 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning degradable hydrogels for orthogonal control of cell microenvironments
Prathamesh M.
Kharkar
a,
Kristi L.
Kiick
*abc and
April M.
Kloxin
*ad
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: kiick@udel.edu; akloxin@udel.edu
bBiomedical Engineering, University of Delaware, Newark, DE 19716, USA
cDelaware Biotechnology Institute, University of Delaware, Newark, DE 19716, USA
dDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
First published on 22nd April 2013
Abstract
Degradable and cell-compatible hydrogels can be designed to mimic the physical and biochemical characteristics of native extracellular matrices and provide tunability of degradation rates and related properties under physiological conditions. Hence, such hydrogels are finding widespread application in many bioengineering fields, including controlled bioactive molecule delivery, cell encapsulation for controlled three-dimensional culture, and tissue engineering. Cellular processes, such as adhesion, proliferation, spreading, migration, and differentiation, can be controlled within degradable, cell-compatible hydrogels with temporal tuning of biochemical or biophysical cues, such as growth factor presentation or hydrogel stiffness. However, thoughtful selection of hydrogel base materials, formation chemistries, and degradable moieties is necessary to achieve the appropriate level of property control and desired cellular response. In this review, hydrogel design considerations and materials for hydrogel preparation, ranging from natural polymers to synthetic polymers, are overviewed. Recent advances in chemical and physical methods to crosslink hydrogels are highlighted, as well as recent developments in controlling hydrogel degradation rates and modes of degradation. Special attention is given to spatial or temporal presentation of various biochemical and biophysical cues to modulate cell response in static (i.e., non-degradable) or dynamic (i.e., degradable) microenvironments. This review provides insight into the design of new cell-compatible, degradable hydrogels to understand and modulate cellular processes for various biomedical applications.
 Prathamesh M. Kharkar | Prathamesh M. Kharkar is currently pursuing a PhD in Materials Science and Engineering at the University of Delaware, where he is developing multi-mode degradable hydrogels for the controlled release of bioactive molecules under the supervision of Professor April Kloxin and Professor Kristi Kiick. He received his BS in Polymers and Coatings from the University Institute of Chemical Technology (UDCT, India), MS in Polymer Science from Joseph Fourier University (France), and subsequently worked as a research engineer under the guidance of Professor Catherine Picart at the Grenoble Institute of Technology. His main research interests include biomaterials for drug delivery and tissue engineering. |
 Kristi L. Kiick | Kristi Kiick is a Professor of Materials Science and Engineering, Professor of Biomedical Engineering, and Deputy Dean of the University of Delaware College of Engineering. She holds a BS from UD and an MS in Chemistry (as an NSF Predoctoral Fellow) from the University of Georgia. She worked as a research scientist at Kimberly Clark Corporation before obtaining a PhD in Polymer Science and Engineering from the University of Massachusetts Amherst after completing her doctoral research as an NDSEG Fellow at the California Institute of Technology. She joined the UD faculty in 2001. Her current research is focused on combining biosynthetic techniques, chemical methods, and bioinspired assembly strategies for the production of advanced multifunctional biomaterials. Kiick's honors have included a Beckman Young Investigator Award, an NSF CAREER Award, a DuPont Young Professor Award, and induction into the College of Fellows of the American Institute for Medical and Biological Engineering. |
 April M. Kloxin | April M. Kloxin, PhD, is an Assistant Professor in the Department of Chemical & Biomolecular Engineering, Department of Materials Science & Engineering, and Biomedical Engineering (affiliate) at the University of Delaware. She obtained her BS (Summa Cum Laude) and MS in Chemical Engineering from North Carolina State University and PhD in Chemical Engineering from the University of Colorado, Boulder, as a NASA GSRP Fellow. She trained as a Howard Hughes Medical Institute post doctoral research associate at the University of Colorado before joining the faculty at the University of Delaware in 2011. Her research group focuses on the design of responsive biomaterials and development of controlled, dynamic models of disease and tissue repair. Kloxin's prior honors include an NSF CAREER Award, the Western Association of Graduate Schools Innovation in Technology Award, the Max S. Peters Outstanding Graduate Research Award, and the ACS Polymer Chemistry Division Excellence in Graduate Polymer Research Award. |
1. Introduction
Cells in vivo interact with biochemical and biophysical cues within their surrounding microenvironment, and such interactions influence cell behavior, function, and fate. The cell microenvironment comprises the extracellular matrix (ECM) proteins, soluble and sequestered bioactive factors, and neighboring cells. Microenvironment biochemical cues, such as receptor binding to ECM proteins or cytokines, and biophysical cues, such as modulus and fibrillar structure, play a vital role in cell fate decisions, from quiescence to activation and progenitor state to terminal differentiation. These fundamental cell–ECM interactions are highly dynamic in nature, as cells interact with and respond to ECM signals and subsequently remodel their surroundings. Understanding and harnessing this bidirectional cross talk between the microenvironment and resident cells is pivotal in strategies to regenerate tissue or regulate disease.Although classic biomaterials, such as metals, ceramics, and synthetic polymers, have been used to successfully replace the mechanical function of tissues, such as teeth or hip and knee joints, their use as ECM mimics for tissue engineering has been limited.1 Given that hydrogels demonstrate many properties similar to those of the ECM, an ever-increasing number of hydrogel-based materials have been developed to study and direct cell behavior.2 Hydrogels comprise hydrophilic crosslinked polymers that contain significant amounts of water and maintain a distinct three dimensional structure.3 The high water content, elasticity, and diffusivity of small molecules in these materials make them attractive candidates for mimicking soft tissue microenvironments as well as serving as reservoirs for water-soluble cytokine and growth factor delivery. Hydrogels also offer great potential to mimic the dynamic, native ECM due to the ease of tailoring their physiochemical and mechanical properties through the incorporation of degradable moieties and orthogonal chemistries.4–6
The building blocks for constructing synthetic, biomimetic microenvironments and manipulating native in vivo microenvironments are rapidly expanding. Synthetic ECMs have been used in vitro to support cells and modulate their behavior and to provide triggered, sustained release of bioactive molecules. Additionally, hydrogels have been increasingly employed for delivering cells and therapeutics within the in vivo microenvironment.7–9 In this review, we aim to provide a comprehensive survey of these building blocks and to overview seminal and recent works utilizing chemistries that are degradable, orthogonal, or both to permit control of biochemical or biophysical signals in the cell microenvironment (Fig. 1). Providing criteria (Section 2) and context for controlling properties in the presence of biological systems, we will summarize (i) natural and synthetic polymers that are commonly employed as the hydrogel base (Section 3), (ii) reactive functional groups for hydrogel formation (Section 4), and (iii) degradable moieties for temporal evolution of physical or biochemical properties (Section 5). We subsequently examine how these degradable groups are being used in conjunction with orthogonal chemistries for probing and regulating cell function in regenerative medicine and integrative biology applications (Section 6).
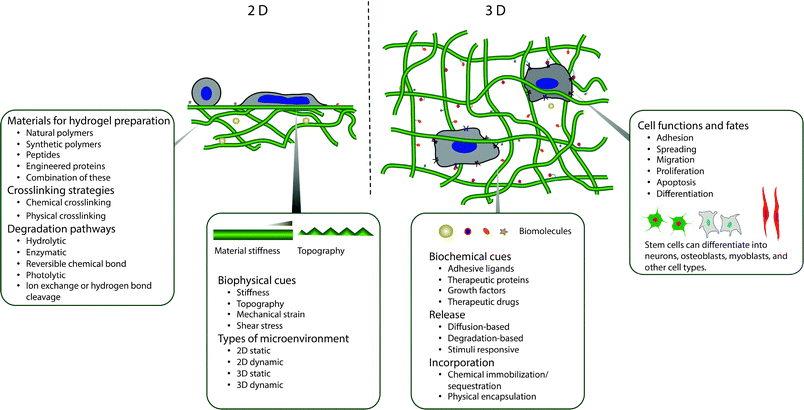 | ||
| Fig. 1 Overview. Degradable hydrogels can be used for orthogonal control of multiple properties in both two- and three-dimensional (2D and 3D) cellular microenvironments. | ||
2. Design considerations
Hydrogels that permit orthogonal control of multiple properties in the cell microenvironment must meet a number of biological and physical design criteria that are dictated by the intended application (Fig. 2). For example, hydrogels for three-dimensional (3D) cell culture or delivery must be crosslinked in presence of cells while maintaining cell viability; additionally, they need to mimic critical aspects of the natural ECM, such as mechanical support and degradation, to enable appropriate and desired cellular functions, such as proliferation and protein secretion.7,10,11 In this section, we will address these challenges and provide perspective on key design criteria for producing cell-compatible hydrogels with properties that can be orthogonally controlled both in space and in time.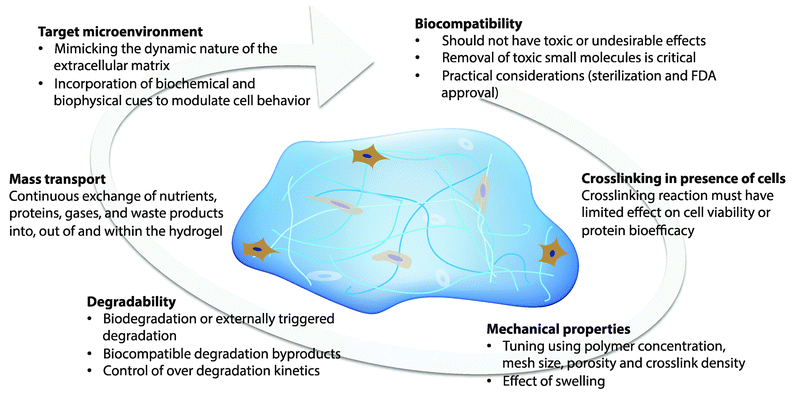 | ||
| Fig. 2 Design considerations. The design of hydrogels for orthogonal property control in cellular microenvironments is dictated by the biocompatibility, crosslinking in presence of cells or proteins, mechanical properties, degradability, mass transport properties, and target microenvironment. | ||
2.1 Biocompatibility
Biocompatibility is the first, and perhaps the most critical, parameter when considering the application of hydrogels in the cellular microenvironment. Biocompatibility is defined as the ability of a biomaterial to perform its desired function without eliciting any undesirable local or systemic side effects.12 The hydrogel must be immunocompatible and not elicit a significant inflammatory response for use within in vivo microenvironments. Various naturally derived polymers (e.g., polysaccharides such as hyaluronic acid) and a few synthetic polymers (e.g., polyethylene glycol) have demonstrated adequate biocompatibility. Removal of small molecules used or generated during hydrogel fabrication (such as unreacted monomer, initiator, and crosslinkers) is essential to consider during material design, as such molecules can be toxic to host cells both in vivo and in vitro. For example, unreacted maleimides, which are widely used in Michael-type addition reactions, are highly potent neurotoxins;13 similarly, photoinitiators, such as 2,2-dimethoxy-2-phenyl-acetophenone used frequently in free-radical polymerization, can be cytotoxic.14In addition, the hydrogel or its base components need to be simple to sterilize and should not undergo any significant functional changes during sterilization. Further, hydrogels for implantation also need to meet appropriate regulatory body (i.e., FDA, EPA) guidelines. Synthetic polymers, such as PEG, PLGA, and PLA, and natural polymers, such as alginate, collagen and fibrin, have been approved for specific clinical applications by the FDA. Kim and Wright recently investigated use of FDA-approved DuraSeal™, a PEG based hydrogel used as a sealant for human spinal fluid leaks.15 In a clinical trial with a total of 158 patients, it was found that DuraSeal™ spinal sealant had a significantly higher rate of intraoperative watertight dural closure (100%) compared to the control (i.e., treated with traditional methods, 65%). In addition, no significant statistical differences were seen in postoperative infection and healing between the PEG hydrogel and the control group. Overall, the PEG hydrogel spinal sealant system was found to be an efficient and safe adjunct to suturing for watertight dural repair. Such biocompatible and clinically tested hydrogels (i.e., DuraSeal™, Evolence®, TachoSil™, Tisseel Artiss™, Tegagel™), which are commercially available, cost effective, easy to use and have a stable shelf life (ranging from 6 months to 36 months) along with well defined in vivo stability, hold potential for bioengineering applications, such as wound healing, tissue engineering, 3D cell culture and vascular surgeries.16
2.2 Crosslinking in presence of cells
The ability to form hydrogels in the presence of cells and cargo molecules is critical for creating three-dimensional, controlled microenvironments in vitro and offers several advantages in vivo, including the ability to mold the gel to the shape of the defect site and delivery in a minimally invasive way. The chemical transformations involved in hydrogel formation, however, can be damaging to cells, and such effects must be considered for both in vitro and in vivo microenvironments. For example, free radicals can cause damage to cell membranes or detrimental loss of the pericellular matrix during cell isolation and encapsulation.17,18 Sudden localized changes in temperature, pH, and free radicals during gelation also can affect the activity of cargo molecules (e.g., oxidation of protein) or cell function or viability.19 However, the incorporation of cells in pre-formed hydrogels is often restricted, since the average mesh size of most hydrogels is much smaller than a cell's diameter; consequently, cells often are introduced within liquid hydrogel precursor solutions.20 By selecting an appropriate gelation mechanism, cells can be encapsulated in hydrogels without significantly altering their viability or activity.21–23 Different chemistries for hydrogel formation in the presence of cells and their cytocompatibility will be discussed in detail within Section 4.2.3 Mechanical properties
The success of cell-compatible hydrogels in a given bioengineering application is usually coupled with achieving appropriate mechanical properties. For example, tissue formation can depend on the mechanical properties of the hydrogel scaffold (e.g., load bearing capability until cells have produced their own functional ECM);24,25 in cell-encapsulation applications, control of the mechanical properties of the hydrogel can determine the therapeutic efficacy of the transplanted cells.26 It is well accepted that these effects are the result of the mechanical properties of the hydrogel substrate influencing cellular responses, including cell migration, proliferation, and differentiation; for example, the seminal work of Discher and coworkers demonstrated that stem cell lineage specification depends on optimal outside-in signaling of hydrogel matrix elasticity.27,28 Polymer concentration, the stoichiometry of reactive groups, and crosslinking density are all commonly used to tune the mechanical properties of cell-compatible hydrogels and accordingly to control the cellular microenvironment.29–31 The mechanism and design considerations associated with mechanotransduction were recently reviewed by Chen and coworkers,32 and relevant examples within the context of degradable cell microenvironments will be presented in Sections 5 and 6.2.4 Degradation
Cell-compatible hydrogels can be designed to degrade via ester hydrolysis, enzymatic hydrolysis, photolytic cleavage or a combination of these mechanisms with varying degrees of control and desired degradation rates depending on the application. In tissue engineering applications, degradation provides space for proliferating cells and allows infiltration of blood vessels.33,34 In controlled 3D cell culture applications, degradation can enable cell proliferation, migration, and synthetic matrix remodeling to better mimic the native ECM and understand in vivo cell behaviors.2 In controlled drug and gene delivery applications, degradation permits spatiotemporal control of the release of cargo molecules.35 Release kinetics are dictated primarily by surface erosion or bulk degradation rates when the hydrogel mesh size is smaller than the hydrodynamic radius of the cargo molecule, and by diffusion when mesh size is larger than the hydrodynamic radius of cargo molecule. For example, Hennink and coworkers demonstrated zero-order release of entrapped proteins from β-cyclodextrin and cholesterol-derivatized PEG hydrogels,36 in which the protein release was controlled by surface erosion and dissolution. Ideally, degradation kinetics are well controlled and stable, and the generated byproducts from degradation are biocompatible without eliciting any potential side effects, such as cytotoxicity, inflammation, or immunological or foreign body responses. An optimum balance between degradability and mechanical properties, such as elastic modulus and matrix integrity, is vital to ensure the proper functionality of the hydrogel within the desired timespan.2.5 Mass transport
Appropriate mass transport properties, matching those of native tissues, are essential for many bioengineering applications. In tissue engineering and cell encapsulation, continuous exchange of nutrients, proteins, gases (i.e., O2 and CO2) and waste products into, out of, or within the hydrogel is vital for survival and proliferation of encapsulated cells. For controlled delivery of bioactive cargo (i.e., therapeutics, proteins) where initial burst is undesirable, restricted free diffusion is essential. Hydrogel matrix permeability is thus an important design parameter, given that mass transport in these materials is controlled primarily by diffusion. The permeability of the scaffold is also correlated with the mechanical properties of the hydrogel network and its swelling properties, and as expected, variation in the permeability is a widely employed strategy for controlling cargo release.37–39 For a comprehensive review of the mass transport and diffusivity of bioactive molecules through hydrogel, readers are referred to reviews by Peppas and coworkers10,40 and Lin and Metters.412.6 Microenvironment
A major, and still mainly unaddressed, challenge in designing cell-compatible hydrogels is the ability to mimic the dynamic nature of the extracellular matrix (ECM). Spatiotemporal control over biological interactions at the material–cell interface, whether that material is native ECM or an engineered hydrogel, mediate cell proliferation, adhesion, migration, and receptor–ligand binding events. The challenge of controlling these interactions is particularly vexing, given that the ECM is a complex environment comprising a plethora of structural ECM proteins (such as collagen, fibronectin, laminin, and elastin), polysaccharides (such as hyaluronic acid, proteoglycans, and glycosaminoglycans), and various growth factors, enzymes, and inhibitors.42 Bidirectional cross talk between the microenvironment and resident cells is termed ‘dynamic reciprocity’,43,44 and opportunities to generate matrices capable of this reciprocity are afforded by multiple strategies, among the most recent being the rational incorporation of degradable chemistries in hydrogel networks (Section 4) and their application in controlled microenvironments (Section 6).Several recent publications have addressed the importance of incorporating ECM components into hydrogel matrices to mimic the native cellular microenvironment for cell survival, proliferation, and differentiation.45–49 For a comprehensive review of engineering hydrogels as extracellular matrix mimics, readers are referred to reviews by Geckill et al.50 and Tibbitt et al.,2 and for reviews of engineering matrices specifically for directing stem cells, readers are directed to Marklein et al.51
3. Materials for hydrogel preparation
Cell-compatible hydrogels have been prepared using a variety of polymeric materials, which can be divided broadly into two categories according to their origin: natural or synthetic.7 Natural polymers such as polysaccharides serve as ideal building blocks for preparing hydrogels that can mimic aspects of the structural and biological properties of the cellular microenvironment. For instance, proteoglycans are one of the vital components of articular cartilage, and use of glycosaminoglycan (GAG) hydrogels, such as those based on hyaluronic acid or chitosan, as a scaffold can be useful for cartilage tissue engineering.52 Moreover, as shown in Table 1, the mechanical properties, water content, and inherent chain flexibility of polysaccharide-based hydrogels help to mimic the natural ECM. In addition, such polymers can be degraded by naturally occurring cell-secreted enzymes in the cellular microenvironment, mimicking the dynamic nature of the ECM. Further, the specific cell–surface receptors for polysaccharides are known and have been extensively studied. For example, in the case of hyaluronic acid (HA), a non-sulfated glycosaminoglycan found in the ECM, both cluster of differentiation (CD) 44 and the receptor for hyaluronan-mediated motility (RHAMM) are known to enable cell adhesion and proliferation on HA.53 However, limited tunability of degradation kinetics, relatively poor mechanical properties, batch-to-batch variations from manufacturers, or potential immunogenic reactions can restrict the application of natural polymer based hydrogels.54 Synthetic polymers afford tunable mechanical properties and a large scope of chemical modification, including the introduction of degradable or biochemical moieties. Commercial availability, coupled with great flexibility in the working range of pH, ionic strength, and chemical conditions, make synthetic polymers excellent candidates for hydrogel preparation. However, purely synthetic materials often exhibit inferior biocompatibility and biodegradability in comparison to naturally derived materials, which may limit their use in applications where targeted and specific biological activity is desired. Hence, many combinations of natural and synthetic polymers have been studied for developing hydrogels with orthogonal property control in the cellular microenvironment. In this section, we will limit the discussion to several widely used natural and synthetic polymer building blocks used in controlled microenvironments.| Feature/function | Natural polymers | Synthetic polymers |
|---|---|---|
| Biocompatibility | Polymer dependent | Polymer dependent |
| Bioactivity (i.e. cell specific receptor) | Possible | Limited |
| Inherent biodegradability | ✓✓ | ✓ |
| Tunability of degradation kinetics | ✓ | ✓✓ |
| Degradation byproducts | Biocompatible | Potentially harmful |
| Flexibility for chemical modification | ✓ | ✓✓ |
| Flexibility of working range (i.e. pH and ionic strength) | ✓ | ✓✓ |
| Tuning of mechanical properties | ✓ | ✓✓ |
| Commercial availability | ✓ | ✓✓ |
| Batch to batch variations | Likely | Controlled |
3.1 Hydrogels from natural polymers
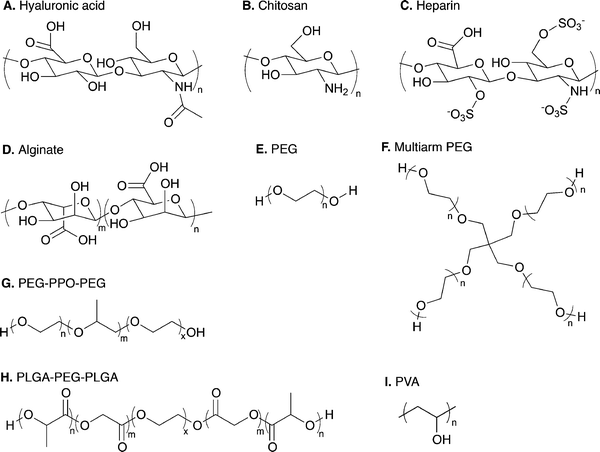 | ||
| Fig. 3 Range of natural and synthetic polymer building blocks. Molecular structures of typical polymer repeat units used for preparation of cell compatible hydrogels: (A) hyaluronic acid, (B) chitosan, (C) heparin, (D) alginate, (E) linear poly(ethylene glycol) (PEG), (F) four-arm PEG, (G) poly(ethylene glycol)-b-poly(propylene oxide)-b-poly(ethylene glycol) (PEG-PPO-PEG), (H) poly(lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly(lactic acid-co-glycolic acid) (PLGA-PEG-PLGA), and (I) poly(vinyl alcohol). | ||
HA can be modified with thiols, haloacetates, dihydrazides, aldehydes, or carbodiimide functional groups to allow crosslinking into hydrogels.63 HA-based hydrogels have shown excellent potential for biomedical engineering applications, such as tissue engineering,64–66 valve regeneration,67,68 controlled delivery,69–72 and controlling stem cell behavior.73,74 For example, Jia and coworkers synthesized HA- and heparin-based spherical hydrogel particles with an inverse emulsion polymerization, creating inherently bioactive delivery vehicles (due to inductive role of HA in chondrogenesis) for controlled growth factor (BMP-2) release (Fig. 4).75 Additionally, Elia et al. used HA-based degradable hydrogels embedded within electrospun silk for sustained release of encapsulated cargo molecules (anti-inflammatory steroid drugs and proteins) over 45 to 400 minutes.72 Such approaches that utilize simple fabrication techniques and tuning of release kinetics make HA hydrogels attractive candidates for tissue regeneration and sustained therapeutic delivery. For a comprehensive overview of HA hydrogels, readers are referred to recent reviews by Burdick and Prestwich63 and by Jia and coworkers.46
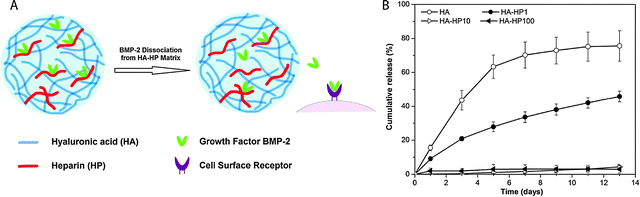 | ||
| Fig. 4 Hyaluronic acid hydrogels for controlled release applications. (A) HA/heparin hydrogel particles were synthesised by inverse emulsion polymerization and amount of heparin in hydrogel particle was varied. BMP-2 was subsequently loaded. (B) The addition of heparin to HA hydrogel particles inflenced the in vitro release of BMP-2 from hydrogels with higher heparin conent, with less than 5% of loaded BMP-2 released over 13 days (HA-HPx, x = micrograms of heparin per milligram in hydrogel particles). Reprinted from Xu et al.75 with permission from Elsevier. Copyright (2011). | ||
The large number of accessible hydroxyl and amine groups in chitosan provide numerous possibilities to create hydrogels via chemical crosslinking.78 These functional groups can react with many bifunctional small molecule crosslinkers, such as glutaraldehyde, formaldehyde, genepin, diethyl squarate and diacrylate, to form chemically crosslinked hydrogels.79 In addition, incorporation of new functionalities along the backbone chain (i.e., those susceptible to the Schiff base reaction, disulfide bonding or Michael-type additions, Section 4) can be used for in situ gel formation. Chitosan-based hydrogels can be used for the controlled delivery of drugs,79,80 proteins,80 and growth factors81 as well as the encapsulation of living cells,81,82 the controlled differentiation of stem cells,83,84 and applications in tissue engineering.85–88 For example, Bellamkonda and coworkers recently reported chitosan-based photocrosslinkable, degradable hydrogels for neural tissue engineering application (Fig. 5).88 Chitosan was functionalized with amino-ethyl methacrylate for network formation via photoinitiated radical polymerization. The cytocompatible hydrogel enhanced differentiation of primary cortical neurons by ∼30% and enhanced dorsal root ganglia neurite extension by about two-fold in 3D in vitro studies, as compared to an agarose-based hydrogel control. In principle, such hydrogels additionally can be used to control cell behavior and lineage specific differentiation by incorporation of growth factors since the gel formation chemistry does not alter the active end groups on chitosan, which allow bioactive molecule binding.
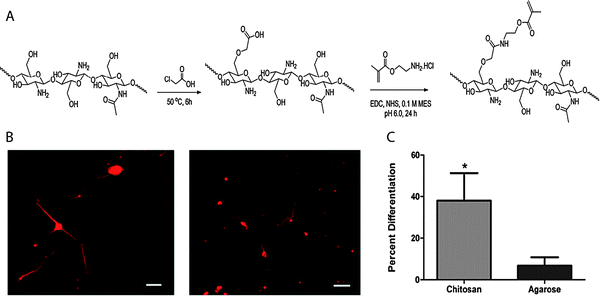 | ||
| Fig. 5 Chitosan-based hydrogels for neural tissue engineering. (A) Schematic of synthesis of methacrylated chitosan. Methacrylated chitosan (0.5 to 2% w/w) hydrogels were crosslinked in the presence of cells by photoinitiated free radical polymerization (Irgacure photoinitiator with 365 nm light). (B) E-18 rat cortical neurons were immobilized within chitosan and agarose (Seaprep®) hydrogels for investigating neuronal survival and differentiation. The cells clumped into groups and displayed extensive neurite outgrowth in chitosan hydrogels (left), as compared to agarose hydrogels (right), indicating enhanced neuron function within the chitosan matrices (scale bar, 50 μm). (C) Neurite outgrowth quantification (p < 0.05). Reprinted from Valmikinathan et al.88 with permission from The Royal Society of Chemistry. Copyright (2012). | ||
Physically and chemically crosslinked heparin-based hydrogels have been employed for the investigation of cell function and fate,96–99 cell encapsulation,100–103 and controlled bioactive molecule delivery.29,104–106 For instance, Kiick and coworkers used heparin-based hydrogels to modulate cell response in a 2D in vitro experiment.96 To modulate cell adhesion and response, hydrogels with different moduli were prepared using the Michael addition reaction between combinations of maleimide-functionalized heparin, thiol functionalized PEG and maleimide functionalized PEG. Such systems, with the ability to tune biochemical and mechanical properties, make heparin based hydrogels promising candidates for controlling adventitial fibroblast remodeling of blood vessels. In another example, Tae and coworkers took advantage of heparin-based hydrogels to stably bind fibrinogen and collagen type I on a hydrogel surface using heparin binding affinity by physisorption.98 The hydrogels were prepared by a Michael-type addition reaction using thiolated heparin and PEG diacrylate. The significant physisorption of proteins on the heparin hydrogel, as compared to a control PEG hydrogel, led to enhanced fibroblast adhesion and proliferation. Such approaches can be used to adhere cells on selective heparin hydrogel surfaces for applications such as biosensors, cell culture, and tissue engineering. Additionally, Werner and coworkers recently reported use of heparin-based hydrogels for cell replacement therapies in the neurodegenerative diseases.99 By tuning the mechanical and biological properties of the PEG-heparin hydrogels, neural stem cell differentiation and axo-dendritic outgrowth were modulated. In vivo stability and excellent histocompatibility make such hydrogel systems attractive candidates for neuronal cell replacement therapies. For a comprehensive overview of heparin hydrogels, readers are referred to a recent book chapter by McGann and Kiick.89
Alginate-based hydrogels have been used for in drug delivery,110–112 tissue engineering,113–115 wound healing,116–118 cell encapsulation,119,120 and as adhesion barriers.121 For instance, recently Kim et al. employed alginate-based hydrogels for delivering differentiated adipogenic cells for adipose tissue engineering.115 Oxidized alginate (susceptible to hydrolysis) was coupled with an adhesion peptide and crosslinked with calcium sulfate to encapsulate cells in vivo. The injected cell-laden hydrogels led to the formation of soft, semitransparent adipose tissue after 10 weeks in male nude mice highlighting the ability of degradable alginate hydrogels to deliver cells and generate living tissue via a minimally invasive injection.
3.2 Hydrogels from synthetic polymers
PEG macromolecules can be functionalized easily via its hydroxyl end groups to yield numerous homofunctional or heterofunctional terminal groups, including thiols,143 vinyl sulfones,144 maleimides,29,145 acrylates146,147 allyls,148 and norbornenes.149,150 The PEG hydrogels have been widely used as blank slates for the presentation of biophysical and biochemical cues in tissue engineering,151–154 cell encapsulation,155–157 controlled stem cell differentiation,158–160 and bioactive molecule delivery applications.152,161–163 For a comprehensive overview of PEG hydrogels, readers are referred to recent reviews by Lin and Anseth35 for controlled delivery applications and by Papavasiliou et al.164 for tissue engineering applications.
A large number of PEG copolymers have been utilized for drug delivery, such as non-biodegradable triblocks of PEG and polypropylene oxide (PPO) (PEG-b-PPO-b-PEG, Pluronics™) and hydrolytically degradable block polymers of PEG, polylactic acid (PLA), and polylactic acid-co-glycolic acid (PLGA), as shown in Fig. 3G. For example, H. Chang et al. investigated the effect on an active form of an antitumor drug, topotecan (TPT), which was encapsulated in an amphiphilic PEGA-PEG-PLGA hydrogel matrix for controlled release.165 Due to the increased pKa of the carboxylate groups as a result of the hydrophobic interactions between the amphiphilic polymer matrix and TPT, the active form content of TPT was increased by about 40%, as compared to free TPT in PBS solution under physiological conditions. Further, the release was sustained for 5 days with only a mild initial burst release.
PVA-based hydrogels can be formed by chemical crosslinking using various chemistries discussed in Section 4, such as click chemistry,168,169 radical polymerization,170–172 and Schiff base reaction.173,174 The hydrogels also can be formed by physical crosslinking via methods such as cryogenic gelation and hydrogen bonding,175–177 and PVA hydrogels formed via these methods have been successfully used for tissue engineering and regenerative medicine applications.176,178–180 For instance, Samal et al. prepared hybrid hydrogels consisting of PVA, chitosan, and multiwalled carbon nanotubes (MWCNT) by the physical freeze-drying method.176 The incorporation of MWCNT improved the mechanical strength, structural coherence, and electrical conductivity of the hydrogel matrix and could influence cell behavior due to biophysical and electrostimulating cues. The hydrogel matrix showed excellent biocompatibility while retaining the inherent properties of PVA, chitosan, and MWCNT, indicating its potential for biomedical applications.
Polyphosphazene, an organometallic polymer with a phosphorous–nitrogen backbone and organic side groups, can degrade under physiological conditions into nontoxic molecules, such as H3PO4 and NH4+. The inorganic backbone undergoes hydrolytic degradation, where the rate of degradation is dictated by the side chain structures.190 Polyphosphazene hydrogels can be prepared via physical crosslinking (i.e., ionic interaction using divalent ions), or chemical crosslinking via glucosyl or glyceryl side groups.191 Readers are referred to a recent review by Allcock for a comprehensive review of polyphosphazene;192 but we note here that polyphosphazene based hydrogels have been used for bioactive molecule delivery and drug delivery.193,194
Polyesters, such as PLA, polyglycolic acid (PGA), and polycaprolactone (PCL), also have been used for the preparation of cell-compatible hydrogels. Polyester-based polymers offer inherent biodegradability due to ester hydrolysis under physiological conditions. Thus, using combinations of polyesters with other synthetic or natural polymers, the rate of hydrogel degradation can be tuned as per application requirements. For a comprehensive overview of polyester-based hydrogels, readers are referred to a review by Tomas and coworkers.195
4. Material functionalization for hydrogel formation
The stable crosslinking of hydrogels is essential to prevent uncontrolled dissolution of macromolecular chains in aqueous cellular microenvironments. Numerous chemical and physical crosslinking strategies have been utilized for the preparation of cell-compatible hydrogels (Fig. 6). Chemical crosslinking strategies covalently couple reactive functional groups for hydrogel formation using chain or step growth reactions, including free radical chain polymerization, click reactions, reactions of Schiff bases, and carbodiimide-mediated activation reactions. Physical crosslinking strategies utilize non-covalent interactions between functional groups, such as ionic interactions, electrostatic interactions, hydrogen bonding, crystallization, hydrophobic interactions, and protein interactions.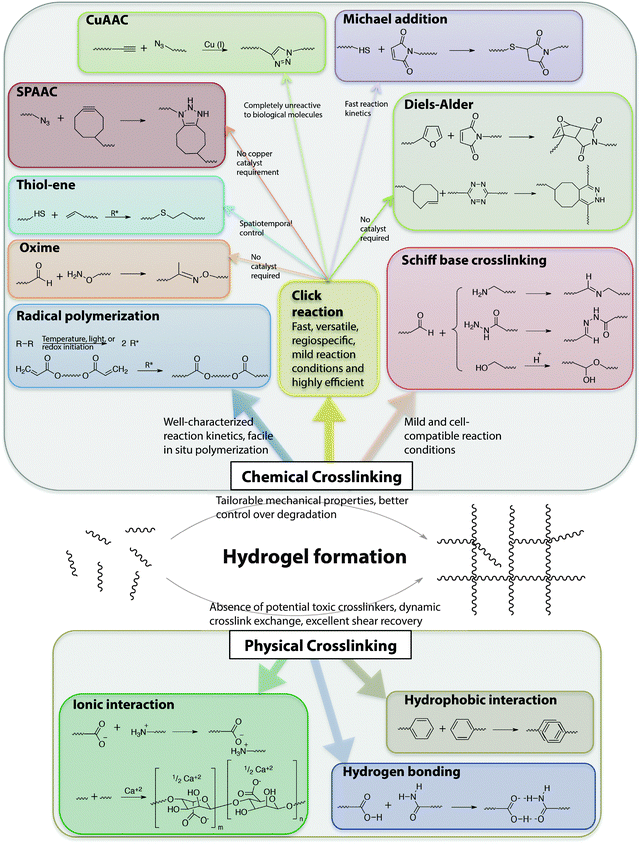 | ||
| Fig. 6 Chemical functional groups for hydrogel formation. A wide range of functional groups is available for either hydrogel formation or modification post-polymerization. Functional group selection depends on several factors related to the application of interest, including the desired initiation mechanism, the specificity and speed of the reaction, and the stability of the resulting bond under various solution conditions. | ||
The crosslink concentration, or density, dictates various physical properties of hydrogels, including elasticity, diffusivity, water content, and mesh size. In addition, the degree of crosslinking influences the hydrogel degradation rate, and hence, precise control over hydrogel crosslinking is highly desirable. Further, for control of the properties of the cell microenvironment, hydrogel formation in the presence of cells or proteins is often required, and it is thus essential to choose a cytocompatible crosslinking method for preparing these applications.
4.1 Chemically crosslinked hydrogels
A significant advantage of radical polymerization methods is that, when used in conjunction with a photoinitiator, they can provide spatiotemporal control over hydrogel formation and in situ properties.14,208 For instance, Guvendiren and Burdick demonstrated short and long-term cellular response to a dynamic microenvironment using methacrylated hyaluronic acid.208 The methacrylated HA was crosslinked with a dithiol via the Michael-type addition, creating a low modulus hydrogel, and subsequently via free radical chain polymerization of the remaining methacrylates, increasing the crosslink density and modulus of the hydrogel at time points of interest. Human mesenchymal stem cells (hMSCs) that were cultured on these hydrogel substrates spread from cell areas of ∼500 to 3000 μm2 and exhibited greater traction over a timescale of hours during stiffening (with E increasing from 3 to 30 kPa). The cell response to matrix stiffening was found to vary over 2 weeks in culture; an increased population of terminally differentiating hMSCs was present over time and was no longer responsive to variations in the mechanical properties of the hydrogel.
Alternatives such as controlled chain polymerization have been employed for hydrogel preparation to provide more control of hydrogel properties;209–211 however, potential cytotoxicity of the unremoved metal catalysts employed during these methods can restrict their use in the cell microenvironment. Free radical step growth polymerization recently has emerged as an alternative hydrogel formation strategy that provides a more homogeneous network structure and enables spatiotemporal control of hydrogel formation;212 recent developments in this area (e.g., thiol–ene click reactions) will be discussed in Section 4.1.2.3.
| Click reactions | Reacting functional groups | Reaction conditions221 | Key features | Applications |
|---|---|---|---|---|
| CuAAC | Azide and alkyne | pH 4–12, reaction time <1 h, Cu catalyst required | – Bioorthogonal | Cell encapsulation and delivery,217 drug delivery,223,224 2D cell culture225 |
| – Reversible | ||||
| – Difficulties with complete removal of cytotoxic Cu | ||||
| SPAAC | Cyclooctyne and azide | pH 7.4, reaction time <1 h | – No catalyst required | Cell encapsulation,230,231 3D cell culture216,218 |
| Diels–Alder | Conjugated diene and substituted alkene | pH 5.5–6.5, reaction time <8 h | – No catalyst required | Cell encapsulation and release,234 controlled cargo delivery235 |
| – Longer reaction time than most of the other click reactions | ||||
| Inverse electron demand Diels–Alder | Dienophile and diene | pH 7.4, reaction time <5 min | – Faster rate of reaction than many other Cu-free click reactions | Live cell imaging,238 drug targeting,239 cell surface protein labeling240 |
| – No catalyst required no catalyst required | ||||
| Thiol–ene | Thiol and unsaturated functional group (radical mediated) | pH 6–8, reaction time <1 h | – Spatiotemporal control possible with select chemistries and using a photoinitiator | Cell encapsulation,149,150 degradable 3D cell culture147,246 |
| Michael addition | Thiol and α,β-unsaturated carbonyl group | pH 6–8, reaction time <30 min | – No catalyst required | Cell encapsulation,157,160,250 controlled cargo delivery29,248 |
| – Reversible | ||||
| Oxime | Aminooxy and aldehyde/ketone | pH 6–8, reaction time <30 min | – No catalyst required | Cell encapsulation,251 protein immobilization253 |
4.1.2.1 Azide–alkyne cycloadditions. Copper(I)-catalyzed azide–alkyne cycloadditions (CuAAC) unite two unsaturated reactants, azides and alkynes, to form triazoles.222 CuAAC click reactions have been extensively used for crosslinking both natural223–225 and synthetic169,217,226 polymer-based hydrogels. One advantage of this class of reactions is that both azides and alkynes are almost completely unreactive toward biological molecules.227 Their limitations include alkyne homocoupling, difficulties removing residual heavy metal catalyst, and the biocompatibility of the resulting 1,2,3-triazoles. In particular, use of toxic and unstable Cu catalysts can limit applicability in cellular microenvironments. Nevertheless, Piluso et al. recently reported the preparation of HA-based hydrogels via CuAAC click crosslinking of alkyne-functionalized HA.225 The elastic modulus of the resulting HA hydrogels was tuned between 0.5 to 4 kPa by varying the stoichiometry, length, and rigidity of an azide-functionalized crosslinker. In this case, limited toxicity was observed with L292 cells encapsulated in these hydrogels, indicating their potential as biomaterials.
Copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) reactions have emerged to address issues with copper toxicity in biological systems.228 Ring strain, as well as electron-withdrawing fluorine substituents in some cases, promotes rapid reaction of cyclooctynes with azides in the absence of the Cu catalyst.229 Owing to the absence of the catalyst, SPAAC click chemistry has been used to crosslink hydrogels in the presence of cells to form controlled cellular microenvironments.216,218,230,231 For instance, Zheng et al. reported use of a SPAAC strategy to create hydrogels by functionalizing PEG with 4-dibenzocyclooctynol.231 The versatility and biocompatibility of this strategy allowed hMSC encapsulation, maintaining their viability as assessed using a live-dead imaging-based cytotoxicity assay (∼90% viability after 24 h). In a broader context, such an approach can be useful for cell delivery, in which cells are hypersensitive to presence of Cu during crosslinking. In another example, DeForest et al. used SPAAC click chemistry for hydrogel formation followed by a thiol–ene reaction for photoaddition of three-dimensional biochemical patterns with micrometer scale resolution and in the presence of fibroblasts (>90% viability at post 24 h encapsulation).218 Specifically, an enzymatically degradable peptide sequence was incorporated into the hydrogel via SPAAC reaction, and the adhesion ligand was incorporated in the hydrogel network via cytocompatible thiol–ene photolithographic patterning. The cells selectively adhered to regions in which the RGD motif was presented and subsequently degraded the hydrogel matrix through cleavage of the enzymatically degradable linker, leading to localized cell proliferation. In principle, such approaches can be used to study cell behavior in spatiotemporally controlled 3D microenvironments.
4.1.2.2 Diels–Alder reactions. The Diels–Alder (DA) reaction is a well-established solution-based reaction that has also been utilized for hydrogel formation. DA reactions involve addition of conjugated dienes to substituted alkenes to form substituted cyclohexenes.215,232 The efficient and facile DA reaction occurs under mild reaction conditions and does not require an initiator, which is advantageous for crosslinking hydrogels in the presence of cells. However, the reactions are slow, which could be a limitation in certain applications. The DA reaction has been utilized for the preparation of various hydrogels for bioengineering applications.233–235
Shoichet and coworkers recently demonstrated the use of a Diels–Alder click reaction to create stable and biocompatible hyaluronic acid hydrogels (Fig. 7).234 The carboxylic acid group of HA was reacted with furfurylamine to create furan-functionalized HA, and the modified HA was crosslinked with a maleimide PEG crosslinker to form a hydrogel. The mechanical and degradation properties of these hydrogels were modulated using the furan to maleimide molar ratio. In vitro studies with a cancer cell line, MDA-MB-231, demonstrated the cytocompatibility of these Diels–Alder HA-PEG hydrogels, and a high level of cell viability was maintained over 2 weeks (>98%, live-dead assay after 14 days). Using a similar approach, Marra and coworkers prepared HA-based hydrogels for controlled release application.235 HA was functionalized with either a maleimide or a furan group and crosslinked in PBS at 37 °C within ∼40 minutes. Insulin (negatively charged) or lysozyme (positively charged) were encapsulated as model proteins within these HA-based hydrogels. The release profiles showed slight or no burst release depending upon the protein, owing to electrostatic interactions. In addition, the hydrogels were cytocompatible and maintained the viability of the entrapped cells. Taken together, these recent examples indicate that the Diels–Alder crosslinking for creating cell-compatible hydrogels is a promising strategy for soft tissue engineering, regenerative medicine and controlled release applications.
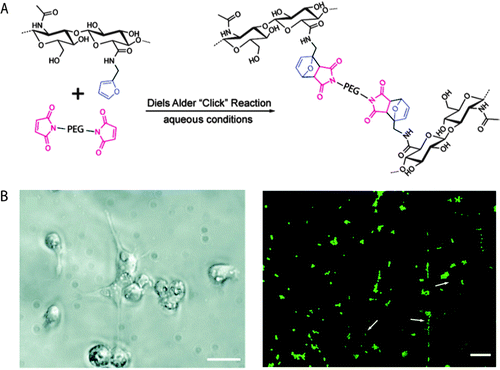 | ||
| Fig. 7 Diels–Alder click reaction for forming degradable hydrogels. (A) Schematic of hydrogel formation using Diels–Alder reaction between furan groups of HA and maleimide groups present on a PEG macromer. (B) Brightfield image of MDA-MB-231 cells (left), which are known to interact with HA via CD 44 receptor. Cells were seeded on HA/PEG hydrogels and after 14 days adopted a flattened or elongated morphology, indicating cell adhesion (scale bar, 20 μm). Cell viability was assessed using a live/dead assay (right, live cells in green, dead cells indicated by arrows) signifying a high level of cell survival (>98%), after 14 days (scale bar, 60 μm). Reprinted from Nimmo et al.234 with permission from American Chemical Society. Copyright (2011). | ||
Fox and coworkers created an inverse-electron-demand Diels–Alder reaction, reacting a trans-cyclooctene with dipyridyltetrazine.236 As compared to any other Cu-free click reaction, the rate of this reaction was an order of magnitude higher (k = 103 M−1 s−1).237 Using a similar approach, reactions of tetrazines with other alkenes such as norbornene238 and cyclobutene239 have also been reported. In principle, such reactions could be valuable for crosslinking cell-compatible hydrogels. Additionally, the inverse-electron-demand Diels–Alder reaction has been used for cell surface protein labeling indicating their bioorthogonality.240
4.1.2.3 Thiol–ene reactions. Thiol–ene reactions typically involve reaction of thiols with unsaturated functional groups, such as unactivated alkenes, maleimides, acrylates, and norbornenes. Thiol–ene reactions can proceed by free radical addition, Michael-type nucleophilic addition, or a combination of these mechanisms depending on the reaction conditions. Thiol–ene reactions share many attributes with classical click reactions: thiol–ene reactions proceed rapidly under mild conditions, have high orthogonality, yield a single regioselective product, and do not yield any byproducts. Hence, reactions that proceed by either mechanism are commonly referred as thiol–ene click reactions. For a comprehensive review of thiol–ene click reactions, readers are referred to recent reviews Hoyle et al.241 and Kade et al.242
Gress et al. were the first to identify the radical-mediated thiol–ene reaction as a click reaction.243 This radical-mediated thiol–ene coupling has since emerged as a highly attractive reaction for hydrogel formation and modification due to its high efficiency, ease of photoinitiation, and orthogonality with numerous functional groups.241,244 The reaction offers advantages, such as spatiotemporal control over crosslinking and the possibility of conducting crosslinking in the presence of cells. Rydholm et al. reported the use of thiol–acrylate mixed mode free radical photopolymerization for the formation of hydrolytically degradable PEG hydrogels.147 The mechanical properties and degradation profiles were modulated with thiol concentration. Use of photoinitiation enables controlled polymerization both spatially and temporally. In addition, thiols and acrylates also can photopolymerize in absence of a photoinitiator, which could prove useful for in situ crosslinking in the presence of cells.241
Fairbanks et al. have utilized a thiol–norbornene reaction to synthesize enzymatically degradable PEG hydrogels.245 Four-arm PEG was functionalized with norbornene end groups, and thiol-containing chymotrypsin- or MMP-degradable peptides were used for crosslinking. The step-growth mechanism ensured homogeneity in the resulting hydrogel network, and the crosslinking reaction did not significantly affect the viability of encapsulated hMSCs. Shih and Lin have recently shown the hydrolytic degradability of similar thiol–norbornene PEG hydrogels via ester hydrolysis under neutral or mildly basic conditions.246 Taken together, degradation properties of these hydrogels can be modulated with the degree of crosslinking and the crosslinking peptide sequence, making them promising for tissue engineering applications in which fine control over degradation is desired.247
Nucleophilic Michael-type addition reactions between thiols and electron deficient ‘ene’s, such as maleimides, methacrylates, α,β-unsaturated ketones, acrylonitrile, and crotonates, are another type of thiol–ene click reaction. Due to the mild reaction conditions, numerous hydrogels have been prepared via Michael-type addition in the presence of cells without significantly altering cell viability.157,160,248–250 For example, Phelps et al. used 4-arm PEG macromers functionalized with maleimide end groups and dithiol-containing protease-cleavable peptides to form hydrogels.157 The mechanical properties of the hydrogels were modulated using appropriate polymer concentrations to mimic the modulus of the native ECM. Further, these PEG hydrogels maintained cell viability during gel formation and promoted the spreading of encapsulated C2C12 cells. Kiick and coworkers have employed Michael-type additions in the production of a variety of hydrogels. In one example, polypeptide-PEG hybrid hydrogels were produced via the reaction of the cysteine (CYS) residues of the polypeptide with vinyl sulfone (VS) functionalized PEG (Fig. 8).250 Resilin-like polypeptides (RLP) were employed owing to the outstanding elastomeric properties of natural resilin for cardiovascular tissue engineering application and to provide bioactivity to inherently inert PEG hydrogels. Depending upon the molecular weight of the RLP and the stoichiometric ratio (CYS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VS), the storage modulus of the hydrogel was modulated from G ∼ 2.6 kPa to 12 kPa. Encapsulated AoAFs adopted a spread morphology over 7 days and maintained their viability within in vitro culture in these hydrogels. These recent examples demonstrate the versatility of Michael-type addition reactions to crosslink hydrogels in presence of cells for soft tissue and cardiovascular tissue engineering.
:VS), the storage modulus of the hydrogel was modulated from G ∼ 2.6 kPa to 12 kPa. Encapsulated AoAFs adopted a spread morphology over 7 days and maintained their viability within in vitro culture in these hydrogels. These recent examples demonstrate the versatility of Michael-type addition reactions to crosslink hydrogels in presence of cells for soft tissue and cardiovascular tissue engineering.
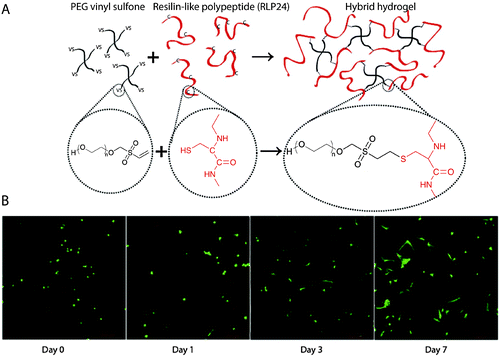 | ||
| Fig. 8 Michael-type addition reaction for hydrogel formation. (A) Schematic of hydrogel formation using the Michael-type addition reaction between vinyl sulfone groups of 4-arm PEG and cysteine residues present on the RLP. (B) Human aortic adventitial fibroblasts (AoAFs) were encapsulated during hydrogel formation and cell viability was evaluated via live/dead staining (fluorescent laser scanning confocal microscopy). AoAFs remained viable throughout the experiment, adopting a spread morphology (scale bar, 200 μm). Image reprinted from McGann et al.250 with permission from John Wiley and Sons publishing. Copyright (2013). | ||
4.1.2.4 Oxime reactions. Oxime reactions between aminooxy and aldehyde or ketone functional groups have recently been classified as click reactions owing to their fast reaction kinetics, orthogonality to various functional groups found in the cell microenvironment, and lack of catalyst. Recently, Grover et al. utilized oxime click reactions to synthesize cytocompatible PEG hydrogels.251 Eight-arm PEG was functionalized with aminoxy groups and crosslinked with glutaraldehyde. By varying the polymer concentration and stoichiometric ratio of aminoxy to aldehyde, hydrogel mechanical properties and water content were modulated. This click reaction permitted encapsulation of murine MSCs, maintaining cell viability and metabolic activity. However, glutaraldehyde has been observed to undergo various structural rearrangements in solution depending on the pH, influencing the reaction mechanisms and potentially influencing the ‘click’ nature of this reaction.252 Maynard and coworkers used oxime click reaction and CuAAC to immobilize different proteins in PEG-hydrogel constructs.253 PEG was functionalized with aminooxy and alkyne groups in order to conjugate ketoamide–myoglobin and azide-modified ubiquintin as model proteins for surface patterning. While the orthogonality of these two reactions is clear, many proteins and cells present free amines in solutions, such as hydrophilic lysines along the backbone of ECM proteins and growth factors; consequently, the specificity of the oxime reaction for orthogonal gel formation should be evaluated based on the protein and application of interest. In principle such an approach can be extended for numerous possible combinations of proteins in adjacent regions of a single plane or in multilayer constructs to modulate cell behavior.
4.2 Physically crosslinked hydrogels
Noncovalent interactions, such as ionic interactions, crystallization, hydrophobic interactions, electrostatic interactions, hydrogen bonding, or combinations of these, can be used for physically crosslinking of macromolecules to obtain cell-compatible hydrogels.255–262 Self-assembled amphiphilic block copolymers, proteins, peptides, and polypeptides typically form hydrogels via physical crosslinking.121,263–266 Physically crosslinked hydrogels afford simple network formation, without the use of any potentially toxic chemical crosslinkers or initiators. In addition, their dynamic crosslink exchange, shear-thinning flow, and excellent shear recovery can be attractive for use as injectable hydrogels for therapeutic delivery.121,261 However, potential limitations include insufficient mechanical strength for some applications due to the weakness of the physical interactions and limited control over their degradation rates, presenting possible challenges for controlled cell culture. Here, physical crosslinking methods used to design cell-compatible hydrogels from ‘off the shelf’ polymers (e.g., alginate, PVA), block copolymers, and peptide–proteins are discussed along with potential applications for orthogonal property control in cellular microenvironments.Ionic interactions have been extensively used to physically crosslink commercially available polysaccharides, such as alginate and chitosan, to form hydrogels.258–260 The use of ionic interactions offers the possibility of biodegradation since ionic species present in cellular microenvironments can competitively bind, leading to dissociation of the hydrogel network. Matyash et al. used physical crosslinking with divalent cations such as Ca2+ to prepare alginate-based hydrogels that were biocompatible and facilitated neurite outgrowth.260 Hydrogels can also be created by the formation of crystallites, which act as physical crosslinks for network formation. As in the example above (Section 3.2.2), PVA can form a highly elastic hydrogel when subjected to a freeze-thawing process to form crystallites, and such hydrogels have been used for various bioengineering applications, such as controlled drug delivery.175,255,262 For example, Abdel-Mottaleb et al. used three cycles of freeze-thawing to prepare PVA hydrogels for topical delivery of Fluconazole within the dermal microenvironment.255 The hydrogels were stable up to 6 months and effective in the topical treatment of skin infections.
Multiblock copolymers or graft copolymers can also be physically crosslinked for hydrogel formation. For example, Hunt et al. developed hydrogels with tunable physical and chemical properties using ionic coacervation upon mixing of two ABA triblock polymers, poly(allyl glycidyl ether-b-ethylene glycol-b-allyl glycidyl ether) with an oppositely charged poly(allyl glycidyl ether)-block, as shown in Fig. 9.257 Non-covalent interactions of the positively charged (ammonium and guanidinium) and negatively charged (sulfonate, carboxylate) ABA triblock copolymers resulted in the formation of polymer-dense coacervate domains leading to network formation. The ionic interactions were efficient, specific, and sensitive to polymer concentration, pH and presence of salt. Such an approach highlights the use of ionic interactions for preparing highly tunable and dynamic physically crosslinked hydrogels with superior mechanical properties and ease of synthesis, which can be potentially used as 3D cell scaffolds.
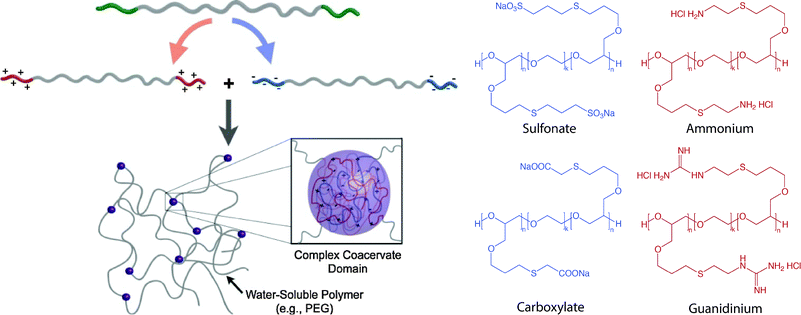 | ||
| Fig. 9 Block copolymer assembly for hydrogel formation. Multiblock copolymer based hydrogels have been prepared with coacervate crosslinking by mixing equimolar dilute solution of negatively charged (sulfonate, carboxylate) and positively charged (ammonium, guanidinium) ionic ABA triblocks. Image reprinted from Hunt et al.257 with permission from John Wiley and Sons publishing. Copyright (2011). | ||
Polypeptides and proteins represent another important class of biocompatible polymers that can be physically crosslinked upon the formation of secondary structures (i.e., α-helix and β-sheet) that drive intermolecular association. Peptide based hydrogels have been synthesized for potential applications in controlled release, 3D cell culture, and tissue regeneration.121,264,267–270 For example, Yan et al. recently prepared β-hairpin peptide-based hydrogels via self-assembly for osteoblast encapsulation.121 The effect of shear flow on the preformed, injectable β-hairpin hydrogel was investigated. The gel that was directly in contact with the syringe wall experienced a velocity gradient, while the central, plug-flow region experienced little to no shear. The study demonstrated that the shear thinning of preformed hydrogels did not significantly affect encapsulated cell viability. Further, Heilshorn and coworkers used tryptophan and proline-rich peptide domains for preparing mixing-induced, two component hydrogels (MITCH) for effective encapsulation of cells within 3D hydrogels.269 In addition to peptide–peptide interactions, specific peptide–polysaccharide interactions also can be utilized for physically crosslinking hydrogels.271
Kiick and coworkers employed noncovalent interactions between heparin-modified PEG polymers and a heparin-binding growth factor (VEGF) to create bioresponsive hydrogels.272 The VEGF–LMWH interactions were confirmed by the increase in hydrogel modulus by addition of VEGF to PEG–LMWH (G′(ω) > 10 Pa in presence of VEGF, ∼1 Pa in absence of VEGF) measured using optical tweezer microrheology. The hydrogels significantly eroded after day 4, and released approximately 80% of VEGF by day 10 in presence of VEGFR-2 (a VEGF receptor), as compared to PBS (∼30% release over same time period). The released VEGF was bioactive, and the hydrogels were biocompatible, as confirmed by in vitro experiments (cell proliferation assay and live-dead staining, respectively). VEGF–LMWH interactions were further studied for their cell-responsive nature employing two different cell types: porcine aortic endothelial (PAE) cells overexpressing VEGFR-2 and PAE cells that were not equipped with VEGFR-2 transcript.273 The hydrogels were eroded by day 4, and VEGF release was greater in presence of VEGFR-2 expressing cells. Such physically crosslinked hydrogels offer novel targeting strategies depending upon cell surface receptor–ligand interactions and could be used for sustained and targeted delivery of VEGF to promote angiogenesis.
5. Engineering degradation
Many cellular processes are influenced by spatiotemporal changes in the cell microenvironment. In hydrogel microenvironments, temporal control of matrix properties is easily achieved through selective incorporation of degradable moieties, enabling examination of how property changes influence cell function and fate. Additionally, as discussed in Section 2, controlled degradation of hydrogels is highly desirable for biomedical applications, including soft tissue engineering to promote cell secretory properties and enable tissue elaboration and therapeutic delivery to allow tunable, controlled release locally or systemically. Degradation can be achieved by forming hydrogels with degradable polymer backbones, degradable crosslinks, degradable pendant groups, or reversible non-covalent interactions. This section will focus on degradation kinetics and modes of degradation.5.1 Controlling degradation rates
The desired rate of network degradation is dictated by the final application of cell-compatible hydrogels. For controlled release of bioactive molecules, rapid degradation can lead to an initial burst or rapid release of cargo, generating large bioactive molecule concentrations which may be desirable, out of a biologically-relevant range, or even toxic depending on the application. For tissue engineering scaffolds and controlled cell culture applications, degradation affects the hydrogel crosslink density and mechanics and hence cell behavior,274 where ideally the rate of degradation should match the rate of new tissue formation. To control degradation and the temporal properties of the cell microenvironment, hydrogel degradation rates can be tuned by careful selection of network chemistries, degradation kinetics, and network connectivity, which influence crosslink density and mass loss. A brief overview of the general ‘handles’ for modulating degradation is provided here while in-depth discussion related to specific degradation chemistries is provided in Section 5.2.Degradation rates are influenced by the chemical nature of the polymer network backbone chain. The number and type of degradable linkages and the local environment surrounding the degradable moieties alter cleavage kinetics. For example, groups present along the polymer backbone or its side chains such as esters, succinimide–thioether linkages, and nitrobenzyl ethers can be degraded via hydrolytic,275–277via retro-Michael reaction249,278 and photolytic143,279,280 degradation mechanisms, respectively. The covalent bond cleavage kinetics will influence the overall rate of hydrogel degradation. For example, Jo et al. studied the effect of adjacent charged amino acids on the hydrolysis rate of ester bonds and the resulting degradation rate of PEG acrylates modified with cysteine-containing oligopeptides.281 The positively charged arginine caused a six-fold increase in ester hydrolysis, as compared to negatively charged aspartic acid, and similarly release of covalently linked bovine serum albumin (BSA) was influenced by the rate of degradation.
Hydrogel degradation rates can be tuned by optimizing network connectivity and mesh size. Increased crosslinking density typically leads to smaller mesh size, increased modulus, and slower degradation, owing to an increased number of cleavable bonds that must be broken for network mass loss and erosion.282 Decreased mesh size also can limit accessibility of the degradable moiety within the hydrogel to larger molecules, such as enzymes, owing to a reduced diffusion rate.162 In such cases, release of cargo molecules will be slower as well due to hindered diffusion.
Encapsulated cells, cell secreted enzymes, and growth media can influence degradation rates for chemically or physically crosslinked hydrogels.283,284 Additionally, the degradation products can influence cell proliferation and differentiation. For instance, Lampe et al. studied the effect of degradable macromer content on neural cell metabolic activity, proliferation and differentiation using PEG and poly(lactic acid) copolymer based hydrogels.285 It was found that the neural cell survival, proliferation and metabolic functions immediately after encapsulation were improved in hydrogels prepared with increasing degradable macromer content, suggesting a beneficial impact of lactic acid released during degradation.
Degradation rates can be investigated using bulk property measurements, such as the in vitro monitoring of hydrogel swelling, mass loss, mechanical properties, or solubilization or the in vivo imaging and analysis of implanted materials. Hydrogel degradation rate also can be studied by monitoring direct bond cleavage or monitoring degradation products (i.e., uronic acid release due to HA degradation).286 Methods for assessing hydrogel degradation rates are well covered within a recent review by Peppas et al.7
5.2 Modes of degradation
Hydrogels can degrade through surface erosion, bulk degradation, or a combination of the two depending upon the type and number of degradable linkages. At high crosslinking density, restricted diffusion of water and enzyme may preferentially lead to surface erosion. Bulk degradation, typically observed in hydrogels owing to their high water content and relatively high diffusivity, occurs when cleavable groups present throughout the bulk as well as on surface degrade simultaneously.Physically crosslinked hydrogels can degrade by processes that reverse the gelation mechanism or disturb the non-covalent interactions of the crosslinks. For example, calcium crosslinked alginate hydrogels are known to degrade in vitro due to ion–exchange processes between Ca2+ ions, present within hydrogel network, and Na+ ions of buffered solutions.108 Further, stereocomplexed hydrogels formed using amphiphilic copolymers of PLA and PEG can be degraded by disruption of the aggregate packing.287
Chemically crosslinked hydrogels can be degraded via several mechanisms, including cleavage of the backbone chain, crosslinker, or pendant groups (Fig. 10). Hydrogels prepared using polymers with degradable functional groups within the backbone chain are degraded into smaller segments of the original polymer depending upon the location of the degradable groups. A large number of hydrogels include degradable crosslinkers, such as peptides, proteins, or polymers that contain chemically labile moieties. Such hydrogel networks degrade into high molecular weight polymer backbone chains and degradation products from the crosslinker. Polymer chains also can be end-capped with degradable functional groups followed by the addition of reactive functionalities, thus creating crosslinkable degradable macromers. After crosslinking and degradation, the hydrogel network is degraded into the components that comprise the polymer network backbone; for example, in the case of PEG-PLA diacrylate hydrogels, the degradation products are PEG, polyacrylate, and lactic acid. Chemically crosslinked hydrogels often are degraded through hydrolysis, enzymatic cleavage, reversible click reactions, or photolytic degradation (Fig. 11). To engineer hydrogel degradability, it is essential to understand the types of cleavable groups and modes of degradation, their byproducts, and factors affecting degradation rates. These modes of degradation are briefly discussed below with respect to their use in cell-compatible hydrogels.
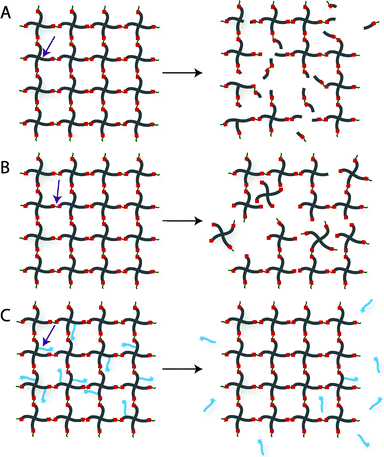 | ||
| Fig. 10 Degradation strategies for controlling hydrogel-based cell microenvironments. Chemically crosslinked hydrogels can be degraded via cleavage of (A) the polymer backbone, (B) crosslinker or (C) pendant group depending upon the chemistry used for hydrogel formation (choice of polymer, crosslinker, and crosslinking mechanism). | ||
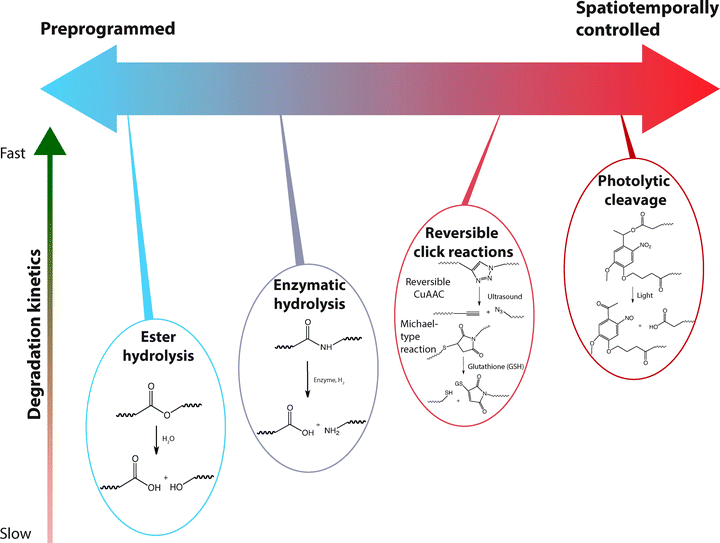 | ||
| Fig. 11 Selection of labile groups to control degradation rates. Chemically crosslinked hydrogels can be engineered to degrade at a preprogrammed, cell-dictated, or user-defined rate with varying degrees of spatiotemporal control. | ||
Enzymatically degradable hydrogels also have been utilized for targeted drug delivery since the concentration of enzyme is dependent upon cell and tissue types, enabling local triggered drug release. For instance, the concentration of hyaluronidase is known to be substantially higher in various carcinomas,289 and enzymatically-degradable HA-based hydrogels can be used as site-specific therapeutic delivery vehicles. HA-based hydrogels degrade in the presence of hyaluronidase, a family of enzymes that catalyze the hydrolysis of C–O, C–N and C–C bonds. Lee et al. prepared a HA-tyramine based injectable hydrogel for protein delivery in which the release of the cargo molecule was partially dependent on hydrogel degradation via hyaluronidase.286 Approximately 70% of the activity of released lysozyme, a model cargo protein, was retained in vitro. In principle, such an approach can be used for sustained, local therapeutic protein release to inhibit tumor growth.
Parameters such as pH, local ionic strength, enzyme concentration, and temperature may change degradation profiles due to their influence on the specificity of enzyme–substrate complex formation. The crosslinking density and pore size of the hydrogel also can influence the hydrogel degradation rate. For instance, Aimetti et al. reported use of a human neutrophil elastase (HNE) sensitive peptide for crosslinking PEG hydrogels using thiol–ene photopolymerization.162 The gels were engineered to degrade via surface erosion by limiting diffusion of HNE inside the hydrogel network via a high crosslink density and small mesh size; upon erosion, a physically entrapped protein was released. Surface degradation was investigated using mass loss and swelling ratio measurements, and the release of the model encapsulated protein, BSA, was modulated by changing peptide kcat values with amino acid substitutions, HNE concentration, and peptide crosslinker concentration.
Incorporation of protein- or peptide-based linkages, which are susceptible to proteases as noted in the example above, is a powerful way to control hydrogel degradation both synthetically and in situ.150,162,290–292 For instance, Patterson and Hubbell prepared PEG hydrogels with protease-sensitive peptides through Michael-type addition reactions.290 When incubated with MMP1 and MMP2, the hydrogel samples degraded via enzymatic hydrolysis with variable rates depending upon the peptide sequence used (MMP1 kcat ∼ 0.1 to 7.9 s−1, MMP2 ∼ kcat 0.30 to 5.6 s−1). Encapsulated fibroblasts showed increased spreading and proliferation when cultured in three-dimensions within hydrogels crosslinked using more rapidly degrading peptides. The results highlighted the possibility of engineering hydrogel degradability in response to specific MMPs that are overexpressed in relevant cell type(s) of interest. For example, endothelial cells predominantly express MMP-2 and MMP-9,293 and thus MMP-2 and MMP-9 sensitive hydrogels can be used to promote endothelial cell invasion for angiogenesis. Further, enzymatically degradable hydrogels have been employed for wound healing294,295 and bone regeneration.296,297
Bielawski and coworkers reported a novel strategy through which the 1,3-dipolar cycloaddition reaction was reversed (Fig. 12).299 2,2′-(1H-1,2,3-triazole-1,4-diyl)diethanol was condensed with 2-bromoisobutyryl bromide for preparing bifunctional initiator; this initiator was used to prepare triazole-centered poly(methyl acrylate) via Cu-mediated single electron transfer living radical polymerization of methyl acrylate. Ultrasound techniques were employed to cause chain scission near the center of the polymer and thus generate the respective azide and alkyne precursors. An optimal polymer molecular weight, triazole location in the chain, and sonication time were determined. The liberated alkyne and azide components subsequently were able to undergo the 1,3-dipolar cycloaddition reaction in the presence of a copper(I) catalyst. Such unclicking approaches could be used to prepare hydrogels capable of degradation under applied mechanical force.
![Ultrasound-induced retro [3+2] cycloaddition of an embedded triazole moiety. Triazole bond formation can be reversed by the application of mechanical force, resulting in azide and alkyne functional groups. The generated azide and alkyne moieties of the functionalized poly(methyl acrylate) (PMA) were subsequently ‘clicked’ (Cu, CH3CN) to form the triazole-based starting material. Image reprinted from Brantley et al.299 with permission from Nature publishing group. Copyright (2011).](/image/article/2013/CS/c3cs60040h/c3cs60040h-f12.gif) | ||
| Fig. 12 Ultrasound-induced retro [3+2] cycloaddition of an embedded triazole moiety. Triazole bond formation can be reversed by the application of mechanical force, resulting in azide and alkyne functional groups. The generated azide and alkyne moieties of the functionalized poly(methyl acrylate) (PMA) were subsequently ‘clicked’ (Cu, CH3CN) to form the triazole-based starting material. Image reprinted from Brantley et al.299 with permission from Nature publishing group. Copyright (2011). | ||
In another example, Baldwin and Kiick recently reported use of a retro click reaction to engineer the degradation rates of heparin-functionalized hydrogels prepared using thiol-based Michael-type addition reactions between multifunctional PEG thiols and maleimide-modified heparin.249,278 Differences in the pKa of the mercaptoacids used to functionalize PEG led to differences in hydrogel degradation rate within a reducing environment (i.e., in the presence of glutathione), owing to differential retro Michael-type cleavage rates of the succinimide–thioether linkage; the more rapid equilibration of an aryl thioether succinimide product with its reactant aryl-thiol modified PEGs and maleimide-functionalized heparin resulted in the capture of the liberated maleimide by the exogenous glutathione (GSH). The choice of mercaptoacid also was used to control the release of bioactive molecules in vitro. The intracellular concentration of GSH, a tripeptide of glutamic acid, cysteine, and glycine, is known to be significantly higher than the extracellular concentration,300 and the GSH content of carcinoma cells also is elevated, owing to the role of GSH in regulating mutagenic mechanisms, DNA synthesis, and growth.301,302 Since the rate of degradation and release of cargo molecules from these gels depend upon the local reducing environment, this degradation strategy is promising for intracellular or site-specific controlled drug delivery.
Another exciting class of reversible click reactions is retro Diels–Alder cycloreversion, which can be an attractive tool to modulate hydrogel degradation. Early examples of incorporating this reversible reaction chemistry within the crosslinks of hydrogels exhibited significant network degradation at temperatures above 60 °C, potentially limiting their translation into controlled cell microenvironments.303,304 However, recent work incorporating furan-functionalized pendant peptides within PEG-maleimide-based hydrogels demonstrates controlled release of these peptide tethers under physiological conditions.305 While higher temperatures (up to 80 °C) increased release, physiological temperature was adequate for significant tether release (∼40%), and dexamethasone released by this mechanism was shown to promote osteogenic differentiation of encapsulated hMSCs.305 This class of reversible click reactions is promising for predictable, tunable control of cell microenvironment properties.
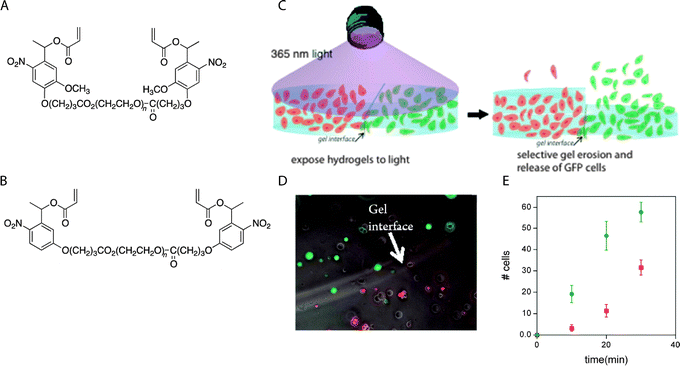 | ||
| Fig. 13 Selective cell release via photodegradation using differences in reactivity of o-nitrobenzyl groups. (A), (B) o-Nitrobenzyl linkers with different degradation kinetics were used to vary the degradation rate of adjacent hydrogels. (C) RFP-expressing hMSCs and GFP-expressing hMSCs were encapsulated within hydrogels made with (A) or (B), where the two hydrogels were in direct contact with each other (RFP = red fluorescent protein, GFP = green fluorescent protein). (D) The interface between hydrogels containing RFP- and GFP-expressing hMSCs was observed using optical microscopy. (E) Gels were exposed to light (10 mW cm−2 at 365 nm, 30 minute total duration), resulting in a biased release of one cell population over another (RGFP/RRFP ∼ 2.4) which was consistence with the degradation rate constants of (A) and (B) (kapp A/kapp B ∼ 2.5). Image reprinted from Griffin et al.280 with permission from American Chemical Society. Copyright (2012). | ||
In a complementary light-mediated approach, Anseth and coworkers used photoinitiators to degrade disulfide-bonded PEG hydrogels.307 When irradiated, the photoinitiator created free radicals through heterolytic decomposition, attacking the disulfide bonds and resulting in hydrogel degradation. In principle, this photoinitiated disulfide bond degradation could be conducted in the presence of cells in conjunction with cytocompatible disulfide gel formation.308 Almutairi and coworkers recently reported synthesis of polymer containing a pendant photocleavable group, 4-bromo7-hydroxycoumarin (Bhc).309 Upon photolysis with cell and tissue compatible near infrared irradiation, the polymer undergoes a triggered cascade of cyclization reactions, leading to degradation of the polymer backbone with potential applications for controlled release in vivo within deep tissues.
6. Orthogonal property control
6.1 Biochemical cues
Bioactive molecules, such as proteins, cytokines, growth factors, and therapeutic drugs, have been extensively used to provide biochemical cues that modulate cell adhesion, migration, differentiation, and proliferation. Spatial and temporal presentation of such biochemical cues can mimic the dynamic nature of the native cellular microenvironment in vitro or enable site-specific regulation of cellular functions within in vivo microenvironments. Significant advances have been made in improving hydrogel properties for spatiotemporal controlled presentation of bioactive molecules. Current technologies have demonstrated the potential of hydrogels for controlled, sustained, and local delivery of bioactive molecules, and the static or dynamic presentation of insoluble biochemical signals, such as immobilized integrin binding peptide sequences or sequestered growth factors. However, some challenges remain for improving the clinical applicability and spatiotemporal functionality of hydrogels for controlled presentation of bioactive molecules. For controlled release, issues with encapsulation efficacy, the stability of encapsulated proteins and sensitive bioactive molecules, and long-term delivery of hydrophobic molecules persist; for insoluble cues, difficulties persist with the dynamic presentation of multiple cues with high spatial resolution.For controlled incorporation or presentation of bioactive molecules, one must consider factors such as the mechanism of release, the triggering mechanism, and the ability to control spatiotemporal presentation or release, in addition to the design considerations discussed in Section 2. The release of cargo molecules can be controlled by diffusion, degradation (surface or bulk erosion), the cleavage of a tether, or a combination of these mechanisms, where the bioactive molecules are chemically immobilized, sequestered or physically encapsulated in the hydrogel network. Here, we present several recent advances in the design and production of cell-compatible hydrogels, including the controlled presentation of bioactive molecules and the manipulation of mechanical and physicochemical properties.
One of the most successful techniques for promoting cell adhesion in cell-compatible hydrogels is to incorporate peptide-based analogs of native ECM components, such as RGD, YIGSR, and IKVAV, into the hydrogel matrix.314–317 The RGD (Arg-Gly-Asp) sequence is found in a number of extracellular proteins, including fibrinogen, fibronectin, vitronectin, and laminin, and binds several integrins with varying strength.318,319 For spatiotemporally controlled presentation of the RGD motif in hydrogels, DeForest and Anseth employed SPAAC and thiol–ene click chemistries (Fig. 14).320 Four-arm cyclooctyne-functionalized PEG was reacted with a bis(azide)-functionalized photodegradable polypeptide using SPAAC reaction for hydrogel formation. Thiol-containing RGD peptide was photopatterned with vinyl functionalities on the polypeptide backbone via a photoinitiated thiol–ene reaction using visible light. Further, fibroblast outgrowth and spreading within a photodegraded channel was directed spatially in the RGD photopatterned region. When incorporating cell-binding domains into hydrogels, it must be recognized that multiple factors, including the bulk density, domain size, and length of the spacer arm to the adhesion ligand, can influence cell adhesion and spreading. Lee et al. studied the effect of the spacer length in RGD-modified substrates for controlling cell phenotype in the 2D and 3D cell microenvironment via the use of GnRGDSP-modified alginate hydrogels.321 From measurements of the cell aspect ratio and projected area, at least four glycines (n = 4) were required for fibroblast adhesion on the GnRGDSP-modified alginate hydrogels.
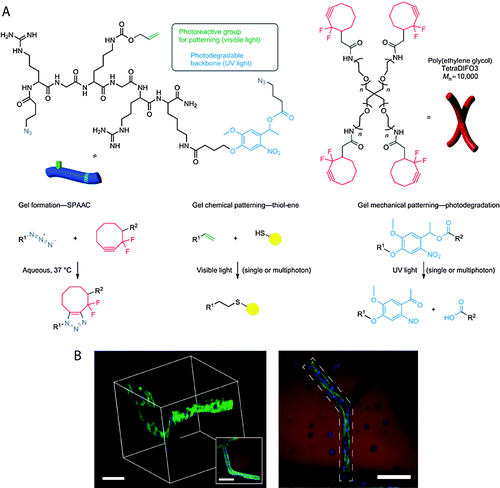 | ||
| Fig. 14 Spatiotemporal control of biochemical cues in 3D microenvironment. (A) Four-arm PEG functionalized with cyclooctyne was reacted with azide di-functionalized polypeptides via SPAAC reaction to form a hydrogel network via step-growth mechanism. A light-mediated thiol–ene reaction (cytocompatible 490–650 nm or 860 nm pulsed laser light) was used to immobilize cell adhesive thiol-functionalized peptides (RGD) using vinyl functionalities present on hydrogel network. Further, 3-D channels were degraded within the hydrogel using pulsed laser light (740 nm) via irreversible cleavage of nitrobenzyl ether moiety. (B) A cell-laden (3T3 fibroblasts) fibrin clot was encapsulated in the hydrogel (3D microenvironment). Biochemical (channel containing RGD noted by dashed polygon) and biophysical cues (photodegraded channel) were added to control 3T3 cell outgrowth in the presence of encapsulated hMSCs (right, top-down projection; left, 3D rendering). (Scale bar, 100 μm, hydrogel shown in red, F-actin in green and cell nuclei in blue) Image reprinted from DeForest et al.320 with permission from Nature publishing group. Copyright (2011). | ||
While incorporation of bioactive peptides into a hydrogel matrix provides an interesting strategy to induce bioactivity and enhance cell adhesion and spreading, understanding the effect of such peptide incorporation on the mechanical and transport properties of the network is essential. Zustiak et al. demonstrated that peptide ligands influence the physical, mechanical and transport properties of PEG hydrogels and that the extent of this influence was dependent on the concentration and the amino acid sequence of a given ligand.322 Incorporation of the peptide RGDS at a concentration of 300 μM in a PEG hydrogel (10% w/v) led to an ∼20% decrease in the hydrogel storage modulus (G′), and accordingly, the calculated average network mesh size (ξ) increased by ∼1 nm. Further, the greatest change in G′ and ξ was observed for hydrogels modified with either IKVAV (10 μM) or YIGSR (100 μM), emphasizing the importance of the amino acid sequence and related ligand–polymer interactions. The pronounced effect of YIGSR ligands on hydrogel properties was hypothesized to arise from the formation of hydrogen bonds between the phenolic OH group of Y and the ether oxygen of the PEG polymer. The incorporation of RGDS, IKVAV or YIGSR at a concentration of 100 μM resulted in a decrease in the diffusivity of encapsulated BSA by approximately 30%.
The process of cell adhesion is mediated through proteins. Hydrogel network hydrophobicity, which promotes protein adsorption, consequently can influence cell adhesion. Ayala et al. recently demonstrated the effect of matrix hydrophobicity on the adhesion, morphology, and differentiation of hMSCs using a hydrogel based on copolymers of select acryloyl amino acids (referred to generally as AA) and acrylamide (Am), as shown in Fig. 15.323 Substrate hydrophobicity was systematically controlled by varying the length of pendant alkyl side chains, as assessed by contact angle measurements, without significantly altering the chemical or mechanical properties of the hydrogel. The adhesion and spreading of hMSCs were found to be non-monotonically dependent on matrix hydrophobicity. A hydrogel equipped with a 5-carbon long alkyl chain (C5) was shown to support improved cell adhesion as compared to those modified with 1–4 (C1 to C4) and 6–10 carbons (C6 to C10). Cell spreading was hypothesized to be greater on the C5-containing hydrogels, as compared to the C1–C4 hydrogels, due to limitations in the accessibility of the AA side chain to fibronectin. For the hydrogels with longer alkyl chains (C6–C10), it was postulated that collapse of the hydrophobic domain into the matrix resulted in limited accessibility of the AA for binding.
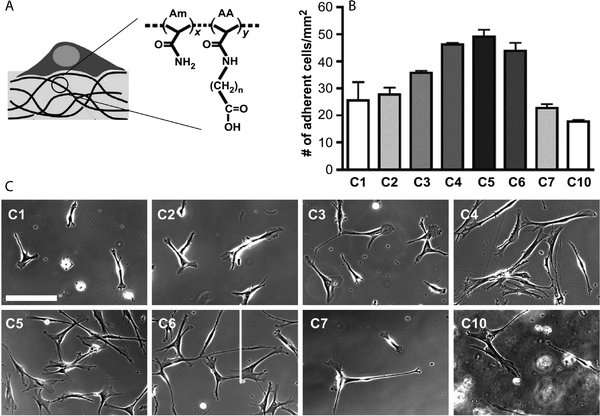 | ||
| Fig. 15 Modulation of cell adhesion and spreading in 2D microenvironment. (A) The hydrogels were prepared by copolymerizing acrylamide (Am) with acryloyl amino acid (AA) using a bis-acrylamide initiator. Depending upon the number of CH2 groups on the AA pendant chain (n = 1 to 10, referred as C1, C2,…,C10), the interfacial hydrophobicity of the hydrogel varied, with water contact angle ranging from 26° to 85° (sessile drop method, 20 °C). (B), (C) Non-monotonic dependence on the monomer side chain length was observed in cell adhesion and spreading of hMSCs on C1–C10 hydrogels (scale bar, 400 μm). Image reprinted from Ayala et al.323 with permission from Elsevier. Copyright (2011). | ||
In addition to controlling cell adhesion, efforts have been made to design cell-compatible hydrogels for highly directed cell migration. Cell migration is a central, highly integrated multistep process required for maintenance and development of numerous physiological processes.324,325 In hydrogel networks, cell migration has been controlled using spatiotemporal gradients of selective cell adhesion ligands. Numerous experiments have shown increased cell migration with increased ligand density up to a critical value, or towards the higher ligand density in a gradient. Cell migration speed is known to have a parabolic response to ligand density.326 In 2D culture, Guarnieri et al. studied the effect of a linear gradient of covalently immobilized RGD within PEG diacrylate-based hydrogels on the migration of fibroblasts (NIH3T3s).327 It was shown that the cells moved preferentially along the direction of increasing concentration of immobilized RGD. Further, the cell migration speed increased with an increase in the magnitude of the gradient. Schwartz et al. studied migration of fibrosarcoma cells (HT-1080) in 3D microenvironments using enzymatically degradable PEG hydrogels.326 MMP degradable peptides and RGD-containing peptides were incorporated inside the PEG hydrogel via thiol–ene reaction between norbornene functionalized 4-arm PEG and cysteine containing peptides. The mesh size of the hydrogel (13 ± 1 nm) was engineered to be much smaller than the size of encapsulated cells (∼10 μm) to limit migration to a proteolytic mechanism. The percentage of migrating fibrosarcoma cells (HT-1080s) was found to have a parabolic response to ligand density. Further, HT-1080s were observed to migrate through a Rho kinase (ROCK)-dependent mechanism with a rounded morphology that quantitatively resembled in vivo migrating cancer cells. In principle, various thiol functionalized biochemical cues can be incorporated into such hydrogels to study cell migration for understanding cancer cell invasive behavior and metastasis.
The ability to promote cell survival and proliferation over desired time periods also is a critical hydrogel design feature and can be achieved by the presentation of growth factors or by controlling cell–ECM and cell–cell interactions. Growth factors are signaling polypeptides that trigger cell responses such as cell survival, migration, differentiation, or proliferation. Precise control over the presentation of proteins and growth factors in hydrogel matrices is critical for mimicking the native cellular microenvironment and promoting cell–substrate and cell–cell interactions for tissue engineering and regenerative medicine applications.328
Growth factor immobilization strategies can take advantage of either covalent tethering or affinity interactions between growth factor(s) and rationally designed hydrogels. For example, Kiick and coworkers have employed heparin-containing PEG-based hydrogels for controlling the release of basic FGF (bFGF, also known as FGF-2), using affinity of the growth factor with heparin for sequestering growth factors.29 Hydrogels were prepared via Michael-type addition chemistry using thiol-functionalized PEG and maleimide-functionalized heparin. The release of FGF-2 could be tuned as a function of polymer weight percent, polymer molecular weight, and initial cargo loading. In addition to local delivery of a single growth factor, simultaneous delivery of multiple growth factors may enhance cell response. For example, Zieris et al. investigated the effect of independent delivery of bFGF and VEGF from PEG-heparin hydrogels, on vascularization (Fig. 16).329 Amine-functionalized PEG was chemically crosslinked with EDC/s-NHS-activated carboxylic acid groups of heparin to form the hydrogel, and growth factors were immobilized post-hydrogel formation via heparin interaction. It was found that the loaded concentration of the growth factor could easily be tuned as a function of the initial concentration of growth factor, and release of the cargo occurred without significant initial burst. The cell number after 3 days, determined indirectly via a MTT metabolic activity assay, was approximately four times higher in hydrogels with growth factors (∼10000–12000 cells cm−2 scaffold area) as compared to a control hydrogel that lacked growth factors (∼2700 cells cm−2 scaffold area), indicating enhanced survival and proliferation in hydrogels with sequestered FGF-2 and VEGF. Quantification of chicken embryo chorioallantoic membrane (CAM) vascularization indicated a significant increase in vascularization in the presence of FGF-2, VEGF or a combination of both (∼20%, 35% and 40%, respectively) as compared to control hydrogel. Overall, the combined delivery of FGF-2 and VEGF resulted in superior pro-angiogenic effects (cell survival, proliferation, differentiation, and migration in vitro and CAM vascularization in vivo) relative to single factor delivery.
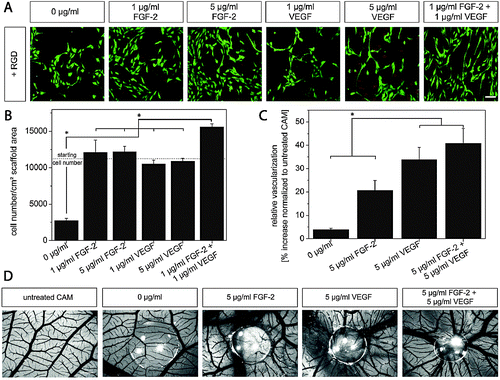 | ||
| Fig. 16 Dual growth factor delivery by sequestration in controlled cell microenvironments. (A) Human endothelial cells from the umbilical cord vein (HUVECs) on RGD-modified hydrogel substrates were presented with varying amounts of basic fibroblast growth factor (FGF-2) and vascular endothelial growth factor (VEGF) via sequestration. The enhanced cell survival and typical spindle-like morphology on the hydrogel substrate comprising the combination of FGF-2 and VEGF highlights the synergistic activity of both growth factors (fluorescence microscopy images after live/dead staining). (B) Further, HUVEC proliferation was enhanced by the dual presentation (MTT assay, day 3). (C) These hydrogels were placed onto the developing chicken embryo chorioallantoic membrane (CAM) from embryonic day 8 until day 12 to study the effect of growth factors on vascularization. An increased number of vessels within the site of gel transplantation was observed; (D) representative images indicate substantial angiogenic response to combined FGF-2 and VEGF delivery. Reprinted from Zieris et al.329 with permission from Elsevier. Copyright (2011). | ||
Cell–cell interactions are important for various cellular processes, including cell survival, proliferation, and differentiation, and hence designing hydrogels to promote cell–cell communication can positively impact or regulate these. Lin and Anseth recently developed functional PEG hydrogels with immobilized cell–cell communication cues in order to enhance the survival of encapsulated pancreatic β-cells.330 A PEG-diacrylate (PEGDA) macromer and thiol-functionalized fusion proteins (EphA5–Fc receptor and ephrinA5–Fc ligand) were polymerized with a cell-compatible photoinitiator via mixed mode thiol–acrylate photopolymerization in the presence of a murine insulinoma cell line (MIN6). EphA–ephrinA binding is known to mediate insulin secretion in pancreatic β-cells and also is linked to several intracellular signaling pathways that influence cell survival.331 The immobilization of these fusion proteins (200 nM) in the hydrogel resulted in more than a 100% increase in cell metabolic activity compared to hydrogels without any immobilized protein. Such an approach could be used to tailor the hydrogel microenvironment via incorporation of appropriate ECM components and using cell–cell communication signals to synergistically enhance cell survival for applications, including soft tissue engineering, controlled 3D cell culture, and cell delivery.
Wylie et al. reported simultaneous patterning of multiple growth factors, sonic hedgehog (SHH) and ciliary neurotrophic factor (CNTF), in three-dimensional hydrogels using orthogonal physical binding.332 Agarose hydrogels containing coumarin-caged thiols yielded reactive thiol groups upon two-photon irradiation; these selectively de-protected thiols subsequently served as sites for the sequential immobilization of maleimide functionalized barnase and streptavidin via a thiol–maleimide reaction. Barstar-SHH and biotin-CNTF were incubated in the hydrogel, for immobilization via their physical binding interactions with barnase and streptavidin, respectively. SHH and CNTF remained bioactive after immobilization. The hydrogel with immobilized growth factors did not show significant cytotoxicity, and in principle, such a simple approach could be used to control the spatiotemporal presentation of multiple growth factors to precisely engineer cell differentiation.
Physical encapsulation of growth factors with stimuli responsive release enables temporal tuning of bioactive molecule concentrations in local microenvironments. The triggering mechanisms used for stimuli responsive hydrogels include pH, temperature, enzymes, externally applied light, or magnetic fields. Garbern et al. designed pH- and temperature-responsive injectable hydrogels using poly(N-isopropylacrylamide-co-propylacrylic acid-co-butyl acrylate) (p[NIPAAm-co-PAA-co-BA]) for sustained and local delivery of bFGF in the acidic microenvironment of ischemic myocardium.337 The (p[NIPAAm-co-PAA-co-BA]) existed as a liquid at room temperature and pH 7.4, but reversibly formed hydrogels at 37 °C and pH 6.8. In vivo studies within a rat model of myocardial ischemia indicated that the bFGF could be highly localized at the site of injection when encapsulated within the responsive hydrogel. The amount of bFGF recovered at day 7 was increased by approximately 10-fold with the hydrogel delivery system as compared to delivery via saline injection. These in vivo studies also indicated an increased microvessel density and improved cardiovascular function (as measured by echocardiography) after 28 days of treatment, indicating the potential for spatiotemporal controlled delivery of the growth factor. Biochemical cues also can be used for regenerative medicine applications. For example, recently Diab et al. reported the use of degradable silk fibroin hydrogels for delivering growth factors, such as bone morphogenetic protein BMP-2 (Fig. 17A and B).338 The hydrogels were prepared using a sonication-induced gelation process in a solution containing BMP-2 and injected at the large femoral segmental defect site. An initial burst was observed and about ∼20–30% of cargo was released by day 4 depending upon hydrogel polymer concentration. An in vivo study demonstrated enhanced bone formation with hydrogels containing BMP-2 compare to control (hydrogels without BMP-2). The histological evaluation after 12 weeks indicated that the silk hydrogel was completely degraded.
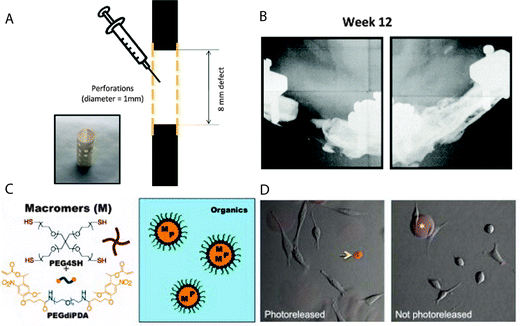 | ||
| Fig. 17 Hydrogels for delivering cargo biomolecules (therapeutic proteins, growth factors, and drugs) with pre-programmed or user-defined release behavior. (A) A degradable silk fibroin hydrogel for pre-programmed release was formed in the presence of BMP-2, using a sonication-induced gelation process, and was injected into a bone defect. (B) Enhanced bone formation was observed in vivo in SASCO Sprague Dawley rats injected with the silk hydrogel containing BMP-2 as compared to the control group (no growth factor) (week 12, X-ray radiography). A, B reprinted from Diab et al.338 with permission from Elsevier. Copyright (2011). (C) Photodegradable hydrogel microparticles were synthesized by an inverse suspension polymerization of a PEG-diphotodegradable acrylate with a PEG tetrathiol (PEG4SH) via base-catalyzed Michael addition. (D) A model cargo protein (Annexin V) was loaded (right), and its release to 3T3 cells (left) was triggered by cytocompatible irradiation (1 min of 13.5 mW cm−2 at 365 nm). C, D reprinted from Tibbitt et al.339 with permission from John Wiley and Sons publishing. Copyright (2012). | ||
For controlled release during cell culture, Anseth and coworkers recently reported photodegradable, PEG based hydrogel microspheres with entrapped cargo proteins that deliver proteins locally upon exposure to selected wavelengths of light (Fig. 17C and D).339 Poly(ethylene glycol) di-photodegradable-acrylate (PEGdiPDA) was copolymerized with poly(ethylene glycol) tetrathiol (PEG4SH) via base-catalyzed Michael-type addition using an inverse suspension polymerization technique. Transforming growth factor beta 1 (TGF-β1), which controls proliferation, differentiation, and apoptosis, was encapsulated inside of the microspheres during hydrogel formation. The o-nitrobenzyl ether moiety in the PEGdiPDA was cleaved using cytocompatible irradiation (at 365 nm for less than 5 minutes), which resulted in network degradation followed by localized release of the cargo molecule. The released TGF-β1 maintained its bioactivity as demonstrated by upregulated luciferase production when applied to a reporter cell line. In principle, such an approach could be used to spatiotemporally control the release of a broad range of cargo molecules, such as growth factors, cytokines, and extracellular matrix components, within 2D and 3D cell microenvironments via multiple wavelengths of light.
Cell differentiation is a commonly occurring process by which a less specialized cell, a stem or progenitor cell, becomes a more specialized cell type. For example, mesenchymal stem cells can differentiate into osteoblasts, chondrocytes, or adipocytes amongst other lineages. Due to their ability to differentiate into a wide variety of cell types, stem cells represent a promising resource for tissue engineering and regenerative medicine. To take maximum advantage of pluripotency for such biomedical applications, it is crucial to understand and control the presentation of cues, such as immobilized factors, ECM signaling molecules, and substrate properties, all which can influence stem cell differentiation. Oh et al. studied the effect of bFGF on the proliferation and osteogenic differentiation of hMSCs encapsulated in collagen hydrogels in a 3D environment.340 Sustained release of encapsulated bFGF was observed up to 30 days (with an initial burst). Enhanced osteogenic differentiation of hMSCs in the bFGF-loaded hydrogels was significant after 14 days in vitro, as detected by gene expression analysis. The combination of natural hydrogel networks with appropriate growth factors, and the resulting control of cell differentiation, is very promising for clinical use in the field of regenerative medicine.
Significant advances in molecular and cell biology have led to the development of increasingly powerful drugs, which can regulate specific cellular activity. To improve the efficacy, stability, and reduce potential side effects of these drugs, hydrogel-based drug carriers have been reported for controlled release applications. For comprehensive review of hydrogels for drug delivery, readers are referred to a review by Hoare and Kohane.341 Other drug carriers such as microspheres and liposomes have also been incorporated in hydrogel matrices to create composite hydrogels. Such approaches can provide superior control over the release profiles of cargo molecules and can enhance biocompatibility of the particular vehicle by its incorporation in the cell-compatible hydrogel. Wei et al. used a dual drug delivery system based on PVA or chitosan hydrogels with encapsulated poly(L-glutamic acid)-b-poly(propylene oxide)-b-poly(L-glutamic acid) micelles that contained aspirin or DOX as the cargo drug molecules, respectively.342 The release of cargo was found to be dependent on pH and temperature with short-term release of aspirin (∼75% release within 3–5 hours) and longer, sustained release of DOX (∼25–75% release within 7 hours). The release of DOX was accelerated at lower pH or higher temperature, indicating the potential for localized delivery of the anti-cancer drugs in carcinoma tissue.
6.2 Biophysical cues
Physical properties of the cellular microenvironment, such as rigidity and topography, also play critical roles in regulating cellular functions and mediating cellular response to a variety of stimuli, such as strain and shear stress. Biophysical cues, whether inherent or applied, have been observed to affect actin cytoskeleton organization, focal adhesion assembly, and cell shape, leading to changes gene and protein expression.343 These signaling processes regulate cell function and fate, such as adult stem cell differentiation. Hydrogel matrices thus can be engineered to control cell behavior by providing appropriate biophysical cues initially and upon degradation. Here, we overview cell-compatible hydrogels that have been used to manipulate cell response by providing appropriate biophysical cues in static and dynamic 2D and 3D microenvironments along with some examples. The static examples are intended to provide motivation and context for the critical microenvironment signals that regulate cellular processes. The dynamic examples highlight recent works that probe how changes in the cell microenvironment influence cell response utilizing orthogonal chemistries and manipulations, degradable moieties, or combinations of these to mimic the native cell microenvironment.Mechanical feedback from the ECM, which is required for integrin clustering and for the subsequent formation of focal adhesions, is critical for stem cell differentiation. Trappmann et al. studied the differentiation of hMSCs and human epidermal stem cells using collagen coated polydimethylsiloxane (PDMS) and polyacrylamide (PAAm) hydrogel surfaces.346 The substrate modulus was varied from 0.1 kPa to 2300 kPa (by modulating polymer-crosslinker ratio) and from 0.5 kPa to 740 kPa for PAAm (by modulating monomer-crosslinker ratio). hMSCs seeded on all PDMS and PAAm substrate with high stiffness differentiated into bone cells, whereas epidermal stem cells differentiated only on soft PAAm substrate. The authors hypothesized that the decreased pore size of PAAm correlated with changes in substrate modulus led to variation in collagen tethering and altered differentiation; to verify this, the authors subsequently varied the collagen tethering. Epidermal cell shape and fate were influenced by the distance between the anchoring points. The absence of stiffness dependent spreading and differentiation on PDMS, apparently contradictory to previous findings of cell differentiation dependence on substrate stiffness, can be explained by the ability of seeded cells to remodel the collagen layer present on viscoelastic PDMS, diminishing their sensitivity to substrate stiffness.347 This study demonstrates the importance of stem cell exerted mechanical forces on substrate-bound ECM, altering the matrix composition on these non-degradable hydrogels, and the influence of traction forces on cell-fate decisions.
Substrate stiffness can have an effect on the phenotypes of numerous other types of cells as well.27,28,348–352 Robinson et al. recently studied the effect of substrate modulus on human vascular endothelial, smooth muscle, and fibroblastic cells using heparinized PEG hydrogels (G′ ∼ 0.3, 5.2, and 13.7 kPa).349 Maleimide-functionalized heparin was reacted with thiol-functionalized PEG to form hydrogels, and bioactivity was ensured by the incorporation of fibronectin and growth factors (bFGF or VEGF, depending upon the cell line). The substrate modulus was varied by changing polymer concentration. Differences in cell behavior (attachment, proliferation and gene expression) were observed and correlated with hydrogel modulus. For example, human vascular smooth muscle cells demonstrated preferential growth on the relatively stiff hydrogel substrate while endothelial cells exhibited preferential growth on the soft substrates. In another example, Murphy and coworkers investigated the effect of substrate stiffness on vascular endothelial cell behavior using polyacrylamide gels with varying modulus (25, 50, and 75 kPa).353 Umbilical vein (HUVEC), aorta (HAEC), saphenous vein (HSaVEC) and dermal microvasculature (HmVEC) endothelial cells were seeded on hydrogels. It was found that the differences in substrate stiffness influenced cell attachment, spreading, proliferation and migration. For example, an increase in modulus from 25 kPa to 75 kPa resulted in an approximately 75% decrease in HUVEC cell attachment after 24 h. Response to substrate stiffness was cell specific, indicating heterogeneity in the response to biophysical cues. Taken together, these examples highlight the need to determine optimal conditions (i.e., range of moduli) for specific cell types in relevant biomedical applications.
In addition to the impact of substrate stiffness, cells can also sense surface topographical features that can impact cellular properties including cell morphology, adhesion, and differentiation. The native ECM exhibits numerous topographical features, including fibers and sheets with micron and submicron dimensions. Two-dimensional hydrogels with topographical features such as grooves and pits thus have been explored as model systems to study such cellular behavior. Poellmann et al. used collagen-coated polyacrylamide hydrogels with a micropatterned array of posts with varied shape and spacing to study the morphologies of murine MSCs.354 The patterned hydrogel was prepared via the polymerization of acrylamide at room temperature with a silicon master pattern floating on top of the prepolymer solution. The patterned posts influenced the cell orientation and the gaps between posts resulted in elongated cell bodies. In another example, Guvendiren and Burdick used micro-scale hydrogel surface wrinkles to modulate hMSC response by changing surface wrinkle size and shapes.355 The hydrogels were prepared using poly(2-hydroxyethyl methacrylate) (PHEMA) and ethylene glycol dimethacrylate and photopolymerized to a PDMS master to induce surface patterning. It was found that hMSCs took the shape of the pattern, and with high aspect ratio patterns, preferentially differentiated into osteoblasts. When seeded in hexagonal patterns, hMSCs exhibited a rounded morphology and differentiated preferentially into adipocytes. This is consistent with findings of others that have shown directed hMSC differentiation via controlled adhesion to micropatterned features356 and suggests the importance of controlled topography to modulate cell behavior, including user-desired lineage specification for regenerative medicine applications.
Frey and Wang have developed hydrogel compositions for 2D culture in which modulus can be decreased with UV irradiation and degradation, providing a method for probing cellular response to changes in substrate rigidity (Fig. 18).360 Photodegradable PAAm hydrogels comprising 4-bromomethyl-3-nitrobenzoic acid (BNBA) and polyacrylamide acryl hydrate (PAAH) were prepared on glutaraldehyde-activated coverslips. UV exposure at a dose tolerated by live cells cleaved the nitrobenzyl group and the network, causing the hydrogel to soften by up to 30% (from 7.2 kPa to 5.5 kPa). Softening of hydrogel network led to reduced area of spread 3T3 fibroblasts; further, localized softening of the substrate underlying the leading edge of the cell resulted in pronounced cell retraction, suggesting that mechanosensing was localized to the anterior of polarized cells. In a complementary study, Wang et al. studied the deactivation of valvular myofibroblasts to dormant fibroblasts using photodegradable hydrogels.358 A photodegradable PEG hydrogel (described in Section 5.2.4) was polymerized with an acrylated adhesion peptide (RGDS), and valvular interstitial cells (VICs) were seeded on this photoresponsive hydrogel. When irradiated with light, the modulus of the hydrogel was reduced from 32 kPa to 7 kPa via in situ photodegradation, leading to de-activation of myofibroblasts to quiescent fibroblasts, assessed by a decrease in cells positive for α-smooth muscle actin stress fibers, negligible apoptosis, and changes in cell proliferation and gene expression. These approaches to dynamically control the 2D microenvironment through degradation are complementary to non-degradable approaches.361,362 In principle, such approaches can be used to probe spatial and temporal response of cells to microenvironment rigidity.
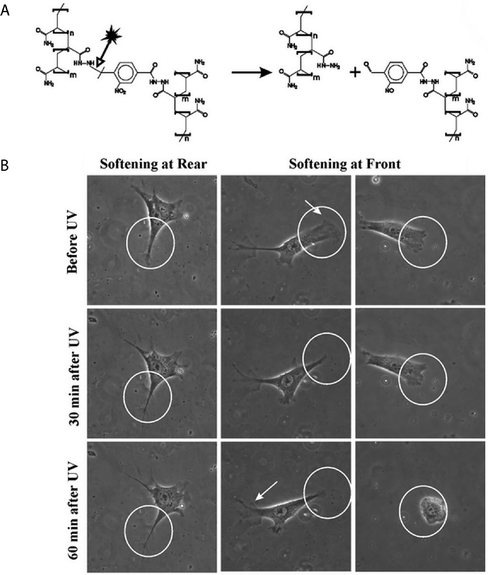 | ||
| Fig. 18 Modulation of substrate stiffness in a 2D dynamic microenvironment. (A) Incorporation of a photodegradable nitrobenzyl moiety in a hydrogel network, prepared using polyacrylamide acryl hydrate (PAAH) and a 4-bromomethyl-3-nitrobenzoic acid (BNBA) crosslinker, enabled gel degradation and softening using cytocompatible doses of UV light (365 nm). (B) Softening of the posterior substratum (rear) did not significantly alter cell spreading (left column). However, softening of the anterior substratum (front) led to reverse polarity (central column) or trapping in the softened region (right column; scale bar, 20 μm). Reprinted from Frey et al.360 with permission from The Royal Society of Chemistry publishing group. Copyright (2009). | ||
To investigate the effect of hydrogel network structure on pericellular and extracellular matrix deposition, Nicodemus et al. prepared PEG hydrogels at different polymer concentrations (10, 15 or 20 wt%) via photopolymerization of a PEG-diacrylate macromer.366 Depending upon the polymer and photoinitiator concentrations, crosslink density was varied to form of hydrogels with compressive moduli varying from 60 to 590 kPa. It was found that glycosaminoglycan production was greater in the lowest crosslinked hydrogels. Further, Collagen II and VI, aggrecan, and decorin were found to be localized in the pericellular region and their presence decreased with an increase in the crosslinking. The study thus indicated that changes in hydrogel crosslinking and matrix stiffness could impact the type of tissue deposited and spatial evolution of the tissue.
Mooney and coworkers have recently studied stem cell response to substrate rigidity in three-dimensional microenvironments.367 Peptide-coupled alginate, agarose, and PEG-dimethacrylate were crosslinked into hydrogel networks (via calcium sulfate, physical crosslinking, and free radical polymerization, respectively) in the presence of murine mesenchymal stem cells (mMSCs). By varying the crosslink density and polymer concentration, it was possible to tune network rigidity (E ∼ 2.5 to 11 kPa) and the density of RGD. The commitment of encapsulated murine mesenchymal stem cells to specific differentiation lineages varied with the rigidity of the hydrogel network (i.e., adipogenic and osteogenic lineage predominantly at 2.5–5 kPa and 11–30 kPa, respectively). Further, it was observed that network stiffness regulated integrin binding and adhesion ligand recognition.
To investigate the effect of a static mechanical stress gradient induced by the material geometry, Ruiz and Chen investigated hMSC differentiation in collagen hydrogel cubes formed using a PDMS mold (Fig. 19).368 Initially, during 2D monolayer culture, cells seeded on the outer edges of an adhesive pattern committed to an osteogenic lineage and interior cells committed to an adipogenic lineage. Using traction force measurements, it was found that the geometry of the pattern induced mechanical stress, which influenced cell fate (high stress regions resulted in osteogenesis, low stress regions resulted in adipogenesis). This finding was translated to 3D hydrogel structures: cells near the edge of 3D cube-shaped collagen constructs differentiated down an osteogenic lineage, and those at the center differentiated down an adipogenic lineage. This study highlights the importance of mechanical patterning for cell differentiation, and hence provides important insight for designing hydrogels for regenerative medicine.
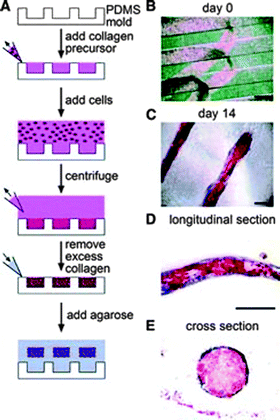 | ||
| Fig. 19 Stress gradients within hydrogels influence cell differentiation in three dimensions. (A) Schematics of the process for creating three-dimensional multicellular hydrogels and encapsulating human mesenchymal stem cells (hMSCs). Briefly, prepolymer type I collagen was added to PDMS molds and hMSCs were suspended, before polymerizing at 37 °C. Liquid agarose was added to the mold to encase the collagen hydrogel at 4 °C. (B) Phase image of hMSCs in three-dimensional structures at day 0. (C) The hydrogel constructs with encapsulated hMSCs were suspended in mixed media and after 14 days, the cells at the edge of the constructs differentiated down an osteogenic lineage (blue) and those at the center underwent adipogenesis (red) (oil droplets, alkaline phosphatase staining) (D) Longitudinal section and (E) cross-sections confirmed the patterning of lineage specification in a tension-dependent manner (scale bar, 250 μm). Reprinted from Ruiz et al.368 with permission from John Wiley and Sons publishing. Copyright (2008). | ||
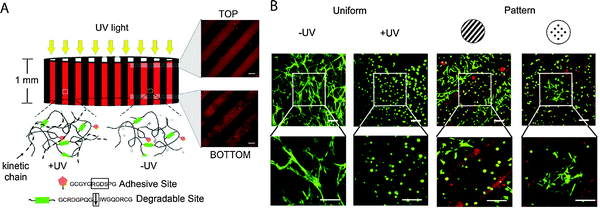 | ||
| Fig. 20 Spatiotemporal manipulation of biophysical cues in 3D cell microenvironments. (A) Hydrogels were prepared by reaction of an acrylated PEG macromer with thiol-functionalized, degradable and cell-adhesive peptides using a Michael-type addition reaction (−UV hydrogel, degradable peptide crosslinks), in the presence of an inactive photoinitiator. Using photolithography (4 min with 10 mW cm−2 at 365 nm), remaining acrylate groups on the PEG macromers in select regions were reacted by photoinitiated free radical polymerization, forming non-degradable covalent crosslinks (+UV hydrogel) (confocal microscopy images show top and bottom surface photopatterned with 250 μm stripes). (B) Encapsulated hMSCs (day 14 stained with calcein) spread only in −UV regions, where the degradable peptide was used as a crosslinker (scale bar, 100 μm). Reprinted from Khetan et al.369 with permission from Elsevier. Copyright (2010). | ||
In the native ECM, the cells can migrate either via localized matrix degradation by matrix metalloproteinases (proteolytic migration) or via local deformation of the ECM (nonproteolytic, or amoeboid migration). Ehrbar et al. took advantage of bioactive PEG hydrogels as an artificial ECM to study the effect of matrix stiffness on migration of encapsulated mouse preosteoblastic cells (MC3T3-E1).370 Using peptide conjugation, the authors synthesized PEG macromers with a glutamine-acceptor substrate (n-PEG-Gln), a lysine-donor substrate containing a MMP-sensitive linker (n-PEG-MMPsensitive-Lys), and a MMP-insensitive linker (n-PEG-MMPinsensitive-Lys). Thrombin-activated factor XIIIa was used to initiate hydrogel formation in presence of cells. By varying the polymer concentration, the modulus of the hydrogel was tuned from ∼100 Pa to 500 Pa. It was found that cell migration correlated with matrix stiffness, with nonproteolytic migration dominating at lower stiffness and proteolytic migration dominating at higher stiffness.
In another example, Guo et al. studied hMSC migration in a controlled 3D hydrogel environment using genetically encoded photoactivatable Rac1 (PA-Rac), a member of the Rho GTPase family that stimulates actin polymerization.371 Channels were photoetched in real-time using a two-photon microscope, and PA-Rac was activated within cells encapsulated in the hydrogel by local exposure to visible light, inducing directional mobility. At an optimum concentration of YRGDS (2.2 mM), the stiffness of the hydrogel was varied from 12 kPa to 50 kPa and the rate of cell migration increased with increased gel stiffness. The rate of cell migration was higher in photodegraded channels as compared to non-degraded hydrogels. The authors demonstrated the ability to modulate migration speed of encapsulated cells by providing appropriate biophysical cues (i.e., matrix stiffness and degradability) in the presence of appropriate biochemical cues (i.e., adhesive peptides and intracellular signaling proteins).
To investigate the role of biophysical cues in the development of branched tissues, such as kidney, lung, and mammary glands, Gjorevski and Nelson used microfabrication of collagen hydrogels to build model mammary epithelial tissue of well-controlled geometries.372 The collagen matrix was further crosslinked by incubation in D-ribose at 37 °C for one week before addition of cells, which lead to an increase in matrix stiffness. The epithelial cells adopted the shape and size of the collagen cavities, fabricated using soft lithography, and formed tubules, which remained dormant until the addition of hepatocyte growth factor. Stiffening of the hydrogel network by modulating D-ribose concentration led to an increase in the magnitude of mechanical stress and enhanced branching from the tubule tips. Further, using a finite element method, branching was determined to occur only at locations where dynamic biochemical and biophysical cues reinforced each other, and the magnitude of mechanical stress at branching sites correlated with the extent of branching. Previously, using a similar a collagen-hydrogel system, Nelson et al. demonstrated that tissue geometry influences the site of mammary branching morphogenesis.373
Recently, Anseth and coworkers developed an enzymatically degradable and photolytically degradable hydrogel platform for spatiotemporally controlling biophysical cues in 3D microenvironments to study critical cues and mechanisms that lead to tissue development and repair.374 The hydrogel was prepared using a click reaction between PEG-tetracycloctyne and photolabile, enzyme-labile diazide peptide along with an azide-functionalized integrin-binding sequence. Cells were seeded within hydrogel microwells created via photolithographic degradation (depth 50–200 μm), and a second hydrogel layer was added to encapsulate the cells within a 3D microenvironment. This platform enabled the geometry or connectivity of the local matrix to be spatiotemporally modulated in the presence of lung epithelial cells using cytocompatible light (740 nm, pulsed laser) and cell morphology and phenotype to be easily assessed over time.
7. Concluding remarks
The building blocks to control biophysical and biochemical cues within artificial cell microenvironments are rapidly expanding. Hydrolytically, enzymatically, or photolytically degradable moieties have been incorporated in water-soluble monomers and polymers to afford tunable cleavage of synthetic covalently crosslinked extracellular matrices or protein-releasing depots in the presence of cells. Additionally, hydrogels that (dis)assemble based on physical interactions offer shorter-term dynamic control of cell microenvironment structure or chemistry. Within either covalently or physically formed hydrogels, orthogonal chemistries are being utilized in conjunction with degradability for controlled presentation of biochemical cues to promote desired cell behaviors, such as adhesion, spreading, or migration.The growing number and combinations of degradable chemistries enables precise and dynamic control of the cell microenvironment. Yet, this added complexity necessitates increased development and utilization of in situ characterization techniques and predictive modeling to fully realize the power of these tools. Innovative approaches are needed to marry in situ hydrogel property characterization with real-time cell response assessment techniques.
Monitoring hydrogel degradation and property evolution in situ and in three-dimensions remains limited but offers great promise in correlating real-time microenvironment changes with dynamic cell response. Enzymatically cleavable peptide sequences containing Förster (or fluorescence) resonance energy transfer (FRET) fluorophore-quencher pairs have been developed to observe hydrogel degradation with confocal microscopy.375 FRET techniques have also been applied to quantitatively analyze interactions at the cell–material interface and assess cell adhesion to integrin-binding peptide sequences.376,377 Increased deployment of degradation models could facilitate the rational design and understanding of the complex degradation profiles that result with cleavage of multiple labile moieties. For example, individual models have been developed to describe mass loss from hydrolytically,378 enzymatically,379 or photolytically374,390 degradable PEG hydrogels, providing insight into the gel structure, degradation mechanisms, and property evolution. As combinations of different labile chemistries are utilized, integration and expansion of these models will aid in material design and correlating property evolution with biological functions, such as the cleavage of multiple variants of photolabile o-nitrobenzyl ether groups for selective cell release.280 Additionally, advances in techniques to monitor matrix modulus, as a measure of crosslink density and degradation, will provide further insight into complex degradation profiles and mechanisms. Recent advances include the pairing of rheometry and microfluidic sample generation plus microrheology (μ2 rheology) to evaluate the full material history over many compositions380 and microparticle barcoding using stop flow lithography to rapidly generate and assess thousands of hydrogel compositions.381 Microrheology has been also used to monitor evolution of the hydrogel modulus in situ during degradation.382
Focusing on monitoring cell response, cell-exerted traction forces, which vary with matrix modulus, allow in-direct monitoring of hydrogel degradation and cell function changes related to dynamic biophysical matrix cues.383,384 Additionally, reporter cell lines allow real-time monitoring of transcriptional or cytoskeletal changes in response to microenvironment stimuli.385 For example, cells can be engineered to produce fluorescent proteins, such as green and red fluorescent protein variants, during transcription of specific gene(s) or fluorescent cytoskeleton fusion proteins for real-time and often high-throughput monitoring of gene expression361,386,387 and signal transduction,388 respectively. Incorporation of reporter chemistries and utilization of in situ property and cell monitoring techniques such as these will advance our understanding of how degradation-induced, evolving microenvironment properties influence cell function and fate.
Last, incorporation of degradable chemistries to control hydrogel properties largely has been focused on covalent hydrogels. Hydrogels formed by physical interactions, which are increasingly designed using de novo principles,389 can uniquely mimic aspects of the native ECM with properties that span multiple size scales. Incorporation of degradable chemistries within these physical gels or assembling peptides within covalent degradable hydrogels could offer additional handles to control the cell microenvironment and enable new experiments to understand and direct cellular processes. Further, utilization of combinations of degradable chemical linkages could afford complementary control over degradation rates and hydrogel properties. In sum, hydrogels are being designed with degradable chemistries to enable in situ property control and dynamically control the cell microenvironment. Degradable materials are promising tools to understand and direct complex biological systems and cell behaviors, such as cell adhesion and spreading, migration, differentiation, proliferation, and apoptosis.
Acknowledgements
The authors gratefully acknowledge support, for related work in their laboratories, from the National Institutes of Health (Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103541), the INBRE program of the National Institutes of Health (P2012A01377), and the NIDCD (R01DC011377A)), the Nemours Research Foundation, and the University of Delaware Research Foundation. Additionally, the authors would like to thank Matthew Rehmann and Lisa Sawicki for feedback on earlier versions of this manuscript.References
- G. Chan and D. J. Mooney, Trends Biotechnol., 2008, 26, 382–392 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- Biomedical Applications of Hydrogels Handbook, ed. R. M. Ottenbrite, K. Park and T. Okano, Springer, 2010 Search PubMed.
- R. J. Wade and J. A. Burdick, Mater. Today, 2012, 15, 454–459 Search PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 Search PubMed.
- M. S. Rehmann and A. M. Kloxin, Soft Matter, 2013 10.1039/c3sm50217a.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- C. Wang, R. R. Varshney and D. A. Wang, Adv. Drug Delivery Rev., 2010, 62, 699–710 CrossRef CAS.
- R. Ulijn, N. Bibi, V. Jayawarna, P. Thornton, S. Todd, R. Mart, A. Smith and J. Gough, Mater. Today, 2007, 10, 40–48 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 Search PubMed.
- D. F. Williams, Biomaterials, 2008, 29, 2941–2953 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- V. A. Liu and S. N. Bhatia, Biomed. Microdevices, 2002, 4, 257–266 CrossRef CAS.
- K. D. Kim and N. M. Wright, Spine, 2011, 36, 1906–1912 Search PubMed.
- S. V. Murphy, A. Skardal and A. Atala, J. Biomed. Mater. Res., Part A, 2013, 101, 272–284 Search PubMed.
- N. E. Fedorovich, M. H. Oudshoorn, D. van Geemen, W. E. Hennink, J. Alblas and W. J. Dhert, Biomaterials, 2009, 30, 344–353 CrossRef CAS.
- C. C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 CrossRef CAS.
- T. P. Kraehenbuehl, L. S. Ferreira, P. Zammaretti, J. A. Hubbell and R. Langer, Biomaterials, 2009, 30, 4318–4324 CrossRef CAS.
- N. C. Hunt and L. M. Grover, Biotechnol. Lett., 2010, 32, 733–742 CrossRef CAS.
- T. Aikawa, T. Konno, M. Takai and K. Ishihara, Langmuir, 2012, 28, 2145–2150 CrossRef CAS.
- S. Sundelacruz and D. L. Kaplan, Semin. Cell Dev. Biol., 2009, 20, 646–655 Search PubMed.
- A. M. Martins, C. M. Alves, F. K. Kasper, A. G. Mikos and R. L. Reis, J. Mater. Chem., 2010, 20, 1638–1645 RSC.
- J. J. Schmidt, J. Rowley and H. J. Kong, J. Biomed. Mater. Res., Part A, 2008, 87, 1113–1122 Search PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS.
- S. A. Bencherif, A. Srinivasan, F. Horkay, J. O. Hollinger, K. Matyjaszewski and N. R. Washburn, Biomaterials, 2008, 29, 1739–1749 CrossRef CAS.
- M. Patenaude and T. Hoare, Biomacromolecules, 2012, 13, 369–378 CrossRef CAS.
- J. Eyckmans, T. Boudou, X. Yu and C. S. Chen, Dev. Cell, 2011, 21, 35–47 Search PubMed.
- J. D. Kretlow and A. G. Mikos, AIChE J., 2008, 54, 3048–3067 CrossRef CAS.
- E. C. Novosel, C. Kleinhans and P. J. Kluger, Adv. Drug Delivery Rev., 2011, 63, 300–311 CrossRef CAS.
- C. C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS.
- F. van de Manakker, K. Braeckmans, N. e. Morabit, S. C. De Smedt, C. F. van Nostrum and W. E. Hennink, Adv. Funct. Mater., 2009, 19, 2992–3001 CrossRef CAS.
- L. M. Weber, C. G. Lopez and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2009, 90, 720–729 CrossRef.
- A. Bertz, S. Wohl-Bruhn, S. Miethe, B. Tiersch, J. Koetz, M. Hust, H. Bunjes and H. Menzel, J. Biotechnol., 2013, 163, 243–249 Search PubMed.
- M. C. Du, W. X. Song, Y. Cui, Y. Yang and J. B. Li, J. Mater. Chem., 2011, 21, 2228–2236 RSC.
- N. A. Peppas and A. R. Khare, Adv. Drug Delivery Rev., 1993, 11, 1–35 CrossRef CAS.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS.
- M. Martins-Green, in Principles of Tissue Engineering, ed. R. Lanza, R. Langer and J. P. Vacanti, Academic Press, 2nd edn, 2000 Search PubMed.
- C. M. Nelson and M. J. Bissell, Annu. Rev. Cell Dev. Biol., 2006, 22, 287–309 CrossRef CAS.
- R. Xu, A. Boudreau and M. J. Bissell, Cancer Metastasis Rev., 2009, 28, 167–176 CrossRef.
- G. D. Prestwich, J. Controlled Release, 2011, 155, 193–199 Search PubMed.
- X. Xu, A. K. Jha, D. A. Harrington, M. C. Farach-Carson and X. Jia, Soft Matter, 2012, 8, 3280–3294 RSC.
- A. M. Ferreira, P. Gentile, V. Chiono and G. Ciardelli, Acta Biomater., 2012, 8, 3191–3200 Search PubMed.
- Engineering Biomaterials for Regenerative Medicine: Novel Technologies for Clinical Applications, ed. S. K. Bhatia, Springer, 2011 Search PubMed.
- Y. Lei, S. Gojgini, J. Lam and T. Segura, Biomaterials, 2011, 32, 39–47 CrossRef CAS.
- H. Geckil, F. Xu, X. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS.
- R. A. Marklein and J. A. Burdick, Adv. Mater., 2009, 22, 175–189 Search PubMed.
- J. K. F. Suh and H. W. T. Matthew, Biomaterials, 2000, 21, 2589–2598 CrossRef CAS.
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS.
- H. G. Garg and C. A. Hales, Chemistry and biology of hyaluronan, Elsevier, Amsterdam, Boston, 2004 Search PubMed.
- T. C. Laurent, The chemistry, biology, and medical applications of hyaluronan and its derivatives, Portland Press, London, Miami, 1998 Search PubMed.
- E. J. Oh, K. Park, K. S. Kim, J. Kim, J. A. Yang, J. H. Kong, M. Y. Lee, A. S. Hoffman and S. K. Hahn, J. Controlled Release, 2010, 141, 2–12 CrossRef CAS.
- D. D. Allison and K. J. Grande-Allen, Tissue Eng., 2006, 12, 2131–2140 CrossRef CAS.
- A. Almond, Cell. Mol. Life Sci., 2007, 64, 1591–1596 Search PubMed.
- L. Liu, Y. Liu, J. Li, G. Du and J. Chen, Microb. Cell Fact., 2011, 10, 99 Search PubMed.
- N. Izawa, M. Serata, T. Sone, T. Omasa and H. Ohtake, J. Biosci. Bioeng., 2011, 111, 665–670 Search PubMed.
- R. Stern, Eur. J. Cell Biol., 2004, 83, 317–325 Search PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS.
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Biomaterials, 2009, 30, 2499–2506 CrossRef CAS.
- R. Jin, L. S. M. Teixeira, A. Krouwels, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Acta Biomater., 2010, 6, 1968–1977 CrossRef CAS.
- X. Jia, Y. Yeo, R. J. Clifton, T. Jiao, D. S. Kohane, J. B. Kobler, S. M. Zeitels and R. Langer, Biomacromolecules, 2006, 7, 3336–3344 CrossRef CAS.
- K. S. Masters, D. N. Shah, L. A. Leinwand and K. S. Anseth, Biomaterials, 2005, 26, 2517–2525 CrossRef.
- D. N. Shah, S. M. Recktenwall-Work and K. S. Anseth, Biomaterials, 2008, 29, 2060–2072 CrossRef CAS.
- R. A. Peattie, E. R. Rieke, E. M. Hewett, R. J. Fisher, X. Z. Shu and G. D. Prestwich, Biomaterials, 2006, 27, 1868–1875 CrossRef CAS.
- J. Patterson, R. Siew, S. W. Herring, A. S. Lin, R. Guldberg and P. S. Stayton, Biomaterials, 2010, 31, 6772–6781 CrossRef CAS.
- A. K. Jha, X. Xu, R. L. Duncan and X. Jia, Biomaterials, 2011, 32, 2466–2478 CrossRef CAS.
- R. Elia, D. R. Newhide, P. D. Pedevillano, G. R. Reiss, M. A. Firpo, E. W. Hsu, D. L. Kaplan, G. D. Prestwich and R. A. Peattie, J. Biomater. Appl., 2013, 27, 749–762 Search PubMed.
- R. A. Marklein and J. A. Burdick, Soft Matter, 2010, 6, 136 RSC.
- O. A. Lozoya, E. Wauthier, R. A. Turner, C. Barbier, G. D. Prestwich, F. Guilak, R. Superfine, S. R. Lubkin and L. M. Reid, Biomaterials, 2011, 32, 7389–7402 Search PubMed.
- X. Xu, A. K. Jha, R. L. Duncan and X. Jia, Acta Biomater., 2011, 7, 3050–3059 Search PubMed.
- Chitosan-based hydrogels: functions and applications, ed. K. Yao, J. Li, F. Yao and Y. Yin, CRC Press, 2012 Search PubMed.
- J. Zhao, in Chitosan-based hydrogels: functions and applications, ed. K. Yao, J. Li, F. Yao and Y. Yin, CRC Press, 2012 Search PubMed.
- M. Prabaharan, J. Biomater. Appl., 2008, 23, 5–36 CrossRef CAS.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS.
- S. Lü, M. Liu and B. Ni, Chem. Eng. J., 2010, 160, 779–787 CrossRef.
- A. Sukarto, C. Yu, L. E. Flynn and B. G. Amsden, Biomacromolecules, 2012, 13, 2490–2502 Search PubMed.
- K. M. Park, S. Y. Lee, Y. K. Joung, J. S. Na, M. C. Lee and K. D. Park, Acta Biomater., 2009, 5, 1956–1965 CrossRef CAS.
- N. D. Leipzig, R. G. Wylie, H. Kim and M. S. Shoichet, Biomaterials, 2011, 32, 57–64 CrossRef CAS.
- S. M. Richardson, N. Hughes, J. A. Hunt, A. J. Freemont and J. A. Hoyland, Biomaterials, 2008, 29, 85–93 CrossRef CAS.
- L. Wang and J. P. Stegemann, Biomaterials, 2010, 31, 3976–3985 CrossRef CAS.
- K. E. Crompton, J. D. Goud, R. V. Bellamkonda, T. R. Gengenbach, D. I. Finkelstein, M. K. Horne and J. S. Forsythe, Biomaterials, 2007, 28, 441–449 CrossRef CAS.
- Y. Zhou, G. Ma, S. Shi, D. Yang and J. Nie, Int. J. Biol. Macromol., 2011, 48, 408–413 Search PubMed.
- C. M. Valmikinathan, V. J. Mukhatyar, A. Jain, L. Karumbaiah, M. Dasari and R. V. Bellamkonda, Soft Matter, 2012, 8, 1964 RSC.
- C. McGann and K. Kiick, in Engineering Biomaterials for Regenerative Medicine: Novel Technologies for Clinical Applications, ed. S. K. Bhatia, Springer, 2011 Search PubMed.
- D. L. Nelson and M. M. Cox, Lehniger Principles of Biochemistry, W. H. Freeman, 4th edn, 2008 Search PubMed.
- D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- J. Turnbull, A. Powell and S. Guimond, Trends Cell Biol., 2001, 11, 75–82 CrossRef CAS.
- S. Alban, in Heparin – A Century of Progress, ed. R. Lever, B. Mulloy and C. P. Page, Springer, 2012, vol. 207 Search PubMed.
- A. Greinacher, Pathophysiol. Haemostasis Thromb., 2006, 35, 37–45 Search PubMed.
- C. Melloni, K. P. Alexander, A. Y. Chen, L. K. Newby, M. T. Roe, N. M. A. LaPointe, C. V. Pollack, Jr., W. B. Gibler, E. M. Ohman and E. D. Peterson, Am. Heart J., 2008, 156, 209–215 Search PubMed.
- T. Nie, R. E. Akins, Jr. and K. L. Kiick, Acta Biomater., 2009, 5, 865–875 CrossRef CAS.
- D. S. Benoit, S. D. Collins and K. S. Anseth, Adv. Funct. Mater., 2007, 17, 2085–2093 Search PubMed.
- M. Kim, Y. J. Kim, K. Gwon and G. Tae, Macromol. Res., 2012, 20, 271–276 Search PubMed.
- U. Freudenberg, A. Hermann, P. B. Welzel, K. Stirl, S. C. Schwarz, M. Grimmer, A. Zieris, W. Panyanuwat, S. Zschoche and D. Meinhold, Biomaterials, 2009, 30, 5049–5060 CrossRef CAS.
- M. Kim, J. Y. Lee, C. N. Jones, A. Revzin and G. Tae, Biomaterials, 2010, 31, 3596–3603 CrossRef CAS.
- S. P. Seto, M. E. Casas and J. S. Temenoff, Cell Tissue Res., 2012, 347, 589–601 Search PubMed.
- R. Wieduwild, M. Tsurkan, K. Chwalek, P. Murawala, M. Nowak, U. Freudenberg, C. Neinhuis, C. Werner and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 2919–2922 Search PubMed.
- G. Tae, Y.-J. Kim, W.-I. Choi, M. Kim, P. S. Stayton and A. S. Hoffman, Biomacromolecules, 2007, 8, 1979–1986 CrossRef CAS.
- A. D. Baldwin, K. G. Robinson, J. L. Militar, C. D. Derby, K. L. Kiick and R. E. Akins, Jr., J. Biomed. Mater. Res., Part A, 2012, 100, 2106–2118 Search PubMed.
- O. Jeon, C. Powell, L. D. Solorio, M. D. Krebs and E. Alsberg, J. Controlled Release, 2011, 154, 258–266 CrossRef CAS.
- G. Bhakta, B. Rai, Z. X. H. Lim, J. H. Hui, G. S. Stein, A. J. van Wijnen, V. Nurcombe, G. D. Prestwich and S. M. Cool, Biomaterials, 2012, 33, 6113–6122 Search PubMed.
- A. Alshamkhani and R. Duncan, J. Bioact. Compat. Polym., 1995, 10, 4–13 CAS.
- C. Gao, M. Liu, J. Chen and X. Zhang, Polym. Degrad. Stab., 2009, 94, 1405–1410 CrossRef CAS.
- T. Boontheekul, H. J. Kong and D. J. Mooney, Biomaterials, 2005, 26, 2455–2465 CrossRef CAS.
- Y. N. Dai, P. Li, J. P. Zhang, A. Q. Wang and Q. Wei, Biopharm. Drug Dispos., 2008, 29, 173–184 Search PubMed.
- E. Josef, M. Zilberman and H. Bianco-Peled, Acta Biomater., 2010, 6, 4642–4649 Search PubMed.
- J. Shi, X. Liu, X. Sun and S. Cao, Polym. Adv. Technol., 2011, 22, 1539–1546 Search PubMed.
- L. Zhao, M. D. Weir and H. H. Xu, Biomaterials, 2010, 31, 6502–6510 CrossRef CAS.
- H. Zhou and H. H. Xu, Biomaterials, 2011, 32, 7503–7513 Search PubMed.
- W. S. Kim, D. J. Mooney, P. R. Arany, K. Lee, N. Huebsch and J. Kim, Tissue Eng., Part A, 2012, 18, 737–743 Search PubMed.
- K. Murakami, H. Aoki, S. Nakamura, S. Nakamura, M. Takikawa, M. Hanzawa, S. Kishimoto, H. Hattori, Y. Tanaka, T. Kiyosawa, Y. Sato and M. Ishihara, Biomaterials, 2010, 31, 83–90 Search PubMed.
- J. O. Kim, J. K. Park, J. H. Kim, S. G. Jin, C. S. Yong, D. X. Li, J. Y. Choi, J. S. Woo, B. K. Yoo, W. S. Lyoo, J. A. Kim and H. G. Choi, Int. J. Pharm., 2008, 359, 79–86 Search PubMed.
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS.
- M. Tang, W. Chen, M. D. Weir, W. Thein-Han and H. H. Xu, Acta Biomater., 2012, 8, 3436–3445 Search PubMed.
- W. H. Tan and S. Takeuchi, Adv. Mater., 2007, 19, 2696–2701 CrossRef CAS.
- C. Yan, M. E. Mackay, K. Czymmek, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Langmuir, 2012, 28, 6076–6087 CrossRef CAS.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3, 1894–1904 Search PubMed.
- R. F. Doolittle, Annu. Rev. Biochem., 1984, 53, 195–229 CAS.
- T. A. Ahmed, E. V. Dare and M. Hincke, Tissue Eng., Part B, 2008, 14, 199–215 Search PubMed.
- D. J. Geer and S. T. Andreadis, J. Invest. Dermatol., 2003, 121, 1210–1216 CrossRef CAS.
- A. J. Man, H. E. Davis, A. Itoh, J. K. Leach and P. Bannerman, Tissue Eng., Part A, 2011, 17, 2931–2942 Search PubMed.
- S. Seetharaman, S. Natesan, R. S. Stowers, C. Mullens, D. G. Baer, L. J. Suggs and R. J. Christy, Acta Biomater., 2011, 7, 2787–2796 Search PubMed.
- H. Hall, Curr. Pharm. Des., 2007, 13, 3597–3607 CrossRef CAS.
- M. Ahearne, C. T. Buckley and D. J. Kelly, Biotechnol. Appl. Biochem., 2011, 58, 345–352 Search PubMed.
- M. E. Kidd, S. Shin and L. D. Shea, J. Controlled Release, 2012, 157, 80–85 Search PubMed.
- C. Scotti, L. Mangiavini, F. Boschetti, F. Vitari, C. Domeneghini, G. Fraschini and G. M. Peretti, Knee Surg Sports Traumatol Arthrosc, 2010, 18, 1400–1406 Search PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS.
- G. Perale, F. Rossi, E. Sundstrom, S. Bacchiega, M. Masi, G. Forloni and P. Veglianese, ACS Chem. Neurosci., 2011, 2, 336–345 Search PubMed.
- K. Numata and D. L. Kaplan, Adv. Drug Delivery Rev., 2010, 62, 1497–1508 CrossRef CAS.
- E. M. Pritchard and D. L. Kaplan, Expert Opin. Drug Delivery, 2011, 8, 797–811 Search PubMed.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- B. D. Cash and B. E. Lacy, Gastroenterol. Hepatol., 2006, 2, 736–749 Search PubMed.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442 RSC.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35, 9–16 CAS.
- C. Fruijtier-Polloth, Toxicology, 2005, 214, 1–38 CrossRef.
- P. K. Working, M. S. Newman, J. Johnson and J. B. Cornacoff, in Poly(Ethylene Glycol): Chemistry and Biological Applications, ed. J. M. Harris and S. Zalipsky, Amer Chemical Soc, Washington, 1997, vol. 680, pp. 45–57 Search PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444 Search PubMed.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, Macromolecules, 2009, 42, 5310–5316 CrossRef CAS.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Polymer, 2007, 48, 4589–4600 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- S. B. Anderson, C. C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS.
- Y. P. Hou, C. A. Schoener, K. R. Regan, D. Munoz-Pinto, M. S. Hahn and M. A. Grunlan, Biomacromolecules, 2010, 11, 648–656 CrossRef CAS.
- J. Kim, T. E. Hefferan, M. J. Yaszemski and L. Lu, Tissue Eng., Part A, 2009, 15, 2299–2307 Search PubMed.
- H. Tan, A. J. DeFail, J. P. Rubin, C. R. Chu and K. G. Marra, J. Biomed. Mater. Res., Part A, 2010, 92, 979–987 Search PubMed.
- F. P. Brandl, A. K. Seitz, J. K. Tessmar, T. Blunk and A. M. Gopferich, Biomaterials, 2010, 31, 3957–3966 Search PubMed.
- C. M. Perez, A. Panitch and J. Chmielewski, Macromol. Biosci., 2011, 11, 1426–1431 CrossRef.
- V. Chan, P. Zorlutuna, J. H. Jeong, H. Kong and R. Bashir, Lab Chip, 2010, 10, 2062–2070 RSC.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. Garcia, Adv. Mater., 2012, 24, 64–70 Search PubMed , 62.
- D. S. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS.
- H. Park, X. Guo, J. S. Temenoff, Y. Tabata, A. I. Caplan, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2009, 10, 541–546 CrossRef CAS.
- G. A. Hudalla, T. S. Eng and W. L. Murphy, Biomacromolecules, 2008, 9, 842–849 CrossRef CAS.
- C. T. Huynh, M. K. Nguyen, J. H. Kim, S. W. Kang, B. S. Kim and D. S. Lee, Soft Matter, 2011, 7, 4974 RSC.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS.
- F. M. Andreopoulos and I. Persaud, Biomaterials, 2006, 27, 2468–2476 CrossRef CAS.
- G. Papavasiliou, S. Sokic and M. Turturro, Synthetic PEG Hydrogels as Extracellular Matrix Mimics for Tissue Engineering Applications, 2012 Search PubMed.
- G. Chang, T. Ci, L. Yu and J. Ding, J. Controlled Release, 2011, 156, 21–27 CrossRef CAS.
- M. I. Baker, S. P. Walsh, Z. Schwartz and B. D. Boyan, J. Biomed. Mater. Res., Part B, 2012, 100, 1451–1457 Search PubMed.
- M. H. Alves, B. E. Jensen, A. A. Smith and A. N. Zelikin, Macromol. Biosci., 2011, 11, 1293–1313 CrossRef CAS.
- M. H. Alves, C. J. Young, K. Bozzetto, L. A. Poole-Warren and P. J. Martens, Biomed. Mater., 2012, 7, 024106 Search PubMed.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- E. Vidović, D. Klee and H. Höcker, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4536–4544 Search PubMed.
- N. Zhang, Y. Shen, X. Li, S. Cai and M. Liu, Biomed. Mater., 2012, 7, 035014 Search PubMed.
- A. Nilasaroya, L. A. Poole-Warren, J. M. Whitelock and P. Jo Martens, Biomaterials, 2008, 29, 4658–4664 CrossRef CAS.
- K. Juntanon, S. Niamlang, R. Rujiravanit and A. Sirivat, Int. J. Pharm., 2008, 356, 1–11 CrossRef CAS.
- L. Wu and C. S. Brazel, Int. J. Pharm., 2008, 349, 144–151 Search PubMed.
- V. I. Lozinsky, L. G. Damshkaln, B. L. Shaskol'skii, T. A. Babushkina, I. N. Kurochkin and I. I. Kurochkin, Colloid J., 2007, 69, 747–764 CrossRef CAS.
- S. Samal, F. Chiellini, C. Bartoli, E. Fernandes and E. Chiellini, in Hydrogels: Biological Properties and Applications, ed. R. Barbucci, Springer, Milan, 2009, pp. 67–78 Search PubMed.
- E. Otsuka and A. Suzuki, J. Appl. Polym. Sci., 2009, 114, 10–16 CrossRef CAS.
- S. Bonakdar, S. H. Emami, M. A. Shokrgozar, A. Farhadi, S. A. H. Ahmadi and A. Amanzadeh, Mater. Sci. Eng., C, 2010, 30, 636–643 Search PubMed.
- K. L. Spiller, J. L. Holloway, M. E. Gribb and A. M. Lowman, J. Tissue Eng. Regener. Med., 2011, 5, 636–647 Search PubMed.
- Z. Y. Peng and Y. Q. Shen, Polym.-Plast. Technol. Eng., 2011, 50, 245–250 Search PubMed.
- J. Michalek, R. Hobzova, M. Pradny and M. Duskova, in Biomedical Applications of Hydrogels Handbook, ed. R. M. Ottenbrite, K. Park and T. Okano, Springer, New York, 2010, pp. 303–315 Search PubMed.
- S. Atzet, S. Curtin, P. Trinh, S. Bryant and B. Ratner, Biomacromolecules, 2008, 9, 3370–3377 CrossRef CAS.
- S. J. Bryant, J. L. Cuy, K. D. Hauch and B. D. Ratner, Biomaterials, 2007, 28, 2978 CrossRef CAS.
- S. M. Paterson, A. M. A. Shadforth, D. H. Brown, P. W. Madden, T. V. Chirila and M. V. Baker, Mater. Sci. Eng., C, 2012, 32, 2536–2544 Search PubMed.
- Y. S. Casadio, D. H. Brown, T. V. Chirila, H.-B. Kraatz and M. V. Baker, Biomacromolecules, 2010, 11, 2949–2959 CrossRef CAS.
- J. T. Zhang, R. Bhat and K. D. Jandt, Acta Biomater., 2009, 5, 488–497 CrossRef.
- K. Varaprasad, S. Ravindra, N. N. Reddy, K. Vimala and K. M. Raju, J. Appl. Polym. Sci., 2010, 116, 3593–3602 Search PubMed.
- J. R. Santos, N. M. Alves and J. F. Mano, J. Bioact. Compat. Polym., 2010, 25, 169–184 Search PubMed.
- X.-Z. Zhang, X.-D. Xu, S.-X. Cheng and R.-X. Zhuo, Soft Matter, 2008, 4, 385 RSC.
- A. K. Andrianov and R. Langer, Polyphosphazenes for Biomedical Applications, John Wiley and Sons, 2009 Search PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef.
- H. R. Allcock, Soft Matter, 2012, 8, 7521–7532 RSC.
- C. Chun, S. M. Lee, C. W. Kim, K.-Y. Hong, S. Y. Kim, H. K. Yang and S.-C. Song, Biomaterials, 2009, 30, 4752–4762 CrossRef CAS.
- T. Potta, C. Chun and S.-C. Song, Biomaterials, 2010, 31, 8107–8120 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, NJ, 2004 Search PubMed.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS.
- A. D. Rouillard, C. M. Berglund, J. Y. Lee, W. J. Polacheck, Y. Tsui, L. J. Bonassar and B. J. Kirby, Tissue Eng., Part C, 2011, 17, 173–179 Search PubMed.
- J. A. Burdick, A. J. Peterson and K. S. Anseth, Biomaterials, 2001, 22, 1779–1786 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- M. D. Brigham, A. Bick, E. Lo, A. Bendali, J. A. Burdick and A. Khademhosseini, Tissue Eng., Part A, 2008, 15, 1645–1653 Search PubMed.
- S. Khetan, J. S. Katz and J. A. Burdick, Soft Matter, 2009, 5, 1601–1606 RSC.
- S. Ekici, P. Ilgin, S. Butun and N. Sahiner, Carbohydr. Polym., 2011, 84, 1306–1313 Search PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- X. Dai, X. Chen, L. Yang, S. Foster, A. J. Coury and T. H. Jozefiak, Acta Biomater., 2011, 7, 1965–1972 Search PubMed.
- A. Morelli and F. Chiellini, Macromol. Chem. Phys., 2010, 211, 821–832 CrossRef CAS.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 Search PubMed.
- K. Matyjaszewski, K. L. Beers, A. Kern and S. G. Gaynor, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 823–830 CrossRef CAS.
- J. A. Yoon, S. A. Bencherif, B. Aksak, E. K. Kim, T. Kowalewski, J. K. Oh and K. Matyjaszewski, Chem.–Asian J., 2011, 6, 128–136 Search PubMed.
- J. A. Yoon, J. K. Oh, W. Li, T. Kowalewski and K. Matyjaszewski, in Hydrogel Micro and Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 169–186 Search PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- C. A. Deforest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS.
- S. Q. Liu, P. L. Ee, C. Y. Ke, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2009, 30, 1453–1461 CrossRef CAS.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS.
- J. F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774 RSC.
- C. M. Nimmo and M. S. Shoichet, Bioconjugate Chem., 2011, 22, 2199–2209 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia and R. Lamanna, Biomacromolecules, 2007, 8, 1844–1850 CrossRef CAS.
- G. Testa, C. Di Meo, S. Nardecchia, D. Capitani, L. Mannina, R. Lamanna, A. Barbetta and M. Dentini, Int. J. Pharm., 2009, 378, 86–92 CrossRef CAS.
- S. Piluso, B. Hiebl, S. N. Gorb, A. Kovalev, A. Lendlein and A. T. Neffe, Int. J. Artif. Organs, 2011, 34, 192–197 Search PubMed.
- M. van Dijk, C. F. van Nostrum, W. E. Hennink, D. T. S. Rijkers and R. M. J. Liskamp, Biomacromolecules, 2010, 11, 1608–1614 CrossRef CAS.
- M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231–1232 RSC.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. Xu, T. M. Filion, F. Prifti and J. Song, Chem.–Asian J., 2011, 6, 2730–2737 Search PubMed.
- J. Zheng, L. A. S. Callahan, J. Hao, K. Guo, C. Wesdemiotis, R. A. Weiss and M. L. Becker, ACS Macro Lett., 2012, 1, 1071–1073 Search PubMed.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- H.-L. Wei, Z. Yang, H.-J. Chu, J. Zhu, Z.-C. Li and J.-S. Cui, Polymer, 2010, 51, 1694–1702 CrossRef CAS.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS.
- H. Tan, J. P. Rubin and K. G. Marra, Macromol. Rapid Commun., 2011, 32, 905–911 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297 CrossRef CAS.
- R. Pipkorn, W. Waldeck, B. Didinger, M. Koch, G. Mueller, M. Wiessler and K. Braun, J. Pept. Sci., 2009, 15, 235–241 CrossRef CAS.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 Search PubMed.
- S. B. Anderson, C. C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS.
- Y. Lei and T. Segura, Biomaterials, 2009, 30, 254–265 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133–143 RSC.
- C. L. McGann, E. A. Levenson and K. L. Kiick, Macromol. Chem. Phys., 2013, 214, 203–213 Search PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 Search PubMed.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790–806 CAS.
- R. M. Broyer, E. Schopf, C. M. Kolodziej, Y. Chen and H. D. Maynard, Soft Matter, 2011, 7, 9972–9977 RSC.
- L. Zhao, L. Zhu, F. Liu, C. Liu, D. Shan, Q. Wang, C. Zhang, J. Li, J. Liu, X. Qu and Z. Yang, Int. J. Pharm., 2011, 410, 83–91 CrossRef CAS.
- M. M. Abdel-Mottaleb, N. D. Mortada, A. A. El-Shamy and G. A. Awad, Drug Dev. Ind. Pharm., 2009, 35, 311–320 Search PubMed.
- H. Hosseinkhani, M. Hosseinkhani, A. Khademhosseini and H. Kobayashi, J. Controlled Release, 2007, 117, 380–386 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS.
- D.-Y. Ji, T.-F. Kuo, H.-D. Wu, J.-C. Yang and S.-Y. Lee, Carbohydr. Polym., 2012, 89, 1123–1130 Search PubMed.
- Y. H. Lin, H. F. Liang, C. K. Chung, M. C. Chen and H. W. Sung, Biomaterials, 2005, 26, 2105–2113 CrossRef CAS.
- M. Matyash, F. Despang, R. Mandal, D. Fiore, M. Gelinsky and C. Ikonomidou, Tissue Eng., Part A, 2012, 18, 55–66 Search PubMed.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2012, 18, 806–815 Search PubMed.
- M. J. Nugent and C. L. Higginbotham, Eur. J. Pharm. Biopharm., 2007, 67, 377–386 CrossRef CAS.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS.
- Z. Li and T. J. Deming, Soft Matter, 2010, 6, 2546 RSC.
- M. J. Glassman, J. Chan and B. D. Olsen, Adv. Funct. Mater., 2012, 23, 1182–1193 Search PubMed.
- S. Koutsopoulos, L. D. Unsworth, Y. Nagai and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef CAS.
- Y. Nagai, L. D. Unsworth, S. Koutsopoulos and S. Zhang, J. Controlled Release, 2006, 115, 18–25 CrossRef CAS.
- C. T. S. W. P. Foo, J. S. Lee, W. Mulyasasmita, A. Parisi-Amon and S. C. Heilshorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22067–22072 CrossRef CAS.
- Y. Liu, B. Liu, J. J. Riesberg and W. Shen, Macromol. Biosci., 2011, 11, 1325–1330 CrossRef CAS.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- N. Yamaguchi, L. Zhang, B. S. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040–3041 CrossRef CAS.
- S. H. Kim and K. L. Kiick, Macromol. Rapid Commun., 2010, 31, 1231–1240 CrossRef.
- D. S. Benoit, A. R. Durney and K. S. Anseth, Tissue Eng., 2006, 12, 1663–1673 CrossRef CAS.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. Xu and J. Ding, Biomaterials, 2011, 32, 4725–4736 CrossRef CAS.
- C. R. Nuttelman, A. M. Kloxin and K. S. Anseth, in Tissue Eng, ed. J. P. Fisher, Springer-Verlag Berlin, Berlin, 2006, vol. 585, pp. 135–149 Search PubMed.
- S. Sahoo, C. Chung, S. Khetan and J. A. Burdick, Biomacromolecules, 2008, 9, 1088–1092 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 Search PubMed.
- Y. S. Jo, J. Gantz, J. A. Hubbell and M. P. Lutolf, Soft Matter, 2008, 5, 440–446 Search PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- P. J. Martens, S. J. Bryant and K. S. Anseth, Biomacromolecules, 2003, 4, 283–292 CrossRef CAS.
- N. C. Hunt, A. M. Smith, U. Gbureck, R. M. Shelton and L. M. Grover, Acta Biomater., 2010, 6, 3649–3656 Search PubMed.
- K. J. Lampe, K. B. Bjugstad and M. J. Mahoney, Tissue Eng., Part A, 2010, 16, 1857–1866 Search PubMed.
- F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS.
- S. J. Buwalda, L. Calucci, C. Forte, P. J. Dijkstra and J. Feijen, Polymer, 2012, 53, 2809–2817 Search PubMed.
- R. S. Ashton, A. Banerjee, S. Punyani, D. V. Schaffer and R. S. Kane, Biomaterials, 2007, 28, 5518–5525 CrossRef CAS.
- J. X. Tan, X. Y. Wang, H. Y. Li, X. L. Su, L. Wang, L. Ran, K. Zheng and G. S. Ren, Int. J. Cancer, 2011, 128, 1303–1315 CrossRef CAS.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS.
- M. C. Giano, D. J. Pochan and J. P. Schneider, Biomaterials, 2011, 32, 6471–6477 CrossRef CAS.
- L. Li, Z. Tong, X. Jia and K. L. Kiick, Soft Matter, 2013, 9, 665–673 RSC.
- D. Seliktar, A. H. Zisch, M. P. Lutolf, J. L. Wrana and J. A. Hubbell, J. Biomed. Mater. Res., Part A, 2004, 68, 704–716 CAS.
- V. W. Wong, K. C. Rustad, M. G. Galvez, E. Neofytou, J. P. Glotzbach, M. Januszyk, M. R. Major, M. Sorkin, M. T. Longaker and J. Rajadas, Tissue Eng., Part A, 2010, 17, 631–644 Search PubMed.
- C. Yang, L. Xu, Y. Zhou, X. M. Zhang, X. Huang, M. Wang, Y. Han, M. L. Zhai, S. C. Wei and J. Q. Li, Carbohydr. Polym., 2010, 82, 1297–1305 Search PubMed.
- J. Patterson, R. Siew, S. W. Herring, A. S. Lin, R. Guldberg and P. S. Stayton, Biomaterials, 2010, 31, 6772–6781 CrossRef CAS.
- J. Kim, I. S. Kim, T. H. Cho, K. B. Lee, S. J. Hwang, G. Tae, I. Noh, S. H. Lee, Y. Park and K. Sun, Biomaterials, 2007, 28, 1830–1837 CrossRef CAS.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 Search PubMed.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS.
- D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg, Jr., V. C. Mody, Jr., R. L. Reed and L. A. Brown, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS.
- J. M. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 Search PubMed.
- R. Franco, O. J. Schoneveld, A. Pappa and M. I. Panayiotidis, Arch. Physiol. Biochem., 2007, 113, 234–258 CrossRef CAS.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- H. L. Wei, J. Yang, H. J. Chu, Z. Yang, C. C. Ma and K. Yao, J. Appl. Polym. Sci., 2011, 120, 974–980 CrossRef CAS.
- K. C. Koehler, D. L. Alge, K. S. Anseth and C. N. Bowman, Biomaterials, 2013, 34, 4150–4158 Search PubMed.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 Search PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 Search PubMed.
- A. Skardal, S. F. Sarker, A. Crabbe, C. A. Nickerson and G. D. Prestwich, Biomaterials, 2010, 31, 8426–8435 Search PubMed.
- N. Fomina, C. L. McFearin, M. Sermsakdi, J. M. Morachis and A. Almutairi, Macromolecules, 2011, 44, 8590–8597 CrossRef CAS.
- C. K. Choi, M. T. Breckenridge and C. S. Chen, Trends Cell Biol., 2010, 20, 705–714 CrossRef CAS.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS.
- A. L. Berrier and K. M. Yamada, J. Cell Physiol., 2007, 213, 565–573 CrossRef CAS.
- L. H. Romer, K. G. Birukov and J. G. Garcia, Circ. Res., 2006, 98, 606–616 CrossRef CAS.
- H. Studenovska, P. Vodicka, V. Proks, J. Hlucilova, J. Motlik and F. Rypacek, J. Tissue Eng. Regener. Med., 2010, 4, 454–463 Search PubMed.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- I. Jun, K. M. Park, D. Y. Lee, K. D. Park and H. Shin, Macromol. Res., 2011, 19, 911–920 Search PubMed.
- M. S. Weiss, B. P. Bernabe, A. Shikanov, D. A. Bluver, M. D. Mui, S. Shin, L. J. Broadbelt and L. D. Shea, Biomaterials, 2012, 33, 3548–3559 Search PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS.
- R. Liddington and M. Ginsberg, J. Cell Biol., 2002, 158, 833–839 Search PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS.
- J. W. Lee, Y. J. Park, S. J. Lee, S. K. Lee and K. Y. Lee, Biomaterials, 2010, 31, 5545–5551 CrossRef.
- S. P. Zustiak, R. Durbal and J. B. Leach, Acta Biomater., 2010, 6, 3404–3414 CrossRef CAS.
- R. Ayala, C. Zhang, D. Yang, Y. Hwang, A. Aung, S. S. Shroff, F. T. Arce, R. Lal, G. Arya and S. Varghese, Biomaterials, 2011, 32, 3700–3711 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- A. J. Ridley, M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons and A. R. Horwitz, Science, 2003, 302, 1704–1709 CrossRef CAS.
- M. P. Schwartz, B. D. Fairbanks, R. E. Rogers, R. Rangarajan, M. H. Zaman and K. S. Anseth, Integr. Biol., 2010, 2, 32–40 RSC.
- D. Guarnieri, A. De Capua, M. Ventre, A. Borzacchiello, C. Pedone, D. Marasco, M. Ruvo and P. A. Netti, Acta Biomater., 2010, 6, 2532–2539 CrossRef CAS.
- P. Tayalia and D. J. Mooney, Adv. Mater., 2009, 21, 3269–3285 CrossRef CAS.
- A. Zieris, K. Chwalek, S. Prokoph, K. R. Levental, P. B. Welzel, U. Freudenberg and C. Werner, J. Controlled Release, 2011, 156, 28–36 Search PubMed.
- C. C. Lin and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6380–6385 CrossRef CAS.
- I. Konstantinova, G. Nikolova, M. Ohara-Imaizumi, P. Meda, T. Kucera, K. Zarbalis, W. Wurst, S. Nagamatsu and E. Lammert, Cell, 2007, 129, 359–370 Search PubMed.
- R. G. Wylie, S. Ahsan, Y. Aizawa, K. L. Maxwell, C. M. Morshead and M. S. Shoichet, Nat. Mater., 2011, 10, 799–806 CrossRef CAS.
- S. E. Kendall, J. Najbauer, H. F. Johnston, M. Z. Metz, S. Li, M. Bowers, E. Garcia, S. U. Kim, M. E. Barish and K. S. Aboody, Stem Cells, 2008, 26, 1575–1586 Search PubMed.
- D. P. Cross and C. Wang, Pharm. Res., 2011, 28, 2477–2489 Search PubMed.
- X. Zhao, S. Jain, H. B. Larman, S. Gonzalez and D. J. Irvine, Biomaterials, 2005, 26, 5048–5063 CrossRef CAS.
- X. Li, X. Liu, W. Zhao, X. Wen and N. Zhang, Acta Biomater., 2012, 8, 2087–2095 CrossRef CAS.
- J. C. Garbern, E. Minami, P. S. Stayton and C. E. Murry, Biomaterials, 2011, 32, 2407–2416 CrossRef CAS.
- T. Diab, E. M. Pritchard, B. A. Uhrig, J. D. Boerckel, D. L. Kaplan and R. E. Guldberg, J. Mech. Behav. Biomed. Mater., 2011, 11, 123–131 Search PubMed.
- M. W. Tibbitt, B. W. Han, A. M. Kloxin and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2012, 100, 1647–1654 Search PubMed.
- S. A. Oh, H. Y. Lee, J. H. Lee, T. H. Kim, J. H. Jang, H. W. Kim and I. Wall, Tissue Eng., Part A, 2012, 18, 1087–1100 Search PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- L. Wei, C. Cai, J. Lin and T. Chen, Biomaterials, 2009, 30, 2606–2613 CrossRef CAS.
- F. Guilak, D. M. Cohen, B. T. Estes, J. M. Gimble, W. Liedtke and C. S. Chen, Cell Stem Cell, 2009, 5, 17–26 CrossRef CAS.
- A. J. Engler, M. A. Griffin, S. Sen, C. G. Bonnemann, H. L. Sweeney and D. E. Discher, J. Cell Biol., 2004, 166, 877–887 CrossRef CAS.
- F. Rehfeldt, A. E. X. Brown, M. Raab, S. S. Cai, A. L. Zajac, A. Zemel and D. E. Discher, Integr. Biol., 2012, 4, 422–430 RSC.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. C. Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642–649 Search PubMed.
- O. Chaudhuri and D. J. Mooney, Nat. Mater., 2012, 11, 568–569 Search PubMed.
- H. N. Chia, M. Vigen and A. M. Kasko, Acta Biomater., 2012, 8, 2602–2611 Search PubMed.
- K. G. Robinson, T. Nie, A. D. Baldwin, E. C. Yang, K. L. Kiick and R. E. Akins, Jr., J. Biomed. Mater. Res., Part A, 2012, 100, 1356–1367 CrossRef.
- B. C. Isenberg, P. A. Dimilla, M. Walker, S. Kim and J. Y. Wong, Biophys. J., 2009, 97, 1313–1322 CrossRef CAS.
- O. V. Sazonova, K. L. Lee, B. C. Isenberg, C. B. Rich, M. A. Nugent and J. Y. Wong, Biophys. J., 2011, 101, 622–630 Search PubMed.
- A. A. Chen, S. R. Khetani, S. Lee, S. N. Bhatia and K. J. Van Vliet, Biomaterials, 2009, 30, 1113–1120 CrossRef CAS.
- J. A. Wood, N. M. Shah, C. T. McKee, M. L. Hughbanks, S. J. Liliensiek, P. Russell and C. J. Murphy, Biomaterials, 2011, 32, 5056–5064 Search PubMed.
- M. J. Poellmann, P. A. Harrell, W. P. King and A. J. W. Johnson, Acta Biomater., 2010, 6, 3514–3523 CrossRef CAS.
- M. Guvendiren and J. A. Burdick, Biomaterials, 2010, 31, 6511–6518 CrossRef CAS.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872–4877 CrossRef.
- J. L. Young and A. J. Engler, Biomaterials, 2011, 32, 1002–1009 CrossRef CAS.
- H. Wang, S. M. Haeger, A. M. Kloxin, L. A. Leinwand and K. S. Anseth, PLoS One, 2012, 7, e39969 Search PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS.
- M. T. Frey and Y. L. Wang, Soft Matter, 2009, 5, 1918–1924 RSC.
- S. Raghavan, R. A. Desai, Y. Kwon, M. Mrksich and C. S. Chen, Langmuir, 2010, 26, 17733 CrossRef CAS.
- M. Guvendiren and J. A. Burdick, Adv. Healthcare Mater., 2013, 2, 155–164 Search PubMed.
- S. S. Rao, S. Bentil, J. DeJesus, J. Larison, A. Hissong, R. Dupaix, A. Sarkar and J. O. Winter, PLoS One, 2012, 7, e35852 Search PubMed.
- J. S. Choi and B. A. Harley, Biomaterials, 2012, 33, 4460–4468 Search PubMed.
- K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf and S. C. Rizzi, Biomaterials, 2010, 31, 8454–8464 CrossRef.
- G. D. Nicodemus, S. C. Skaalure and S. J. Bryant, Acta Biomater., 2011, 7, 492–504 Search PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS.
- S. A. Ruiz and C. S. Chen, Stem Cells, 2008, 26, 2921–2927 Search PubMed.
- S. Khetan and J. A. Burdick, Biomaterials, 2010, 31, 8228–8234 CrossRef CAS.
- M. Ehrbar, A. Sala, P. Lienemann, A. Ranga, K. Mosiewicz, A. Bittermann, S. C. Rizzi, F. E. Weber and M. P. Lutolf, Biophys. J., 2011, 100, 284–293 CrossRef CAS.
- Q. Guo, X. Wang, M. W. Tibbitt, K. S. Anseth, D. J. Montell and J. H. Elisseeff, Biomaterials, 2012, 33, 8040–8046 Search PubMed.
- N. Gjorevski and C. M. Nelson, Integr. Biol., 2010, 2, 424–434 RSC.
- C. M. Nelson, M. M. Vanduijn, J. L. Inman, D. A. Fletcher and M. J. Bissell, Science, 2006, 314, 298–300 CrossRef CAS.
- A. M. Kloxin, K. J. Lewis, C. A. Deforest, G. Seedorf, M. W. Tibbitt, V. Balasubramaniam and K. S. Anseth, Integr. Biol., 2012, 4, 1540–1549 RSC.
- S. H. Lee, J. J. Moon, J. S. Miller and J. L. West, Biomaterials, 2007, 28, 3163–3170 CrossRef CAS.
- H. J. Kong, T. R. Polte, E. Alsberg and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4300–4305 CrossRef CAS.
- N. D. Huebsch and D. J. Mooney, Biomaterials, 2007, 28, 2424–2437 CrossRef CAS.
- A. T. Metters, C. N. Bowman and K. S. Anseth, J. Phys. Chem. B, 2000, 104, 7043–7049 CrossRef CAS.
- M. A. Rice, J. Sanchez-Adams and K. S. Anseth, Biomacromolecules, 2006, 7, 1968–1975 CrossRef CAS.
- K. M. Schultz and E. M. Furst, Soft Matter, 2012, 8, 6198–6205 RSC.
- D. C. Appleyard, S. C. Chapin, R. L. Srinivas and P. S. Doyle, Nat. Protoc., 2011, 6, 1761–1774 Search PubMed.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, ACS Macro Lett., 2012, 1, 706–708 Search PubMed.
- C. Franck, S. A. Maskarinec, D. A. Tirrell and G. Ravichandran, PLoS One, 2011, 6, e17833 Search PubMed.
- N. Gjorevski and C. M. Nelson, Biophys. J., 2012, 103, 152–162 Search PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS.
- S. Alimperti, P. Lei, J. Tian and S. T. Andreadis, Gene Ther., 2012, 19, 1123–1132 Search PubMed.
- M. W. Tibbitt, A. M. Kloxin, K. U. Dyamenahalli and K. S. Anseth, Soft Matter, 2010, 6, 5100 RSC.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |