and the physical properties, e.g. particle size and density, hydrophobicity and mechanical robustness of the immobilisate, i.e. the immobilised enzyme as a whole (enzyme + support). The choice of immobilisate is also strongly dependent on the reactor configuration used, e.g. stirred tank, fixed bed, fluidised bed, and the mode of downstream processing. Emphasis is placed on relatively recent developments, such as the use of novel supports such as mesoporous silicas, hydrogels, and smart polymers, and cross-linked enzyme aggregates (CLEAs).
 Roger A. Sheldon | Roger Sheldon is a recognised authority on Green Chemistry and Catalysis and Professor Emeritus of Biocatalysis and Organic Chemistry at Delft University of Technology. He is widely known for developing the concept of E factors for assessing the environmental footprint of chemical processes. He has a PhD in organic chemistry from the University of Leicester (UK). Prior to joining Delft University in 1991, he was Vice President for Research and Development at DSM/Andeno from 1980 to 1990 and with Shell Research Amsterdam from 1969 to 1980. He is currently CEO of CLEA Technologies BV, a biotech company. |
 Sander van Pelt | Sander van Pelt studied Chemical Technology and Bioprocess Technology at the Technical University of Delft. After obtaining a masters degree in 2005, during which he was an intern at Degussa and Codexis, a PhD position was taken in the group of Roger Sheldon. This research resulted in 2010 in a thesis entitled: “The Application of Nitrile Hydratases in Organic Synthesis”. In 2010 he joined CLEA Technologies BV, where he is currently Chief Technical Officer. |
Key learning points(1) The importance of immobilisation for improving the operational performance and, hence, cost-effectiveness of enzymes in sustainable biocatalytic processes.(2) The advantages and limitations of different approaches to immobilising enzymes, i.e.adsorption on prefabricated supports (carriers), encapsulation and carrier-free cross-linking, e.g. as cross-inked enzyme aggregates (CLEAs). (3) Clarification of the terminology of enzyme immobilisation, e.g. immobilisation yield and efficiency, activity recovery and enzyme loading. (4) The effects of various physical parameters, e.g. particle size and density, porosity, hydrophilicity–hydrophobicity, on the properties of immobilised enzymes. (5) Methods for preparing ‘smart’ immobilised enzymes, e.g. using thermoresponsive polymers as supports or magnetic nanoparticles for producing magnetisable immobilisates such as the magnetic cross-linked enzyme aggregates. |
1. Introduction
Enzymes are Nature's sustainable catalysts. They are biocompatible, biodegradable and are derived from renewable resources. Enzymatic processes are conducted under mild conditions (close to ambient temperature, atmospheric pressure and physiological pH) in water, with high rates and selectivities. Furthermore, the use of enzymes generally obviates the need for functional group protection and/or activation, affording synthetic routes that are more step economic, generate less waste and are more energy efficient than conventional organic syntheses. In short, enzymatic processes are more environmentally friendly, more cost-effective and, ultimately, more sustainable. Consequently, in the last two decades biocatalysis has emerged as an important technology for meeting the growing demand for green and sustainable chemicals manufacture,1,2 particularly in the synthesis of pharmaceuticals, flavour and fragrances, vitamins and other fine chemicals.3,4Thanks to advances in biotechnology and protein engineering it is now possible to produce most enzymes for commercially acceptable prices and to manipulate them such that they exhibit the desired properties with regard to, inter alia, substrate specificity, activity, selectivity, stability and pH optimum.5,6
Notwithstanding all these advantages, industrial application of enzymes is often hampered by a lack of long-term operational stability and difficult recovery and re-use of the enzyme. These drawbacks can generally be overcome by immobilisation of the enzyme.7–10 In addition to more convenient handling of the enzyme, as a solid rather than a liquid formulation, it provides for its facile separation from the product, thereby minimising or eliminating protein contamination of the product. Moreover, an immobilised enzyme cannot easily penetrate the skin and, therefore, exhibits low or no allergenicity. Immobilisation also facilitates the efficient recovery and re-use of the enzyme, thus enabling its cost-effective use in, for example, continuous, fixed-bed operation. A further benefit is generally enhanced stability, under both storage and operational conditions, e.g. towards denaturation by heat or organic solvents or by autolysis. Improved enzyme performance and repeated re-use is reflected in higher catalyst productivities (kg product per kg enzyme) which, in turn, determine the enzyme costs per kg product.
2. Types of immobilisation
Basically, methods of enzyme immobilisation can be divided into three categories, binding to a support (carrier), entrapment (encapsulation) and cross-linking (Fig. 1):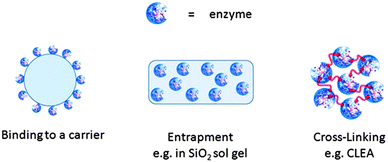 | ||
| Fig. 1 Different methods for immobilising enzymes. | ||
(i) Binding to a support (carrier) can be physical (such as hydrophobic and van der Waals interactions), ionic, or covalent in nature.11 However, physical binding is generally too weak to keep the enzyme fixed to the carrier under rigorous industrial conditions of high reactant and product concentrations and high ionic strength. Ionic binding is generally stronger and covalent binding of the enzyme to the support would generally prevent the enzyme from leaching from the surface. On the other hand, covalent bonding to the enzyme has the disadvantage that if the enzyme is irreversibly deactivated, both the enzyme and the (often costly) support are rendered unusable. Typical supports for enzyme immobilisation are synthetic resins, biopolymers, such as polysaccharides, or inorganic solids such as (mesoporous) silicas or zeolites.
(ii) Entrapment via inclusion of an enzyme in a polymer network, typically organic or inorganic polymer matrices, such as polyacrylamide and silica sol–gel, respectively, or a membrane device such as a hollow fiber or a microcapsule. The physical restraints generally are too weak, however, to prevent enzyme leakage entirely. Hence, additional covalent attachment is often required. The difference between entrapment and support binding is often not clear. For the purpose of this tutorial review we define support binding as the binding of an enzyme to a prefabricated support (carrier) irrespective of whether the enzyme is situated on the external or internal surface. Entrapment generally requires the synthesis of the polymeric matrix in the presence of the enzyme. For example, when an enzyme is immobilised in a prefabricated mesoporous silica the enzyme may be situated largely in the mesopores but this would not be entrapment. On the other hand when the enzyme is present during the synthesis of a silica sol–gel the enzyme is entrapped.
(iii) Cross-linking of enzyme aggregates or crystals, employing a bifunctional reagent, is used to prepare carrierless macroparticles. The use of a carrier inevitably leads to ‘dilution of activity’, owing to the introduction of a large portion of non-catalytic ballast, ranging from 90% to >99%, which results in lower space-time yields and productivities. This is not alleviated by using higher enzyme loadings as this leads to loss of activity owing to difficult accessibility of some of the enzyme molecules when they consist of multiple layers on the surface of the carrier or are situated deeply within the carrier pores, inaccessible to substrate. The optimum situation, from a specific activity viewpoint, is a monolayer of enzyme molecules adsorbed on the surface of the carrier. Consequently, there is an increasing interest in carrier-free immobilized enzymes, such as cross-linked enzyme crystals (CLECs),12 and cross-linked enzyme aggregates (CLEAs).13 This approach offers clear advantages: highly concentrated enzyme activity in the catalyst, high stability and low production costs owing to the exclusion of an additional (expensive) carrier.
3. Terminology and general considerations
The terminology of immobilisation is often inconsistent and/or confusing. The three terms most often used to determine the success of enzyme immobilisation are the immobilisation yield, the immobilisation efficiency and the activity recovery. The immobilisation yield should be used to describe the percentage of total enzyme activity from the free enzyme solution that is immobilised:| Yield (%) = 100 × (immobilised activity/starting activity) |
The “activity that is immobilised” can only be correctly determined by measuring the total residual enzyme activity that remains in the enzyme solution after immobilisation and by subtracting this activity from the total starting activity. In some cases a parallel blank experiment should be carried out to compensate for free enzyme deactivation under the immobilisation conditions. Sometimes protein measurements are used to determine the immobilisation yield. This could be misleading, especially when a crude protein mixture is used for immobilisation, as the different proteins can have different immobilisation yields. It can however be useful to monitor both enzyme activity and protein concentration in the supernatant, to rule out any deactivation of the free enzyme and to determine the protein and/or enzyme loading (wt%) of the immobilised biocatalyst.
The second term often used to describe the success of immobilisation is the immobilisation efficiency. The immobilisation efficiency describes the percentage of bound enzyme activity that is observed in the immobilisate:
| Efficiency (%) = (observed activity/immobilised activity) |
In theory one can have an immobilisation yield of 100% and an immobilisation efficiency of 0% when all of the enzyme in solution is immobilised but no activity is found in the immobilisate because the enzyme was deactivated or became inaccessible for some reason upon immobilisation.
The third term to describe the success of immobilisation is the activity recovery. Activity recovery is the immobilisation yield multiplied by the immobilisation efficiency, which in one number gives you an idea of the success of the total immobilisation process. With activity recovery, the activity of the immobilisate is compared to that of the total starting activity of the free enzyme:
| Activity recovery (%) = (observed activity/starting activity) |
Needless to say, all the terms above have to be calculated by using total activities (units, i.e. μmol min−1) and not by using specific activities (i.e. U mL−1, U mg−1). Furthermore, the exact same activity assay conditions should be used to determine all of the activities.
For example: A lipase is immobilised by hydrophobic adsorption on a bead-like carrier. 1 gram of beads is incubated in an enzyme solution containing a total of 100 units of lipase activity and 50 mg of protein. After 24 hours the beads are filtered and washed. Total lipase activity left in the enzyme solution and wash water is 20 units and the total left-over protein concentration is 10 mg. The washed beads are assayed for activity and the total activity of the beads is found to be 40 units. In this case the immobilisation yield would be 80%, the immobilisation efficiency 50% and the activity recovery 40%. Protein loading on the beads would be 4 wt%.
The observed activity in the immobilisate relative to the activity of the free enzyme (immobilisation efficiency and immobilisation yield) can be highly dependent on the activity assay used (i.e. type of substrate, substrate concentration, pH and temperature) and on the physical properties of the immobilised biocatalyst (i.e. particle size, hydrophobicity–hydrophilicity and pore size). This dependency is most often caused by mass transfer limitations of substrate and/or product in the immobilisation matrix, leading to varying immobilisation yields and activity recoveries.
For example, an enzyme immobilised in a dense polar matrix will have a higher immobilisation yield and activity recovery when the activity assay is carried out with a small polar substrate at high concentration than when a big apolar substrate at low concentration is used, assuming the original free enzyme is equally active on both compounds.
Although the difference in the example above is quite obvious, smaller less evident differences can have a large influence on the results. It is therefore vital for the economics of the immobilisation process to design the immobilised biocatalyst for a specific application and to carry out activity assays as close as possible to the final application in which the immobilised enzyme will be applied.
For use in organic solvents the calculation of activity recoveries becomes more complicated, since the comparison of suspensions of a free enzyme powder and the enzyme in immobilised form in an organic solvent is not always easy. In general, one would expect an immobilised enzyme to give higher rates, owing to a better accessibility of the individual enzyme molecules, when they are neatly spread out on a surface compared to the bulk enzyme powder which, to make matters worse, often contains additives such as (poly)saccharides. Direct comparisons of activities in these cases often lead to activity recoveries higher than 100%.
In short, one has to distinguish between intrinsic loss of activity by deactivation and (apparent) loss of activity owing to inaccessibility of a fraction of the enzyme molecules in the immobilisate. This can be determined using active-site titration.14 The latter technique enables measurement of the amount of active enzyme by employing compounds that irreversibly inhibit the enzyme, by binding successfully to the active-site of the enzyme, while at the same time releasing a compound that can be easily detected. Knowing the amount of active enzyme available in an immobilised biocatalyst is very useful to understand the immobilisation process, diffusional limitations, and how the enzyme responds to a certain material. It could be used, for example, to determine whether the inherent catalytic activity of an enzyme changes as a result of conformational changes caused by interaction with the support, such as the so called “hyper activation” phenomenon observed with lipases (see later).
4. Immobilisation on prefabricated supports
The properties of supported enzyme preparations are governed by the properties of both the enzyme and the carrier material. The interaction between the two provides an immobilised enzyme with specific chemical, biochemical, mechanical and kinetic properties. The support (carrier) can be a synthetic organic polymer, a biopolymer or an inorganic polymer.4.1 Synthetic organic polymers
Various porous acrylic resins, such as Amberlite XAD-7, are used to immobilise enzymes via simple adsorption. For example, the widely used enzyme C. antarctica lipase B, (CaLB),15 is commercially available in immobilised form as Novozym 435 which consists of the enzyme adsorbed on a macroporous acrylic resin. A disadvantage of immobilisation in this way is that, because it is not covalently bound, the enzyme can be leached from the support in an aqueous medium or in the presence of substrates and/or products with surfactant-like properties.In order to suppress leaching, covalent attachment to surface-functionalised acrylic resins, such as Eupergit® C, a macroporous copolymer of N,N′-methylene-bi-(methacrylamide), glycidyl methacrylate, allyl glycidyl ether and methacrylamide, is widely used for immobilisation of enzymes.16 Eupergit C is highly hydrophilic and stable, both chemically and mechanically, over a pH-range from 0 to 14, and does not swell or shrink even upon drastic pH changes in this range. The average particle size is 170 μm and the pore diameter is 25 nm. Protein binding involves reaction of surface oxirane moieties with the free amino groups of the enzyme to form covalent bonds which have long-term stability within a pH range of pH 1 to 12 (see Fig. 2). The remaining epoxy-groups can be quenched with a variety of reagents, such as mercaptoethanol, ethanolamine and glycine. Due to the high density of oxirane groups on the surface of the beads enzymes are immobilised at various sites on their surface. This “multi-point-attachment” is largely responsible for the high operational stability of enzymes bound to Eupergit® C.
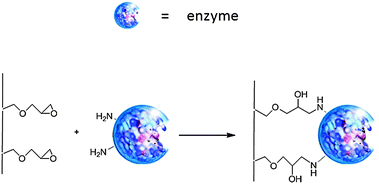 | ||
| Fig. 2 Covalent binding of an enzyme to a functionalised support. | ||
Immobilisation by covalent attachment to Eupergit C has been successfully applied in a variety of industrial settings. Penicillin amidase on Eupergit C, for example, maintained 60% of its initial activity over >800 cycles.17 Sepabeads FP-EP consist of a polymethacrylate-based resin functionalised with oxirane groups and exhibit characteristics similar to Eupergit C. In a comparison of the immobilisation of various lipases on supports with varying hydrophobicity, in the esterification of oleic acid with 1-butanol in isooctane, the highest activity was observed with sepabeads containing octadecyl chains18 which was attributed to the hydrophobic nature of the support facilitating opening of the hydrophobic lid of the lipase.
Alternatively, an enzyme immobilised on a prefabricated support, by simple adsorption, can be stabilised towards leaching and mechanical stress by deposition of a silicone coating formed from inexpensive readily available raw materials.19 For example, Novozyme 435 was coated with a silicone polymer obtained in a hydrosilylation reaction. The silicone was not only deposited as an external layer but also permeated into the porous carrier. The resulting silicone coated Novozyme 435 exhibited high mechanical strength with excellent stability towards leaching. Moreover, the high activity retention (92%) indicated that no significant diffusion limitations were caused by the silicone coating.
4.2 Natural polymers
A variety of naturally occurring polymers, mainly water-insoluble polysaccharides such as cellulose, starch, agarose and chitosan20 and proteins such as gelatin and albumin have been widely used as supports for immobilising enzymes. Indeed, the Tanabe process,21 for the production of L-amino acids by resolution of racemic acylamino acids (Fig. 3), commercialised more than 40 years ago, employs a fixed bed of aminoacylase from Aspergillus oryzae immobilised by ionic adsorption on DEAE-Sephadex (cellulose modified with diethylaminoethyl functionalities).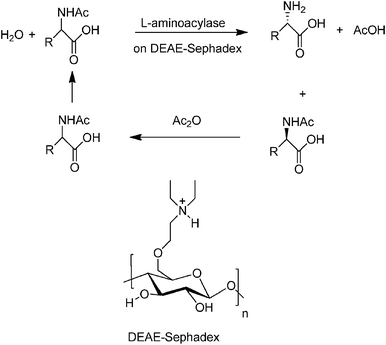 | ||
| Fig. 3 Tanabe aminoacylase process using a packed bed of biocatalyst. | ||
4.3 Inorganic polymers
A variety of inorganic supports are used for the immobilisation of enzymes, e.g., silica,22zeolites23 and mesoporous silicas24 such as MCM-41, and SBA-15. One of the simplest and most inexpensive methods to immobilise an enzyme is by silica granulation.15 It is used in detergent formulations which release the enzyme into the washing liquid during washing. Granulation technology was used to immobilise CaLB lipase on silica granules, by first adsorbing the lipase on silica powder followed by agglomeration.15 Granulates are only suitable for use in organic media. In an aqueous medium the lipase is desorbed and the particle slowly disintegrates. However, the CaLB silica granules can be used in a direct ester synthesis if the water is removed by e.g.evaporation under vacuum. Applying the granules in packed bed reactors also minimises the contact time with high water concentrations. The CaLB silica granules exhibited a similar activity to Novozyme 435 in the direct synthesis of the skin emollient, isopropyl myristate. In order to maintain stability in an aqueous environment the enzyme needs to be covalently attached to a functionalised silica support.Mesoporous silicas, nowadays often referred to as nanosilicas, have several advantages as supports. They have uniform pore diameters (2–40 nm), very high surface areas (300–1500 m2 g−1) and pore volumes (ca. 1 ml g−1), and are inert and stable at elevated temperatures. Moreover, the surface can be easily functionalised. The large pores and cages of these materials can accommodate relatively small enzymes. Whether the enzyme is situated inside the pores or cages or on the outer surface can be determined by comparing immobilisation on calcined and non-calcined material (i.e. the latter still contains the template). If these values are roughly the same then most of the enzyme is on the outer surface whereas when the calcined material adsorbs much more enzyme this indicates that most of the enzyme resides in the pores.
Covalent binding of α-chymotrypsin (EC 3.4.21.2) to a mesoporous sol–gel glass, which had been modified by reaction of surface hydroxyls with 3,3,3-trimethoxypropanal, afforded an immobilised catalyst with a half-life one thousand times that of the free enzyme.25 Similarly, immobilization of Mucor javanicus lipase (EC 3.1.1.3) on functionalized silicananoparticles resulted in enhanced thermal stability and a high retention of activity over a wider pH range.26
Another approach to preventing the leaching of immobilised enzymes from mesoporous hosts is to form physical aggregates of enzyme molecules by precipitation in the nanopores and cages of the host. Subsequent addition of a cross-linker results in the formation of cross-linked enzyme aggregates (CLEAs; see later) entrapped in the nanoscale channels while still allowing accessibility of substrates to the active sites.23
4.4 Protein-coated microcrystals (PCMCs)
So-called protein-coated microcrystals (PCMCs) comprise a novel immobilisation of enzymes on an inorganic support.27 It is based on the fact that lyophilised enzyme powders can be stabilised through the addition of carbohydrates or inorganic salts. PCMCs are prepared by mixing an aqueous solution of the enzyme with a concentrated solution of a salt such as potassium sulphate, an amino acid or a sugar. The resulting solution is added dropwise with vigorous mixing to a water-miscible solvent such as isopropanol, whereupon micron-sized crystals, containing the enzyme on the surface, are formed. A major advantage of the technique is that the enzyme molecules are dehydrated by a mechanism that leaves the majority of the enzymes in an active conformation and minimises denaturation. The PCMCs can be separated and stored or used as a suspension in an organic solvent. Obviously in an aqueous medium they dissolve to liberate the free enzyme. In a transesterification of N-acetyl tyrosine ethyl ester with isopropanol (Fig. 4) PCMCs of subtilisin Carlsberg (EC 3.4.21.62) exhibited an activity three orders of magnitude higher than that of the lyophilised powder.28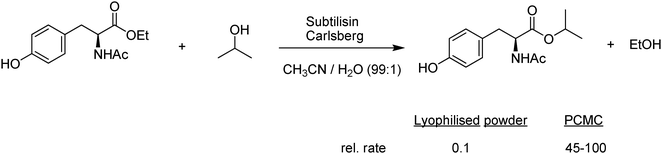 | ||
| Fig. 4 Transesterification over protein-coated microcrystals of subtilisin Carlsberg. | ||
4.5 Smart polymers
A novel approach to immobilisation of enzymes is via covalent attachment to stimulus-responsive or ‘smart polymers’ which undergo dramatic conformational changes in response to small changes in their environment, e.g. temperature, pH and ionic strength.29 The most well-known example is the thermo-responsive and biocompatible polymer, poly-N-isopropylacrylamide (polyNIPAM). Aqueous solutions of polyNIPAM exhibit a critical solution temperature (LCST) around 32 °C, below which the polymer readily dissolves in water while, above the LCST it becomes insoluble owing to expulsion of water molecules from the polymer network. Hence, the biotransformation can be performed under conditions where the enzyme is soluble, thereby minimising diffusional limitations and loss of activity owing to protein conformational changes on the surface of a support. Subsequently, raising the temperature above the LCST leads to precipitation of the immobilised enzyme, thus facilitating its recovery and reuse. An additional advantage is that runaway conditions are avoided because when the reaction temperature exceeds the LCST the catalyst precipitates and the reaction shuts down.Two methods are generally used to prepare the enzyme–polyNIPAM conjugates: (i) introduction of polymerisable vinyl groups into the enzyme followed by copolymerisation with NIPAM or (ii) reaction of NH2 groups on the surface of the enzyme with a copolymer of NIPAM containing reactive ester groups (Fig. 5) or the homopolymer containing an N-succinimide ester function as the end group.
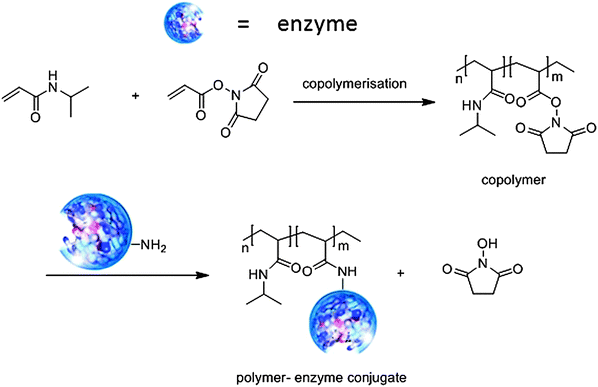 | ||
| Fig. 5 Polymer–enzyme conjugates as thermoresponsive biocatalysts. | ||
For example, penicillin G amidase was immobilised by condensation with a copolymer of NIPAM containing active ester groups.30 The resulting enzyme–polymer conjugate exhibited hydrolytic activity close to that of the free enzyme and was roughly as effective in the synthesis of the semi-synthetic cephalosporin, cephalexin, an important beta lactamantibiotic, by reaction of D-phenylglycine amide with 7-ADCA (Fig. 6).
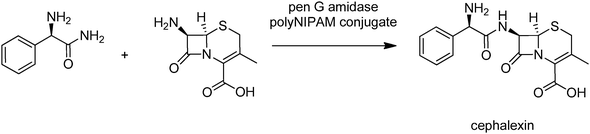 | ||
| Fig. 6 Cephalexin synthesis with a thermoresponsive polymer–penicillin G amidase conjugate. | ||
4.6 Smart immobilisation: enzyme–magnetic nanoparticle hybrids
Immobilisation of enzymes on solid carriers allows for their separation by filtration or centrifugation. For good filterability the particles should be relatively large but increasing the particle size can result in loss of activity owing to diffusion limitations, i.e. slow diffusion of the substrate through the large particles. This means that in practice a compromise has to be made where the particles are large enough to facilitate filtration or centrifugation but not too large that diffusion limitations become an issue. This is even more so in the production of larger volume products where processes are often conducted in continuous operation over a packed bed of (bio)catalyst. In this case it is important to have relatively large particles in order to avoid a pressure drop over the column. Here again, a compromise has to be struck to avoid the pressure drop without decreasing activity as a result of diffusion limitations. In contrast, very small (micron- or even nano-size) particles can be successfully used in a so-called fluidised bed, whereby the liquid feed is passed through the bed of solid catalyst at high enough velocities to suspend the solid and cause it to behave as though it were a fluid. The particles should be small but relatively dense to avoid them being blown out of the column.Alternatively, enzymes can be immobilised by attaching them to functionalised magnetic nanoparticles (MNPs) which can be separated from the reaction mixture by magnetic decantation or used in magnetically stabilised fluidised bed reactors.31 Functionalised MNPs have become commercially available in the last decade driven by various biomedical and biotechnological applications. They consist of an iron oxide core (Fe3O4) coated with, for example, silica containing pendant amine or carboxyl moieties. The latter can be employed to bind the enzyme to the MNP.
5. Entrapment
In the case of enzyme immobilisation by entrapment the support is not prefabricated. It is formed in the presence of the enzyme whereby the latter becomes entrapped inside rather than on the support. In practice, the technique is used more with whole cell biocatalysts rather than with free enzymes.5.1 Silica sol gels
Enzymes can be immobilised by entrapment in silica sol gels prepared by hydrolytic polymerisation of tetraethoxysilane. The morphologies of the silica sol–gels depend on the method of drying.32 Drying by evaporation affords so-called xerogels in which capillary stress causes a shrinkage of the nano cages and pores. When alkyl siloxanes, RSi(OR)3 are used together with Si(OR)4 the surface of the sol–gel is more densely populated by the hydrophobic alkyl groups and the capillary stresses which operate during evaporation are largely attenuated, affording a so-called ambigel in which there is no contraction of the nano cages. Alternatively, drying with supercritical carbon dioxide affords so-called aerogels in which the delicate pore structure and accompanying high porosity is maintained.Entrapment of lipases in sol–gels derived from Si(OEt)4 afforded immobilisates with disappointingly low activities in the esterification of lauric acid by 1-octanol.33 In contrast, entrapment in a sol–gel prepared from a mixture of Si(OMe)4 and RSi(OMe)3 afforded a more hydrophobic matrix exhibiting rate enhancements of 2–8 fold compared with the corresponding lyophilised powder. This method has been widely used for the immobilisation of enzymes.34,35 An interesting elaboration involves the addition of porous supports such as Celite during the sol–gel process to bind the lipase-containing gels. This “double immobilisation” afforded materials with higher thermal stability and activity.36 Lipases from Burkholderia cepacia and Candida antarctica were entrapped in silica aerogels, prepared from mixtures of Si(OMe)4 and MeSi(OMe)3 and reinforced with silicaquartz fibre felt to improve their mechanical properties.37
5.2 Hydrogels
Enzymes can also be immobilised in natural or synthetic hydrogels or cryogels. Polyvinylalcohol (PVA) cryogels, for example, have been widely used for immobilisation of whole cells.38 Partial drying of PVA hydrogels (3–5 mm diameter and 200–400 μm thickness) at room temperature afforded lens-shaped hydrogels, so-called Lentikats, exhibiting good mechanical stability, easy separation and stability towards degradation.39 Lentikats are useful for the entrapment of whole cell biocatalysts. In principle free enzymes can also be entrapped in Lentikats by mixing them directly with the liquid precursor of the Lentikat. Unfortunately the dimensions of most enzymes are not large enough to prevent them leaching from the hydrogel network in an aqueous environment. In order to prevent this the reaction should be performed in non-aqueous media or the size of the enzyme should be increased, e.g. by cross-linking. For example, Gröger and coworkers40 entrapped an (R)-hydroxynitrile lyase in a Lentikat PVA hydrogel by cross-linking it using a mixture of glutaraldehyde and chitosan (Fig. 7). The resulting immobilised biocatalyst had a well-defined particle size of 3–5 mm and showed no leaching during the enantioselective hydrocyanation of benzaldehyde in a biphasic aqueous buffer–organic solvent system. It could be recycled 20 times without loss of yield or enantioselectivity.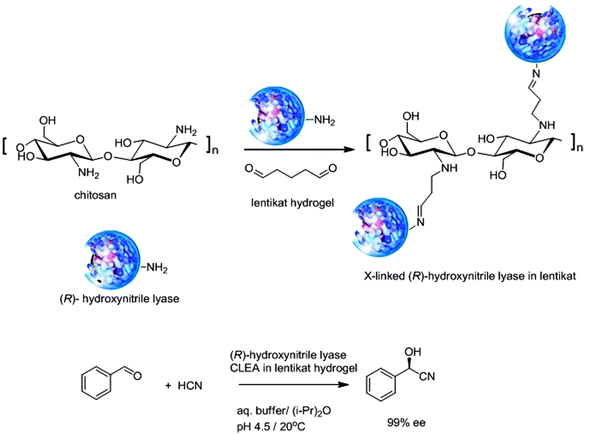 | ||
| Fig. 7 Immobilisation of (R)-hydroxynitrile lyase as a CLEA in a lentikat. | ||
6. Carrier-free immobilisation by cross-linking
In the early 1960s, studies of solid phase protein chemistry led to the discovery that cross-linking of dissolved enzymes via reaction of surface NH2 groups with a bi-functional chemical cross-linker, such as glutaraldehyde, afforded insoluble cross-linked enzymes (CLEs) with retention of catalytic activity. However, this methodology had several drawbacks: low activity retention, poor reproducibility, low mechanical stability, and difficulties in handling the gelatinous CLEs. Mechanical stability and ease of handling could be improved by cross-linking the enzyme in a gel matrix or on a carrier but this led to a disadvantageous dilution of activity. Consequently, in the late 1960s, emphasis switched to carrier-bound enzymes, which became the most widely used methodology for enzyme immobilisation for the following three decades.6.1 Cross-linked enzyme crystals (CLECs)
Cross-linked enzyme crystals (CLECs) are prepared by allowing the enzyme to crystallise from aqueous buffer at the optimum pH and then adding a bifunctional reagent, usually glutaraldehyde, to cross link the crystals. The resulting CLECs are robust, highly active immobilisates of controllable particle size, varying from 1 to 100 μm, depending on the enzyme![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cross-linker ratio and the cross-linking time. The use of CLECs as industrial biocatalysts was commercialised by Altus Biologics in the 1990s.41 The method was broadly applicable, the only requirement being that the enzyme could be crystallised. CLECs are significantly more stable to denaturation by heat, organic solvents and proteolysis than the corresponding soluble enzyme or lyophilised powder. Their operational stability and ease of recycling, coupled with their high catalyst and volumetric productivities, renders them ideally suited for industrial biotransformations. However, an inherent drawback of CLECs is the need to crystallise the enzyme, an often laborious procedure requiring enzyme of high purity. In practice this translates to prohibitively high costs for many applications. To our knowledge CLECs are no longer commercially available and have now been superseded by the closely related CLEAs (see next section).
:cross-linker ratio and the cross-linking time. The use of CLECs as industrial biocatalysts was commercialised by Altus Biologics in the 1990s.41 The method was broadly applicable, the only requirement being that the enzyme could be crystallised. CLECs are significantly more stable to denaturation by heat, organic solvents and proteolysis than the corresponding soluble enzyme or lyophilised powder. Their operational stability and ease of recycling, coupled with their high catalyst and volumetric productivities, renders them ideally suited for industrial biotransformations. However, an inherent drawback of CLECs is the need to crystallise the enzyme, an often laborious procedure requiring enzyme of high purity. In practice this translates to prohibitively high costs for many applications. To our knowledge CLECs are no longer commercially available and have now been superseded by the closely related CLEAs (see next section).
6.2 Cross-linked enzyme aggregates (CLEAS®)
A simpler, and less expensive, alternative to crystallisation is precipitation. The addition of salts, or water miscible organic solvents or non-ionic polymers, to aqueous solutions of proteins leads to their precipitation as physical aggregates of protein molecules, held together by non-covalent bonding without perturbation of their tertiary structure, that is without denaturation. Subsequent cross-linking of these physical aggregates renders them permanently insoluble while maintaining their pre-organised superstructure, and, hence their catalytic activity. This led to the development of a new technology for immobilising enzymes as so-called cross-linked enzyme aggregates (CLEA®) (Fig. 8). Since precipitation from an aqueous medium, by addition of ammonium sulfate or polyethylene glycol, is often used to purify enzymes, the CLEA methodology essentially combines purification and immobilisation into a single unit operation that does not require a highly pure enzyme. It can be used, for example, for the direct immobilisation of an enzyme from a crude fermentation broth.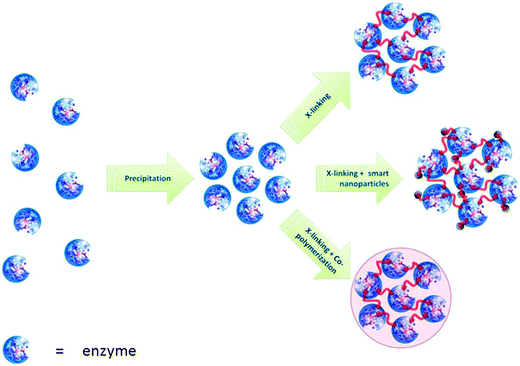 | ||
| Fig. 8 Formation of cross-linked enzyme aggregates (CLEAs). | ||
The CLEA can be modified by performing the cross-linking in the presence of a monomer that undergoes (co)polymerisation under these conditions. This affords CLEA–polymer composites with tunable physical properties. For example, if the cross-linking is performed in the presence of a siloxane, e.g. Si(OMe)4 or RSi(OMe)3, the latter undergoes simultaneous polymerisation to afford a CLEA–silica composite.13 The latter is fundamentally different to the sol gel encapsulated free enzymes formed by polymerisation of an alkoxysilane in the presence of a free enzyme (see Section 5.1). The silica–CLEA composites can be produced with much higher enzyme loadings, and are much less susceptible to enzyme leaching, than the corresponding sol gel encapsulated enzymes. The hydrophobic–hydrophilic properties and particle size of the silica–CLEA composites can be tailored by manipulating the structure of the siloxane used. In an elaboration of this concept, ‘smart’ magnetic CLEAs were prepared by conducting the cross-linking in the presence of functionalised magnetic nanoparticles.42 These mCLEAs can be separated by magnetic decantation or can be used in a magnetically stabilised fluidised bed reactor, affording novel combinations of bioconversions and downstream processing. Another variation on the theme of cross-linked enzyme aggregates are the so-called spherezymes, prepared by addition of precipitant and a cross-linker to water-in-oil emulsions of, inter alia, lipases.43
The first examples of CLEAs were derived from penicillin G amidase an industrially important enzyme used in the synthesis of semi-synthetic penicillin and cephalosporinantibiotics (see earlier). The free enzyme exhibits limited thermal stability and a low tolerance to organic solvents, making it an ideal candidate for stabilisation by immobilisation. Indeed, penicillin G amidase CLEAs, proved to be effective catalysts for the synthesis of beta lactamantibiotics, such as the semi-synthetic penicillin, ampicillin, in organic media (Fig. 9).44
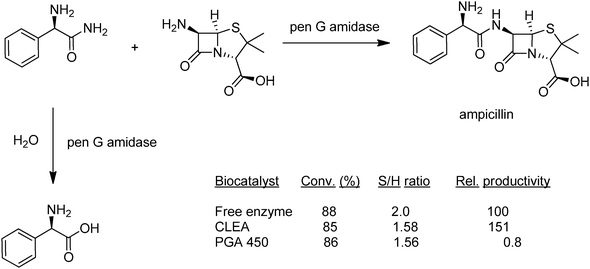 | ||
| Fig. 9 Ampicillin synthesis using penicillin G amidase–CLEA. | ||
Glutaraldehyde is generally the cross-linker of choice as it is inexpensive and readily available in commercial quantities. However, other cross-linkers, such as dextran polyaldehyde, have been used successfully in cases where glutaraldehyde gave poor results.45 Cross-linking involves reaction of the primary amino groups of lysine residues on the enzyme surface with dialdehydes resulting in reversible Schiff's base formation. Subsequent reduction with, e.g.sodium borohydride, to form the corresponding amine, renders the cross-linking irreversible. However, this is generally not necessary with glutaraldehyde as cross-linker because reaction of the latter with the enzyme is more complicated than simple Schiff's base formation.
Since cross-linking largely involves reaction of the amino groups of lysine residues on the external surface of the enzyme, every enzyme can be expected to perform differently. For example, electronegative enzymes contain a paucity of lysine residues on their surface and, hence, cross-linking is expected to be less effective. One way of compensating for this lack of surface amino groups is to coprecipitate the enzyme with a polymer containing numerous free amino groups, e.g. poly-L-lysine,46 polyethylene imine47 or a second protein containing multiple lysine residues48 such as bovine serum albumin (BSA) as a so-called “proteic feeder”.
CLEAs have several benefits in the context of industrial applications. There is no need for highly pure enzyme; they can be prepared from very crude enzyme preparations, even directly from crude cell-free extracts obtained from fermentation broth. Since they are carrier-free they avoid the costs associated with the use of (often expensive) carriers. They exhibit high catalyst productivities (kgs product per kg biocatalyst) and facile recovery and recycle. They generally have improved storage and operational stability with regard to denaturation by heat, organic solvents and autolysis and are stable towards leaching in aqueous media. Another benefit of the CLEA technology is that it is an excellent method for stabilising the quaternary structures of multimeric enzymes, a structural feature encountered with many industrially important enzymes, such as alcohol dehydrogenases, oxidases, peroxidases and nitrile hydratases (see later).49
An important property of CLEAs, from the point of view of large scale applications, is their particle size which obviously has a direct effect on mass transfer limitations and filterability. The particle size is generally in the region of 5–50 μm and filtration or, better still, centrifugation does not pose a problem. If necessary the particle size can be tuned by, inter alia, varying the enzyme/cross-linker ratio and cross-linking time.
The CLEA technology has broad scope and has been applied to an increasingly wide selection of hydrolases, oxidoreductases and lyases.13 The majority of the CLEAs that have been reported to date involve hydrolases, inter alia proteases, lipases, esterases, amidases, nitrilases and glycosidases, mainly because they are the enzymes that have the most industrial applications and are often the simplest enzymes to work with.
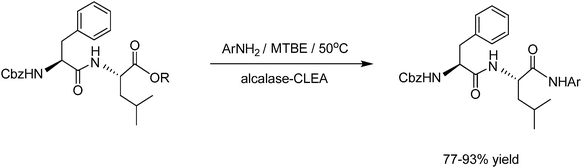
CLEAs have been prepared from a variety of proteases. A pertinent example is the alcalase-CLEA prepared from the Bacillus licheniformis alkaline protease (E.C.3.4.21.62), a laundry detergent enzyme. Alcalase-CLEA has been widely used in amino acid and peptide biotransformations in organic media. For example, under nearly anhydrous conditions, alcalase-CLEA catalysed the mild and cost-efficient synthesis of C-terminal arylamides of amino acids and peptides by aminolysis of the corresponding free carboxylic acid, or the methyl or benzyl ester, with aromatic amines (Fig. 10).50 The products were obtained in high chemical and enantio- and diastereomeric purities. In contrast to with state of the art chemical methods, no racemisation was observed with the enzymatic method.
The same group described51 an elegant, fully enzymatic procedure for the synthesis of peptidesvia a novel C-terminal ester interconversion catalysed by alcalase-CLEA (Fig. 11). This fully enzymatic elongation strategy via C-terminal ester interconversion was successfully applied to the synthesis of biologically active peptides up to the pentamer level.
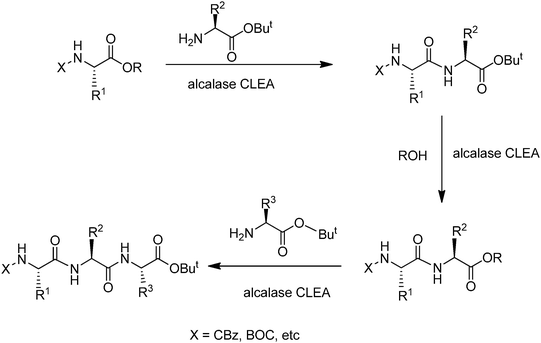 | ||
| Fig. 11 Alcalase-CLEA catalysed peptide synthesis. | ||
In the example shown in Fig. 12 the alcalase-CLEA was used to catalyse the enantioselective hydrolysis of racemic N-protected 2-chlorophenylglycine methyl ester, affording the S-acid in 34% isolated yield and 98% ee.52 The product is an intermediate in the synthesis of the anti-thrombotic drugClopidogrel (Plavix).
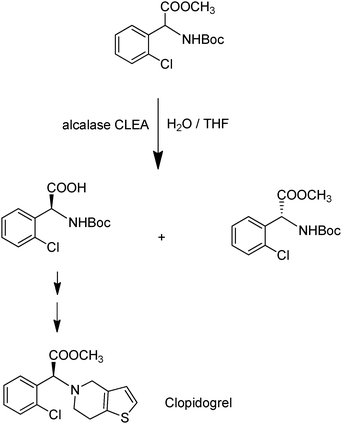 | ||
| Fig. 12 Alcalase-CLEA catalysed hydrolysis of a protected amino ester. | ||
CLEAs have been successfully prepared from a wide variety of lipases (EC 3.1.1.3).13 In one study53 hyperactivation of certain lipases was observed by coprecipitation with additives, such as surfactants and crown ethers, that are known to have an activating effect on lipases. Subsequent cross-linking of the enzyme aggregates, can ‘lock’ the enzyme in a more favourable conformation and, since it is not covalently bonded to the enzyme, the additive can subsequently be washed from the CLEA with an appropriate organic solvent to leave the immobilised enzyme locked in the favourable confirmation. The experimental procedure was further simplified by combining precipitation, in the presence or absence of additives, with cross-linking into a single operation.
Initial studies of CLEAs derived from the popular Candida antarctica lipase B (CaLB) revealed that the excellent performance observed in water, compared to that of the standard immobilised form, Novozyme 435 (CaLB immobilised on a macroporous acrylic resin), could not be directly translated to organic media. In contrast, when the procedure was modified to produce a more lipophilic CLEA a dramatic improvement in activity was observed in the enantioselective acylation of 1-phenethylamine in diisopropyl ether as solvent.13
Recyclable CLEAs were also prepared from a variety of oxidoreductases, e.g. an alcohol dehydrogenase (EC 1.1.1.1), chloroperoxidase (CPO; E.C.1.11.1.10), glucose oxidase (EC 1.1.3.4), galactose oxidase (EC 1.1.3.9) and laccase (EC 1.10.3.2). Laccase, in particular, has many potential applications, e.g. for bleaching in the pulp and paper or textile industries, aqueous effluent treatment and, in combination with the stable radical TEMPO, for the catalytic aerobic oxidation of alcohols, diols and polyols.54
Similarly, CLEAs have been prepared from a variety of lyases. For example, Fe-and Co-dependent nitrile hydratases (NHases; E.C. 4.2.1.84) catalyse the addition of water to nitrile moieties, a reaction of considerable industrial relevance.55 NHases are multimeric enzymes that are generally used as whole-cell biocatalysts because of the limited stability of the isolated enzymes outside the cell, probably owing to dissociation of tetramers resulting in the loss of activity. CLEA formation affords a dramatic increase in operational stability and recyclability, presumably by holding the catalytically active multimer together, analogous to that observed with other multimeric enzymes.49
CLEAs have been successfully prepared from various C–C bond forming lyases, notably the R- and S-specific hydroxynitrile lyases (EC 4.1.2.10) which catalyse the enantioselective hydrocyanation of aldehydes.13 For example, a CLEA prepared from the (R)-specific oxynitrilase from almonds, Prunus amygdalis (PaHnL) was highly effective in the hydrocyanation of aldehydes under microaqueous conditions and could be recycled ten times without loss of activity.56 CLEAs were similarly prepared from the (S)-specific oxynitrilases from Manihot esculenta and Hevea brasiliensis.13 These hydroxynitrile lyase CLEAs perform exceptionally well in organic solvents, affording higher enantioselectivities than those observed with the free enzymes owing to the essentially complete suppression of competing non-enzymatic hydrocyanation.57
6.3 Combi-CLEAs and catalytic cascade processes
The ultimate in environmental and economic efficiency is to combine atom efficient, catalytic steps into a one-pot, catalytic cascade process without the need for separation of intermediates. Catalytic cascade processes have numerous potential benefits: fewer unit operations, less reactor volume, and higher volumetric and space-time yields, shorter cycle times and less waste generation. Furthermore, by coupling steps together unfavourable equilibria can be driven towards product. In principle, this can be achieved by co-precipitation and cross-linking of two or more enzymes in ‘combi CLEAs’. For example, combi CLEAs have been prepared from catalase in combination with glucose oxidase or galactose oxidase. The catalase serves to catalyse the rapid degradation of the hydrogen peroxide formed in the aerobic oxidation of glucose and galactose, respectively, catalysed by these enzymes.A combi CLEA containing an S-selective hydroxynitrile lyase from Manihot esculenta and a nonselective nitrilase from Pseudomonas fluorescens, catalysed the smooth, one-pot conversion of benzaldehyde to S-mandelic acid (Fig. 13)58 in di-isopropyl ether–water (9:1 v/v) at pH 5.5. Enantioselectivity is provided by the hydroxynitrile lyase and in situ conversion by the nitrilase serves to drive the equilibrium of the first step towards product formation. Interestingly, the combi-CLEA was more effective than a mixture of the two separate CLEAs. A possible explanation for this observation is that the close proximity of the two enzymes inside the combi-CLEA is more favourable, compared to the case with two separate CLEAs, for transfer of the product of the first step to the active site of the enzyme for the second step.
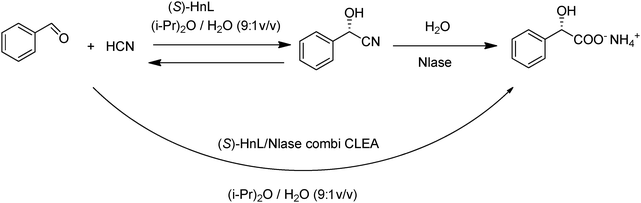 | ||
| Fig. 13 Conversion of benzaldehyde to (S)-mandelic acid with a combi-CLEA. | ||
7. Enzyme-immobilised microchannel reactors: process intensification
Process intensification through the use of microchannel reactors (microfluidic devices) has many advantages compared with traditional batch process technologies: rapid mass and heat transfer and large surface area to volume ratios. These are attractive features for conducting catalytic reactions in microreactors containing the enzyme immobilised on the inner walls of the microchannels, as an enzyme–polymer membrane, for example.59 Thus, a solution of α-chymotrypsin in aqueous buffer was mixed with glutaraldehyde and formaldehyde as cross-linkers in commercially available polytetrafluoroethylene (PTFE) tubing (inner diameter 500 μm). In this way a CLEA membrane was formed on the inner walls of the tubing. With electronegative enzymes coprecipitation of the enzyme in the presence of poly-L-lysine was used to realise fast and efficient CLEA formation.46 The use of such enzyme immobilised microchannel reactors clearly has considerable potential for the design of green and sustainable biotransformations.Littlechild and coworkers60 employed a different strategy. They prepared CLEAs from a thermophilic L-aminoacylase from Thermococcus litorali, which had been overexpressed in E. coli and subsequently mixed them with controlled pore glass before packing them in a capillary reactor fitted with a silica frit to contain them in the reactor. The CLEA microchannel reactor retained activity for at least two months during storage at 4 °C.
8. Conclusions and prospects
Enzyme immobilisation continues to be a subject of immense interest, in both industry and academia. The commercial viability of industrial biotransformations stands or falls with the cost contribution of the enzyme. Immobilisation is an enabling technology that, in addition to providing an active and stable biocatalyst, should be a relatively simple operation not requiring a highly pure enzyme preparation or an expensive support that may not be commercially available. Immobilisation as silica granulates, for example, meets all these criteria but the methodology is not compatible with aqueous environments (see earlier). Cross-linked enzyme aggregates (CLEAs) would appear to have considerable industrial potential based on their high activity retention and stability coupled with ease of preparation from crude enzyme samples and no requirement for a support. Because they consist mainly of active catalyst they also display high catalyst productivities and space time yields. It is also clear that every enzyme is different and, consequently, there is no all-encompassing, ‘one size fits all’ solution to enzyme immobilisation. Driven by the industrial and societal need for sustainable chemical products and processes, and the attractive features of biocatalysis in this context, we expect that interest in improving the operational performance of enzymes by effective immobilisation will continue unabated in the future.References
- J. Tao and R. J. Kazlaukas, Biocatalysis for Green Chemistry and Chemical Process Development, Wiley, Hoboken, New Jersey, 2011 Search PubMed.
- R. Wohlgemuth, Curr. Opin. Biotechnol., 2010, 21, 713–724 CrossRef CAS.
- D. Munoz Solano, P. Hoyos, M. J. Hernaiz, A. R. Alcantara and J. M. Sanchez-Montero, Bioresour. Technol., 2012, 115, 196–207 CrossRef CAS.
- C. M. Clouthierz and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS.
- A. Illanes, A. Cauerhff, L. Wilson and G. R. Castro, Bioresour. Technol., 2012, 115, 48–57 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- D. N. Tran and K. J. Balkus, Jr., ACS Catal., 2011, 1, 956–968 CrossRef CAS.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- L. Cao, Carrier-bound Immobilized Enzymes, Principles, Applications and Design, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. J. Roy and T. E. Abraham, Chem. Rev., 2004, 104, 3705–3721 CrossRef CAS.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS; R. A. Sheldon, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef.
- M. H. A. Janssen, L. M. van Langen, S. R. M. Pereira, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2002, 78, 425–432 CrossRef CAS.
- O. Kirk and M. W. Christensen, Org. Process Res. Dev., 2002, 6, 446–451 CrossRef CAS.
- E. Katchalski-Katzir and D. M. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef CAS.
- A. I. Kallenberg, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2005, 347, 905–926 CrossRef CAS.
- M. Petkar, A. Lali, P. Caimi and M. Daminati, J. Mol. Catal. B: Enzym., 2006, 39, 83–90 CrossRef CAS.
- L. O. Wiemann, P. Weisshaupt, R. Nieguth, O. Thum and M. B. Ansorge-Schumacher, Org. Process Res. Dev., 2009, 13, 617–620 CrossRef CAS.
- B. Krajewska, Enzyme Microb. Technol., 2004, 35, 126–139 CrossRef CAS.
- T. Tosa, T. Mori, N. Fuse and I. Chibata, Enzymologia, 1966, 31, 214 CAS.
- A. Petri, P. Marconcini and P. Salvadori, J. Mol. Catal. B: Enzym., 2005, 32, 219–224 CrossRef CAS.
- J. Diaz and K. J. Balkus, J. Mol. Catal. B: Enzym., 1996, 2, 115–126 CrossRef CAS.
- Z. Zhou and M. Hartmann, Top. Catal., 2012, 55, 1081–1100 CrossRef CAS.
- P. Wang, S. Dai, S. D. Waezsada, A. Y. Tsao and B. H. Davison, Biotechnol. Bioeng., 2001, 74, 249–255 CrossRef CAS.
- M. I. Kim, H. O. Ham, S. D. Ho, H. G. Park, H. N. Chang and S. H. Choi, J. Mol. Catal. B: Enzym., 2006, 39, 62–68 CrossRef CAS.
- M. Kreiner and M.-C. Parker, Biotechnol. Lett., 2005, 27, 1571–1577 CrossRef CAS.
- M. Kreiner, B. D. Moore and M.-C. Parker, Chem. Commun., 2001, 1096–1097 RSC.
- Smart Polymers for Bioseparation and Bioprocessing, ed. I. Y. Galaev and B. Mattiasson, Taylor and Francis, London, 2004 Search PubMed.
- A. E. Ivanov, E. Edink, A. Kumar, I. Y. Galaev, A. F. Arendsen, A. Bruggink and B. Mattiasson, Biotechnol. Prog., 2003, 19, 1167–1175 CrossRef CAS.
- H. H. P. Yiu and M. A. Keane, J. Chem. Technol. Biotechnol., 2012, 87, 583–594 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4265 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Angew. Chem., 1995, 107, 373–376 CrossRef.
- M. T. Reetz, P. Tielmann, W. Wiesenhöfer, W. Könen and A. Zonta, Adv. Synth. Catal., 2003, 345, 717–718 CrossRef CAS.
- A. C. Pierre, Biocatal. Biotransform., 2004, 22, 145–170 CrossRef CAS.
- M. T. Reetz, A. Zonta, J. Simpelkamp and W. Könen, Chem. Commun., 1996, 1397–1398 RSC.
- O. Orçaire, P. Buisson and A. C. Pierre, J. Mol. Catal. B: Enzym., 2006, 42, 106–113 CrossRef.
- V. I. Lozinski and F. M. Plieva, Enzyme Microb. Technol., 1998, 23, 227–242 CrossRef.
- M. Jekel, A. Buhr, T. Willke and K.-D. Vorlop, Chem. Eng. Technol., 1998, 21, 275–278 CrossRef CAS.
- H. Gröger, E. Capine, A. Barthuber and K.-D. Vorlop, Org. Lett., 2001, 3, 1969–1972 CrossRef.
- A. L. Margolin and M. A. Navia, Angew. Chem., Int. Ed., 2001, 40, 2204 CrossRef CAS.
- R. A. Sheldon, M. J. Sorgedrager and B. Kondor, WO2012/023847A2, 2012, to CLEA Technologies B.V Search PubMed.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS.
- L. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS.
- T. Honda, M. Miyazaki, H. Nakamura and H. Maeda, Adv. Synth. Catal., 2006, 348, 2163–2171 CrossRef CAS.
- F. Lopez-Gallego, L. Betancor, A. Hidalgo, N. Alonso, R. Fernandez-Lafuenta and J. M. Guisan, Biomacromolecules, 2005, 6, 1839–1842 CrossRef CAS.
- S. Shah, A. Sharma and M. N. Gupta, Anal. Biochem., 2006, 351, 207–213 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- T. Nuijens, C. Cusan, J. A. W. Kruijtzer, D. T. S. Rijkers, R. M. J. Liskamp and P. J. L. M. Quaedflieg, J. Org. Chem., 2009, 74, 5145–5151 CrossRef CAS.
- T. Nuijens, C. Cusan, T. J. G. M. van Dooren, H. M. Moody, R. Merkx, R. J. A. W. Kruijtzer, D. T. S. Rijkers, R. M. J. Liskamp, P. J. L. M. Quaedflieg and J. L. M. Peter, Adv. Synth. Catal., 2010, 352, 2399–2404 CrossRef CAS.
- P. Ferraboschi, M. De Mieri and F. Galimberti, Tetrahedron: Asymmetry, 2010, 21, 2136–2141 CrossRef CAS.
- P. Lopez-Serrano, L. Cao, F. van Rantwijk and R. A. Sheldon, Biotechnol. Lett., 2002, 24, 1379–1383 CrossRef CAS.
- A. Diaz-Rodriguez, I. Lavandera, S. Kanbak-Aksu, R. A. Sheldon, V. Gotor and V. Gotor-Fernandez, Adv. Synth. Catal., 2012, 354, 3405–3408 CrossRef CAS.
- S. van Pelt, F. van Rantwijk and R. A. Sheldon, Chim. Oggi, 2008, 26(3), 2 CAS.
- L. M. van Langen, R. P. Selassa, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2005, 7, 327–329 CrossRef CAS.
- C. Roberge, F. Fleitz, D. Pollard and P. Devine, Tetrahedron Lett., 2007, 48, 1473 CrossRef CAS.
- C. Mateo, A. Chmura, S. Rustler, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2006, 17, 320–323 CrossRef CAS.
- M. Miyazaki and H. Maeda, Trends Biotechnol., 2006, 24, 463–470 CrossRef CAS.
- A. M. Hickey, L. Marle, T. McCreedy, P. Watts, G. M. Greenway and J. A. Littlechild, Biochem. Soc. Trans., 2007, 35, 1621–1623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cs60075k. |
| This journal is © The Royal Society of Chemistry 2013 |