 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilisation of hydroxynitrile lyases
Ulf
Hanefeld
*
Gebouw voor Scheikunde, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl; Fax: +31 15 278 1415; Tel: +31 15 278 9304
First published on 29th January 2013
Abstract
Hydroxynitrile lyases are a versatile group of enzymes that are applied both in the laboratory and on an industrial scale. What makes them particularly interesting is that to date five structurally unrelated categories of hydroxynitrile lyases have been discovered. Given their great importance they have often been immobilised utilising many different methodologies. Therefore the hydroxynitrile lyases are ideally suited to compare different immobilisation methods and their dependence on the structural features of the enzyme in question, since the activity is the same in all cases. This review examines all the different immobilisation methods applied to hydroxynitrile lyases and draws conclusions on the effect of the approach.
 Ulf Hanefeld | Ulf Hanefeld was born in 1966 in Köln, Germany, and grew up in then West Berlin and London. In 1993 he received his PhD from the Georg-August-Universität zu Göttingen, having performed the research both in Göttingen (Prof. H. Laatsch) and Seattle (Prof. H. G. Floss). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes, enzyme immobilisation and heterogeneous catalysis in organic synthesis. |
1. Introduction
Enzymes as versatile tools for organic chemistry have already been discovered a long time ago. Indeed, the first enantioselective synthesis ever described is the application of emulsin for the preparation of enantiopure mandelic acid. In 1908 Rosenthaler utilised this crude hydroxynitrile lyase (HNL; also known as oxynitrilase) from almonds (Prunus amygdalus) for the stereospecific addition of cyanide to benzaldehyde.1 In reversing the natural direction of the reaction the first enantioselective preparation of a stereocentre was a fact. The conversion of the unstable mandelonitrile into the stable corresponding acid then enabled the determination of the absolute stereochemistry. This milestone of organic chemistry was immediately followed by a search for more HNLs by Rosenthaler and others.2,3 Krieble shortly afterwards reconfirmed the results and the first enantioselective catalyst was thus introduced into chemistry (Scheme 1).4,5 | ||
| Scheme 1 First enantioselective synthesis: PaHNL catalysed addition of cyanide to benzaldehyde and subsequent acid catalysed hydrolysis to (R)-mandelic acid. | ||
With their application the quest for immobilisation of enzymes also immediately arose. The targets were twofold: separation and improvement. By enabling straightforward separation of the enzyme, a pure product is readily obtained and recycling of the enzyme becomes possible. By immobilisation the stability of enzymes is often improved, but remarkably also the activity and selectivity can be positively influenced.6–14
1.1. Hydroxynitrile lyases (HNLs)
After the initial rush hydroxynitrile lyases were neglected until their rediscovery by Pfeil 50 years ago.15–18 The enzyme from almonds Prunus amygdalus PaHNL was purified, crystallised and characterised. It was shown to contain a flavine co-factor and to be highly enantioselective in the synthesis of (R)-cyanohydrins. This led to the application of this enzyme by the groups of Kyler,19 Brussee20 and Effenberger21 and to the search for HNLs with different enantioselectivities. From 1990 onwards many new enzymes were identified and their structures elucidated.22–25 This revealed a fascinating aspect of these enzymes.The Prunus HNLs are evolved from flavine dependent dehydrogenase/oxidase structures and are in general (R)-selective and glycosylated.26,27 From Sorghum bicolor SbHNL was isolated and shown to be (S)-selective;28,29 structurally it is related to serine carboxypeptidase with an α/β-hydrolase fold. Similarly several other HNLs, namely Hevea brasiliensis HbHNL,27Manihot esculenta MeHNL,30Arabidopsis thaliana AtHNL,31,32Xylella fastidiosa XfHNL33 and Baliospermum montanum BmHNL,34 have an α/β-hydrolase fold; they are related to the esterase/lipase superfamily. Interestingly, of these 5 structurally closely related HNL's some are (R)- and others (S)-selective. The generally (R)-selective Linum usitatissimum LuHNL35 is structurally closely related to Zn-dependent alcohol dehydrogenases and is equally Zn-dependent. The very recently described cupin superfamily based HNLs PsmHNL and BpHNL are the newest members of the hydroxynitrile lyases.36 Structurally these barrel shaped enzymes are very different from all other HNLs but their catalytic activity is the same.
Clearly HNLs are a prime example of convergent evolution (Fig. 1) with five different ancestors and four different protein scaffolds for one catalytic reaction. All these enzymes are stable at around pH 7 and catalyse the enantioselective synthesis of cyanohydrins.26,27 The plant derived Prunus, Sorghum, Hevea, Manihot and Linum HNLs in nature are all part of a defence mechanism, catalysing the release of toxic HCN. Interestingly, most of these plants form part of the human diet.37–39 For the AtHNL, XfHNL, BmHNL, PsmHNL and BpHNL no natural substrates are known26 and the cupin based HNLs are much better catalysts for the synthesis than the breakdown of cyanohydrins.36 For six of these structurally diverse HNLs X-ray structures have been reported (Fig. 1) and many of them are commercially available.
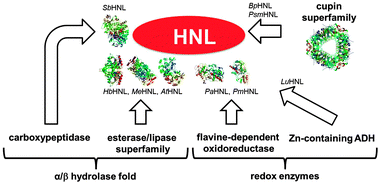 | ||
| Fig. 1 Convergent evolution of hydroxynitrile lyases (HNL). The X-ray structures have the Protein Data Bank ID: SbHNL: 1gxs, HbHNL: 3c6x, MeHNL: 1eb9, AtHNL: 3dqz, PaHNL: 3gdn, PmHNL: 3red. The cupin depicted is a decarboxylase with ID: 2uy9. | ||
1.2. Cyanohydrin chemistry
When the initial experiments with HNLs were reported, it was noted that the reproducibility of the results was a significant problem. The optical rotation values changed over the course of the reaction and in 1921 it was demonstrated that the results were highly pH dependent.5 The chemical, racemic reaction is base catalysed and also proceeds at pH 7. Only at lower pH values, preferably below 5, it is suppressed (Scheme 2). At the same time most enzymes do not function optimally at this low pH, some are even inactive. It is therefore essential to find reaction conditions under which the HNLs are stable and active while the base catalysed racemic reaction is suppressed.26,40–42 Several different approaches have been developed: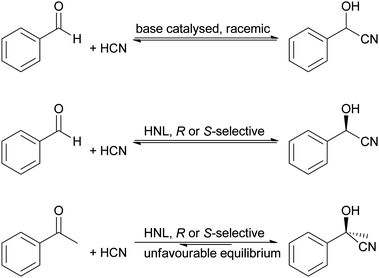 | ||
| Scheme 2 The base catalysed cyanohydrin synthesis and the HNL catalysed cyanohydrin synthesis compete with each other. Reaction conditions that suppress the base catalysed reaction are essential for the application of HNLs. Moreover, the reaction equilibrium is not always in favour of the cyanohydrin formation. | ||
1. Work in an aqueous phase with a low pH to guarantee a large difference in rate between the HNL and the chemical reaction. Immobilisation often stabilises the HNLs under these conditions. Equally lower temperatures have this effect, but less pronounced.
2. Work in a biphasic system with a low pH in the aqueous phase. Since the substrate and product concentrations are now lower in the aqueous phase, the chemical reaction is slowed down. In the organic layer the chemical reaction virtually does not proceed.
3. In a further development of method 2 it is possible to work in emulsions to overcome diffusion limitations of a biphasic system.
4. Since the chemical reaction virtually does not proceed in an organic phase, it is possible to work in organic solvents. For this purpose the HNL has to be immobilised.
Immobilisation is highly important for at least two of the approaches that ensure a high enantiopurity of the desired cyanohydrin. From the very beginning of HNL catalysed reactions, this has therefore been carefully investigated.
Next to the pH value, a key point is the equilibrium of the reaction. In particular for ketone derived cyanohydrins a large excess of cyanide, four to five fold, has to be applied (Scheme 2).26,40–42In situ derivatisation of the cyanohydrin would enable shifting of the equilibrium. This, however, can only be achieved under water-free reaction conditions. Immobilisation might enable this in future, by helping to create HNLs that are active in dry organic solvents.
1.3. Immobilisation
Immobilisation has, during the past decades, been the key to success for several industrial, enzyme catalysed processes. Many different approaches have been developed, consequently there is a large choice of methods available. These have been classified in many different ways, but in general one can distinguish between three approaches, which can then again be subdivided. Other ways of classification have been used and are equally valid, however, for ease of discussion for the HNL case this grouping is chosen (Fig. 2).6–14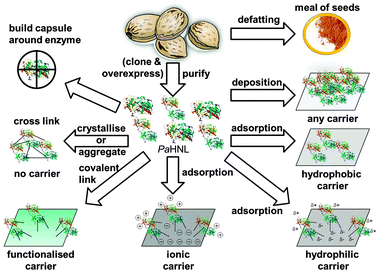 | ||
| Fig. 2 Enzymes can be immobilised in many different ways, by deposition, adsorption, covalent binding (with and without a carrier) and by encapsulation. To illustrate all these methods PaHNL is depicted here. This enzyme has not been immobilised by all these methods. | ||
1. Non-covalent immobilisation on a carrier: this can be subdivided into adsorption via hydrophobic interactions, via hydrogen bonds and via ionic interactions and also includes immobilisation by deposition of the enzyme on the carrier material.
2. Covalent immobilisation: this is based on a chemical reaction of functional groups of the enzyme with a linker/tether that binds the enzyme to a carrier or, in the case of carrier-free immobilisation, binds the enzymes together. This second case yields essentially pure enzyme as a cross-linked insoluble solid.
3. Encapsulation or entrapment: here the enzyme itself remains essentially unaltered, it is however physically locked up in a porous system. This encapsulation can range from really encapsulating single enzymes per capsule or container to an entrapment by a membrane. It can even be argued that in a biphasic system the enzyme is entrapped in the water layer, and thus immobilised, however, here only entrapment and encapsulation in a carrier are considered as immobilisation.
Given the wide range of applications of the HNLs essentially all different methods of immobilisation have been applied. Since many of these methods have also been applied to different HNLs including those from different superfamilies, the HNLs are an ideal test-case for comparing these methods.
In addition to all the mentioned immobilisation methods the simplest way of applying HNLs as immobilised catalysts is by using the crude plant preparation, possibly after defatting or by employing whole cells. These systems and the purified dissolved enzymes form the reference point with which all immobilisation efforts should be compared.
2. Crude enzyme systems
The first experiments with PaHNL were performed with emulsin, a soluble, crude enzyme preparation first described in 1837 by Wöhler and Liebig as the active principle that releases HCN from amygdalin (glycosylated mandelonitrile).43 In 1991 Brussee et al. replaced this by ground, defatted almond meal, a solid.44 In this almond meal the enzyme is essentially immobilised in its natural cellular environment and it proved its value in the synthesis of enantiopure, aromatic cyanohydrins. In 1992 the almond meal approach was followed by Huuhtanen and Kanerva, applying it successfully also for the preparation of a wide range of aliphatic cyanohydrins (Scheme 3).45 In the second case acetone cyanohydrin was used as a cyanide source, i.e. the PaHNL first releases cyanide from the ketone based cyanohydrin and then utilises it in the synthesis reaction. This transcyanation19 is possible due to the differences in the equilibrium constants of ketone and aldehyde based cyanohydrins, mentioned above (Section 1.2 and Scheme 2). Since then many variations of the almond meal approach – also from other plant kernels – have been described. The beauty of this system lies in its simplicity. The plant material is ground and defatted with an organic solvent and then is ready for use. In particular for the screening of plant materials for HNL activity this has proven its value.24,46,47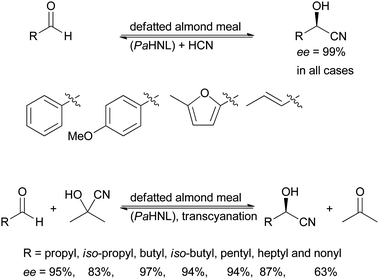 | ||
| Scheme 3 Defatted almond meal is a powerful tool for cyanohydrin synthesis, both with HCN and acetone cyanohydrin as a cyanide source. | ||
Most of the described reactions were performed in biphasic reaction systems. A modified approach was introduced by reducing the aqueous phase significantly. In the “micro-aqueous” approach the water layer is essentially the water attached to the defatted almond, loquat or peach meal. The real problem here is, however, that while data on the water content of the organic solvent (diisopropyl ether) are given, the water content of the kernel meal is unknown. A true control of the water content is thus impossible; it seems to be close to saturation.48–50
The application of almond meal in diisopropyl ether (DIPE) was carefully investigated for two industrially relevant intermediates: o-chlorobenzaldehyde (for clopidogrel synthesis)51 and hydroxy-pivalaldehyde (for pantolactone synthesis).52 These substrates are known to be difficult for PaHNL (Scheme 4). Modest to good results were obtained for o-chlorobenzaldehyde, for hydroxy-pivalaldehyde the enantioselectivity was disappointing. No recycling experiments were performed, it thus remained unclear whether one of the targets of immobilisation – separation and improvement of the enzyme – was achieved. In tert-butyl methyl ether (TBME) and toluene the almond meal methodology was successfully employed as part of complex natural product syntheses, vittatalactone44,53,54 and sugar cyanohydrin esters.55 In none of these cases any recycling studies were performed.
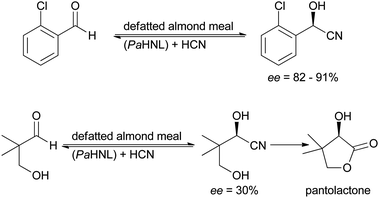 | ||
| Scheme 4 Application of almond meal (PaHNL) for the synthesis of industrially relevant intermediates. | ||
Very interestingly a variation of this approach has recently been described by Pohl for the acid sensitive AtHNL. The isolated enzyme is inactive at pH values below 5 and therefore it is difficult to suppress the racemic chemical reaction when applying it in aqueous media (see above Section 1.2.). AtHNL was overexpressed in E. coli and the whole cells were used rather than a plant meal. When applying them in a buffer saturated solvent (“micro aqueous” conditions) the chemical reaction was suppressed and excellent results were obtained. Remarkably a significant difference between wet cells and lyophilised cells was observed, the dry cells giving higher enantioselectivity (Scheme 5). In this case recycling studies were performed and the enzyme preparation showed good stability over three cycles, both for activity and enantioselectivity.56
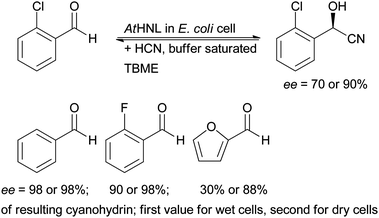 | ||
| Scheme 5 Whole cells of E. coli expressing AtHNL employed under “micro aqueous” conditions. The first value for the ee is derived from the reaction with wet cells, the second from the reaction with lyophilised cells. | ||
A cascade of two reactions was realised in a whole cell system by cloning both MeHNL and arylacetonitrilase from Pseudomonas fluorescens into one expression system. Initially Pichia pastoris was used, then E. coli whole cells were shown to give the better whole cell catalysts. These whole cell duo catalysts proved to be excellent for the conversion of benzaldehyde into (S)-mandelic acid in a single process step. Reactions were performed in aqueous systems and in combination with ionic liquids. Enantioselectivities of greater than 94% were obtained (Scheme 6).57–59
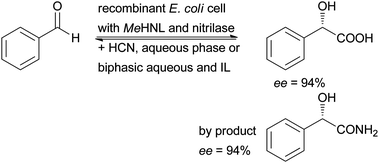 | ||
| Scheme 6 Recombinant E. coli whole cells containing MeHNL and aryl acetonitrilase from Pseudomonas fluorescens efficiently catalyse the formation of (S)-mandelic acid and (S)-mandelic acid amide as by product. | ||
3. Non-covalent immobilisation
HNLs are soluble enzymes that are not membrane bound; additionally the Prunus HNLs tend to be glycosylated. Consequently they all have polar and also charged groups on their surfaces and cannot be immobilised via hydrophobic interactions like lipases (Fig. 2). All other non-covalent immobilisation methods work, as will be discussed here.The first example for the successful immobilisation was described in 1965 by Pfeil, utilising ECTEOLA cellulose, a cellulose treated with epichlorohydrin and subsequently with triethylamine. The carrier was first charged with chloride ions and then pure, salt-free PaHNL was absorbed. The enzyme is thus immobilised via ionic interactions. Using salt-free 50% aqueous MeOH as the reaction medium, any desorption of the enzyme is avoided. This allowed for the application in a packed bed reactor with a continuous flow, demonstrating that one of the targets of immobilisation – separation – can be achieved very readily (Scheme 7). Although no details on stability and time on stream are provided this separation enabled a continuous reaction.60,61
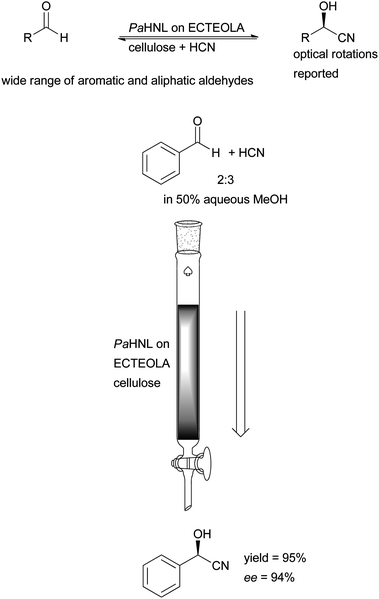 | ||
| Scheme 7 PaHNL immobilised on ECTEOLA cellulose for the continuous production of (R)-mandelonitrile, this enzyme preparation can be used for a wide range of aldehydes. | ||
Taking up the earlier work Effenberger et al. in 1987 compared the application of PaHNL solution with PaHNL immobilised on Avicel, a crystalline cellulose.21 After pre-treatment of this cellulose with buffer (pH = 5.4) the enzyme solution was adsorbed. The immobilisation thus occurred via hydrogen bonds. This wet enzyme preparation was utilised in buffer saturated ethyl acetate and compared with a biphasic system with aqueous enzyme solution and an organic layer. The immobilised enzyme was less active – possibly due to diffusion limitations – but gave much better enantioselectivities. The improvement in enantiopurity of the products is due to the reduced aqueous layer (see Section 1.2). In a later work the group could demonstrate that by lowering the pH value of the reaction, both for the aqueous phase and for the Avicel immobilisation approach, the enantioselectivity could be improved even further.62–64 The notoriously difficult ketones as substrates were converted by this Avicel PaHNL with excellent results (Scheme 8). Clearly, the Avicel has no significant influence on the stability, activity or selectivity of the enzyme. The main advantage of this immobilisation is that it enables the application in organic solvents. Dispersing the enzyme on a carrier suppresses lumping of the enzyme and thus diffusion limitations are reduced. The organic solvents have to be buffer saturated; in many ways this is similar to the method that was labelled “micro aqueous” for the kernel meal and whole cell approach.
 | ||
| Scheme 8 Lower pH values enable higher ee values for ketone based cyanohydrins, independent of immobilisation. | ||
Avicel immobilised PaHNL was then compared with Celite immobilised enzyme. Celite (kieselguhr, diatomaceous earth) are the silicate skeletons of diatoms that can be mined at several locations.6,8 There are many different Celites with very different structures but in most cases the literature is unclear about the nature of the Celite. The immobilisation on this material does in all cases, however, take place via hydrogen bonds. The material bares no charges and consists mainly of silica. Both the Avicel and the Celite immobilised PaHNL gave good but not excellent ee = 87–89% for the conversion of hydroxy-pivalaldehyde, an intermediate for the production of pantolactone (Scheme 4).65 The results with Celite as a carrier were confirmed independently in another comparative study.52 Nonetheless, both carriers clearly give much better results than the almond meal approach (see Section 2), most likely because the almond meal was used at a high pH value (5.5).52
The real breakthrough for the production of enantiopure pantolactone was achieved by expressing a single isoenzyme of PaHNL. This isoenzyme PaHNL5 could be applied at very low pH = 2.5 in an aqueous phase. These conditions were compared with the same enzyme on Celite in a micro aqueous system in DIPE. While the ee in both cases was 97%, the immobilised enzyme gave much lower yields. No explanations for this were given.66,67
When the first synthetic application of MeHNL was described it was immobilised on nitrocellulose and the authors state that Avicel was not suitable.68 Nitrocellulose is a charged carrier with zwitter ionic nitro groups. It thus enables an ionic interaction with the enzyme which seems to be essential for MeHNL. Immobilisation was again achieved by soaking the support with a buffer with a low pH value (here 3.3) and then adding the enzyme dropwise. The immobilised enzyme performed very well in buffer saturated solvents, both with aldehydes and ketones, aromatic and aliphatic.
It should be noted that most of the immobilised HNLs up to this point were not tested for recyclability or improved stability. The target of immobilisation in all cases except the first was to apply the enzyme in a wet organic solvent rather than in aqueous or biphasic systems. In a very systematic study Wehtje et al. immobilised PaHNL by adsorption on controlled pore glass, Sephadex G-25 and Celite.69 In dry DIPE no chemical reaction was observed but also the enzyme was inactive. Full activity was achieved with 2% water and Celite was the best carrier. It has to be noted that Celite can be used to control water activities and keep them constant.8 It might therefore be the case that Celite creates the most constant water concentration for the enzyme, ensuring optimal working conditions. In a packed bed reactor with a continuous flow of benzaldehyde and HCN in DIPE close to 90% yield could be obtained and the enzyme remained stable for at least 50 h (similar to Scheme 7). To maintain activity it was essential to add enough water to the substrate stream.
Adlercreutz beautifully demonstrated the importance of water in several further studies, by immobilising HbHNL on Avicel (crystalline cellulose), Celite and the modestly hydrophobic Accurel EP-700 polyamide. Again Celite was the best carrier and water had to be added. In addition to HbHNL PaHNL, SbHNL and MeHNL were also immobilised on Celite. HbHNL displayed the highest enantioselectivity and all enzymes required water to be active.70–72HbHNL on Celite was then studied in many different solvents and was co-immobilised with a series of additives. While sugars had no effect on the enzyme, polyethylenimine (PEI), different PEG’s and albumin improved the enzyme half-life. PEI had the most pronounced stabilising effect. So clearly Celite immobilisation enables separation and continuous reactions and in addition also the improvement of an enzyme.73 Both initially defined targets of immobilisation are thus achieved.
Improvement of the enzyme was also attained for AtHNL. This (R)-selective enzyme is very acid sensitive, which limits its application. By immobilising it on Celite R-633 and drying it its application in buffer saturated TBME became possible (results similar to Scheme 5).74 Interestingly, the enzyme also remained active with less water, displaying activity when all other HNL's were already inactive. Recycling studies showed that the enzyme preparation could be reused several times, exhibiting excellent enantioselectivity for mandelonitrile with ee >98% and minor loss of activity. For this enzyme both targets of enzyme immobilisation were accomplished, improved pH stability and thus high enantioselectivity and easy separation and hence recycling of the AtHNL.
A very systematic study of carriers was performed for MeHNL. Out of 15 carriers, including Avicel, several zeolites, alumina, ZrO2 and TiO2, Microbead silica gel 300A was shown to be most suitable with 90% adsorption rate. The pH range optimal for immobilisation was 4.8–6.8. The enzyme was tested in a 20 L reactor with buffer saturated TBME in the synthesis of (S)-mandelonitrile from benzaldehyde and HCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5). 22 batches were performed and overall 23.3 kg benzaldehyde were converted into 28.6 kg cyanohydrin (98% yield) with an ee = 98.9%.75 A recent patent by the same authors indicates that this type of process is implemented by Nippon Shokubai with PaHNL for the production of (R)-3-chloro-5-difluoromethoxymandelonitrile with an ee = 99% (Scheme 9), again in a batch reactor.76
:1.5). 22 batches were performed and overall 23.3 kg benzaldehyde were converted into 28.6 kg cyanohydrin (98% yield) with an ee = 98.9%.75 A recent patent by the same authors indicates that this type of process is implemented by Nippon Shokubai with PaHNL for the production of (R)-3-chloro-5-difluoromethoxymandelonitrile with an ee = 99% (Scheme 9), again in a batch reactor.76
 | ||
| Scheme 9 Immobilisation on Microbead Silica Gel 300A enables the application of different HNLs in a batch reactor. Excellent activity and selectivity is reported for recycling in at least 22 batches. | ||
Finally, a critical point on the way of adsorption has to be made. In all these studies except for the one by Pfeil, the carrier is soaked with an aqueous enzyme solution and left to dry, some studies from Effenberger mention centrifugation before drying but the other studies do not. This means that all the enzyme in solution is sucked into the porous carrier and then the solvent – water – is evaporated. Whether the enzyme is then immobilised by hydrogen bonds or ionic interactions or just deposited is often unclear. In all those cases where the immobilised enzyme is then used in an organic solvent in which it cannot dissolve the nature of the non-covalent attachment is fortunately not relevant. To really be sure about the nature of the interactions much more rigorous studies need to be performed.
4. Covalent immobilisation
Enzymes can be immobilised by formation of covalent bonds. On the surface of the enzyme amino groups from lysine can react with aldehydes to form imines (which can then be reduced to amines), they can attack highly reactive epoxides and form an amine link or they react with (activated) acids to form amides. Equally acid groups on the surface can react with amines or alcohols to form amides or esters. Glycosylated enzymes can additionally be immobilised via the sugar groups. Thus a wide range of reactions is available to form covalent bonds with the enzyme (Scheme 10). As illustrated in Fig. 2 two different strategies can be applied: the covalent link can tie the enzyme to a carrier (carrier-based immobilisation) or to another enzyme (carrier-free immobilisation).6–14,77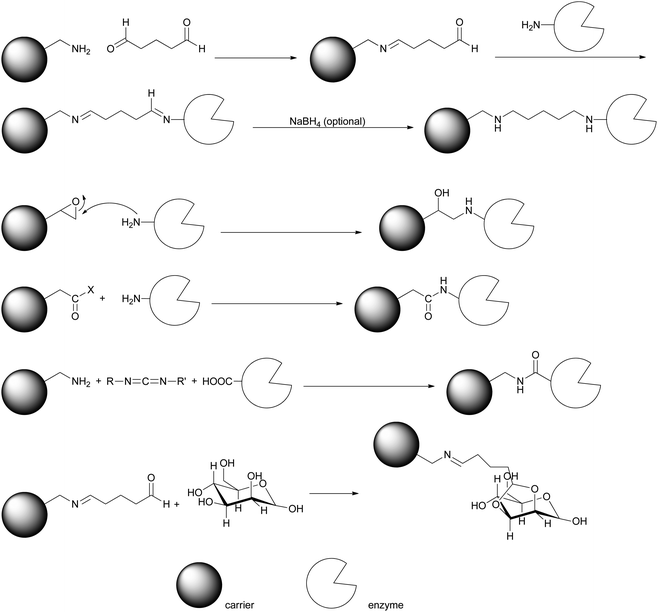 | ||
| Scheme 10 Covalent enzyme immobilisation is commonly achieved by reacting amino- or acid groups on the surface of the enzyme with a suitable activated carrier. Equally sugar rests of glycosylated enzymes can be derivatised. | ||
4.1. Carrier-based covalent immobilisation
Wehtje et al. immobilised PaHNL covalently (in addition to his non-covalent studies see 3) on silica beads and controlled pore glass. The silica beads performed much better and three different linker systems were tested.78 The silica was treated with aminopropyl triethoxysilane and this amino tether was then extended with glutaraldehyde (Scheme 10). After binding of the PaHNL the diimine was reduced with Na(CN)BH3. Alternatively the silica beads were functionalised with glycidoxypropyl trimethoxysilane, thus introducing an epoxide group, which was then hydrolysed to a diol. This can be treated with periodate, again generating an aldehyde group that reacts with an amino group of the enzyme with subsequent reduction. As a third approach the diol can be activated with tresyl chloride and then treated with the enzyme, leading to an SN2 reaction.The first method with the glutaraldehyde immobilised all enzymes with good activity and high loadings were attained. With the periodate approach much less enzyme was immobilised but with excellent activity, the third line led to almost complete deactivation. Therefore the glutaraldehyde route was used and the immobilised PaHNL was tested in a packed bed reactor with acetate buffer pH = 5.4 and 25% MeOH, similar to Becker and Pfeil (Scheme 7).61 With a benzaldehyde to HCN ratio of 1:2 the reactor was run for 100 h continuously and a constant yield of 95% (R)-mandelonitrile with ee = 95% was achieved. Clearly PaHNL can be immobilised both covalently and non-covalently and used for prolonged reactions under continuous conditions. The strategies are equally suitable for industrial conditions.
Kula et al. then described the immobilisation of LuHNL on Eupergit.79 This is a copolymer of methacrylamide, N,N′-methylen-bis(acrylamide) and a monomer with an epoxide group. The immobilisation is based on the nucleophilic attack of an enzyme amino group, ring opening the epoxide (Scheme 10). When applying this LuHNL in buffer (pH = 4.0) saturated TBME the enzyme was very stable, while the native enzyme degraded rapidly. It could be recycled and used for several days, demonstrating the improvements made to the enzyme by immobilisation. The enantioselectivity equalled that of the best results with native enzyme (ee = 88%) for the notoriously difficult substrate butanone. When scaled to gram size the ee value did however drop to 77% (Scheme 11). Nonetheless this immobilisation demonstrated that both targets of immobilisation are achieved: separation and improvement of the enzyme.
 | ||
| Scheme 11 Immobilisation on Eupergit stabilises LuHNL for use under acidic conditions. | ||
Extending this Eupergit work Tükel et al. reported that Prunus pseudoarmeniaca PpHNL could be immobilised under the same conditions and be applied also at low pH = 4 in buffer saturated TBME.80 20 cycles were performed without loss of activity or enantioselectivity, proving the versatility of this carrier for HNLs with very different structures.
An unusual immobilisation was described by Gröger et al. with chitosan as a carrier. This polymer of amino sugars was activated with glutaraldehyde and encapsulated in a polyvinyl alcohol hydrogel (Lentikat). Then the PaHNL was covalently attached via imine bonds. The enzyme was tightly attached and could be recycled 20 times without loss of activity or enantioselectivity. A biphasic reaction mixture with buffer pH = 4.5 and TBME–hexane proved to be most suitable for the production of (R)-mandelonitrile.81 A complimentary study showed that immobilisation via this method yielded very little attached enzyme, leaving room for improvement.82
4.2. Carrier-free covalent immobilisation
Immobilisation of the neat enzyme by cross linking it has the great advantage that the preparation is essentially only enzyme.83 No unnecessary ballast is introduced but the enzyme is ideally stabilised by the cross linking (Fig. 3). When crystals of MeHNL were cross linked with glutaraldehyde to obtain Cross Linked Enzyme Crystals (CLEC) it was however found that the glutaraldehyde could also deactivate the enzyme. With 1% glutaraldehyde concentration more than 90% of the MeHNL activity was lost. The obtained CLEC was then very stable. When only 0.1% aldehyde was used 25% activity was kept but the CLEC was not robust. Overall this approach was disappointing.84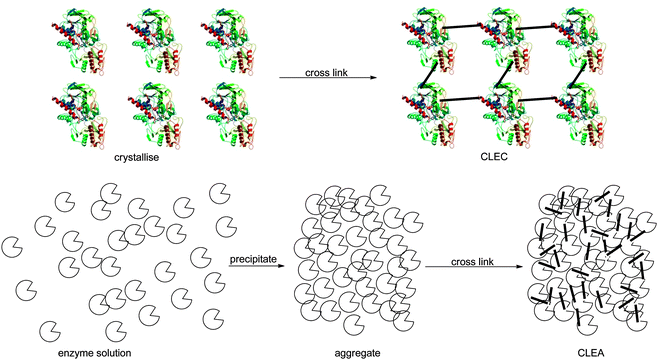 | ||
| Fig. 3 Carrier-free immobilisation of HNLs was performed with CLEC and CLEA methodology. | ||
Much better results were obtained with Cross Linked Enzyme Aggregates (CLEA; Fig. 3), introduced by Sheldon et al.85,86PaHNL was aggregated with 1,2-dimethoxyethane and cross linked with glutaraldehyde. When applying this CLEA in DIPE saturated with pH = 5.5 buffer the undesired chemical reaction could be suppressed and an ee of 95–99% could be obtained for several different aldehydes (benzaldehyde, o-chlorobenzaldehyde, cinnamon aldehyde). Additionally the enzyme could be recycled 10 times without loss of activity. Extending these studies to MeHNL it was found that here ammonium sulphate was the best precipitant.87,88 The MeHNL CLEA worked best under micro aqueous conditions, i.e. buffer saturated solvents. Interestingly the structurally related HbHNL89 and also AtHNL90 did not yield CLEAs with a high activity. A very detailed study on the working conditions of PaHNL and MeHNL CLEAs showed that utilising buffer saturated solvents gave the best results for both enzymes, conditions that were then employed to test the advantages of immobilisation.91 By working in a wet solvent and simply filtering off the enzymes the filtrate could be used for a follow-up step. Base catalysed TMS protections, acid catalysed hydrolysis of the nitrile group and Ritter reactions were possible with good step economy (Scheme 12). Clearly these CLEAs achieved one of the two targets of enzyme immobilisation: separation.
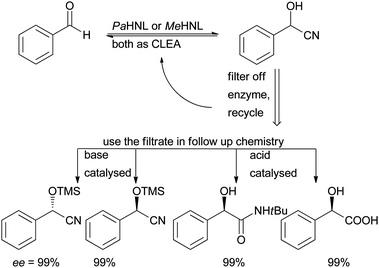 | ||
| Scheme 12 CLEA immobilisation of HNLs enabled their application in buffer saturated organic solvents and after a straightforward filtration step the next step could immediately be performed. | ||
LuHNL was also immobilised as a CLEA.92 Just like the carrier bound LuHNL on Eupergit79 it was tested in the butanone conversion (Scheme 11). The enzyme could be recycled 4 times, but some loss of activity was observed. The enantioselectivity remained, however, high and the ee of the cyanohydrin was 79–81%, a very small improvement on the Eupergit immobilised LuHNL.
Until recently there was some confusion about the water content of the reaction mixture necessary for HNLs to work well. Adlercreutz et al. had shown that different HNLs were inactive in dry organic solvents but regained the activity when placed in aqueous reaction mixtures.73 On the other hand some CLEAs displayed activity in dry solvents. This was therefore rigorously studied. The reaction did indeed proceed in dry solvents with MeHNL as the catalyst. However, analysis at the end of the reaction revealed that the reaction mixture was essentially water saturated. It was found that MeHNL, but also PaHNL CLEAs consist of more than 30% of water.93 They create their own “micro aqueous” conditions. To date no HNL is active under completely dry conditions. Only AtHNL on Celite does display low activity at very low water concentrations.74
A combi CLEA was prepared from MeHNL and a nitrilase.94 The work preceded the co-expression of the enzymes discussed above (Scheme 6 and Section 2)57–59 and demonstrated that the application of the CLEA immobilisation approach is not limited to one single enzyme.
Overall HNL CLEAs help to achieve the targets of immobilisation, separation and improved stability towards organic solvents.
5. Encapsulation
The inclusion of an enzyme in a structured material offers several advantages over the above-described immobilisation methods.6–14 The enzyme is not modified and if it is kept in solution it should behave as in homogeneous solution. The polarity of the carrier might influence the enzyme and equally the diffusion of the substrate and the product. In addition the pore size strongly influences the diffusion of the substrate, large carrier structures limiting it while small carrier particles can actually lead to an increased surface area and thus particularly easy access to the enzyme.In a biphasic liquid crystal system HbHNL was immobilised and investigated in the synthesis of (S)-mandelonitrile. In this exploratory work it was shown that good to excellent space time yields (100 mmol l−1 × h) and ee = 80–95% could be achieved, however only with a 5-fold excess of HCN.95
A more successful approach is the sol–gel encapsulation of enzymes.8,12–14 In this approach a suitable silica gel precursor is chosen, such as tetramethoxylsilane (TMOS). By adding alkylated silicon derivatives for instance methyltrimethoxysilane (MTMS) or dimethyldimethoxysilane (DMDMS) the polarity of the gel can be tuned. It can vary between the hydrophilicity of silica gel with many silanol groups up to the hydrophobicity of reverse phase silica which is completely alkylated on its surface. Acid is used to induce hydrolysis and the precursors of the sol–gel form monomeric and then oligomeric silicates. The partly hydrolysed, partly condensed oligomers are soluble and called sol. Further condensation leads to the gel, a highly porous and amorphous structure filled with water and the alcohol released during the reaction, the aqua-gel. For the HNLs it was noticed that the alcohol could be detrimental for the enzyme.96 In those cases it can be removed from the sol under reduced pressure.
The enzyme can be added to the sol stage and it is then encapsulated in the growing aqua-gel. Drying by lyophilisation or just under vacuum will lead to the Xero-gel with shrunk pores due to capillary forces. If the water in the aqua-gel is exchanged against acetone and that against supercritical CO2, it is possible to avoid the shrinking of the gel and an aero-gel is obtained (Scheme 13). Since all HNLs do not survive this treatment and are only active in the presence of water HNLs are immobilised in aqua-gels.
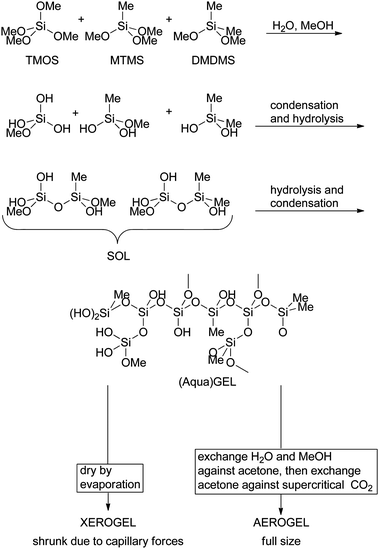 | ||
| Scheme 13 HNLs can be encapsulated in sol–gels. Many of them are sensitive to MeOH so this needs to be removed under vacuum from the sol. Subsequent addition of the enzyme in neutral buffer ensures the encapsulation in the aqua-gel. | ||
Utilising a hydrophobic sol–gel PaHNL was immobilised and compared with Avicel adsorbed enzyme. The sol–gel immobilised enzyme showed higher activity and it was also more stable, however, no data on the enantioselectivity were given.97
PaHNL, MeHNL, AtHNL and HbHNL were then immobilised in an aqua-gel and tested in a range of conversions.74,89,96 Excellent enantioselectivity and activity were observed, only for the bulky m-phenoxy benzaldehyde the reaction was slowed down, most likely due to diffusion problems.96 While some of the HbHNL was lost during the encapsulation the obtained gel enabled a heterogeneous reaction and no activity leached during the reaction. All reactions were performed with aqua-gels that were filled with a suitable buffer in an organic solvent, TBME or similar. The HbHNL aqua-gel could be recycled several times without loss of enantioselectivity (ee = 98%) but the activity decreased.96 This was shown to be due to the brittle structure of the aqua-gels. The capsules containing the enzyme seem to break and release the enzyme during the washing between cycles. This problem was also observed for AtHNL.74 With these properties the sol–gels are not really recyclable and hence the target separation is only partly achieved.
The most remarkable finding for the aqua-gel immobilised HNLs was the improved activity, in particular for AtHNL (10 fold).74 In the case of lipases it was hypothesised that a similar increase in activity was due to conformational changes, i.e. the permanent opening of the lid.8,10,11 This cannot be the case for the HNLs. It is much more likely that the highly increased surface area plays a key role. The HNLs in the aqua-gel are surrounded by water. Due to the gel the surface area thereof is large and the substrate can readily diffuse from the organic layer into the water layer in the gel. This is then similar to the advantages of an emulsion.
6. Comparison
Comparing all the heterogeneous enzyme preparations (Table 1) two points can be noted:| Enzyme | |||||||
|---|---|---|---|---|---|---|---|
| Method | PaHNL | PpHNL | HbHNL | MeHNL | AtHNL | SbHNL | LuHNL |
| Homogeneous |
Aqueous;
biphasic |
Biphasic |
Aqueous;
biphasic |
Aqueous;
biphasic |
Aqueous;
biphasic |
Aqueous;
biphasic |
Aqueous;
biphasic |
| Separation | No | No | No | No | No | No | No |
| Improvement | — | — | — | — | — | — | — |
| Meal/whole cell |
Yes
Meal |
No | No | No |
Yes
Whole cell |
No | No |
| Separation | Filtration |
3 × recycle
pH stability; |
|||||
| Improvement | Org. layer only | Org. layer only | |||||
| Non-covalent | Yes | No | Yes | Yes | Yes | Yes | No |
| On carrier |
ECTEOLA cellulose;
Avicel; Celite |
Celite |
Nitrocellulose;
microbead silica gel 300A |
Celite | Celite | ||
| Separation | Continuous reaction with Celite and ECTEOLA cellulose | — | 22 × recycle | 5 × recycle | — | ||
| Improvement |
Very stable
Org. layer only |
Org. layer only
Very stable |
pH stability;
Org. layer only |
pH stability;
Org. layer only |
pH stability;
Org. layer only |
||
| Covalent | Yes | Yes | No | No | No | No | Yes |
| On carrier | Silica beads with glutaraldehyde; chitosan + glutaraldehyde + Lentikat | Eupergit | Eupergit | ||||
| Separation | Continuous react. with silica beads/glutaraldehyde; 22 × recycle with modif. Lentikat | 20 × recycle | Several × recycle | ||||
| Improvement | Both very stable | Very stable, improv. temp. stab. |
pH stability;
org. layer only |
||||
| Covalent | Yes | No | Yes | Yes | Yes | No | Yes |
| Carrier-free | CLEA | CLEA | CLEA/ CLEC | CLEA | CLEA | ||
| Separation | 10 × recycle | — | Several × recycle | — | 4 × recycle | ||
| Improvement | Improv. stability; org. layer only | Moderately active | Improv. stability; org. layer only (CLEA); CLEC: loss of activity |
Loss of activity |
Improv. stability;
org. layer only |
||
| Encapsulation |
Yes
Different sol–gels |
No |
Yes
Liquid crystal Sol–gel |
Yes
Sol–gel |
Yes
Sol–gel |
No | No |
| Separation | Filtration | 4 × recycle | Filtration | 3 × recycle | |||
| Improvement | Higher activity due to larger surface area | Higher activity due to larger surface area | Higher activity due to larger surface area | Higher activity due to larger surface area | |||
1 The success of a method is not linked to the enzyme structure; i.e. the CLEA method works beautifully for PaHNL, MeHNL and LuHNL. But for HbHNL and AtHNL little activity could be immobilised although they are structurally similar to MeHNL (Fig. 1). On the other hand non-covalent immobilisation on Celite and the similar Microbead silica gel works for all four structure types of HNLs tested so far.
2 The key improvement made is that the enzymes can be used in almost pure organic solvents, allowing much more concentrated reactions (no water phase) and suppressing the undesired racemic reaction.
It is also clear that the two targets of immobilisation namely separation and improvement of the enzyme have been attained for the HNLs, in particular with the Celite immobilisation and the covalent immobilisation on Eupergit and similar carriers.
The reference point to define the real success of an immobilisation is the homogeneous enzyme in solution and to a lesser extent the crude and solid enzyme preparation, the kernel meal (Section 2). The ideal conditions for HNL catalysed cyanohydrin synthesis were discussed under Section 1.2. The best conditions are a low pH value for the buffered aqueous layer in a biphasic system. Low temperatures are advantageous since they also help to suppress the racemic base catalysed reaction and they prevent volatile HCN from evaporating. The organic layer in which most of the starting material and product reside also helps to suppress the racemic reaction, since no reaction occurs in it. It does however introduce diffusion limitations, which have to be overcome by rapid stirring. Griengl et al. improved this even further by performing the reactions in emulsion, reducing the conversion time to minutes.98–100
Several homogeneous HNL catalysed reactions have been scaled and are used industrially, in particular by DSM.101 A clear trend is visible, typically the biphasic system with low pH and temperature is used (Scheme 14).102 Only for one example an emulsion is reported which has to be broken by filtering over Celite, i.e. by immobilising the enzyme after the reaction.103 The first large scale success was the HbHNL catalysed synthesis of m-phenoxy-benzaldehyde cyanohydrin pioneered by Griengl (Scheme 15). More examples followed, most on a smaller scale. It has to be noted that in many cases the key to success was a genetic modification of the enzyme. In this way improved enantioselectivity was introduced. The details for all processes are shown Scheme 15. All these processes are batch processes and are based on wild type or mutant enzymes that were expressed in microorganisms, ensuring cheap and abundant supply of the HNLs. A micro-reactor system that allows continuous processes has been described. It gives similar results as the batch processes on a μl scale.104,105
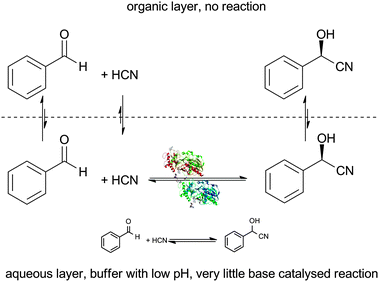 | ||
| Scheme 14 When the homogeneous HNL catalysed reaction is performed in a biphasic system no reaction takes place in the organic layer. The undesired racemic chemical reaction can be suppressed with a low pH in the aqueous layer. | ||
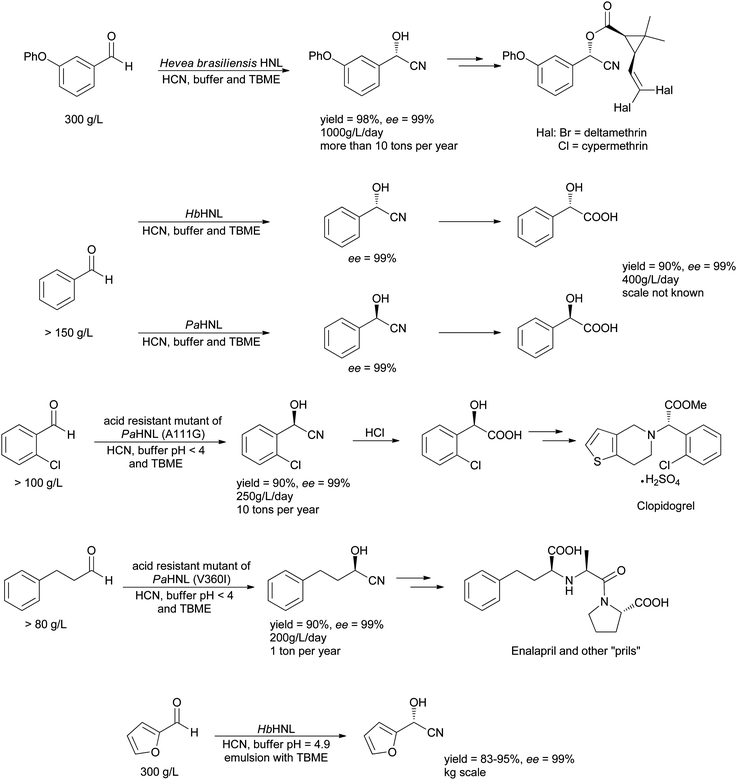 | ||
| Scheme 15 A number of HNL catalysed reactions are performed on an industrial scale. | ||
The main differences between the homogeneous and the immobilised HNLs are that the aqueous layer is reduced and that the solid HNLs can be filtered off. This is equally achieved with the crude enzymes. The reduced aqueous layer helps to suppress the undesired racemic reaction that proceeds only in water (Scheme 14), demonstrating the benefit of immobilising the enzymes. In addition the immobilised HNLs have the distinct advantage that they can be recycled more than 20 times without significant loss of activity or selectivity when used in a batch reactor. In a packed bed reactor a continuous reaction was stable for 100 h proving this reactor concept beyond doubt.
7. Conclusions and outlook
The immobilisation of enzymes has two targets: to enable separation, thus ensuring recycling and continuous reactions as well as pure products, and to improve the enzymes immobilised (stability, activity). Both targets were achieved with widely applicable immobilisation techniques that are straightforward to utilise. Adsorption on Celite or silica beads and covalent immobilisation on Eupergit worked in all attempted cases. It can therefore be expected that these techniques will in future be applied even more widely, easing HNL application in the laboratory and industry.Acknowledgements
The author thanks all the co-workers, students and colleagues in- and outside Delft, past and present, who have contributed to the immobilisation work of the laboratory.Notes and references
- L. Rosenthaler, Biochem. Z., 1908, 14, 238–253 CAS.
- L. Rosenthaler, Arch. Pharm., 1913, 251, 56–84 CrossRef CAS.
- V. K. Krieble, J. Am. Chem. Soc., 1913, 35, 1643–1647 CrossRef CAS.
- V. K. Krieble, J. Am. Chem. Soc., 1915, 37, 2205–2213 CrossRef CAS.
- V. K. Krieble and W. A. Wieland, J. Am. Chem. Soc., 1921, 43, 164–175 CrossRef CAS.
- L. Cao, Carrier-bound Immobilized Enzymes, Wiley-VCH, Weinheim, 2005 Search PubMed.
- L. Cao, Immobilized Enzymes, in Bioreactions and Bioreactor Operation, ed. M. Moo-Young, Elsevier, Comprehensive Biotechnology, 2nd edn, 2011, vol. 2, pp. 461–476 Search PubMed.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- L. Cao, Curr. Opin. Chem. Biol., 2005, 9, 217–226 CrossRef CAS.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- R. C. Rodrigues, A. Berenguer-Murcia and R. Fernandez-Lafuente, Adv. Synth. Catal., 2011, 353, 2216–2238 CrossRef CAS.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844–857 RSC.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef CAS.
- K. Ariga, Q. Ji and J. P. Hill, Adv. Polym. Sci., 2010, 229, 51–87 CrossRef CAS.
- W. Becker, U. Benthin, E. Eschenhof and E. Pfeil, Angew. Chem., Int. Ed. Engl., 1963, 2, 44–45 CrossRef.
- W. Becker and E. Pfeil, Naturwissenschaften, 1964, 51, 193 CrossRef CAS.
- W. Becker, U. Benthin, E. Eschenhof and E. Pfeil, Biochem. Z., 1963, 337, 156–166 CAS.
- W. Becker and E. Pfeil, Biochem. Z., 1966, 346, 301–321 CAS.
- V. I. Ognyanov, V. K. Datcheva and K. S. Kyler, J. Am. Chem. Soc., 1991, 113, 6992–6996 CrossRef CAS.
- J. Brussee, E. C. Roos and A. van der Gen, Tetrahedron Lett., 1988, 29, 4485–4488 CrossRef CAS.
- F. Effenberger, T. Ziegler and S. Förster, Angew. Chem., Int. Ed. Engl., 1987, 26, 458–460 CrossRef.
- A. Pratush, M. Sharma, A. Seth and T. C. Bhalla, J. Biochem. Technol., 2011, 3, 274–279 CAS.
- Y. Asano, K. Tamura, N. Doi, T. Ueatrongchit, A. H-Kittikun and T. Ohmiya, Biosci., Biotechnol., Biochem., 2005, 69, 2349–2357 CrossRef CAS.
- A. Solis, M. Solis-Oba, H. I. Perez, N. Manjarrez and J. Cassani, Biosci., Biotechnol., Biochem., 2011, 75, 985–986 CrossRef CAS.
- Y. Fukuta, S. Nanda, Y. Kato, H. Yurimoto, Y. Sakai, H. Komeda and Y. Asano, Biosci., Biotechnol., Biochem., 2011, 75, 214–220 CrossRef CAS.
- M. Dadashipour and Y. Asano, ACS Catal., 2011, 1, 1121–1149 CrossRef CAS.
- K. Gruber and C. Kratky, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 479–486 CrossRef CAS.
- U. Niemeyer and M. R. Kula, Angew. Chem., Int. Ed. Engl., 1990, 29, 386–387 CrossRef.
- E. Smitskamp-Wilms, J. Brussee, A. van der Gen, G. J. M. van Scharrenburg and J. B. Sloothaak, Recl. Trav. Chim. Pays-Bas, 1991, 110, 209–215 CrossRef CAS.
- J. Hughes, F. J. P. De C. Carvalho and M. A. Hughes, Arch. Biochem. Biophys., 1994, 311, 496–502 CrossRef CAS.
- J. Andexer, J. van Langermann, A. Mell, M. Bocola, U. Kragl, T. Eggert and M. Pohl, Angew. Chem., Int. Ed., 2007, 46, 8679–8681 CrossRef CAS.
- J. N. Andexer, N. Staunig, T. Eggert, C. Kratky, M. Pohl and K. Gruber, ChemBioChem, 2012, 13, 1932–1939 CrossRef CAS.
- C. Sulzbacher Caruso, R. de Fatima Travensolo, R. de Campus Bicudo, E. Gertrudes de Macedo Lemos, A. P. Ulian de Araujo and E. Carrilho, Microb. Pathog., 2009, 47, 118–127 CrossRef CAS.
- M. Dadashipour, M. Yamazaki, K. Momonoi, K. Tamura, K. Fuhshuku, Y. Kanase, E. Uchimura, G. Kaiyun and Y. Asano, J. Biotechnol., 2011, 153, 100–110 CrossRef CAS.
- L. L. Xu, B. K. Singh and E. E. Conn, Arch. Biochem. Biophys., 1988, 263, 256–263 CrossRef CAS.
- Z. Hussain, R. Wiedner, K. Steiner, T. Hajek, M. Avi, B. Hecher, A. Sessitsch and H. Schwab, Appl. Environ. Microbiol., 2012, 78, 2053–2055 CrossRef CAS.
- N. Bjarnholt and B. Lindberg Møller, Phytochemistry, 2008, 69, 1947–1961 CrossRef CAS.
- D. A. Jones, Phytochemistry, 1998, 47, 155–162 CrossRef CAS.
- D. Kadow, K. Voß, D. Selmar and R. Lieberei, Ann. Bot., 2012, 109, 1253–1262 CrossRef CAS.
- J. Holt and U. Hanefeld, Curr. Org. Synth., 2009, 6, 15–37 CrossRef CAS.
- J. von Langermann, J.-K. Guterl, M. Pohl, H. Wajant and U. Kragl, Bioprocess Biosyst. Eng., 2008, 31, 155–161 CrossRef CAS.
- J. N. Andexer, J. von Langermann, U. Kragl and M. Pohl, Trends Biotechnol., 2009, 27, 599–607 CrossRef CAS.
- F. Wöhler and J. Liebig, Ann. Pharm., 1837, 22, 1–24 CrossRef.
- P. Zandbergen, J. van der Linden, J. Brussee and A. van der Gen, Synth. Commun., 1991, 21, 1387–1391 CrossRef CAS.
- T. T. Huuhtanen and L. T. Kanerva, Tetrahedron: Asymmetry, 1992, 3, 1223–1226 CrossRef CAS.
- E. Kiljunen and L. T. Kanerva, Tetrahedron: Asymmetry, 1997, 8, 1551–1557 CrossRef CAS.
- R. J. H. Gregory, S. M. Roberts, J. V. Barkley, S. J. Coles, M. B. Hursthouse and D. E. Hibbs, Tetrahedron Lett., 1999, 40, 7407–7411 CrossRef CAS.
- S. Han, G. Lin and Z. Li, Tetrahedron: Asymmetry, 1998, 9, 1835–1838 CrossRef CAS.
- G. Lin, S. Han and Z. Li, Tetrahedron, 1999, 55, 3531–3540 CrossRef CAS.
- P. Chen, S. Han, G. Lin, H. Huang and Z. Li, Tetrahedron: Asymmetry, 2001, 12, 3273–3279 CrossRef CAS.
- L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Org. Process Res. Dev., 2003, 7, 828–831 CrossRef CAS.
- L. Synoradzki, T. Rowicki and M. Włostowski, Org. Process Res. Dev., 2006, 10, 103–108 CrossRef CAS.
- Y. Schmidt, K. Lehr, U. Breuninger, G. Brand, T. Reiss and B. Breit, J. Org. Chem., 2010, 75, 4424–4433 CrossRef CAS.
- F. Effenberger and S. Gaupp, Tetrahedron: Asymmetry, 1999, 10, 1765–1775 CrossRef CAS.
- A. Hietanen and L. T. Kanerva, Eur. J. Org. Chem., 2012, 2729–2737 CrossRef CAS.
- K. E. Scholz, D. Okrob, B. Kopka, A. Grünberger, M. Pohl, K.-E. Jaeger and U. Krauss, Appl. Environ. Microbiol., 2012, 78, 5025–5027 CrossRef CAS.
- S. Rustler, H. Motejadded, J. Altenbuchner and A. Stolz, Appl. Microbiol. Biotechnol., 2008, 80, 87–97 CrossRef CAS.
- O. Sosedov, K. Matzer, S. Bürger, C. Kiziak, S. Baum, J. Altenbuchner, A. Chmura, F. van Rantwijk and A. Stolz, Adv. Synth. Catal., 2009, 351, 1531–1538 CrossRef CAS.
- S. Baum, F. van Rantwijk and A. Stolz, Adv. Synth. Catal., 2012, 354, 113–122 CrossRef CAS.
- W. Becker, H. Freud and E. Pfeil, Angew. Chem., Int. Ed. Engl., 1965, 4, 1079 CrossRef CAS.
- W. Becker and E. Pfeil, J. Am. Chem. Soc., 1966, 88, 4299–4300 CrossRef CAS.
- T. Ziegler, B. Hörsch and F. Effenberger, Synthesis, 1990, 575–578 CrossRef CAS.
- F. Effenberger, B. Hörsch, F. Weingart, T. Ziegler and S. Kühner, Tetrahedron Lett., 1991, 32, 2605–2608 CrossRef CAS.
- F. Effenberger and S. Heid, Tetrahedron: Asymmetry, 1995, 6, 2945–2952 CrossRef CAS.
- F. Effenberger, J. Eichhorn and J. Roos, Tetrahedron: Asymmetry, 1995, 6, 271–282 CrossRef CAS.
- B. Pscheidt, M. Avi, R. Gaisberger, F. S. Hartner, W. Skranc and A. Glieder, J. Mol. Catal. B: Enzym., 2008, 52–53, 183–188 CrossRef CAS.
- B. Pscheidt, Z. Liu, R. Gaisberger, M. Avi, W. Skranc, K. Gruber, H. Griengl and A. Glieder, Adv. Synth. Catal., 2008, 350, 1943–1948 CrossRef CAS.
- S. Förster, J. Roos, F. Effenberger, H. Wajant and A. Sprauer, Angew. Chem., Int. Ed. Engl., 1996, 35, 437–439 CrossRef.
- E. Wehtje, P. Adlercreutz and B. Mattiasson, Biotechnol. Bioeng., 1990, 36, 39–46 CrossRef CAS.
- D. Costes, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 1999, 25, 384–391 CrossRef CAS.
- M. Persson, D. Costes, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 2002, 30, 916–923 CrossRef CAS.
- U. Hanefeld, A. J. J. Straathof and J. J. Heijnen, J. Mol. Catal. B: Enzym., 2001, 11, 213–218 CrossRef CAS.
- D. Costes, G. Rotcenkovs, E. Wehtje and P. Adlercreutz, Biocatal. Biotransform., 2001, 19, 119–130 CrossRef CAS.
- D. Okrob, M. Paravidino, R. V. A. Orru, W. Wiechert, U. Hanefeld and M. Pohl, Adv. Synth. Catal., 2011, 353, 2399–2408 CrossRef CAS.
- H. Semba, Y. Dobashi and T. Matsui, Biosci., Biotechnol., Biochem., 2008, 72, 1457–1463 CrossRef CAS.
- Y. Tsuchihashi, Manufacture of optically active mandelic acid derivative using hydroxynitrile lyase, Nippon Shokubai Co, Ltd, Japan, Jp. Pat. 2011205986 A, CAN 155:560651, 2011 Search PubMed.
- R. A. Sheldon, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef CAS.
- E. Wehtje, P. Adlercreutz and B. Mattiasson, Appl. Microbiol. Biotechnol., 1988, 29, 419–425 CrossRef CAS.
- J. Albrecht, I. Jansen and M. R. Kula, Biotechnol. Appl. Biochem., 1993, 17, 191–203 CAS.
- S. S. Tükel, D. Yildirim, D. Alagöz, Ö. Alptekin, G. Yücebilgic and R. Bilgin, J. Mol. Catal. B: Enzym., 2010, 66, 161–165 CrossRef.
- H. Gröger, E. Capan, A. Barthuber and K.-D. Vorlop, Org. Lett., 2001, 3, 1969–1972 CrossRef.
- E. Capan, U. Jahnz and K.-D. Vorlop, Landbauforsch. Voelkenrode, 2002, 241, 151–153 CAS.
- L. Cao, L. van Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS.
- D. Costes, E. Wehtje and P. Adlercreutz, J. Mol. Catal. B: Enzym., 2001, 11, 607–612 CrossRef CAS.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS.
- L. M. van Langen, R. P. Selassa, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2005, 7, 327–329 CrossRef CAS.
- A. Chmura, G. M. van der Kraan, F. Kielar, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2006, 348, 1655–1661 CrossRef CAS.
- C. Roberge, F. Fleitz, D. Pollard and P. Devine, Tetrahedron Lett., 2007, 48, 1473–1477 CrossRef CAS.
- F. L. Cabirol, U. Hanefeld and R. A. Sheldon, Adv. Synth. Catal., 2006, 348, 1645–1654 CrossRef CAS.
- D. Okrob, Optimierung der Hydroxynitril-Lyase aus Arabidopsis thaliana für die enantio-selektive Synthese von (R)-Cyanhydrinen, PhD thesis, 2012, available online: http://www.fz-juelich.de/zb/juwel Search PubMed.
- F. L. Cabirol, A. E. C. Lim, U. Hanefeld and R. A. Sheldon, Org. Process Res. Dev., 2010, 14, 114–118 CrossRef CAS.
- F. L. Cabirol, P. L. Tan, B. Tay, S. Cheng, U. Hanefeld and R. A. Sheldon, Adv. Synth. Catal., 2008, 350, 2329–2338 CrossRef CAS.
- M. Paravidino, M. J. Sorgedrager, R. V. A. Orru and U. Hanefeld, Chem.–Eur. J., 2010, 16, 7596–7604 CrossRef CAS.
- C. Mateo, A. Chmura, S. Rustler, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2006, 17, 320–323 CrossRef CAS.
- M. Boy and H. Voss, J. Mol. Catal. B: Enzym., 1998, 5, 355–359 CrossRef CAS.
- L. Veum, U. Hanefeld and A. Pierre, Tetrahedron, 2004, 60, 10419–10425 CrossRef CAS.
- I. Gill and A. Ballesteros, J. Am. Chem. Soc., 1998, 120, 8587–8598 CrossRef CAS.
- N. Klempier, U. Pichler and H. Griengl, Tetrahedron: Asymmetry, 1995, 6, 845–848 CrossRef CAS.
- H. Griengl, N. Klempier, P. Pöchlauer, M. Schmidt, N. Shi and A. A. Zabelinskaja-Mackova, Tetrahedron, 1998, 54, 14477–14486 CrossRef CAS.
- M. Bauer, H. Griengl and W. Steiner, Enzyme Microb. Technol., 1999, 24, 514–522 CrossRef CAS.
- D. J. Ager and O. May, Spec. Chem. Mag., 2008, 28, 30–32 CAS.
- T. Purkarthofer, W. Skranc, C. Schuster and H. Griengl, Appl. Microbiol. Biotechnol., 2007, 76, 309–320 CrossRef CAS.
- T. Purkarthofer, T. Pabst, C. van den Broek, H. Griengl, O. Maurer and W. Skranc, Org. Process Res. Dev., 2006, 10, 618–621 CrossRef CAS.
- K. Koch, R. J. F. van den Berg, P. J. Nieuwland, R. Wijtmans, M. G. Wubbolts, H. E. Schoemaker, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Eng. J., 2008, 135S, S89–S92 CrossRef.
- K. Koch, R. J. F. van den Berg, P. J. Nieuwland, R. Wijtmans, H. E. Schoemaker, J. C. M. van Hest and F. P. J. T. Rutjes, Biotechnol. Bioeng., 2008, 99, 1028–1033 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |