 Open Access Article
Open Access ArticlePhoto-induced covalent cross-linking for the analysis of biomolecular interactions
George W.
Preston
ab and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK. E-mail: A.J.Wilson@leeds.ac.uk; Fax: +44 (0)113 3436565; Tel: +44 (0)113 3431409
bAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
First published on 11th February 2013
Abstract
Photo-induced cross-linking (PIC) is a powerful strategy for generating information on biomolecular interactions. In PIC, the utility of traditional cross-linking methods is supplemented by the temporal control of photo-activation, enabling the study of non-covalent kinetic intermediates and heterogeneous mixtures. This tutorial review will introduce the photochemistry of activation, reactive intermediates, methods for the functionalisation of biomolecules and the installation of additional functionalities (e.g., affinity tags). In doing so, we shall illustrate the wealth of data that can be obtained using this approach, ranging from the identification of interacting partners and structural data to temporal information. Alongside a discussion of the strengths and weaknesses of the various approaches, their applicability to different types of biological system will be described.
 George W. Preston | George Preston received his BSc from Durham University in 2007 and an MSc from the University of Leeds in 2008. He remained at Leeds for his doctoral studies where, under the supervision of Prof. Andrew Wilson, Prof. Alison Ashcroft and Prof. Sheena Radford, he explored the use of photo-crosslinking chemistry for probing peptide supramolecular structures. George is currently undertaking postdoctoral research with Prof. David Phillips at King's College, London. His research interests are focused on protein chemistry and bioanalytical methods. |
 Andrew J. Wilson | Andy Wilson joined the University of Leeds in 2004 and was promoted to full professor in 2012. He currently serves as Deputy Director of the Astbury Centre and co-director of PPI-net. His research is concerned with (a) modulating protein–protein interactions, (b) developing fundamental approaches and building blocks for self-assembly and (c) mechanistic studies of self-assembly using photo-crosslinking. He completed a PhD at Warwick University supervised by Prof. David Leigh FRS before postdoctoral research with Prof. Andrew Hamilton FRS at Yale University and with Prof. E. Meijer and Prof. Rint Sijbesma at Technische Universiteit Eindhoven. |
Key learning points1. Different PIC methodologies are based on different underlying principles, and selection of the correct method is key to extracting useful data.2. An understanding of the mechanistic aspects of PIC (photochemistry, reactive intermediate chemistry, cross-link stability) is essential for interpreting the results of a PIC experiment. 3. Where PIC functionalities are to be incorporated into biomolecules, a wealth of strategies are available, and each has advantages and limitations. 4. Multifunctionality can be highly effective in assisting the separation and analysis of cross-linked products. 5. PIC can be used to obtain information on supramolecular structure in a temporal manner and with varying degrees of resolution (ranging from low resolution information on quaternary biomolecular structure to domain, residue or even atomistic resolution). |
Introduction
The aim of an analytical cross-linking experiment is to encode information about supramolecular structure in the system under observation. Supramolecular connectivity, which may be transient and/or weak, is converted to covalent connectivity, which should be much more stable. This allows transient and/or weak interactions to be captured and analysed under conditions that might normally result in disassembly (Fig. 1).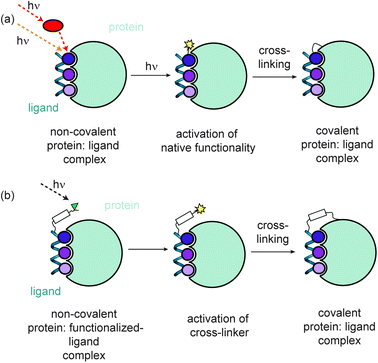 | ||
| Fig. 1 Strategies for PIC using (a) an exogenous reagent (red) or direct excitation (orange) to promote cross-linking of native functionality, and (b) a site-specifically incorporated functionality. | ||
Additionally, cross-linking may be useful in extracting information from heterogeneous systems that defy conventional biophysical structure determination, including polydisperse mixtures, membrane systems and nanostructures. Finally, in photo-induced cross-linking (PIC) methodologies,1 the use of light to stimulate the formation of cross-links is potentially very powerful because it can provide strict temporal control over the capture of interactions. The approaches developed to date can be divided into two main categories depending on whether the complexes of interest are wholly native, or whether they have been engineered to contain PIC-functionalised components (Fig. 1). This review will describe the activation and reactive intermediate chemistry and, where appropriate, methods of incorporation pertinent to widely used PIC strategies, including PICUP, benzophenone, azide and diazirine. Alongside the fundamental chemistry needed to understand and use this approach, we will illustrate the power of PIC through selected examples that highlight its ability to provide detailed temporal and structural information on biologically relevant interactions, assemblies and assembly processes.
Experimental considerations associated with cross-linking experiments
The extent or quality of information encoded as a result of a cross-linking experiment can range from subunit composition of a complex, to specific non-covalent contacts between interacting partners. The choice of cross-linking strategy should depend on the type of information that is required: for low resolution information on quaternary biomolecular structure, an approach that maximises the number of different cross-links may be favoured (i.e., in order to capture as many interactions as possible); whereas for information at the domain, residue or atom levels, an approach that minimises the number of different cross-links is desirable (i.e., in order to obtain more analytically tractable products). In this context, most effective implementations of PIC exploit a photo-reactive functionality that is attached to an interacting partner and activated in situ; owing to the fast kinetics of the photochemical steps, the temporal resolution of the experiment is not limited by diffusion of an external cross-linking reagent to its site of reaction. Of course, to achieve rapid cross-linking, the reaction via which the covalent cross-links form must also be rapid. In this regard, another advantage uniquely conferred by using light as a stimulus is the ability to unmask highly reactive intermediates (e.g., carbenes, nitrenes and radicals). In short, acquisition of useful cross-linking results depends on (i) aspects of experimental design (e.g., choosing whether to maximise or minimise the number of cross-links), (ii) the photochemistry of the PIC group, and (iii) the chemistry of the reactive intermediate.Approaches based on native functionality
Some approaches to PIC of protein complexes involve the use of native functionality or external photo-activated reagents, which react with functional groups that are naturally present on biomacromolecule surfaces. Direct PIC of native functionalities (e.g., to capture protein–RNA interactions) tends to rely on high-energy irradiation, and is beyond the scope of this review. Indirect PIC of native functionalities (i.e., via an external reagent) is a two-step process, whereby irradiation generates an external reactive intermediate that confers its reactivity to a native protein. This type of strategy is discussed below.The PICUP approach
In 1999, Fancy and Kodadek reported a protocol for the cross-linking of protein complexes via the photo-induced generation of monomer radicals in situ.2 This report has served as the basis for analyses of discrete protein complexes during the last decade.3 The approach, referred to as Photo-Induced Cross-linking of Unmodified Proteins (PICUP),2 confers reactivity from a photo-oxidised metal complex, which is generated from a chemically benign precursor by irradiation with visible light. As its name suggests, the key advantage of PICUP is that functional groups from which the radicals derive, as well as those to which they subsequently couple, are all native. The potential for any distortion to supramolecular structure during the reaction is therefore minimised. Furthermore, use of visible light can also be considered advantageous because wavelengths in the visible range are typically not damaging to biological samples. In a generalised PICUP experiment, buffered protein solution is mixed with the metal complex and an appropriate electron acceptor, then irradiated with visible light (λ > 380 nm). Protein radicals are generated in two principal steps: firstly, photo-oxidation of the metal complex occurs via transfer of an electron from a photo-excited state to the electron acceptor; secondly, the oxidised metal complex abstracts an electron from an amino acid side chain to form the protein radical. In their original experiments, Fancy and Kodadek reported cross-linking with tris-bypyridyl ruthenium(II) (Ru(Bipy)32+) and persulphate as an electron acceptor. Fig. 2a and b shows their structures and a general mechanism for photo-activation.![(a) Representative metal complex utilised in PICUP; (b) mechanism for the photo-oxidation of a generalised metal complex. [MLn] denotes a complex comprising a metal (M) co-ordinated by n ligands (L); the reduced starting form of the complex is denoted [MLn]red; the oxidised product is denoted [MLn]ox (c) plausible route for formation of a Tyr-Tyr cross-link in a PICUP experiment.](/image/article/2013/CS/c3cs35459h/c3cs35459h-f2.gif) | ||
| Fig. 2 (a) Representative metal complex utilised in PICUP; (b) mechanism for the photo-oxidation of a generalised metal complex. [MLn] denotes a complex comprising a metal (M) co-ordinated by n ligands (L); the reduced starting form of the complex is denoted [MLn]red; the oxidised product is denoted [MLn]ox (c) plausible route for formation of a Tyr-Tyr cross-link in a PICUP experiment. | ||
Using this approach, radicals generated by oxidation of certain amino acid side chains will be more thermodynamically stable than others, and the same is true of the radical products arising from the coupling step. As a consequence, both reactions display pronounced regioselectivity towards the side chains of Tyr, Trp, Cys and Met residues. This has been demonstrated indirectly by irradiating Ru(Bipy)32+ in the presence of free amino acids, whereupon free Tyr, Trp, Cys and Met strongly inhibited cross-linking.2b A notable feature is that detection of cross-links is possible as a result of characteristic spectroscopic properties of the Tyr-Tyr fluorophore. A plausible mechanism for the formation of the cross-link has been suggested (Fig. 2c).2a The coupling step might also be expected to proceed via nucleophilic attack of an amine or a thiol on the initially-formed radical. Thus, a reasonable diversity of cross-links is possible but, in each case, the likelihood of cross-link formation will be strictly governed by mechanism.
In their original reports, Kodadek and co-workers demonstrated the ability of PICUP to cross-link multimeric protein complexes using a natively hexameric protein (bacteriophage T4 UvsY).2a Specificity was then demonstrated by the cross-linking of a transcription factor (Saccharomyces cerevisiae TATA binding protein) to a transcriptional activator (Gal4 acidic activation domain, fused to glutathione-S-transferase), but not to a protein with which it does not associate (glutathione-S-transferase). In subsequent work, PICUP has been used in studies of varied biomolecular interactions of interest (e.g., interactions involved in protein targeting4 and interactions between constituents of the Escherichia coli translocon).3
Implementation of PICUP
In PICUP, neither the site from which a cross-link originates, nor the site to which it forms are predetermined, except by the moderate regioselectivity of the radical propagation steps. Thus, the strength of a PICUP experiment tends not to be in its ability to localise individual non-covalent contacts (which should be severely limited by the heterogeneity of the cross-links); but rather in its ability to form extensive networks of cross-links, which could aid in the characterisation of multimeric complexes. To date, PICUP has been implemented predominantly for studying homomeric protein assemblies.2a,5 There have been a few studies in which the method was used to study heteromeric complexes,3,6 but the observed cross-linking was rarely extensive enough to identify more than two of the interacting molecules. The report from Denison and Kodadek, however, has suggested that PICUP may yet prove useful for determining the composition of heteromeric multi-protein complexes.6 These authors outlined an elegant strategy involving the mapping of protein complexes based on mass spectrometry (MS) of proteolysed cross-linked proteins. This strategy does not rely on localising cross-links, but rather on the identification of proteolytic fragments derived from the interacting monomers that become cross-linked and are isolated using gel electrophoresis. Analysis was based on the principle that digestion should yield tryptic peptides that are diagnostic of the constituent monomers; thus for a dimer AB, the detection of unique tryptic peptides of A and B could be taken as evidence that the proteins from which they derive had been in non-covalent contact. Clearly, the information derived from such an experiment would render only a low resolution model of the protein complex in question, but there are nevertheless many multiprotein complexes for which this kind of data would be considered very valuable.Perhaps the most successful application of PICUP has been in analysing assembly intermediates in amyloid formation. Non-covalent peptide structures that are intermediate between monomer and mature amyloid fibrils (in terms of size, but also in the kinetic sense) appear to be very significant in the molecular basis of disease.7 Information about the supramolecular organisation of these intermediate structures is very valuable, but is difficult to obtain on account of two characteristics: metastability and heterogeneity.7 PIC is therefore a highly appropriate strategy for analysing these systems, mainly because of the temporal control that it affords. In addition, the covalent stabilisation of components in a mixture means that the investigator does not have to rely on the kinetic stability of an isolated component when conducting separation and analysis. Using PICUP, Teplow, Bitan and co-workers have probed the early stages of amyloid formation extensively (Fig. 3).5 Although these authors focused mainly on the amyloid-beta peptide (Aβ), it has been demonstrated that PICUP is equally applicable to studies of other self-assembling peptides (e.g., α-synuclein).8 Teplow and co-workers' original study involved the cross-linking of Aβ1–40 (residues 1–40 of the Aβ peptide) in the lag phase of its assembly (i.e., before nucleation had taken place). Direct cross-linking of dissociated and chromatographically separated peptide suggested that Aβ1–40 exists as a mixture of monomer and small oligomers (≤7-mer) prior to nucleation.9 By comparing the results obtained for Aβ1–40 with (i) results obtained for other aggregating and non-aggregating molecules, and (ii) simulated results for dissociated monomers, it was shown that the cross-linked products should indeed represent the oligomer size distribution in solution.9 A particularly noteworthy study reported pronounced differences in the oligomerisation of Aβ1–40 and Aβ1–42 (residues 1–42 of the Aβ peptide):10 whilst Aβ1–40 generated an equilibrium mixture of monomers through tetramers, Aβ1–42 was mainly monomer in equilibrium with pentamers and hexamers. It was thus shown that, on account of its extended C-terminus, Aβ1–42 follows an altered assembly pathway that gives rise to oligomers with altered structure and function.
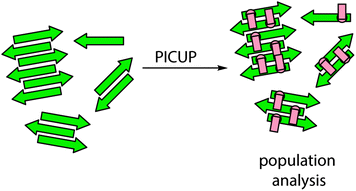 | ||
| Fig. 3 Schematic depicting generalized PICUP strategy for study of aggregation processes; the supramolecular ensemble is subjected to cross-linking at any given time point and cross-linked species characterized to produce a population distribution. | ||
Approaches based on unnatural functionality
In characterising the interactions of a protein of interest, some studies involve the profiling of its interacting partners in a biological medium, whilst others will seek more specific information about the parts of the proteins that are involved in the interactions. Clearly, a PIC-based strategy would suit these types of investigation, but the use of an external cross-linking reagent (e.g., in PICUP) may not be convenient or appropriate.In localising specific supramolecular contacts, for example, it is imperative that cross-link heterogeneity is minimised. In a PIC experiment, one way of doing this is site-specific incorporation of the photo-reactive functional group: covalent bonds form between a known site (the site of incorporation, chosen by the investigator) and an unknown site. Analytical methods used to localise the cross-link need therefore only consider one variable, i.e., the site to which the cross-link was formed. In forming the bond, the most useful results will be obtained using a cross-linker with indiscriminate reactivity; when chemical affinity is at a minimum, any cross-link heterogeneity should be solely attributable to the structural dynamics of the complex.
For site-specific incorporation and non-selective cross-linking, a range of PIC functionalities have been developed. These will now be discussed in terms of (i) photochemistry and (ii) reactive intermediate chemistry.
Chemistry of modern PIC functionalities
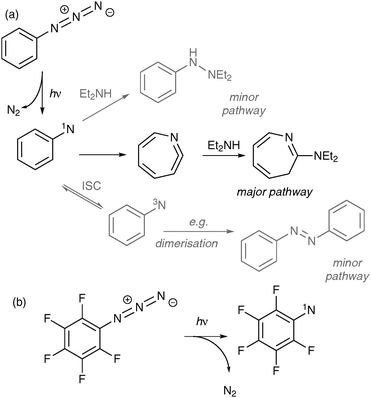 | ||
| Fig. 4 Photochemistry of aryl azides exemplified by photolysis of PA and pentafluoro-PA in diethylamine. (a) PA is photolysed to form the singlet nitrene (1N), which mostly undergoes ring expansion to 1,2-didehydroazepine, to which nucleophiles (e.g., diethylamine) can add. Other pathways, via direct reaction of the singlet nitrene with diethylamine, or via intersystem crossing (ISC) to triplet state (3N), are considered to be suppressed at ambient temperature (but note, however, that oxidation products are attributed to reactions of 3N with molecular oxygen); (b) the preferred reaction pathway of fluorinated PA is via direct insertion of the singlet nitrene. | ||
Given that its practical utility is limited by (i) a low λmax of activation, and (ii) intramolecular rearrangement of the singlet state, much effort has been expended on tuning the properties of PA to make it more suitable for PIC. Most significantly, Platz and co-workers experimented with fluorination of the ring. These authors systematically evaluated different fluorination patterns and discovered that a double substitution at both positions ortho to the azide group had a pronounced inhibitory effect on the kinetics of ring expansion.13 By slowing down the ring expansion, the lifetime of the nitrene became sufficient for intermolecular reactions to become competitive. The differences in reactivity between unfurnished and polyfluorinated aryl azides are conveniently summarised by photolysis experiments carried out in diethylamine.13 The products of these reactions are indicative of the mechanisms by which they form: unfurnished PA forms 1,2-didehydroazepine (see above), which undergoes nucleophilic addition to render azepine; fluorinated PA undergoes insertion into the N–H bond of diethylamine to render hydrazine. Interestingly, the results obtained by Platz and co-workers indicated that the mechanism by which fluorinated nitrenes react might be strongly dependent on environment. On the basis of solvent effects, the authors postulate that hydrophobic environments should promote reactions of the singlet state, whilst polar environments should promote those of the triplet. In practice, the impact of these nuances in a biological labelling study may be difficult to discern, but nevertheless, they should be borne in mind when interpreting the results.
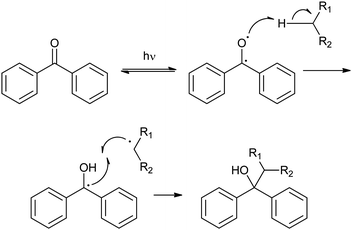 | ||
| Fig. 5 Mechanism of PIC by BP exemplified by its reaction with a methylene group. | ||
Upon irradiation at the relevant wavelength, BP generates a triplet ketyl biradical that can react with protein functional groups via a sequential abstraction–recombination mechanism (Fig. 5).17 The photochemical basis for formation of the biradical is absorption of a quantum (λ ∼ 350 nm) by the BP chromophore, which promotes a non-bonding electron on the carbonyl oxygen into the carbonyl π* orbital. The triplet excited state that is formed can abstract hydrogen by way of the resulting electron deficiency on the ketyl oxygen. Singlet to triplet intersystem crossing takes place with BP, contributing to its effectiveness as a photoactive group. It should also be noted that excitation of BP is a reversible process and, in the event that hydrogen abstraction does not take place during the lifetime of the excited state, BP may return to the ground state. The ground state is then available for re-excitation. The mechanism by which cross-links form is dependent on the substrate, and has been discussed in terms of two possibilities:18 aliphatic hydrogens (e.g., the methylene hydrogen of Gly) and hydrogens α to hydroxyl groups (if reactive) must be abstracted directly, whereas amines with α-hydrogen can form charge-transfer complexes that may aid the hydrogen abstraction step.18 The product of either process, irrespective of the mechanism, will almost certainly be a carbon radical that can recombine with the substrate to form a C–C cross-link. This cross-link is unlikely to be destroyed during subsequent analysis, but the presence of a hydroxyl group (formed when the ketyl oxygen abstracts hydrogen) means that it is susceptible to acid-catalysed dehydration.19
The efficient cross-linking of BP to the α-carbon of Gly originally observed by Printz and co-workers14 was perhaps fortuitous, given that other radical-based coupling reactions involving hydrogen abstraction exhibit a marked preference for Gly.20 This property is attributed to the high stability of α-centred Gly radicals.20 Other properties of BP that determine its reactivity centre on (i) thermodynamic stability of the radical formed at the site of hydrogen abstraction,21 (ii) stereoelectronic effects particular to the substrate (e.g., amines with α-hydrogen)18 and (iii) The geometry of BP triplet (approximately planar) and the way it presents the relevant orbitals and substituents. A further point regarding the reactivity of BP is that it may not be chemically inert in the dark, as the carbonyl is available for imine formation with biological amines. Schultz's group, for example, observed the specific covalent attachment of a BP-containing cyclic peptide to a Lys residue of HIV protease.22 Thus, it appears that BP does not fulfil the criterion of indiscriminate chemical reactivity particularly well because there are protein functional groups that should be preferred. This is evidenced by fact that BP triplet is essentially unreactive towards water.23 Although this is generally regarded as advantageous, it could be argued that the inability of BP to cross-link to water compromises its ability to report on molecular environment, since this environment may consist of water molecules.
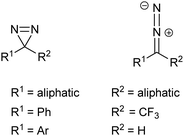 | ||
| Fig. 6 The diazirine ring, its linear diazo isomer and commonly-encountered substitutions thereof. | ||
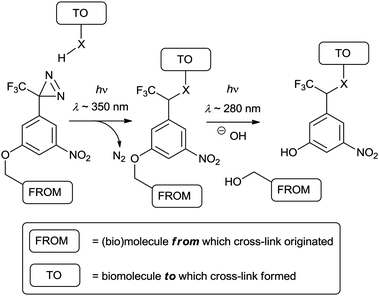
Diazirines have largely superseded diazo compounds as PIC reagents on account of their improved chemical stability in the dark and their photochemical responsiveness.1 Nevertheless, the diazo group remains relevant to discussions of modern PIC reagents because it is an undesired by-product of diazirine photolysis.24 The tendency for diazo compounds to form diazonium ions (via protonation at carbon), and subsequently carbocations (via light-independent loss of N2) means that these isomers also tend to be unwanted and can lead to unwanted cross-linking reactions.24 The exclusive labelling of protein nucleophilic groups by an aliphatic diazirine has been presented as evidence of this problem.25 Unfortunately, the conversion of a diazirine to its diazoisomer is stochastic, and cannot be completely avoided.24,26 Consequently, efforts have been directed towards stabilising the diazoisomer and therefore disfavouring diazonium formation; sufficient stabilisation should render an isomer that exists only as a benign spectator in subsequent reactions. In addressing this issue, Richards and co-workers prepared and evaluated compounds containing the 3-trifluoromethyl-3-aryldiazirine (TFMD) group.27 It was found that the combination of trifluoromethyl and aromatic substituents conferred stability to the diazoisomer whilst maintaining the favourable photochemical properties of the diazirine ring, although Preston et al. observed acid mediated decomposition.24 The TFMD group is consequently favoured over earlier diazirine-containing PIC functionalities, and it is noteworthy that if cross-linking yield is important this can be “boosted”, by irradiation at a second wavelength (302 nm), which leads to efficient conversion of the residual diazoisomer to the carbene.26
In order to render a reactive carbene intermediate, the diazirine must extrude molecular nitrogen from an excited state (cf. PA). In this process, the two σ bonds that connect the sp3 carbon atom to the azo group are severed and their bonding electrons are redistributed between the newly-formed divalent carbon atom and the departing molecule of nitrogen. The mechanism by which this occurs can be understood as follows: the highest occupied molecular orbitals of the diazirine ring are the p-orbitals of the azo group, each of which contain two non-bonding electrons.28 Absorption of a quantum (λ ∼ 350 nm) causes transitions of these electrons to anti-bonding orbitals of the ring, eventually resulting in a partially dissociated excited state.29 Decay of the excited state leads to complete dissociation of molecular nitrogen and formation of a singlet carbene at what was formerly the sp3 carbon atom of the ring.29 It is noteworthy that pathways to the triplet state are also theoretically plausible, and that it may interconvert with the singlet state.30
The reactivity of the TFMD-derived singlet carbene is conveniently illustrated by Richards and coworkers' original experiments, in which a diazirine was cross-linked to solvents.27 In these experiments, irradiation of diazirine in methanol or cyclohexane gave methyl ether and a cyclohexyl derivative, respectively (Fig. 7). These are typical products arising from the reaction of singlet carbenes with X–Y groups (e.g., C–H of cyclohexane or O–H of methanol). Whereas the singlet state reacts via a concerted mechanism, the triplet state should do so via a sequential radical abstraction–recombination mechanism (cf. BP triplet). As a result, it is recognised that singlet and triplet states will exhibit altered regioselectivity, which could influence the distribution of cross-linked products. This is exemplified by considering their reactions with a simple alcohol: the singlet state should preferentially react at the O–H bond, whereas the triplet should do so at one of the C–H bonds. In practice, the triplet state is often evidenced by the formation of ketones from reaction of the carbene with molecular oxygen, but its role in cross-linking of a biomolecule may not be so clear. In addition to O–H and aliphatic C–H groups, TFMD has also been shown to cross-link two other biologically-relevant functional groups, namely the amino group and the benzene ring.31 Where the former is concerned, amine adducts readily eliminate to the corresponding enamine/imine which then may undergo hydrolysis (Fig. 8).
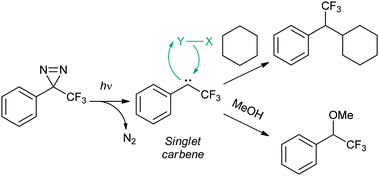 | ||
| Fig. 7 Photogeneration of a singlet carbene and products arising from insertion reactions with cyclohexane or methanol. A mechanism is given for the insertion reaction, featuring the generalised substrate X–Y (typically, Y = hydrogen). | ||
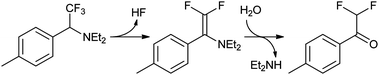 | ||
| Fig. 8 Products arising from the photolysis of a TFMD compound in diethylamine. The diethylamine adduct eliminates a molecule of hydrofluoric acid to form enamine, followed by facile hydrolysis. | ||
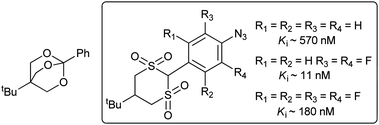 | ||
| Fig. 9 Compounds employed by Goeldner and co-workers in their comparative PIC study.33 | ||
Despite the Ki varying over two orders of magnitude, the authors were able to use the data to ensure that the binding site occupancies of all three analogues were identical. This allowed direct comparison of the efficiencies with which each ligand could be covalently cross-linked to the receptor. The authors observed efficient cross-linking of the fluorinated ligands but not the unfurnished aryl azide, highlighting how the chemistry of the reactive intermediate can have a pronounced effect on the result of the experiment: the authors speculate that the inability of the simple aryl azide to cross-link to the receptor is indicative of a hydrocarbon-rich binding site, with which the ring-expanded form could not react (cf.Fig. 4). In a different system Bartholomew and co-workers observed the opposite behaviour by comparing the cross-linking efficiencies of different photo-reactive DNA constructs to yeast RNA polymerase III. They found that a certain subunit of the polymerase was always cross-linked more efficiently using a simple aryl azide (versus fluorinated aryl azide, TFMD or BP).34
Comparative studies on the fundamental cross-linking chemistry have been performed using simple solvents as model reactants. Weber and Beck-Sickinger synthesised analogues of the pentapeptide thymopentin (RKDVY), in which either (i) the C-terminal Tyr residue was substituted with a PA- or TFMD-containing unnatural amino acid (UAA), or (ii) the N-terminus was functionalised with a photo-reactive acyl group (p-benzoylbenzoyl or 2-diazo-3,3,3-trifluoropropionyl).23 Reactivity towards media of varying hydrophobicity was assessed by photolysing the thymopentin analogues in water, n-propanol or a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of these solvents. When peptides were uniformly irradiated at wavelengths >300 nm, all but the diazo analogue were photolysed. Benzophenone exhibited a low affinity for water, instead preferring to form unspecified intramolecular rearrangement products. In contrast, the TFMD derivative formed a product distribution that more accurately reflected the environment of the cross-linker. Reactions of the aryl azide proceeded less cleanly.
:1 (v/v) mixture of these solvents. When peptides were uniformly irradiated at wavelengths >300 nm, all but the diazo analogue were photolysed. Benzophenone exhibited a low affinity for water, instead preferring to form unspecified intramolecular rearrangement products. In contrast, the TFMD derivative formed a product distribution that more accurately reflected the environment of the cross-linker. Reactions of the aryl azide proceeded less cleanly.
Beyond fundamental comparisons of cross-linking efficiency and reactivity, it is rare that the site of covalent attachment has been characterised in atomic detail, although an indication is often available from proteolytic digestion and tandem mass spectrometry (MS/MS; cf. localisation of a post-translational modification35). For instance, Chassaing and co-workers carried out degradation analysis in a study of peptide–lipid interactions.36 BP and TFMD-functionalised analogues of a cell penetrating peptide were irradiated in the presence of phospholipid (dimyristoylphosphatidylglycerol) vesicles. Following hydrolysis to remove the lipid constituents (i.e., fatty acid and/or glycerol and/or phosphate) that were not cross-linked, MS revealed that each analogue had become covalently attached to both fatty acid and glycerol, however the experiments could not differentiate between the two glycerol moieties of the phospholipid (Fig. 10) or the extent to which the cross-linking results reflected the true structure of the peptide–lipid complex.
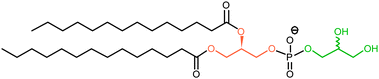 | ||
| Fig. 10 Structure of dimyristoylphosphatidylglycerol. The two glycerol moieties are highlighted in different colours. | ||
To summarise, a limited number of comparative studies of PIC reagents have been reported. The most extensive have involved (i) some indication of the extent to which the modification has perturbed the system relative to the unmodified case, and (ii) an analysis of the site of cross-linking. A comparison of the sites to which the different PIC functionalities have become covalently attached would be highly informative, but has been rarely considered.
Site-specific incorporation of PIC functionality into peptides and proteins
Site-specific incorporation of unnatural functionalities has been the focus of considerable recent effort: of the functionalities that have successfully introduced, considerable emphasis has been placed on PIC reagents because of their potential role in elucidating protein–protein and protein–peptide interactions.37 This section will briefly review methods for the site-specific incorporation of PIC functionalities into peptides and proteins. Although focused on PIC reagents, most concepts are applicable to the introduction of any unnatural functionality carrying masked or bio-orthogonal reactivity.Ligation onto pre-synthesised polypeptides
Native polypeptides contain naturally reactive groups that can be exploited for the direct ligation of unnatural functionalities.38 Primary amines (i.e., N-terminal α-amine; ε-amine of Lys) can be reacted with, for example, activated esters or isothiocyanates to yield amides and thioureas, respectively; thiols (i.e., β-thiol of Cys) can be reacted with maleimides or iodoacetamides to yield thioethers, or exchanged with disulphides. Although convenient, these methods can become restrictive when site-specific incorporation is required because (i) in the first instance, distribution of reactive Cys or Lys in the protein of interest is unlikely to be suitable for the experiment, and (ii) modification of amines or thiols can alter the functional properties (e.g., isoelectric point) of the protein. Generally, the most effective way of achieving site specificity using a ligation strategy is to position a single reactive Cys at the required site by mutagenesis.Recent work by Hamachi and co-workers has exploited the specific binding of a ligand for regioselective attachment of a PIC group onto a protein surface; the ligand-directed labelling method.39 This methodology utilised a labelling reagent, which comprises a ligand moiety and a PIC group connected by an electrophilic tosylate ester linkage (Fig. 11a). Attack of a nucleophilic group in the vicinity of the ligand binding site results in displacement of the ligand moiety and covalent attachment of the PIC group to the protein surface (Fig. 11b). By optimising the linker moiety, covalent attachment could be restricted to Glu, His or Tyr residues of FK506-binding protein that were proximal to the site at which a synthetic ligand moiety was bound. The aliphatic diazirine group that had become covalently attached was used to cross-link interacting proteins within living HeLa cells, and inclusion of a biotin tag facilitated the isolation and analysis of cross-linked proteins.
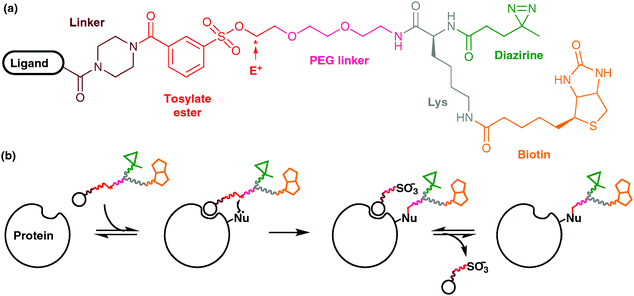 | ||
| Fig. 11 Regioselective covalent attachment of a PIC functionality using the method of Hamachi and co-workers: (a) the multifunctional labelling reagent. E+ denotes the electrophilic carbon of the tosylate ester linkage; (b) cartoon representation of the strategy, in which a nucleophilic group (Nu) attacks the tosylate ester to render a covalently-modified protein featuring diazirine and biotin groups. | ||
Solid-phase peptide synthesis
The most versatile method for introduction of unnatural groups, from a chemical diversity point of view, is solid-phase peptide synthesis (SPPS). SPPS has been used to incorporate PIC functionality in two ways: firstly, by selective acylation of the α-amine prior to cleavage of the peptide from the resin. Although limited in scope, this route may be favoured because acylating reagents tend to be synthetically accessible or commercially available. Secondly, functionality may be incorporated at a side chain site via an appropriately N-protected unnatural amino acid (UAA). N-Protected PIC UAAs bearing PA,40 fluorinated PA,41 BP,42 aliphatic diazirine43 and TFMD24 photo-reactive groups (1–7), have been incorporated into peptides for PIC studies (Fig. 12). Some of these UAAs bear a strong structural resemblance to their natural counterparts and therefore can be substituted conservatively into polypeptide sequences.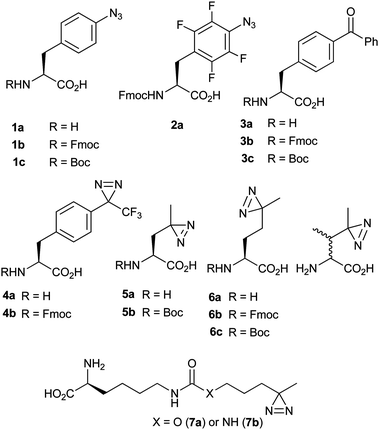 | ||
| Fig. 12 PIC UAAs and reported N-protected derivatives that have been incorporated within peptides/proteins. | ||
A recent example where SPPS was employed for site-specific incorporation of a diazirine in a cross-linking study focused on amyloidogenic peptides. Preston et al. used modified Aβ16–22 peptides to provide information on the exact position of cross-links within a fibril aggregate providing information on supramolecular organisation (Fig. 13).24 Such work complements that described by the Teplow group where PICUP was used to determine aggregate size in a temporal manner (see earlier).5
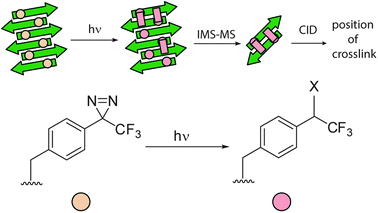 | ||
| Fig. 13 Schematic depicting generalised approach for determination of supramolecular structure in peptide aggregates. Photo-excitation of a site-specifically introduced diazirine results in covalent cross-linking. Cross-linked adducts are isolated by ion-mobility spectrometry (IMS) – mass spectrometry (MS) then sequenced by collision induced dissociation (CID) to identify cross-link position. | ||
Semi-synthesis
SPPS is a reliable method for polypeptide assembly, but there is a limit to the degree of polymerisation that can be achieved reliably; semi-synthetic approaches exploit the versatility of SPPS whilst circumventing this limitation. In a typical semi-synthesis, the UAA is incorporated into a peptide segment by SPPS, whilst the remainder of the protein is prepared, in tandem, as one or more (recombinant) polypeptide segments. The segments are then assembled sequentially using a chemical (expressed) ligation strategy, for which appropriate functional groups are required at the termini (e.g., a C-terminal thioester for reaction with an N-terminal Cys residue in native chemical ligation). Muir and co-workers' use of semi-synthesis represents a particularly appropriate example because the tandem incorporation of a second non-proteinogenic amino acid (phosphoserine) alongside the PIC functionality was sought.43 Tandem incorporation of the aliphatic diazirine and phosphoserine (both in a single synthetic pentapeptide) allowed the authors to covalently capture homomeric protein complexes whose assembly specifically depended on the phosphorylation of serine.Biochemical methods
For incorporation of non-native functionality, biochemical UAA incorporation represents a powerful alternative to total or semi-synthesis. Biochemical incorporation of PIC UAAs was first achieved in the 1980s and has since been implemented in numerous studies of protein interactions.32 Throughout the development of translation systems for UAA incorporation, the desire to incorporate PIC UAAs can be considered one of the driving forces for the development of biosynthetic methods.Early efforts involved incorporating the UAA via a chemically mis-aminoacylated tRNA. One of the first biosynthetic incorporations of a PIC UAA was achieved by acylating the ε-amino group of lysyl-tRNALys with a derivative of PA and introducing the resulting construct into a Lys-free in vitro translation system.44 Although this and more recent studies exploiting the “feed and flash strategy”45 (whereby the translational machinery of living mammalian cells is harnessed to generate proteins with diazirine groups at multiple sites) represent powerful demonstrations of the possibilities, incorporation of the PIC UAA using these approaches is not orthogonal. Subsequent methods addressed this issue by exploiting amber suppression, whereby one of the STOP codons (TAG; the amber codon) is erroneously read as if it codes for Tyr.46 This property is observed in certain E. coli mutants, and was shown to result from the presence of a tRNATyr with an altered anticodon (the amber suppressor tRNA, tRNASup). By including (i) a gene containing appropriately placed amber codons and (ii) tRNASup aminoacylated with the UAA of interest, an expression system can be expanded by one amino acid enabling UAAs to be incorporated in a controlled, site-specific fashion.
Implementation of methodologies based on amber suppression initially depended on chemically aminoacylated tRNASup which, as in the previous example, had to be synthesised and added to a cell-free translation system. Using this method, the site-specific incorporation of diazirine into E. coli preprolactin, and of benzophenone into bacteriophage T4 lysozyme were achieved.47 More recently, the pioneering work of Schultz has enabled the incorporation of UAAs using in vivo methods that do not require any in vitro manipulations of tRNASup. These methods necessitate a more sophisticated expression system, in which the aminoacylation of tRNASup is orthogonal to charging of the expression host's endogenous tRNAs. Specifically, this type of system requires an aminoacyl-tRNA synthetase (AAtRS) that binds the UAA selectively, and then uses it to selectively aminoacylate tRNASup.47 Schultz and co-workers went on to demonstrate the power of this methodology by site-specific incorporation of azide, benzophenone and diazirine based PIC UAAs.47 More recently, PIC-based pyrrolysine analogues have been incorporated in both prokaryotic and eukaryotic cell types.48
The development of this methodology has enabled the study of complex supramolecular systems, of which various examples have been summarised by Kohler and co-workers.32 Very recent work using genetically-incorporated PIC UAAs in situ has been carried out by Chen and co-workers.48b In their study of protein interactions in the periplasm of acid-stressed E. coli, these authors expressed the chaperone HdeA with genetically-encoded diazirine based pyrrolysine analogues at several sites.48b Irradiation of the bacteria at low pH, followed by proteomic analysis of cross-linked products, allowed conditional interactions of HdeA to be profiled.
Multifunctional cross-linkers
In combining a PIC group with a second, complementary functional group, there is scope for generating powerful analytical reagents. For studies in which PIC is used to profile protein–protein interactions, additional functionality can be employed to facilitate the isolation and identification of the cross-linked products. For experiments in which a PIC-functionalised bait protein is used to capture interacting partners, an affinity tag can be helpful for the isolation step. The inclusion of a tag such as biotin (e.g. in Hamachi's modified ligand,39 see Fig. 11), can enable pull-down of photolysed bait protein, along with any covalently-linked interacting partners. Representative examples are given in Fig. 12 and include diazirines conjugated to biotin 849 and radiolabelled diazirines 9–10.50Although modifications of this nature facilitate identification of interacting protein partners, pinpointing individual covalent cross-links remains challenging. This is because, typically, part of the protein from which the covalent bond originated (e.g., the bait) will still be attached to the peptide of interest, which can make the results of degradation analyses (e.g., tandem MS) complicated.24 One solution to this problem is the use of label transfer, whereby the biomolecule from which the cross-link originated is detached to leave a small diagnostic covalent modification. This can be achieved by introducing a linkage between the PIC functionality and the biomolecule which is stable throughout preparation and cross-linking, but which can be cleaved subsequently using mild reagents. The most advanced implementations of this strategy have used disulphide bonds, which can be both generated and cleaved by disulphide exchange. In this way, Khorana and co-workers were able to use a tritiated PIC reagent 9 (Fig. 14) to study interactions of rhodopsin with its heterotrimetic binding partner, transducin (αβγ).50a PIC generated a covalently-stabilised complex, whose disulphide bond could be cleaved with 1,4-dithiothreitol to yield radiolabelled transducin. The radiolabelled subunit was identified by separating α, β and γ subunits using denaturing gel electrophoresis and identifying the radioactive band (Fig. 15).
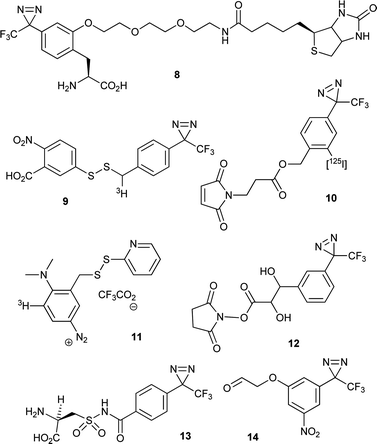 | ||
| Fig. 14 Multifunctional PIC reagents described in the text. | ||
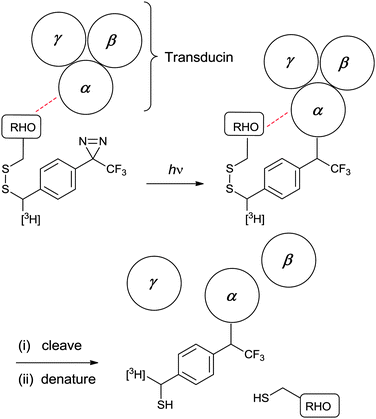 | ||
| Fig. 15 Label transfer strategy employed by Resek et al.50a to identify interactions between specific sites on rhodopsin (RHO) and subunits of transducin (α, β and γ). Photo-reactive rhodopsin constructs were prepared by treating different Cys mutants with a tritiated cross-linker. Following PIC, the disulphide bond was cleaved with 1,4-dithiothreitol and the sample was denatured. Gel electrophoresis with autoradiography enabled detection of the subunit(s) to which the label had been transferred. | ||
Kessler and co-workers employed a similar strategy using a radiolabelled aryl diazonium PIC functionality 11 (Fig. 14)51 to study the binding of Naja nigricollis α-neurotoxin to a nicotinic acetylcholine receptor. The aryl diazonium and disulphide groups were used to perform sequential cross-linking and cleavage/label transfer reactions, respectively. Impressively, it was demonstrated that the sites of labelling within a specific subunit could be identified using a combination of gel electrophoresis, in-gel cyanogen bromide cleavage and Edman sequencing.
A different cleavage strategy was reported by Bochkariov and Kogon, who reacted the N-hydroxysuccinimide ester of a diazirine based reagent 12 (Fig. 14) with the α-amine of phenylalanyl-tRNAPhe and, following PIC, cleaved its 1,2-diol linkage with periodate.52 Benzyl esters (Fig. 14), which are cleavable under alkaline conditions, have also been identified as candidate reagents for label transfer strategies.50b Two cleavable TFMD reagents were prepared by Brunner and co-workers e.g.10,50b but there are no reports of them having been used for label transfer. More recently, Hatanaka and co-workers reported the synthesis and site specific peptide incorporation of a diazirine containing a cleavable acylsulphonamide linkage 13.53 Upon activation by N-alkylation, the linkage becomes highly susceptible to alkaline hydrolysis. Perhaps the only drawbacks to using this construct are (i) the negative charge carried by the acylsulphonamide at physiological pH, and (ii) that the alkylation step might modify other parts of the molecule. Finally, Nakanishi and co-workers have outlined a promising label transfer strategy that uses light to effect both cross-linking and cleavage.54 Multifunctional constructs for this purpose have been prepared which feature a nitro substituent on the aromatic ring of TFMD.54 The modified TFMD group 14 is connected to a biomolecule or ligand of interest via an ether linkage (Fig. 16). Owing to the photochemical properties imparted by the nitro group, the ether linkage can be cleaved by irradiation at 280 nm in alkaline solution. This allows for sequential photolysis and then cleavage of the PIC group using different wavelengths of light (i.e., 350 nm followed by 280 nm).
 | ||
| Fig. 16 Label transfer method outlined by Nakanishi and co-workers that uses light to effect both cross-linking and cleavage. The result is a modified biomolecule carrying a 219 Da tag. | ||
Conclusions
In conclusion, a range of PIC functionalities is available for experiments involving the covalent cross-linking of biomolecules, and these functionalities can be incorporated in various different ways. Each of the different types of cross-linking functionality posses different advantages and disadvantages and have been employed in a plethora of settings, however true comparative studies are rare; thus whilst it may appear that the properties of TFMD in particular are consistent with those of an idealised PIC functionality, cross-linking studies might best be undertaken with a range of probes that can afford different information. PIC UAAs have been demonstrated to be useful reagents, particularly because they can be used to install PIC functionality using the cellular protein synthesis machinery. Despite the availability of accurate cross-linkers (e.g., TMFD, fluorinated PA) and methods with which to incorporate them, however, the potential for PIC to generate detailed structural information about biomolecular interactions has been realised only rarely. Application of the considerations described herein should allow cross-linking as a technique to extend beyond simple identification of interacting partners to the simultaneous acquisition of high-quality structural and temporal information from complex biomolecular systems.Acknowledgements
We thank the European Research Council [ERC-StG-240324] and the British Biotechnology and Biological Sciences Research Council (BBSRC) for a studentship for G.W.P. The authors wish to thank Prof. Sheena E. Radford and Alison E. Ashcroft for comments on early drafts of this manuscript and insightful discussions.Notes and references
- H. Bayley, Photogenerated Reagents in Biochemistry and Molecular Biology, Elsevier, Amsterdam, 1983 Search PubMed.
- (a) D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS; (b) D. A. Fancy, C. Denison, K. Kim, Y. Xie, T. Holdeman, F. Amini and T. Kodadek, Chem. Biol., 2000, 7, 697–708 CrossRef CAS.
- K. Deville, V. A. M. Gold, A. Robson, S. Whitehouse, R. B. Sessions, S. A. Baldwin, S. E. Radford and I. Collinson, J. Biol. Chem., 2011, 286, 4659–4669 CrossRef CAS.
- E. M. Clérico, A. Szymańska and L. M. Gierasch, Biopolymers Pept. Sci., 2009, 92, 201–211 CrossRef.
- G. Bitan and D. B. Teplow, Acc. Chem. Res., 2004, 37, 357–364 CrossRef CAS.
- C. Denison and T. Kodadek, J. Proteome Res., 2004, 3, 417–425 CrossRef CAS.
- R. Kodali and R. Wetzel, Curr. Opin. Struct. Biol., 2007, 17, 48–57 CrossRef CAS.
- K. Ono, T. Ikeda, J.-i. Takasaki and M. Yamada, Neurobiol. Dis., 2011, 43, 715–724 CrossRef CAS.
- G. Bitan, A. Lomakin and D. B. Teplow, J. Biol. Chem., 2001, 276, 35176–35184 CrossRef CAS.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS.
- W. T. Borden, N. P. Gritsan, C. M. Hadad, W. L. Karney, C. R. Kemnitz and M. S. Platz, Acc. Chem. Res., 2000, 33, 765–771 CrossRef CAS.
- M. S. Rizk, X. Shi and M. S. Platz, Biochemistry, 2005, 45, 543–551 CrossRef.
- K. A. Schnapp, R. Poe, E. Leyva, N. Soundararajan and M. S. Platz, Bioconjugate Chem., 1993, 4, 172–177 CrossRef CAS.
- R. E. Galardy, L. C. Craig, J. D. Jamieson and M. P. Printz, J. Biol. Chem., 1974, 249, 3510–3518 CAS.
- M. Krishnamurthy, A. Dugan, A. Nwokoye, Y.-H. Fung, J. K. Lancia, C. Y. Majmudar and A. K. Mapp, ACS Chem. Biol., 2011, 6, 1321–1326 CrossRef CAS.
- A. L. Stokes, S. J. Miyake-Stoner, J. C. Peeler, D. P. Nguyen, R. P. Hammer and R. A. Mehl, Mol. BioSyst., 2009, 5, 1032–1038 RSC.
- G. Dorman and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef CAS.
- S. G. Cohen, A. Parola and G. H. Parsons, Chem. Rev., 1973, 73, 141–161 CrossRef CAS.
- G. F. Egnaczyk, K. D. Greis, E. R. Stimson and J. E. Maggio, Biochemistry, 2001, 40, 11706–11714 CrossRef CAS.
- F. Kotzybahibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef CAS.
- V. A. Burgess, C. J. Easton and M. P. Hay, J. Am. Chem. Soc., 1989, 111, 1047–1052 CrossRef CAS.
- T. S. Young, D. D. Young, I. Ahmad, J. M. Louis, S. J. Benkovic and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11052–11056 CrossRef CAS.
- P. J. A. Weber and A. G. Beck-Sickinger, J. Pept. Res., 1997, 49, 375–383 CrossRef CAS.
- G. W. Preston, S. E. Radford, A. E. Ashcroft and A. J. Wilson, Anal. Chem., 2012, 84, 6790–6797 CrossRef CAS.
- M. R. Ziebell, S. Nirthanan, S. S. Husain, K. W. Miller and J. B. Cohen, J. Biol. Chem., 2004, 279, 17640–17649 CrossRef CAS.
- M. Hashimoto and Y. Hatanaka, Anal. Biochem., 2006, 348, 154–156 CrossRef CAS.
- J. Brunner, H. Senn and F. M. Richards, J. Biol. Chem., 1980, 255, 3313–3318 CAS.
- P. L. Mueller-Remmers and K. Jug, J. Am. Chem. Soc., 1985, 107, 7275–7284 CrossRef CAS.
- N. Yamamoto, F. Bernardi, A. Bottoni, M. Olivucci, M. A. Robb and S. Wilsey, J. Am. Chem. Soc., 1994, 116, 2064–2074 CrossRef CAS.
- D. Bethell, G. Stevens and P. Tickle, J. Chem. Soc., Chem. Commun., 1970, 792–794 RSC.
- M. Platz, A. S. Admasu, S. Kwiatkowski, P. J. Crocker, N. Imai and D. S. Watt, Bioconjugate Chem., 1991, 2, 337–341 CrossRef CAS.
- Y. Tanaka, M. R. Bond and J. J. Kohler, Mol. BioSyst., 2008, 4, 473–480 RSC.
- I. Kapfer, P. Jacques, H. Toubal and M. P. Goeldner, Bioconjugate Chem., 1995, 6, 109–114 CrossRef CAS.
- J. J. Tate, J. Persinger and B. Bartholomew, Nucleic Acids Res., 1998, 26, 1421–1426 CrossRef CAS.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS.
- C. Y. Jiao, I. D. Alves, V. Point, S. Lavielle, S. Sagan and G. Chassaing, Bioconjugate Chem., 2010, 21, 352–359 CrossRef CAS.
- A. Sinz, Mass Spectrom. Rev., 2006, 25, 663–682 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- T. Tamura, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 2216–2226 CrossRef CAS.
- R. Schwyzer and M. Caviezel, Helv. Chim. Acta, 1971, 54, 1395–1400 CrossRef CAS.
- J. E. Redman and M. R. Ghadiri, Org. Lett., 2002, 4, 4467–4469 CrossRef CAS.
- J. C. Kauer, S. Erickson-Viitanen, H. R. Wolfe and W. F. DeGrado, J. Biol. Chem., 1986, 261, 10695–10700 CAS.
- M. Vila-Perello, M. R. Pratt, F. Tulin and T. W. Muir, J. Am. Chem. Soc., 2007, 129, 8068–8069 CrossRef CAS.
- U. C. Krieg, P. Walter and A. E. Johnson, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8604–8608 CrossRef CAS.
- M. Suchanek, A. Radzikowska and C. Thiele, Nat. Methods, 2005, 2, 261–268 CrossRef CAS.
- H. M. Goodman, J. Abelson, A. Landy, S. Brenner and J. D. Smith, Nature, 1968, 217, 1019–1024 CrossRef CAS.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS.
- (a) S. M. Hancock, R. Uprety, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 14819–14824 CrossRef CAS; (b) M. Zhang, S. Lin, X. Song, J. Liu, Y. Fu, X. Ge, X. Fu, Z. Chang and P. R. Chen, Nat. Chem. Biol., 2011, 7, 671–677 CrossRef CAS.
- M. Hashimoto, Y. Hatanaka, Y. Sadakane and K. Nabeta, Bioorg. Med. Chem. Lett., 2002, 12, 2507–2510 CrossRef CAS.
- (a) J. F. Resek, D. Farrens and H. G. Khorana, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 7643–7647 CrossRef CAS; (b) T. Weber and J. Brunner, J. Am. Chem. Soc., 1995, 117, 3084–3095 CrossRef CAS.
- P. Kessler, R. Thai, F. Beau, J.-L. Tarride and A. Menez, Bioconjugate Chem., 2006, 17, 1482–1491 CrossRef CAS.
- D. E. Bochkariov and A. A. Kogon, Anal. Biochem., 1992, 204, 90–95 CrossRef CAS.
- N. B. Bongo, T. Tomohiro and Y. Hatanaka, Bioorg. Med. Chem. Lett., 2009, 19, 80–82 CrossRef CAS.
- K. Fang, M. Hashimoto, S. Jockusch, N. J. Turro and K. Nakanishi, J. Am. Chem. Soc., 1998, 120, 8543–8544 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |