 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent adaptable networks: smart, reconfigurable and responsive network systems
Christopher J.
Kloxin
a and
Christopher N.
Bowman
*b
aDepartment of Materials Science and Engineering & Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St., Newark, DE 19716, USA. E-mail: cjk@udel.edu; Fax: +1 302 831 4545; Tel: +1 302 831 8670
bDepartment of Chemical and Biological Engineering, University of Colorado, JSCBB C121, Boulder, CO 80309, USA. E-mail: christopher.bowman@colorado.edu; Fax: +1 303 492 4825; Tel: +1 303 492 3247
First published on 12th April 2013
Abstract
Covalently crosslinked materials, classically referred to as thermosets, represent a broad class of elastic materials that readily retain their shape and molecular architecture through covalent bonds that are ubiquitous throughout the network structure. These materials, in particular in their swollen gel state, have been widely used as stimuli responsive materials with their ability to change volume in response to changes in temperature, pH, or other solvent conditions and have also been used in shape memory applications. However, the existence of a permanent, unalterable shape and structure dictated by the covalently crosslinked structure has dramatically limited their abilities in this and many other areas. These materials are not generally reconfigurable, recyclable, reprocessable, and have limited ability to alter permanently their stress state, topography, topology, or structure. Recently, a new paradigm has been explored in crosslinked polymers – that of covalent adaptable networks (CANs) in which covalently crosslinked networks are formed such that triggerable, reversible chemical structures persist throughout the network. These reversible covalent bonds can be triggered through molecular triggers, light or other incident radiation, or temperature changes. Upon application of this stimulus, rather than causing a temporary shape change, the CAN structure responds by permanently adjusting its structure through either reversible addition/condensation or through reversible bond exchange mechanisms, either of which allow the material to essentially reequilibrate to its new state and condition. Here, we provide a tutorial review on these materials and their responsiveness to applied stimuli. In particular, we review the broad classification of these materials, the nature of the chemical bonds that enable the adaptable structure, how the properties of these materials depend on the reversible structure, and how the application of a stimulus causes these materials to alter their shape, topography, and properties.
 Christopher J. Kloxin | Professor Christopher Kloxin is an Assistant Professor in the Departments of Materials Science and Engineering and Chemical and Biomolecular Engineering at the University of Delaware. He received his PhD in Chemical Engineering from North Carolina State University in 2006. He began his post-doctoral training under the advisement of Christopher Bowman at the University of Colorado, where he designed covalent adaptable networks for applications ranging from self-healing materials to low polymerization-stress dental composites. In 2011, he started at the University of Delaware, where his group utilizes a combination of click and reversible chemistries towards creating self-healing and self-assembling macromolecular structures as well as spatioselectively controlling polymer network topography and physiochemical properties. |
 Christopher N. Bowman | Professor Christopher Bowman is a Distinguished Professor and the James and Catherine Patten Chair in Chemical and Biological Engineering as well as the Director of the Materials Science and Engineering Program at the University of Colorado at Boulder. He received his BS and PhD in Chemical Engineering from Purdue University, leaving in 1992 to start at Colorado. Since that time, Professor Bowman has focused his research on polymeric materials, particularly polymer networks derived from photopolymerization reactions. He seeks to integrate molecular design, monomer and polymer synthesis, and polymer and polymerization analysis to create smart, responsive polymeric materials that achieve unique behavior and capabilities. |
Key learning points• Critical differences between thermosets, thermoplastics, and covalent adaptable networks.• Bond-breaking, bond-forming versus bond-forming, bond-breaking network strand rearrangement sequence. • Behavior and mechanical properties derived from reversible addition/condensation and exchange crosslinking. • Design and synthesis of covalent networks with reversible structures. • Triggers enabling activation of covalent adaptable network. |
Introduction
Covalently crosslinked polymer systems, i.e., thermosets, are appropriate for numerous applications in polymer science due to their mechanical and thermal integrity, often ideal optical performance, and their capability to be used in various high technology and biomedical applications. Thermosets represent a highly functional class of covalently crosslinked polymeric materials used in coatings, adhesives, biomedical materials, and structural applications. The presence of the covalent crosslinks in the material renders these materials to be largely elastic in their response to strain deformations. Essentially, the presence of the crosslinked structure makes the shape and nature of the polymer permanent and largely intractable. Whether epoxies, photopolymerized (meth)acrylic networks, or thermally cured polymers formed by reaction injection molding, these networks, by their very definition, are “set” when the crosslinking and/or polymerization reaction is complete, and the polymer networks thus formed become intractable, non-meltable, and unable to achieve any significant flow – they essentially will always “remember” the shape in which they were initially formed. While this behavior is highly attractive from the desirable perspectives associated with mechanical strength and high modulus, it has severe disadvantages in a lack of recyclability, stresses that develop during the initial polymerization, and a general inability of the polymeric material thus formed to change its shape or structure. Ultimately, the same crosslinked structure that is the origin of the desirable thermo-mechanical network properties subsequently leads to the most severe of the material's limitations.Various creative implementations of covalently crosslinked networks have been widely utilized as stimuli responsive materials. The broadest class of polymer networks used as stimuli responsive materials is swollen gels, most commonly hydrogel materials, which respond to a change in a condition such as pH, temperature or ionic strength to trigger a change in equilibrium degree of swelling. Further, work has also been done on polymer networks that function as shape memory materials where a strained polymer network is most frequently trapped in a non-equilibrium state by quenching the material below its glass transition temperature. Upon subsequent heating above the glass transition temperature, the network relaxes to its equilibrium shape and this associated actuation has been used in numerous applications. Despite these innovative implementations in stimuli responsive materials, the irreversible nature of the covalently crosslinked structure remains as a significant limitation and barrier to broader implementation of these materials.
The absence of crosslinks in thermoplastics, i.e., polymer chains without permanent covalent links between chains, has dramatic consequences in regards to the characteristics and behavior of these materials as compared to thermosets. Thermoplastic materials may have a glass transition and/or a crystalline transition; above these transitions, the material is capable of macroscopic flow or plastic deformation that permanently alters the equilibrium shape of the material without altering the molecular structure. Thermoplastic materials can readily be reprocessed and are often recyclable.
As almost a hybrid material that contrasts to networks formed from covalent bonds and crosslinks, physical networks are formed from linear or branched polymers that contain reversible, physical bonds such as hydrogen bonds, pi–pi stacking or ionic interactions. These materials represent an interesting class of materials that are able to adapt to changing conditions, including applied stresses, depending on the timescale of the response – if the stress is applied for much longer than the lifetime of the non-covalent crosslink, then the material will flow like a thermoplastic whereas if the stress is applied for a timescale that is much shorter than the lifetime of the crosslink, then the material will be elastic and behave largely like a thermoset. Unfortunately, physical gels often do not have sufficient mechanical integrity to withstand the demands of many thermosetting applications such as those presented above. However, these physical gels clearly have guided the thinking and direction of a great deal of research in polymer networks that has sought to combine the best attributes of covalently crosslinked networks with the ability for these materials to be molecularly reformed and macroscopically reshaped, as is critical to enable a broader spectrum of stimuli responsive applications. Thus, the desirable combination of a covalently crosslinked polymer structure with the capability for a triggerable reversion of the crosslinked polymer structure has recently been coined a Covalent Adaptable Network (CAN). In particular, CANs are polymer networks in which the crosslinking strands within the polymer material can undergo reversible rearrangement reactions. These molecular rearrangements provide a microscopic mechanism for achieving macroscopic flow and stress relaxation in a class of materials for which this was previously impossible. Further, the activation of the reversible structures within the network can be coupled to an external or environmental stimulus, enabling these materials to be smart, responsive materials. These CANs have all the mechanical benefits of a traditional thermoset while their unique chemical bond structures enable them to respond selectively to applied stimuli to alter their structure, properties, and even their shape. The unique chemical structures and material property sets of these CAN materials places them in a category of materials not previously implemented – as seen in Fig. 1, the powerful molecular structures of these materials essentially enable them to form a bridge across the chasm that previously existed between the independent thermoset and thermoplastic structures and behavior with various stimuli being used to trigger the transition from conventional thermoset behavior to thermoplastic behavior.
 | ||
| Fig. 1 Covalent adaptable networks (CANs) exhibit many of the properties of thermosets, while also possessing the triggerable processability and relaxation behavior of thermoplastics. CANs represent a unique class of materials that not only combine the desirable behaviors of each of these materials, but also enable the transition from one set of material properties (i.e., thermoset-like) to the other (i.e., thermoplastic-like) either upon application of an external stimulus, such as light or temperature, or simply by considering the material behavior on different timescales that are intimately linked to the timescales of network rearrangement. | ||
The focus of this tutorial review is then on these CAN materials, their formation and chemical structures, their unique molecular structure-dependent behavior, and their implementation in stimuli responsive applications in which a selective trigger is used to cause the network to undergo viscoelastic solid to viscoelastic liquid transition that is fundamentally unique to these materials. We will introduce two different motifs that are used to create CANs: reversible addition and condensation and reversible bond exchange processes. Further, we will address how the mechanism by which the network is formed has a pronounced impact on how the rearrangement dynamics and polymer structure change the material properties. The relationship between the kinetics and equilibrium extent of the reactions that form (and break) these crosslinks have a pronounced and unique influence on the material properties that will be explored. Finally, the various mechanisms for triggering the viscoelastic solid to viscoelastic liquid transition (essentially behaving like a transition from a predominantly elastic material to a plastic material) will be discussed as will their implementation in forming stimuli responsive materials.
The recent emergence of CANs in the literature is spawned from the interest in new stimuli-responsive and materials having a dynamic range of mechanical properties. The field of CANs has a rich history dating back to the development of vulcanized rubbers. These materials contain disulfide (and polysulfide) crosslinks between hydrocarbon polymer chains, resulting in a material with enhanced mechanical properties, which are more broadly associated with thermosets as presented above. It was noticed that these and other polysulfide-containing polymer networks, such as thiokols, exhibited the characteristic creep or continuous deformation under stress. In the 1940s, Green and Tobolsky postulated that the disulfide crosslinks are in fact at equilibrium, breaking and reforming, enabling non-degradative bond rearrangement or ‘permanent set’.1 These concepts and others developed in the next several decades have essentially laid the foundation for all reversible covalent networks.2,3
In the last decade there have been several high profile papers that have created and described various CANs, with each network type possessing reversible covalent crosslinks but exhibiting dramatically different stress relaxation and bond rearrangement mechanisms. As an illustration, we compare CANs utilizing Diels–Alder (DA) crosslinks with those utilizing transesterification crosslinks, as presented by Chen et al.4 and Montarnal et al.,5 respectively. In the former system, the thermorereversible Diels–Alder cycloaddition reaction between multifunctional maleimide and furan monomers is used to form a network. Upon heating, the material undergoes the retro-Diels–Alder reaction, enabling network rearrangement (Fig. 2, top). In the latter system, an irreversible reaction between epoxy and carboxylic acid monomers is used to form the initial network, which contains ester crosslinks and pendent alcohol functional groups. With both the addition of a catalyst and the application of heat, the pendent alcohol and ester crosslink undergo a rapid transesterification reaction, shuffling the bonds and enabling network rearrangement (Fig. 2, bottom). While both networks exhibit stress relaxation, reprocessability and self-healing at elevated temperatures, they were each formed using different polymerization strategies: one from a reversible DA reaction and the other from an irreversible epoxide ring-opening reaction. Moreover, these CAN systems employ two fundamentally different reversible bond rearrangement mechanisms. In subsequent sections, we will discuss the advantages and disadvantages of designing a CAN with the various reversible chemistries and network formation strategies and how their distinct chemistries and mechanisms are related to the mechanical properties of the material.
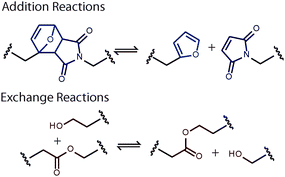 | ||
| Fig. 2 Reversible bond rearrangement reactions employed by Chen et al.4 (top) and Montarnal et al.5 (bottom). Reversible addition reactions (top), such as the Diels–Alder cycloaddition, result in breaking and reforming of the polymer backbone. In contrast, reversible exchange reactions (bottom), such as the transesterification reaction, result in bond rearrangement or shuffling without a disruption in connectivity. | ||
Adaptable network design
There are two basic considerations when designing and synthesizing a covalent adaptable network: the reversible crosslinking chemistry and the network formation strategy. The reversible crosslinking reaction dictates the dynamics of the network strand rearrangement, which determines the relaxation behavior of the material. The network formation strategy ultimately dictates the molecular architecture. In this section, we will discuss how the components of the reversible molecular architecture control the material's complex behavior, enabling one to decide which network formation strategy and reversible crosslinking type is best suited for a particular application.Reversible crosslink type
While there are many reversible covalent crosslinking reactions, we will broadly group these into two categories based on their rearrangement mechanism. Reversible addition and condensation reactions (Fig. 3) exhibit similar bond rearrangement dynamics and follow a bond-breaking, bond-forming reaction sequence. The rearrangement of network strands necessarily and primarily involves a transition of decreased network connectivity and crosslink density, as for example in the DA-based CANs (Fig. 4, top). In contrast, reversible exchange reactions (Fig. 3) follow a bond-forming, bond-breaking sequence (e.g., addition–fragmentation), without a decrease in connectivity or crosslink density during the transition, as for example seen with the transesterification-based CANs (Fig. 4, bottom). This difference in mechanism is critical, and it is this rearrangement mechanism sequence that primarily determines the mechanical behavior of a CAN in its activated state.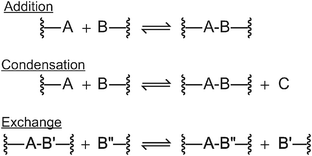 | ||
| Fig. 3 Reversible reaction types: addition, condensation, and exchange reactions (top to bottom). In both the addition and condensation reaction approaches, it is worth noting that the network connectivity and the number of elastically active network chains are altered based on the relative extents of the forward and reverse reactions. In the exchange reaction system, the specific molecular connectivity is changed by the forward and reverse reactions; however, the average number of elastically active chains in the network is not altered. This key difference in the fundamental mechanism of the reaction yields dramatic differences in the temperature dependent properties of these materials as well as the dependence of crosslink density, modulus and other critical characteristics of the material when these reactions are triggered. | ||
 | ||
| Fig. 4 Connectivity transition difference for the reversible addition (top) and reversible exchange (bottom) crosslinking reactions. Based on these differences in connectivity, it is clear that both the equilibrium state of these two reactions as well as the dynamic exchange, i.e., the kinetics, between the various states are important. | ||
Before continuing, we note that while reversible addition and condensation crosslinking reactions have similar dynamic behavior, condensation crosslinking reactions involve the production of a small molecule byproduct. The reversibility of materials incorporating condensation crosslinks is therefore controlled by the presence or absence of the condensate (via Le Chatelier's principle). In this context, the condensate can be viewed as a trigger to instigate bond rearrangement and dynamic material behavior. Nevertheless, it is often difficult to control the condensate concentration, particularly within the network, since it is typically a small molecule, such as water, and is susceptible to evaporation. For simplicity, this tutorial review will use two specific reversible covalent chemistries as examples of these two broad classes of materials, reversible addition and exchange crosslinking reactions, recognizing that all others exhibit similar dynamic properties depending on whether they follow bond-breaking, bond-forming or bond-forming, bond-breaking reaction sequence mechanisms.
The effect of temperature on the rearrangement reaction rate is an essential consideration for selecting the appropriate reversible crosslinking reaction. In the two CAN examples above, both rearrangement reactions are accelerated by temperature and follow an Arrhenius dependency. From a practical point of view, in principle, these crosslinking reactions should be at or near equilibrium at the application temperature. In polymeric materials below their glass transition temperature, however, the molecular motion becomes extremely limited and often prohibits reactions from achieving equilibrium. Further, the bond exchange reactions also may become non-Arrhenius in the glassy state. Bond formation and breaking in the glassy state will also not introduce as much stress relaxation given the restrictions on rearrangement of the polymer network strands.
Presuming that the reversible DA crosslinking reaction is in dynamic equilibrium, both the crosslinking density and kinetics are affected by temperature: specifically, increasing temperature favors the endothermic reaction and shifts the reaction toward the reactants in the DA system, thereby decreasing the number of network bonds and the crosslink density. In contrast, once the ester crosslinks are formed in the transesterification-type CAN, changes in temperature primarily affect only the kinetics. This behavior is simply understood by noting the reactants and products in the transesterification reaction are chemically identical, thus on this basis alone, there is not a thermodynamic driving force for affecting crosslinking. Fig. 5 summarizes the equilibrium and kinetic constant relationships for these various reaction types.
 | ||
| Fig. 5 Temperature and concentration dependent equilibrium and reaction rates for each of the different reaction mechanisms. For the condensation and addition reactions reaction, both the reaction rate and extent, based on the equilibrium, are strongly dependent on temperature and functional group concentrations because of differences in reactants and products. In contrast, for the exchange reactions the reactants and products are the same and the equilibrium constant is a temperature independent unity; only the rate of exchange is temperature dependent rather than the reaction extent. Thus, for all reaction types the rate of bond breaking and reformation is strongly temperature dependent but only in reversible addition and condensation reactions is the extent of reaction, and hence the crosslink density, dependent on temperature. | ||
Thus, a distinguishing characteristic between reversible addition- and reversible exchange-type CANs is that the crosslink density in the former is adjustable. The DA network and others similar to it are readily heated to promote depolymerization. At high enough temperatures the material will even undergo a gel-to-sol transition. At this point the material relaxes stress through flow rather than by bond rearrangement. Nevertheless, it should be emphasized that the material is still capable of undergoing stress relaxation at temperatures well below the gel-to-sol temperature transition through network strand rearrangement (see Fig. 2). As with the alcohol–ester transesterification exchange reaction, the presence of excess pendent functional groups facilitates this exchange process. In the DA network, the presence of excess pendent furan and/or maleimide functional groups (i.e., diene and dienophile, respectively) is almost always the case, since the network conversion is lower than 100% as dictated by the equilibrium thermodynamics of the reaction. While the DA cycloaddition is not an exchange reaction, the net effect of network strand rearrangement is an exchange process, where the reactants and the products have different connectivity but are chemically identical.
Reversible covalent chemistry
In the last several decades, there has been a renewed interest in reversible or dynamic covalent chemistry (DCC)6–9 for a number of applications, from dynamic combinatorial libraries to supramolecular chemistry. There is a tremendous opportunity to discover new materials with dynamic properties by incorporating these chemistries and concepts into a polymer network framework. It is not the purpose of this tutorial review to list all reversible covalent reactions, but rather to facilitate their organization for an efficient a priori design of CANs. Fig. 6 shows but a few examples of the reactions in addition and exchange reactions that have been employed in CANs. Beyond these covalent chemistries and not reviewed here, there is also a rich literature and abundance of examples of non-covalent chemistries that in many respects and at appropriate timescales, temperatures and other conditions qualitatively would share characteristics and behavior with these CANs. These reversible, non-covalent “lock-and-key” motifs have become abundant and range from oligonucleotide hybridization to multiple hydrogen bond formation to metal–ligand interactions.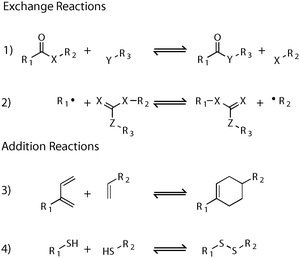 | ||
| Fig. 6 A selection of reversible covalent chemistries that have been incorporated into a polymer network to produce a CAN: (1) transacylation; (2) reversible addition–fragmentation; (3) the Diels–Alder cycloaddition; and (4) disulfide formation. Adapted from ref. 10. | ||
Trans-X reactions (i.e., transesterification,5,11trans-‘siloxanificiation’,12,13 transimination,14etc.) are prototypical exchange reactions, which proceed through a bond-forming, bond-breaking mechanism (Fig. 6, entry 1). CANs utilizing these chemistries generally require a catalyst to promote the exchange reaction and yield reaction kinetics that are sufficiently rapid at appropriate temperatures to achieve macroscopic stress relaxation of the material over application-relevant time-scales. Leibler and coworkers recently5 established this paradigm with their outstanding example of transesterification-based networks. However, given the robustness and vast range of reaction chemistries, kinetics, and material properties that are achievable via this approach, this route to the formation of CANs remains largely unexplored and ripe with opportunities.
Addition–fragmentation chain transfer exchange reactions, commonly associated with low polydispersity linear and block copolymerpolymerization (i.e., RAFT), follow a radical-based mechanism and thereby undergo rapid reaction kinetics (Fig. 6, entry 2) though they have the simultaneous advantage and disadvantage of necessitating radicals to mediate the exchange process. Epoxy,15 methacrylate16,17 and Michael addition18 reactions have all been used to form the AFT-containing networks; however, the implementation of AFT-functional groups has been limited to allyl sulfides and trithiocarbonates. This limited scope and the present limitation that materials employing these reactions are primarily held back by the method and number of radicals generated afford great opportunities for development. Research in the development of chemical structures that undergo bond exchange reactions catalyzed by other active centers or other catalysts that are formed in response to an applied stimulus would be of great advantage in moving this field forward and expanding the range of materials and properties achievable.
Cycloaddition reactions, specifically the [2+4] Diels–Alder cycloaddition, are among the best examples of reversible addition crosslinks, as they tend to be orthogonal to a wide range of functional groups that may be present in the greater network (Fig. 6, entry 3).4,19–22 While we have selected the work of Chen et al.5 as the example of DA-based CANs, the discovery and application goes back at least to the 1960s, where the DA reaction was used to crosslink linear polymers.23 An additional example of the cycloaddition reaction approach, but one that is photoactivated, is the [2+2] photoinduced cycloaddition reaction, commonly involving cinnamate chemistry.24 This reaction was historically used to crosslink linear polymers and has been used to form photosensitive CANs in which the extent of crosslinking can be reversibly controlled by the irradiation wavelength. Though slow and limited to optically thin films, the photosensitive nature of this process affords the advantage of forming a thermally stable material that can be heated without reversing the crosslinked structure. Disulfide formation and cleavage are among some of the oldest studied reversible bonds incorporated into a polymer network, though historically they were considered more of nuisance, affecting the thermal stability of vulcanized rubbers (Fig. 6, entry 4). The facile oxidation and reduction of the disulfide bond, makes this reversible chemistry an excellent candidate to fabricate a pH responsive material.9,25–27 Moreover, the disulfide bond is readily cleaved with long wavelength UV light to produce two thiyl radicals, which readily recombine after the irradiation is ceased. In this manner, the thermal behavior of the DA-based CANs is readily translated to light or chemical sensitivity in the disulfide-based networks.
Here, we emphasize that it is through the bond reversibility mechanism by which the reaction is categorized, not by the chemical functional group(s) themself. Many functional groups blur the lines between the addition and exchange reaction categories. For example, while homolytic cleavage of the disulfide leads to two thiyl radicals, the formed radical is subsequently capable of an addition–fragmentation chain transfer reaction with another disulfide bond.28–30 One might argue that the initial homolytic cleavage was simply a radical initiation event for the exchange reaction; however, the initial cleavage is both reversible and results in the disruption of connectivity. The mechanism, therefore, dictates that this is a hybrid reversible addition–exchange reaction sequence. Whichever mechanism is most dominate with respect to the rate of bond-forming–bond-breaking sequences, will ultimately determine the macroscopic behavior of the CAN.
Network formation strategies
The selection of the reversible crosslink chemistry and its incorporation into a polymer network must be considered simultaneously. The polymerization scheme will determine the distribution of reversible crosslinks and network architecture, both of which play a critical role in the relaxation behavior of a CAN. Moreover, the polymerization of the network must not interfere with the reversible crosslinking chemistry. Fundamentally, with the appropriate selection of the chemistry through which the crosslinked structure is reversed, CANs and their reversibility are suitable for essentially all mechanisms of network formation, whether it is by a step- or chain-growth polymerization mechanism, whether it is a radical or epoxy–amine reaction, or whether it is assembled from monomers or prepolymers.One facile method to create a CAN is to use the reversible chemistry as the crosslinking reaction. This strategy was utilized by Chen et al.4 to fabricate DA networks, where multifunctional furan and maleimide monomers were used. This strategy has the main advantage of simplicity, since one does not have to employ a separate orthogonal polymerization scheme; however, it cannot be applied to reversible exchange crosslinking reactions, such as transesterification, since these reactions do not produce new bonds. In the transesterification example above, Montarnal et al.5 cleverly created the reversible ester bond via the irreversible addition of an epoxide with an alcohol.
An alternative, and perhaps more versatile, strategy is to form networks with monomers that possess reversible bonds within their backbone flanked by polymerizable functional groups. This enables a broad range of polymerization reaction chemistries and mechanisms to be employed, allowing a diverse set of the network architectures to be created. If the polymerizable functional groups undergo a step-growth mechanism (e.g., a thiol–ene or epoxy–amine reaction), then the network will contain reversible crosslinked small molecular segments, or reversible monomers (Fig. 7). In contrast, if the polymerization proceeds by a chain-growth mechanism (e.g., a radical-initiated vinyl reaction or anionic ring-opening reaction), the resulting networks will consist of reversibly crosslinked polymer chains, or reversible macromers (Fig. 7). These two materials exhibit significantly different mechanical properties and relaxation behavior when activated.
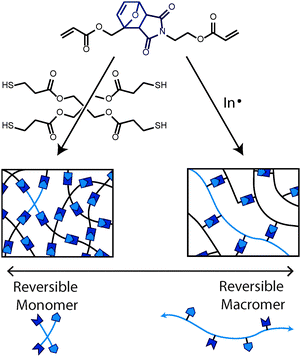 | ||
| Fig. 7 Two polymerization strategies of the same CAN-capable monomer resulting in two different network architectures. If stoichiometric amounts of the multifunctional thiol and acrylate monomers are copolymerized via a base-catalyzed Michael addition reaction, the resulting network will contain small reversible monomers (left). If an excess of the same acrylic monomer is polymerized using a free-radical initiator, the network would be constituted with reversible macromers (right). The length of the reversible macromer segments is readily controlled by the addition of a chain transfer agent, such as a thiol. | ||
In the subsequent section, the mechanical properties of the network, beyond simply the modulus, will be discussed in terms of the CAN dynamics. The advantages of utilizing reversible exchange versus reversible addition crosslinking reactions or a step- versus a chain-growth mechanism are most evident in the final mechanical properties of the polymer network. Nevertheless, tuning the dynamics and architecture of the network is simply reduced to understanding and manipulation of the mechanism of the reversible crosslinking reaction and polymerization, respectively.
Mechanical properties
Polymeric materials are conventionally categorized as thermosets or thermoplastics, which, as the name implies, is based on whether the material flows at elevated temperatures. A more elegant observation, though related, is that a thermoplastic under stress will deform continuously at a sufficiently long time-scale. The macroscopic behavior of a thermoplastic is derived from the capability of polymer chains to move past one another, and is only temporarily constrained by weak physical bonds, entanglements and occupied volume. Given a long enough time, the temporary physical bonds will break and reform. For linear polymers of sufficient molecular weight (above their entanglement molecular weight), the macromolecules will move via curvilinear diffusion (i.e., reptation). The time it takes a molecule or molecular segment to move roughly its own size is defined as the relaxation time; thus, the largest species in a thermoplastic has a finite relaxation time. In contrast, polymers chains in thermosets are confined by the chemical crosslinks and large scale polymer diffusion is not possible. By definition, covalently crosslinked networks are a single molecule with infinite molecular weight, and thus, thermosets have a non-finite relaxation time.A common method of assessing the linear mechanical properties (i.e., at small deformation) is to monitor the stress of the material as a function of an applied oscillatory strain (i.e., dynamic mechanical analysis). By changing the frequency of the applied strain, one observes the response of the material at different time-scales. At sufficiently high frequencies, both thermosets and thermoplastics are unable to relax on anything but the very smallest length-scales (i.e., side-group bond rotations), which are associated with glassy dynamics. Upon decreasing frequency, both materials are able to relax at size-scales related to segmental motion. For thermosets, the material cannot relax at length-scales larger than the molecular dimension between crosslinks, and therefore at lower frequencies they exhibit a modulus plateau that is a function of crosslink density. Thermoplastics comprised of polymers of sufficient molecular weight also exhibit a plateau upon decreasing frequency, since the polymer diffusion is constrained by the presence of other chains or ‘entanglements’, which act as effective crosslinks. The only mode for relaxation is for the polymer to undergo curvilinear diffusion as if it were in a tube. The plateau for a thermoplastic is related to the molecular weight between entanglements and the ultimate relaxation time is related to the length of the polymer. Thus, both thermosets and thermoplastics exhibit plateau behavior but for different underlying molecular reasons.
Both the similarity and contrast in the dynamic mechanical properties between thermosets and thermoplastics are additionally observed in CANs. Moreover, it is the molecular interpretation of the plateau and ultimate relaxation time that defines a CAN. Specifically, the plateau of a CAN is defined by the crosslink density, as would be expected for a thermoset; however, when activated, CANs also possess a finite relaxation time that is related to the reversible network rearrangement dynamics.
Prediction of network plateau modulus
Rubber elasticity theory is one of the most simple, yet powerful molecular structure–property theories developed in the last several decades, and it has proven to be a robust, valuable prediction of the mechanical behavior of covalently crosslinked thermosets. The entropic penalty for deforming a polymer chain between crosslinks is related to the modulus of the material. In its simplest form, the elastic modulus, G (specifically shear elastic modulus where T ≫ Tg), is linearly related to the crosslink density, ρx, as G = ρxRT, where R and T are the gas constant and absolute temperature, respectively. There are many assumptions associated with this expression (e.g., network architecture and defects, entanglements, deformation extent, etc.), but it is remarkably accurate to a first approximation and serves as a good polymer network design equation.The modulus predicted by the rubbery elasticity theory approximates the plateau modulus in CANs (Fig. 8). The modulus can be tuned by controlling or rather selecting the number of functional groups per monomer molecular weight. Thermoreversible addition networks, such as the DA network, are additionally dependent on the equilibrium of the reversible crosslink. For a given temperature and at a specific instant in time, a population of the DA monomers exists in the non-crosslinked state. In contrast, reversible exchange networks, such as the transesterification network, are not susceptible to this effect, simplifying the network design considerations.
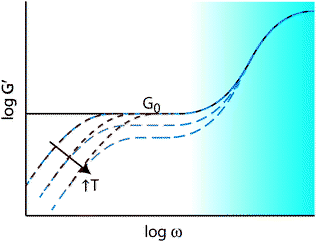 | ||
| Fig. 8 Illustration of G′ versus frequency for an ideal thermoset (black solid line) and thermally activated addition- and exchange-type CANs (blue, dashed line and red, dotted line, respectively) as a function of temperature. All polymeric materials experience a rapid increase in modulus at high frequency related to the small length-scale glassy dynamics. Additionally, all covalently crosslinked materials have a modulus, G0, that is related to crosslink density through the theory of rubber elasticity. Both exchange- and addition-type CANs exhibit liquid-like stress relaxation at low frequency (long time-scales), which, for thermoreversible CANs, increases to higher frequency upon increasing temperature. A critical difference between the two materials is the decrease in modulus as a function of temperature experienced by addition-type CANs owing to the decrease in crosslink density. | ||
A convenient method for tuning the crosslink density is to employ copolymerizations of monomers with a different number of functional groups. For example, the crosslink density of step-growth copolymerization of a tetrathiol and diacrylate (e.g., see Fig. 7) is controlled by either changing the stoichiometric ratio of the monomers (ratio of thiol to diacrylate) or by introducing a third monomer with a different number of functional groups, such as a dithiol. In the latter strategy, adding a monomer with fewer functional groups decreases the modulus while increasing the number of reversible functional groups per crosslink. Systematic variations in functionality, monomer molecular weight, stoichiometric ratio, and other aspects of the monomer such as core stiffness, are all useful in controlling the Tg, crosslink density and stimuli responsiveness. Increases in both mobility and likelihood of network strand rearrangement enhance the ability of the network to undergo stress relaxation. Thus, it would seem there is a compromise between producing a high modulus material and having efficient stress relaxation. As will be discussed in the subsequent sections, the kinetics of the reversible crosslinking reaction is a non-linear handle enabling tuning of the relaxation behavior of the material.
Gel-point
In addition to the transient modulus behavior in reversible addition-networks, a critical demarcation in polymer network formation is the point at which the material transitions from liquid-like behavior to solid-like behavior. The chemical functional group conversion at which a crosslinking material makes this transformation is often characterized by the presence of a single sample-spanning molecule that arises first at the gel-point conversion. The Flory–Stockmayer criterion for gelation translates this molecular argument to the theoretical definition of the gel point being the conversion at which the weight average molecular weight diverges. While this provides a theoretical definition of the gel-point, its measurement and analytical determination are much less straightforward and have been the subject of great debate. Dynamic mechanical analysis (or rheometry) is often employed to measure the viscous (G′′) and elastic (G′) moduli. It is assumed as a simple, practical definition that the gel-point conversion occurs where the elastic modulus exceeds the viscous modulus (i.e., the cross-over point); however, these moduli are generally a function of frequency, and thus the cross-over may occur at different frequencies for different conversions. Therefore, this definition is insufficient, since the gel-point conversion is obviously not a function of the applied frequency. The Winter–Chambon criterion defines the gel-point as the conversion where tan(δ) = 1 for all frequencies (i.e., tan(δ) = G′′/G′). Another way of expressing the Winter–Chambon expression is that the viscous and elastic moduli exhibit similar frequency scaling at the gel-point conversion (i.e., G′′ ∼ G′ ∼ ωΔ). This criterion reflects the self-similar dynamics at the gel-point, which is presumed to extend from the length-scale of the (Kuhn) monomer up to the incipient gel. It should be noted that while the Winter–Chambon criterion is often viewed as the rigorous metric for gel-point determination, the frequency dependent G′ and G′′ cross-over is often in the vicinity of the gel-point as defined by the Winter–Chambon criterion.The Winter–Chambon gel-point criterion for a covalent crosslinking reaction are equally valid for CANs, but the latter has a lower frequency bound, corresponding to the bond reversibility timescale, that breaks the frequency scaling of G′ and G′′. Fig. 9 shows the frequency dependent G′ and G′′ behavior for the evolution of a DA polymer network through its gel-point. At elevated temperatures, the retro-DA reaction dominates and the material exhibits liquid-like behavior for all time-scales (with the exception of those related to high frequency glassy dynamics). The material goes through a liquid-like to solid-like transition as the temperature is decreased, which is delineated by the self-similar scaling behavior related to the gel-point temperature. Curiously, the low frequency, long-time scaling dynamics of the material exhibits a viscoelastic liquid relaxation (i.e., G′ ∼ ω2 and G′′ ∼ ω1) even below its gel-point temperature (above its gel-point conversion), which is unique for a covalently crosslinked network and characteristic of a CAN.
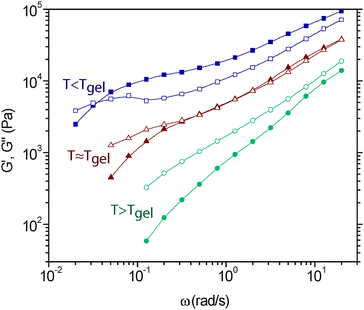 | ||
| Fig. 9 Frequency dependent elastic (G′, filled symbols) and viscous (G′′, open symbols) moduli for a DA-network as a function of temperature in the vicinity of the gel-point. At temperatures higher than Tgel (95 °C, circles), the material exhibits liquid-like properties, whereas temperatures below Tgel (87 °C, squares) the material is solid-like for intermediate frequencies. Neat the gel-point (91 °C, triangles) the elastic and viscous moduli exhibit similar scaling with frequency, indicative gel-point dynamics. At low frequencies, the material exhibits a viscoelastic-liquid relaxation, even for materials that are above their gel-point. Adapted from ref. 32. Copyright (2008) American Chemical Society. | ||
For thermally sensitive reversible addition and condensation systems such as the DA system, it is readily apparent that there is a temperature above which the gel phase no longer exists on any timescale. When the equilibrium extent of reaction becomes insufficient to cause gelation, the material is able to flow, enabling macroscopic rearrangement of the material shape.31 Thus, the classical gel-point conversion can readily be translated into a gel-point temperature, Tgel. It is important to note that this behavior only occurs for reversible bonds that are thermally activated. Addition–fragmentation, transesterification and other reversible exchange systems do NOT have a reversible Tgel. In systems that do exhibit this behavior, above Tgel the polymer does not contain an infinite molecular weight macromolecule and the material is a liquid for all intents and purposes. Below Tgel the network exhibits the dynamic behavior characteristics. The gel-point conversion, according to the Flory–Stockmayer criterion for a step-growth co-polymerization of multifunctional monomers (e.g., as in the DA-network) is
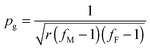
where p represents the equilibrium conversion and c0 represents the initial concentration of the stoichiometric functional group mixture prior to any reaction. Solving this relationship for Tgel at which p = pg,
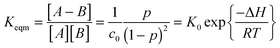
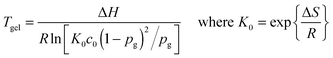
The ability to reverse the gelation behavior in these materials represents one of the most enabling aspects of the CANs materials, enhancing the capability for recycling, healing, reforming and various other aspects of processing generally regarded as being limited to thermoplastics. Above the Tgel all these properties can be realized though limitations clearly exist as high temperatures have the ability to promote undesirable side reactions as well.
Stress relaxation
Low-frequency, viscoelastic liquid scaling in a covalently crosslinked network in its gelled state is indicative of bond rearrangement. In our DA network example (Fig. 9) the reversible bond half-life (ln(2)/kr, where kr is reverse kinetic constant) is on the same order of magnitude as the rheologically determined longest relaxation time. The direct relationship between the bond kinetics and the relaxation behavior is achieved by applying the Semenov–Rubinstein (SR) model for the dynamics of an associating polymer to a DA-crosslinked network.33 By contemplating clusters within the network that are made up of dynamic connectivity, a scaling model is developed that ultimately relates the longest relaxation time to the bond lifetime and kinetics. The network connectivity must be taken into account, and is adequately done so by using Miller–Macosko formalism.34 The molecular picture of the gel-point is that at intermediate time-scales, the behavior is set by a single sample spanning macromolecule that constitutes the incipient gel. At longer time-scales the connectivity is broken thereby enabling stress relaxation. However, since the material is dynamic, the incipient gel is reformed. Thus at the gel-point and at any instant in time, there exists at least one sample-spanning macromolecule.The DA-network, which employs reversible addition crosslinks, is able to relax by both flow, below its gel-point conversion, and bond-rearrangement, above its gel-point conversion. This behavior is most evident in the solid-like, low temperature DA-network (i.e., T < Tgel), which exhibits viscoelastic liquid relaxation behavior (see Fig. 9). As mentioned above, transesterification-based networks do not depolymerize and thus only relax via bond rearrangement.
The relaxation behavior for exchange-type CANs is similar to addition-type CANs except the plateau modulus is unchanged. Fig. 10 shows the stress relaxation behavior of the transesterifcation-network, examined by Montarnal et al.5 (note that the abscissa is time whereas Fig. 9 is frequency). The relaxation is significantly shifted toward shorter time-scales upon increasing temperature, which enhances the rearrangement reaction kinetics. As further evidence, they measured the rearrangement time-scale in model compounds to find the relaxation time-scale in the polymer network to be similar. The inset of Fig. 10 demonstrates that the modulus is unchanged, indicating that the underlying crosslink density is constant (the slight increase owes to the entropic contribution to the modulus, i.e., E′ ∼ ρxT). Thus, this material experiences a low temperature glassy region, followed by an intermediate temperature rubbery region whose magnitude is defined by the crosslink density and relaxes on the time-scale of the rearrangement kinetics.
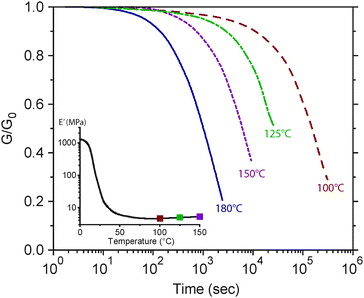 | ||
| Fig. 10 Normalized stress relaxation behavior for a transesterification-network as a function of temperature. The time-scale for relaxation is decreased owing to the increase in the rate of rearrangement kinetics. The dynamic mechanical analysis shown in the inset (at 1 Hz) reveals that the modulus is relatively unchanged aside for the entropic temperature dependency captured in the theory of rubber elasticity (i.e., E′ ∼ ρxT). Figure adapted from ref. 5. Reprinted with permission from AAAS. | ||
Stimuli-responsive CANs
Thus far, we have examined the basic concepts of the CAN design, from the mechanism of reversible strand rearrangement to the macroscopically observed stress relaxation behavior. Throughout, we have illustrated the adaptability of CANs by highlighting and contrasting the unique attributes of the bond-breading, bond-forming DA-based network with the bond-forming, bond-breaking transesterification-based network. In both systems the rate of bond rearrangement is enhanced by increasing temperature, where the DA-system additionally may undergo a solid-to-liquid transition. In these cases, temperature is the primary control variable, which tunes the relaxation time of the material. The ability of these covalently crosslinked materials to undergo stress relaxation is a fundamental property of all CANs; however, it is often necessary for a material to be static at one time and dynamic at another. In these cases, it is necessary to decouple the properties until some activation event occurs. In the introduction, we presented CANs as the bridge from thermoset to thermoplastic behavior. Here, we explore different triggering mechanisms that will transform the static, dormant CAN into a dynamic, activated CAN.Thermally-activated CANs
As presented above, both the DA- and transesterification-based networks are ostensibly thermally-triggered CANs, but there is one critical consideration that has yet to be addressed: the application. If a particular application requires that material to be static at room temperature and dynamic at 150 °C, then there are a large number of suitable thermoreversible chemistries available. However, there are fewer options if the temperature range over which the CAN needs be activated is smaller.One non-chemical means of controlling activation is to employ the effect of vitrification. In the glassy state, molecular mobility is significantly decreased, which inhibits strand rearrangement in a CAN. If the glass transition is sufficiently narrow, it can be used as an effective thermal trigger of stress relaxation. The temperature and range over which the glass transition occurs is controlled by the network architecture, where homogeneous crosslinking leads to a narrow transition, which is often achieved using a step-growth polymerization reaction scheme.
Triggering a static-to-dynamic transition thermally is often complicated by practical considerations, such as heat transfer and how one is to heat geometrically large materials uniformly. In a laboratory, materials are readily heated in an oven or in a non-swelling solvent bath. This approach or one similar to it can be used to reprocess materials via processes such as elevated temperature extrusion (Fig. 11b).5 Unfortunately, it is often impractical in large-scale applications to heat the material globally. One other approach is to embed dyes or photothermal particles in to the polymer network that emit heat upon being irradiated.35,36 The external triggering method has the advantage of being spatioselective, wherever the CAN is irradiated is where it is activated and transitions from thermoset to thermoplastic. Another approach is to embed nano-sized conducting particles to make a composite that is susceptible to heating via an alternating electromagnetic (EM) field (i.e., Joule heating). Additionally, the temperature can be precisely tuned by utilizing ferromagnetic particles that rapidly convert the energy of the EM field to heat via hysteresis heating, which is limited to temperatures below ferromagnetic particle's Curie temperature (the temperature above which the magnetic domains within the ferromagnetic material become randomized and hysteresis heating is no longer effective). Adzima et al. utilized this phenomenon in a DA-network and demonstrated breaking and reforming of the material (i.e., healing) over multiple cycles without degradation in mechanical properties (Fig. 11c).
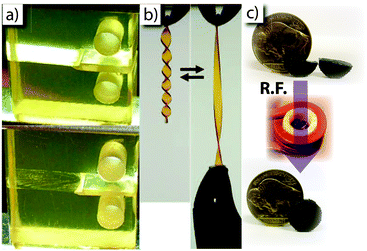 | ||
| Fig. 11 Examples of healing, remolding, and remending of thermoreversible CANs. (a) Using a DA network, Chen et al. demonstrated the healing of a crack upon thermal treatment of the sample (adapted from ref. 4. Reprinted with permission from AAAS); (b) thermal treatment of a transesterification-base network enabled Montarnal et al. to manipulate the equilibrium shape of the material (adapted from ref. 5. Reprinted with permission from AAAS); and (c) utilizing CrO2nanoparticles embedded in a DA network, Adzima et al. demonstrated the EM-field triggered healing of the fractured material (adapted from ref. 31). | ||
Photo-activated CANs
Throughout this tutorial review, we have highlighted the specific reversible chemistries of the DA and transesterification reaction which are not photo-catalyzed – though it is conceivable one could create a photoreversible catalyst that would affect the rearrangement kinetics. Nevertheless, there are several covalent chemistries that are photoreversible and have been utilized within a polymer network construct.Among the photoreversible addition reactions, there are two types: one that cleaves with one wavelength and reforms with a different wavelength and one that forms effectively stable radicals (long-lived) that will recombine over time. The former type of reaction tends to be a cycloaddition, such as the [2+2]24,37,38 and [4+4]39cycloaddition, which forms a bond at one wavelength and undergoes photoscission at a longer wavelength (e.g., see Fig. 12b). It should be noted that these crosslinking reactions do not undergo rearrangement unless the sample is irradiated with both the bond-breaking and bond-forming wavelengths simultaneously. An example of the latter reaction is the arylbenzofuranone dimer, which forms stable (i.e., long-lived) radicals upon being irradiated.40 Both of these photoreversible addition reactions are single-photon, single-reaction processes in which high irradiation intensities and doses are required to activate a photoreversible CAN. An example where photoreversible bond cleavage leads to multiple reactions is the disulfide bond. As mentioned above, disulfide-based networks are the classical CAN, which undergo both photoreversible dimer cleavage (i.e., reversible addition reaction) and subsequent addition–fragmentation chain transfer reaction (i.e., reversible exchange reaction).28–30
![Demonstrations of photo-induced healing, shape change, and surface topography change in photo-induced CANs. (a) Using thiuram disulfide crosslinks, Amamoto et al. created a polymer network that is capable of photo-induced healing with visible light (initial, fracture, after photo-healing – top to bottom) [adapted from ref. 45. Reprinted by permission from Wiley-VCH]; (b) Lendlein et al. utilized a secondary photoreversible cinnamate crosslinks to create a photo-induce shape memory material (initial, stretched and photofixed at λ > 260 nm, photocleaved at λ < 260 nm – left to right) [reprinted by permission from Macmillan Publishers Ltd: Nature (24), copyright 2005]; and (c) Kloxin et al. irradiated an optically thick, biaxially stretched specimen containing allyl sulfide crosslinks to demonstrate mechanophotopatterning on an elastomer (biaxial stretch, irradiate at 365 nm, release – top to bottom) [adapted from ref. 18. Reprinted by permission from Wiley-VCH].](/image/article/2013/CS/c3cs60046g/c3cs60046g-f12.gif) | ||
| Fig. 12 Demonstrations of photo-induced healing, shape change, and surface topography change in photo-induced CANs. (a) Using thiuram disulfide crosslinks, Amamoto et al. created a polymer network that is capable of photo-induced healing with visible light (initial, fracture, after photo-healing – top to bottom) [adapted from ref. 45. Reprinted by permission from Wiley-VCH]; (b) Lendlein et al. utilized a secondary photoreversible cinnamate crosslinks to create a photo-induce shape memory material (initial, stretched and photofixed at λ > 260 nm, photocleaved at λ < 260 nm – left to right) [reprinted by permission from Macmillan Publishers Ltd: Nature (24), copyright 2005]; and (c) Kloxin et al. irradiated an optically thick, biaxially stretched specimen containing allyl sulfide crosslinks to demonstrate mechanophotopatterning on an elastomer (biaxial stretch, irradiate at 365 nm, release – top to bottom) [adapted from ref. 18. Reprinted by permission from Wiley-VCH]. | ||
The incorporation of an addition–fragmentation chain transfer crosslink enables a cascading exchange reaction to occur upon exposure to radicals. Thus, in the presence of a radical generator, such as a photoinitiator, the material is able to undergo a static-to-dynamic network transition upon light exposure with the 4D (i.e., spatiotemporal) control afforded by typical photoinitiation processes. Networks utilizing an allyl sulfide addition–fragmentation chain transfer functional group were among the first materials to be recognized as an exchange-type CAN.41 Networks utilizing other addition–fragmentation chain transfer groups, such as the trithiocarbonate functional groups, have demonstrated photo-activated CANs behavior, opening the door to an entire library of functional groups that are typically associated with RAFT polymerization.42–44
Conclusions and outlook
As indicated, the CANs class of materials provides a customizable approach that bridges the gap that has classically existed between thermoset and thermoplastic materials. This tutorial review has provided a broad discussion and tutorial on the types of materials and specific molecular chemistries that enable a network to behave as a CAN while also broadly reviewing their material behavior and mechanical properties. The two mechanisms associated with reversible addition or condensation crosslinking and reversible bond exchange each afford distinct advantages, particularly when coupled with the ability to control the network topology. One must consider not only the reversible chemistry but the molecular architecture of the polymer network into which the reversibility is incorporated to properly design a CAN for a specific application.To date, several distinctive approaches have been pioneered for creating networks that achieve the desired CAN behavior; however, their practical implementation and broader scale development have only been nascent. In particular, each of these distinctive approaches based on reversible reactions such as cycloaddition reactions as exemplified by the DA reaction, trans-X reactions as exemplified by the transesterification reaction, and the addition–fragmentation reaction represent broad classes of reversible addition or reversible exchange reactions. As such, great developments in the types and variability of chemistries available within each of these classes are likely to be achieved as are the developments of additional classes of reversible chemistry. In particular, mechanochemical approaches to reversibility represent an outstanding opportunity for the future. Recently, extensive work has been done to incorporate mechanically sensitive molecular structures into polymer networks,46–49 and it will be exciting to see the combination of these approaches with network reversibility and implementation in CANs. Additionally, an obvious downside of CANs, particularly thermally activated CANs, is the inability to have the networks become permanent. If the networks always respond to temperature increases by undergoing viscoelastic solid to viscoelastic liquid transition, that limits their applications. Techniques that can be used for permanently fixing the chemical structure at some point have value in a number of applications.50
The nature of polymer networks with reversible crosslinks is that they can be affected by a range of stimuli. Temperature remains the most common stimulus for inducing transformations and bond reversibility. Temperature changes can be induced conventionally to act as a stimulus; however, coupling the temperature rise to other processes such as radiofrequency field exposure or using photothermal effects also has advantages. More conventional and direct photoinduced processes enable 4D control of the viscoelastic solid to viscoelastic liquid transition and have been used to activate a number of distinct physical transformations including shape memory, actuation, and topography control.
Molecular sensing as a means to induce responses in CAN-based materials has not been widely implemented and remains an exciting opportunity for future developments. Activation of a catalyst, by for example the presence or absence of a co-catalyst or through pH sensitivity, for example, for reversible addition or reversible exchange would be a means for enabling the presence or absence of a target molecule to induce the thermoset to thermoplastic transition. Further, while we have intentionally avoided reversible condensation-based crosslinking materials, they are readily formed and depolymerized in the presence of their condensate. The condensate necessarily drives the reversible reaction to the uncrosslinked state (i.e., reactants). Interestingly, the mere act of swelling a network can also induce depolymerization in reversible addition networks, since swelling effectively dilutes the A + B ⇔ A–B reaction, shifting it towards the depolymerized state. The presence of solvents has also been used recently to control the crosslink density and structure in reversible exchange systems,51 acting as a trigger to change the crosslink density and affect the equilibrium swelling. It is noted the dynamics of all types of CANs are likely to be affected by the presence of a solvent, as observed in many small molecule reactions.
In this manuscript we have attempted to provide a framework within which CANs materials can be understood, designed, and implemented with common understanding. The universal drive towards customizable, responsive materials will push this field further towards continued growth in molecular structures suitable for enabling CANs, in architectures within which to form the desired networks, in the development of triggers that render the materials more adaptable and responsive, and in their application. The potential for these materials is bright with vast developments and implementation awaiting.
Notes and references
- M. S. Green and A. V. Tobolsky, J. Chem. Phys., 1946, 14, 80–92 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS.
- L. P. Engle and K. B. Wagener, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1993, C33, 239–257 CrossRef CAS.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- J. M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- C. J. Kloxin, in Healable Polymer Systems, ed. W. Hayes and B. W. Greenland, RSC Publishing, 2013 Search PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS.
- A. R. Gilbert and S. W. Kantor, J. Polym. Sci., 1959, 40, 35–58 CrossRef CAS.
- P. W. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS.
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS.
- C. M. Moorhoff, W. D. Cook, F. Chen, D. Nghiem, C. Braybrook, S. H. Thang, J. Z. Sun, T. F. Scott and C. N. Bowman, Aust. J. Chem., 2011, 64, 1083–1093 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS.
- H. Y. Park, C. J. Kloxin, A. S. Abuelyaman, J. D. Oxman and C. N. Bowman, Macromolecules, 2012, 45, 5640–5646 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, H. Y. Park and C. N. Bowman, Adv. Mater., 2011, 23, 1977–1981 CrossRef CAS.
- E. B. Murphy, E. Bolanos, C. Schaffner-Hamann, F. Wudl, S. R. Nutt and M. L. Auad, Macromolecules, 2008, 41, 5203–5209 CrossRef CAS.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J. M. Lehn, Chem.–Eur. J., 2009, 15, 1893–1900 CrossRef CAS.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- H. Laita, S. Boufi and A. Gandini, Eur. Polym. J., 1997, 33, 1203–1211 CrossRef CAS.
- J. M. Craven, US Pat., 3,435,003, 1969 Search PubMed.
- A. Lendlein, H. Y. Jiang, O. Junger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS.
- K. N. Plunkett, M. L. Kraft, Q. Yu and J. S. Moore, Macromolecules, 2003, 36, 3960–3966 CrossRef CAS.
- G. H. Deng, F. Y. Li, H. X. Yu, F. Y. Liu, C. Y. Liu, W. X. Sun, H. F. Jiang and Y. M. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- A. V. Tobolsky, W. J. Macknight and M. Takahashi, J. Phys. Chem., 1964, 68, 787–790 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolay, Y. Z. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS.
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS.
- R. J. Sheridan and C. N. Bowman, Macromolecules, 2012, 45, 7634–7641 CrossRef CAS.
- D. R. Miller and C. W. Macosko, Macromolecules, 1976, 9, 206–211 CrossRef CAS.
- A. B. S. Bakhtiari, D. Hsiao, G. X. Jin, B. D. Gates and N. R. Branda, Angew. Chem., Int. Ed., 2009, 48, 4166–4169 CrossRef CAS.
- S. Yamashita, H. Fukushima, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Langmuir, 2011, 27, 14621–14626 CrossRef CAS.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3077 CrossRef CAS.
- Y. Imai, T. Ogoshi, K. Naka and Y. Chujo, Polym. Bull., 2000, 45, 9–16 CrossRef CAS.
- Y. J. Zheng, F. M. Andreopoulos, M. Micic, Q. Huo, S. M. Pham and R. M. Leblanc, Adv. Funct. Mater., 2001, 11, 37–40 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- B. J. Adzima, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Macromol. Rapid Commun., 2012, 33, 2092–2096 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 478–481 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |