 Open Access Article
Open Access ArticleImmobilisation and application of lipases in organic media
Patrick Adlercreutz*
Dept. of Biotechnology, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: Patrick.Adlercreutz@biotek.lu.se; Tel: +46 46 2224842
First published on 12th February 2013
Abstract
Different methods of preparing lipases for use in organic media are critically reviewed. Solid lipase preparations can be made by typical immobilisation methods such as adsorption, entrapment, covalent coupling or cross-linking. Immobilisation is especially attractive for lipases because, in addition to the normal benefits of enzyme immobilisation, it can also lead to a considerable increase in catalytic activity, probably caused by conformational changes in the lipase molecules. Activation can be achieved, for example, using hydrophobic support materials or surfactants during the immobilisation procedure. Surfactants can also be used to solubilise lipases in organic media via the formation of hydrophobic ion pairs, surfactant-coated lipase or reversed micelles. Lipase preparation methods are discussed with regard to potential lipase inactivation and activation effects, mass transfer limitations, lipase stability and other features important for applications. The practical applications of lipases in organic media reviewed include ester synthesis, modification of triacylglycerols and phospholipids, fatty acid enrichment, enantiomer resolution, biodiesel production and acylation of carbohydrates and bioactive compounds.
 Patrick Adlercreutz | Patrick Adlercreutz has a MSc degree (1979) in Chemical Engineering and a PhD degree (1985) in Biotechnology. The topic of his doctoral studies was “Oxygen supply to immobilised cells”. Thereafter he focussed on biocatalysis in organic media and built up a research group. Since 1999 he has been Professor of Biotechnology at Lund University, Sweden. He has published about 200 peer-reviewed papers. His research interests include fundamentals of biocatalysis in organic media as well as the use of all kinds of biocatalysts in synthesis, especially to produce lipids, carbohydrates, peptides, surfactants and chiral building blocks. |
1. Introduction
The natural function of lipases is to hydrolyse the ester bonds of triacylglycerols. These substrates are poorly soluble in water, and the reaction thus normally takes place at an organic–aqueous interface at which lipases work very well. In fact, most of them express higher catalytic activity under such conditions than in aqueous solution with the dissolved substrate molecules; a phenomenon called interfacial activation.1–3 The generally accepted hypothesis for the mechanism of interfacial activation is that the active site of the lipase in aqueous solution is covered by a flexible region of the enzyme molecule, often called the lid. Interaction with a hydrophobic phase can cause opening of the lid to make the active site accessible. This hypothesis is supported by crystal structures of lipases, some of which show a closed form of the lipase,4,5 while others show an open form (Fig. 1).6 Since interfacial activation can cause a dramatic increase in catalytic activity, it is of profound importance for all applications of lipases, and should always be considered when developing lipase immobilisation methods. It should be pointed out that there are individual variations among the lipases; there are reports on a lipase having two lids7 while others lack the typical lid and show no interfacial activation.8,9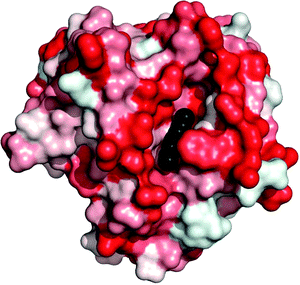 | ||
| Fig. 1 Rhizomucor miehei lipase (RML, PDB structure 4TGL). This form of the lipase is “open” and the co-crystallised inhibitor (black) is therefore visible in the active site. The intensity of red colour indicates hydrophobicity according to Eisenberg et al.10 The large hydrophobic area around the active site can interact with a hydrophobic phase, thus stabilising this form of the enzyme. The figure was constructed using PyMOL. | ||
In general, lipases have very broad substrate specificity and are therefore used in a wide range of applications, both in research laboratories and in industry.11–13 This review will focus on lipases of microbial origin, since this is the most widely used group for practical applications. While some mammalian lipases need colipases under certain conditions,11 microbial lipases do not need any coenzymes. The use of lipases in organic media has expanded dramatically since the mid 1980s, allowing the efficient use of lipases in esterification and transesterification reactions, in addition to the traditional hydrolysis reactions. A major reason for this important development is that it is easier to achieve high activity and stability of lipases in organic media than most other enzymes. This review will focus on the use of lipases in organic reaction media, with organic solvents as a typical example. These may be “normal”, non-ionic solvents or ionic liquids. Furthermore, solvent-free mixtures, in which the substrates also act as the solvent, are attractive for many applications. Other reaction media which will not be covered in great detail are supercritical fluids. All these media have the common feature that the water content is low, and similar methodologies can be used to prepare enzymes for use in all of them.
Immobilisation is attractive for all types of enzymes, but for enzymes in organic media, in particular lipases, immobilisation often provides the extra advantage of an increase in catalytic activity compared to the non-immobilised enzyme. The literature on immobilised lipases is extensive and this review will focus on examples of particular interest rather than aiming for completeness.
1.1 Commonly used lipases
Although many different lipases have been described, most of the literature on the immobilisation and application of lipases deals with a rather limited number of enzymes. The most commonly used lipases and the abbreviations used for them in this review are listed in Table 1. It should be noted that some of the lipases originate from microorganisms that have been re-classified and, therefore, they have different names in the literature. Literature reports on CRL should be read with special care since this lipase can contain at least 5 isoenzymes14 of varying proportions, which are seldom specified in the enzyme preparations described in the literature. These isoenzymes have different substrate specificity and stability.15 CALB has been used extensively as a catalyst in organic synthesis,16 while TLL is used in large amounts in detergents and various synthetic applications.17 RML is used for many applications in the conversion of fats and oils,18 as well as in chemical processes.19| Abbreviation | Source organism | Comments, including other names |
|---|---|---|
| BCL | Burkholderia cepacia | Formerly Pseudomonas cepacia; Lipase PS |
| CALB | Candida antarctica | Novozym 435 is a commonly used immobilised form. C. antarctica also contains another lipase (lipase A) |
| CRL | Candida rugosa | Formerly C. cylindracea. Contains isoenzymes! |
| RML | Rhizomucor miehei | Formerly Mucor miehei. Lipozyme RM IM is a commonly used immobilised form |
| ROL | Rhizopus oryzae | Several Rhizopus lipases are almost identical |
| TLL | Thermomyces lanuginosus | Formerly Humicola lanuginosa. Lipozyme TL IM is a commonly used immobilised form |
In addition to these commonly used lipases, many others have been described. Those isolated from microorganisms tolerant to organic solvents have the potential to be especially useful for applications in organic media.20,21
1.2 Lipase-catalysed reactions
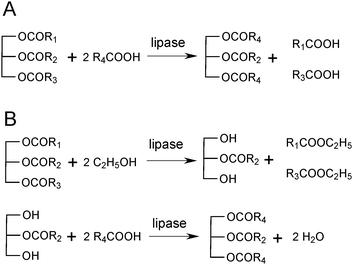
The natural reaction of lipases is to hydrolyse ester bonds. This reaction involves nucleophilic attack of the active site serine on the carbonyl carbon of the ester bond, forming an acyl-enzyme intermediate, and the first product, which is an alcohol (Scheme 1). The acyl-enzyme intermediate is subsequently hydrolysed by water, forming the second product, which is a carboxylic acid. Because of the broad substrate specificity of lipases, many different compounds can act as acyl donors. Many esters are excellent acyl donors, but free carboxylic acids can also act in this way. Likewise, many nucleophiles, in addition to water, can deacylate acyl-enzyme intermediates; hydroxyl-containing compounds being the most important examples. This leads to esterification and transesterification reactions, which can become dominant in organic media. It is often so that many reactions occur simultaneously. The potential acyl donors present compete for reaction with the native enzyme, and the nucleophiles present compete for deacylation of the acyl-enzyme. In reaction mixtures with substrates containing different carboxylic acids and/or their esters, all of these can normally react to form their respective acyl-enzymes.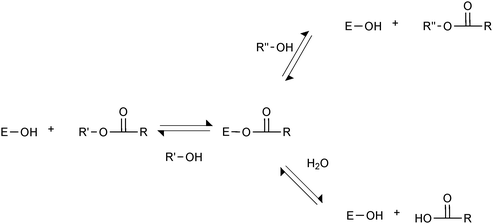 | ||
| Scheme 1 Lipase-catalysed reactions. The lipase with its active site serine hydroxyl group (E–OH) can react with esters or carboxylic acids to form an acyl-enzyme, which in turn can react with various nucleophiles, such as alcohols and water. | ||
In addition to the reactions mentioned above, lipases are able to catalyse several other reactions. Amines are important alternative nucleophiles which cause the formation of amide products. Furthermore, lipases are excellent examples of “promiscuous” enzymes, catalysing reactions quite different from the normal ones.22 Lipase-catalysed aldol addition,23 racemisation24 and epoxidation25 constitute a few interesting examples.
1.3 Effects of reaction conditions on lipase-catalysed reactions
The observed catalytic activity of enzymes in organic solvents is sometimes several orders of magnitude lower than in water, making practical applications unattractive. However, the situation can be drastically improved by choosing the appropriate conditions for biocatalysis in organic solvents, with the first step being to determine the cause of the differences in activity. Klibanov and co-workers carefully examined the factors behind the difference in activity in water and organic solvents, and found the following to be of prime importance:26• Method of enzyme preparation (immobilisation, etc.)
• The quantity of water
• The kind of solvent
• The pH
Immobilisation and other ways of making enzyme preparations suitable for use in organic solvents, which is the main topic of this review, are discussed in Section 2. The effects of water are very important for lipases and other hydrolytic enzymes, and are discussed in Section 3. The effects of the solvent and pH are outside the scope of this review, but a few references are given below.
The solvent influences the rate of lipase-catalysed reactions in several ways. As in other enzyme-catalysed reactions, solvation of the substrate can have a profound influence on the observed reaction rate. Good solvation of the substrate in the bulk solvent decreases its free energy and, therefore, slows down its conversion.27 However, even after correction for these effects, considerable differences remain between rates in different solvents.28,29 These differences may be due to direct interactions between the solvent molecules and enzyme molecules. Molecular dynamics simulations have shown that the solvent has a profound effect on the flexibility of the lipase studied (CALB), which is likely to cause differences in catalytic activity.30 In addition, molecular modelling studies have shown that opening of the lid can be induced by organic solvents,7,31,32 and it is possible that the efficiency in the interfacial activation is different in different solvents. Another possible interaction between a lipase and a solvent is that the solvent can act as a competitive inhibitor in the reaction catalysed. Some ketones have been found to be inhibitors of CALB, while hydrocarbons did not inhibit that enzyme.33 In addition, solvents can cause complete inactivation of enzyme molecules, either due to solvent molecules dissolved in water or to the presence of an organic–aqueous interface, although the latter mechanism is generally not applicable to lipases, which, instead, often are activated by interfaces.34,35 Stabilisation of CALB towards hydrophilic organic solvents has been achieved by rational design involving the creation of new internal hydrogen bonds in the lipase.36
While choosing a solvent for a specified application, many aspects should be considered, including enzyme activity and stability, substrate solubility, equilibrium position, boiling point and toxicity. When all relevant characteristics of a solvent are not known, computational methods can give approximate values, quite useful for preliminary screening of solvents.37
The effect of pH on enzymatic reactions in organic solvents is seldom studied in detail. It is known that the protonation state of an enzyme is of vital importance for its catalytic activity in aqueous media and, most probably, also in organic solvents. Most researchers assume that controlling the pH in the aqueous solution used for enzyme preparation (immobilisation, etc.) is sufficient to keep it in an active protonation state when added to an organic medium. However, problems are known to occur in reactions involving acidic or basic substrates and products. These problems can sometimes be overcome using buffers adapted for use in organic media, such as organo-soluble buffers or solid-state buffers.38–40
2. Lipase immobilisation
Lipases can be immobilised using most of the methods developed for enzyme immobilisation in general.41,42 The major types of immobilisation are illustrated in Fig. 2, and will be discussed below. It should be pointed out that a combination of immobilisation methods can be quite useful. One such example is adsorption followed by cross-linking of the adsorbed lipase to reduce the risk of enzyme leakage from the support.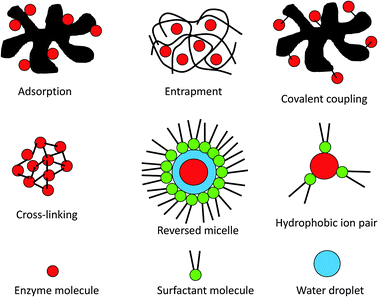 | ||
| Fig. 2 Schematic presentation of enzyme immobilisation methods and methods of using surfactants to solubilise enzymes in organic media. | ||
Immobilisation facilitates enzyme reuse and product purification, thus dramatically improving process economy. However, immobilisation can also cause inactivation of the enzyme and, as in all cases of heterogeneous catalysis, mass transfer limitations can cause a decrease in the observed specific activity of the enzyme (moles of substrate converted per unit enzyme per unit time). The enzyme molecules in the middle of the immobilised enzyme particles are exposed to lower substrate concentration than in the bulk solution due to substrate concentration gradients, leading to a lower reaction rate. Mass transfer limitations are more severe, the larger the biocatalyst particles and the faster the reaction.43
2.1 Evaluation of immobilised lipase preparations
A large part of this review will be devoted to the discussion of different types of immobilised lipase preparations. To evaluate their performance critically, the following parameters will be considered.• Catalytic activity – it is usually important to achieve a high specific activity, so that the enzyme is used as efficiently as possible. This parameter is influenced to a large extent by the method of immobilisation and is thus of special importance in this review.
• Final yield – this is an important parameter for practical applications. However, it is seldom influenced to any high degree by the immobilisation procedure, and is therefore not so relevant for the evaluation of immobilised preparations in most cases.
• Operational stability – operational stability is often of crucial importance, especially for large-scale applications. Good stability is required to achieve high productivity, i.e., the amount of product that can be produced per unit weight of enzyme.
In this review, most attention will be devoted to the catalytic activity of the immobilised lipases, and other results will be mentioned when they are of special interest.
| Reaction | Most common detection method | Medium |
|---|---|---|
| Triacylglycerol hydrolysis | Titration, pH stat | Aqueous or emulsion |
| p-Nitrophenyl ester hydrolysis | Spectrophotometric | Aqueous, emulsion or organic |
| Esterification | Chromatographic (GC/HPLC) | Organic |
| Transesterification | Chromatographic (GC/HPLC) | Organic |
2.2 Adsorption
Physical adsorption is perhaps the simplest method of enzyme immobilisation. An enzyme solution, usually aqueous, is simply brought into contact with a supporting material and, provided the enzyme–support interactions are strong enough, adsorption will occur. In the case of lipases, hydrophobic interaction is most common, but ionic interactions with ion-exchange materials, etc., can also be useful.47 Because lipases adsorb spontaneously from aqueous solutions onto hydrophobic surfaces to a higher degree than most other proteins, this method can be used for simultaneous purification and immobilisation of lipases.48–50 Interestingly, adsorption on the support often leads to enhancement of the catalytic activity of the lipase.49 This is interpreted as being due to hydrophobic interactions between the support and part of the lipase molecule causing opening of the lid to yield the active conformation, in analogy with the interfacial activation caused by a hydrophobic substrate phase.The surface properties of mesoporous silica can be tailored using various silanes as building blocks or derivatisation agents. For lipases, it is of special interest to use silanes containing hydrophobic groups. The contact angle formed between the material and water has been demonstrated to be a useful parameter to characterise the surface hydrophobicity.56 A series of mesoporous silica materials containing different functional groups has been prepared and evaluated with respect to contact angle and the catalytic activity of the adsorbed lipases. The most hydrophobic material, which contained phenyl groups, had a contact angle of about 110 degrees and provided the highest catalytic activity for both CALB and BCL (denoted PCL in the figure) (Fig. 3).57 Interestingly, the trend of increasing activity with increasing contact angle was found to be similar in a hydrolysis reaction in water and in a transesterification reaction in hexane.57 In a previous study, controlled pore glass derivatised with various organo-silanes was used as a support for RML catalysing the esterification of oleic acid with 1-octanol, and the highest catalytic activity was observed with the most hydrophobic material, containing octadecyl groups.55 Several other studies have confirmed an increase in catalytic activity with increasing hydrophobicity of the support. Among mesoporous organo-silicas, the most hydrophobic ones, containing phenyl groups, led to the highest activity of TLL in a transesterification reaction between 1-butanol and vinyl propionate in hexane.58 Compared to pure silica, the introduction of methylene groups increased the activity of adsorbed ROL, catalysing a transesterification reaction between (S)-glycidol and vinyl butyrate in isooctane.59 Likewise, functionalisation of mesoporous silica with octyl groups increased the catalytic activity of adsorbed BCL in a transesterification reaction between 1-phenylethanol and vinyl acetate in hexane.60 Furthermore, octyl groups were found to provide a good microenvironment for CALB on a mesoporous silica in the acylation of ethanolamine with lauric acid in acetonitrile.61
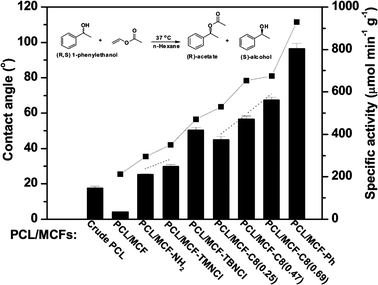 | ||
| Fig. 3 Surface hydrophobicity and specific activity of the adsorbed lipase (denoted PCL in the figure). A series of mesoporous silica materials (MCFs) was prepared with different functional groups. The hydrophobicity of the supports was characterised in terms of the contact angle of water (■). The specific lipase activity in the model reaction given at the top of the figure is depicted by bars. The specific activity clearly increased with increasing hydrophobicity (contact angle) of the surface. Reprinted with permission from Jin et al. Langmuir 2011, 27, 12016–12024. Copyright (2011) American Chemical Society.57 | ||
In order to create pores of the desired size and shape in the mesoporous silica, template molecules, such as surfactants, are used in the preparation procedure. Normally, these are later removed by extraction with an organic solvent and/or by calcination at high temperature. However, it has been shown that it can sometimes be advantageous to leave some of the template in the structure. The catalytic activity of BCL adsorbed on mesoporous silica was seen to increase with increasing amount of template (P123, a polyethylene oxide–polypropylene oxide block copolymer).62 It thus seems that the remaining template provided a suitable microenvironment for the lipase in the otherwise non-derivatised silica, leading to activation in analogy with the hydrophobic groups in the derivatised silicas mentioned above.
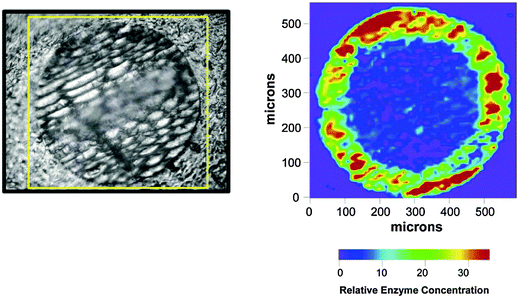 | ||
| Fig. 4 Visible light (left) and infrared (right) images of Novozym 435 (immobilised CALB) showing the uneven distribution of the lipase in the particles. Reprinted with permission from Mei et al. Biomacromolecules 2003, 4, 70–74. Copyright (2003) American Chemical Society.66 | ||
Supports used for lipase adsorption are normally relatively hydrophobic materials, but adsorption of BCL on alginate has been shown to provide a preparation with good catalytic activity and low enzyme leakage, even in aqueous media.67 Furthermore, RML adsorbed on a strong anion-exchange resin expressed quite high activity. In this case, the lipase was adsorbed in the presence of a surfactant, which activated it, and the high catalytic activity remained when the surfactant was removed. This was interpreted as being due to fixing of the lipase in the open conformation by multipoint ionic interaction with the support.68
PCMCs of lipases have been found to express activities up to 200 times higher than the same enzymes in powder form.71 It has also been shown that CALB-PCMCs lost only 10% of their activity after storage as a suspension in propanol with 1% water for one year, while the corresponding preparation of a Pseudomonas lipase lost about 50% of its activity during the same time.72 When different excipients and precipitating solvents were evaluated in the preparation of PCMCs from BCL, it was found that potassium sulphate and acetone provided the catalysts with the highest activity in the ethanolysis of a vegetable oil.73 In most reported studies, the enzyme molecules in PCMCs were not cross-linked. However, cross-linking with glutaraldehyde has been evaluated, yielding cross-linked, protein-coated microcrystals. This procedure could be expected to provide a biocatalyst that is more resistant to enzyme leakage but, surprisingly, the catalytic activity has been observed to be higher than the corresponding PCMC without cross-linking for both CRL and BCL in four different organic solvents.74
It is of interest to compare the reaction rates obtained using adsorbed lipases in organic media with those observed in emulsions, since this could provide information on the extent to which interfacial activation can be achieved by adsorption on a suitable support. Hydrolysis of tributyrin was chosen as a model reaction for such a comparison because it can be carried out either in a water–tributyrin emulsion with free lipase as catalyst, or in neat tributyrin with adsorbed lipase. CALB adsorbed on porous polypropene expressed 49% of the activity in the emulsion system, while for TLL only 9.2% of the activity in emulsion was obtained with the adsorbed enzyme.80 CALB is a lipase that does not show typical interfacial activation, and it is clear that it can be made to work well in the adsorbed form in an organic medium. The relatively poor activity of polypropene-adsorbed TLL and the fact that almost all the competent active sites remain after this immobilisation method77 indicate that rather incomplete interfacial activation was achieved. Thus, there seems to be room for further improvement of lipase adsorption methodology to achieve more efficient interfacial activation.
2.3 Entrapment
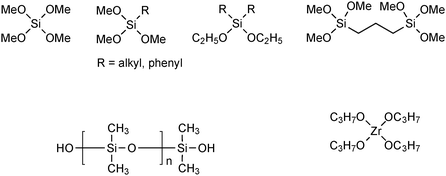 | ||
| Fig. 5 Building blocks for sol–gel materials. | ||
The sol–gel immobilisation method has been further developed. Considerable activation of several lipases has been achieved using more efficient additives, such as Celite, Tween 80 or a crown ether.84 A further variant of the method is to carry out the sol–gel process in a supercritical fluid. Immobilisation of ROL in a silica-based sol–gel provided 5–7 times higher esterification activity when prepared in supercritical carbon dioxide, than when the corresponding material was prepared in aqueous solution.86 It is difficult to judge whether this difference was due to differences in the number of competent active sites, different degrees of interfacial activation or to differences in particle morphology, i.e., particle size, etc.
Zirconia-based sol–gels have been tested as an alternative to the silica-based ones, with the motivation that the former can be prepared under milder conditions.87 However, the observed specific activity of the immobilised Rhizopus lipase was only 3.8% of that of the free enzyme in a hydrolysis reaction. One explanation of this may be that the hydrophobicity of the material was not sufficiently high since no alkyl groups were present in the building blocks.
Another way to entrap enzymes in organic polymer networks is to use electrospinning. Using this approach, Rhizopus arrhizus lipase was entrapped in poly(vinyl alcohol) fibres.89 The fibres were quite small, having a diameter of about 1 μm, and were used to catalyse a transesterification reaction in a flow-through reactor.
Yet another way to entrap enzymes in organic polymers is to use a thermosensitive polymer such as poly-N-isopropylacrylamide. In this method, pre-formed polymer particles were mixed with the aqueous enzyme solution, followed by an increase in temperature (typically to 50 °C) causing shrinkage of the polymer and entrapment of the enzyme. CALB has been immobilised in micron-sized poly-N-isopropylacrylamide hydrogel particles, and the particles were transferred to propanol and subsequently to hexane.90 The shrinkage was reversible, so in water the enzyme could leak out of the particles, while the enzyme remained in the particles in the organic medium as it was not soluble in the reaction medium. Considerable inactivation was observed compared to a procedure involving only low temperature, but since CALB is quite thermostable it is likely that the increased temperature itself did not cause the inactivation and that the procedure could be modified to improve the activity retention and still achieve efficient immobilisation.
2.4 Covalent coupling
Covalent coupling to a solid support is the most typical way of immobilising enzymes, and a wide variety of such methods has been described.41 Many methods described for other enzymes should also be applicable to lipases. Covalent immobilisation frequently causes a limited degree of inactivation of the enzyme, but inactivation can be severe if functional groups in the enzyme of critical importance for catalysis are covalently modified. When immobilising a new enzyme, it is usually a good strategy to try a few different immobilisation methods to ascertain which one is suitable for that particular enzyme. Immobilisation is sometimes carried out in the presence of substrates, substrate analogues or inhibitors, which interact reversibly with the active site, thereby preventing covalent modification in this region. In the case of lipases it might be an advantage to carry out immobilisation under conditions favouring the closed form to protect the active site, and after immobilisation convert it to the open form. However, when immobilisation is carried out by multipoint attachment, the immobilised enzyme cannot change conformation and therefore it should be in the open form during immobilisation, although there are risks of inactivation.There are several commercially available activated support materials intended for covalent enzyme immobilisation. Some of them contain epoxy groups which are intended to react mainly with the amino groups of the enzyme to be immobilised. This can be done by mixing the material directly with an aqueous solution of the lipase. This straightforward way of immobilising CRL on Eupergit supports (acrylate-based materials) containing epoxy groups has been compared with two more complex ways of using the same supports (Scheme 2).91 In both these methods, the support was pretreated with 1,2-diaminoethane to provide amino groups on the support surface. In one of them, this treatment was followed by glutaraldehyde treatment before coupling to the enzyme, and in the other, the carbohydrate groups of the enzyme were first oxidised by periodate to generate aldehyde groups, which could react with the amino groups of the pretreated support. The best retention of activity (about 43%) was observed after immobilisation on Eupergit C according to the latter method.91
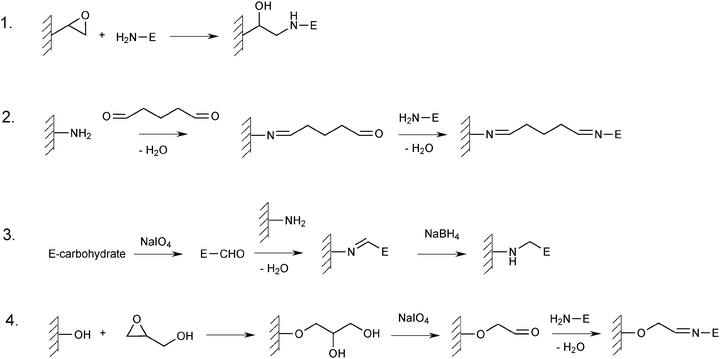 | ||
| Scheme 2 Covalent enzyme immobilisation methods. (1) Direct coupling to epoxy-activated support. (2) Glutaraldehyde-mediated immobilisation on a support containing amino groups. (3) Oxidation of the carbohydrate part of a glycosylated enzyme, and subsequent coupling to a support containing amino groups. (4) Glyoxyl activation of a hydroxyl group containing support and subsequent immobilisation of the enzyme. | ||
Agarose-based materials have been widely used for enzyme immobilisation, and their activation using cyanogen bromide is a classical method. This can be carried out under mild conditions, with high activity retention for CRL, although lower than that obtained following adsorption on a hydrophobic support.92 Immobilisation on glyoxyl-activated materials93 has been used to achieve multipoint attachment, which can be very useful in obtaining high enzyme stability (Scheme 2). Coupling of CRL using this method provided a preparation with high thermostability, although the activity directly after immobilisation was lower than with the other methods.92
A clear advantage of covalent coupling compared to adsorption is that the enzyme remains on the support even under harsh conditions, such as in the presence of surfactants. Covalently immobilised TLL, which had been inactivated by heat or organic solvent, could thus be reactivated by treatment with a CTAB (cetyltrimethylammonium bromide) solution without enzyme leakage from the support.94
Interesting new developments in the area of covalent lipase immobilisation include immobilisation in nanoparticles. Immobilisation in a polyacrylamide nanogel was achieved by covalent acrylation of CRL using N-acryloxysuccinimide as reagent, followed by the addition of acrylamide and the induction of polymerisation.95 The lipase-containing nanogel obtained had much better stability than native lipase in polar organic solvents, and this made it possible to use the biocatalyst for the acylation of dextran with vinyl decanoate in dimethyl sulfoxide.95 In a variant of this method, glycidyl methacrylate was used to acrylate CRL, leading to higher catalytic activity than with the original method.96 The nanogel particles typically contained a single lipase molecule,95 although aggregation of particles occurred under certain conditions.96
Covalent modification of enzymes is not only carried out to achieve immobilisation. Small molecules have been covalently coupled to enzymes to bring about stability of the enzyme towards organic solvents, as illustrated by the modification of BCL with pyromellitic anhydride.97 In this case, chemical modification was followed by the formation of protein-coated microcrystals (see Section 2.2.5). Covalent modification with PEG has been used to achieve solubility of the modified enzymes in organic solvents.98 However, it has been shown that, at least in some cases, the PEG-modified enzymes formed relatively large aggregates in organic solvents.99 Recently, a comb-shaped PEG was used to covalently modify CRL to make it soluble in ionic liquids.100 Interestingly, the activity in a transesterification reaction with vinyl acetate was much higher in ionic liquids than in other organic solvents. The water content in the ionic liquid system affected the catalytic activity to a large extent, with maximal activity around water saturation,100 and thus a water activity close to 1, while CRL usually has a lower optimal water activity (see Section 3.1).
2.5 Cross-linking
Typical cross-linked preparations contain the enzyme as the main constituent, but cross-linking is also used in combination with other immobilisation methods, such as adsorption, to prevent enzyme leakage. As in the case of covalent coupling to supports, it is in most cases the amino groups of the enzyme that participate in bond formation, and glutaraldehyde is the most common cross-linking reagent. Hydration, oligomerisation and aldol condensation reactions occur in an aqueous solution of glutaraldehyde,101 and it is important to bear in mind that simple Schiff's base formation between amino groups and the aldehyde groups of glutaraldehyde is not usually the dominating mechanism, but rather coupling involving the hydrated or oligomerised forms. As a consequence, treatment with sodium borohydride or similar reagents to reduce Schiff's bases is usually not needed to stabilise the bonds after cross-linking with glutaraldehyde.The most common agent used for cross-linking of enzyme crystals is glutaraldehyde, but several other reagents have also been used.104 In the case of lipases, it has been observed that treatment with surfactants can considerably increase the catalytic activity of CLECs.106 It was shown that the main increase in activity was not due to differences in water content. Instead, it is likely that the surfactants caused activation due to at least partial conversion to the open conformation of the lipases (CRL and BCL), as has been observed in other types of preparations in several later studies (see Section 2.6). CALB-CLECs, about 20 μm in size, have been used for the resolution of racemic 1-phenyl ethanol with vinyl acetate in supercritical carbon dioxide.107 No mass transfer limitations were observed and the biocatalyst was used in three consecutive batches. The decrease in conversion to about 50% in the third batch indicates problems, but these were probably due to difficulties in maintaining the enzyme at constant water activity.107
Typical precipitants in CLEA production are salts such as ammonium sulphate, and organic solvents, and the most common cross-linking agent is glutaraldehyde. The catalytic activity of CLEAs prepared from an enzyme varies considerably depending on the precipitation and cross-linking procedures. In a comparison of different methods for the preparation of CLEAs from different lipases, it was found that ammonium sulphate was the best precipitant for some enzymes, and 1,2-dimethoxyethane in other cases.110 In several cases, the presence of surfactants, such as sodium dodecyl sulphate or Triton X-100, during the procedure increased the catalytic activity of the CLEAs, probably due to interfacial activation of the lipases. In another study involving several commercially available lipases, PEG200 was found to be the precipitant giving the most active CLEAs, but it should be pointed out that in a couple of examples (including TLL) simple acetone powders obtained without cross-linking were more active than the CLEAs.111 As with other covalent enzyme modification processes, glutaraldehyde cross-linking can cause enzyme inactivation. In a study of CRL-CLEAs it was shown that CLEAs with maximal catalytic activity were obtained using 25 mM glutaraldehyde for cross-linking.112 Considerable inactivation occurred when a higher glutaraldehyde concentration was used, while poor performance was observed at a lower concentration, probably because of insufficient cross-linking. In a study of BCL-CLEAs it was found that increased glutaraldehyde concentration caused increased thermal stability, but decreased the initial activity.113 It has also been shown that glutaraldehyde cross-linking can cause a reduction in the number of competent active sites of penicillin acylase, and, the milder cross-linker dextran polyaldehyde can be used instead to avoid this.114 In this case, Schiff's bases were formed between the enzyme and the reagent, and were reduced by sodium borohydride to increase the stability of the binding.
For enzymes with only a few lysine residues at their surface, such as CALB, cross-linking can be inefficient. In such cases, cross-linking can be increased by the addition of a polyamine109 or another protein, such as BSA.115
CLEA–silica composites can be prepared by introducing silanes in the CLEA production procedure.109 This opens the way for the introduction of hydrophobic groups, which is of special interest for lipase immobilisation. It is thus possible that lipase preparations with even higher activity could be prepared, analogous to the promising results obtained with mesoporous silicas and sol–gel entrapment (see Sections 2.2.2 and 2.3.1).
2.6 Surfactant-based lipase preparations
In the sections above, it has been mentioned that surfactants can activate lipases in different immobilisation procedures, probably by increasing the fraction of the open, active lipase conformation. In addition, surfactants can be used as the main agent for the preparation of lipases for use in organic media. Because of their high potential in lipase applications, these methods will be discussed here, although some of them do not involve immobilisation.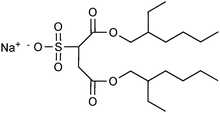 | ||
| Fig. 6 The surfactant Aerosol OT, frequently used together with lipases in organic media to make hydrophobic ion pairs, surfactant-coated lipases and microemulsions. | ||
The most critical issue in the preparation of surfactant-coated lipases is the choice of surfactant, and many different ones have been tried. It should be noted that some of them contain ester bonds, which means that there is a risk that they will be degraded by the lipases used. From this point of view, surfactants without ester bonds are a safer choice, especially when the reaction is lengthy.
Sorbitan esters have frequently been used to prepare surfactant-coated lipase, sorbitan monostearate (Span 60, Fig. 7) being the most popular one. Sorbitan esters with shorter fatty acids have also been tried, but they provided lipase complexes with lower catalytic activity.124 The very hydrophobic sorbitan tri-esters with stearic acid or oleic acid (Span 65 and Span 85) have also been used. In the coating of CRL, Span 85 provided the biocatalyst with the highest esterification activity of nine surfactants tested, but Span 60 was not included in the comparison.125 Other commercially available surfactants used to form complexes with lipases include Tween 80, Triton X-100126 and propylene glycol stearate.127
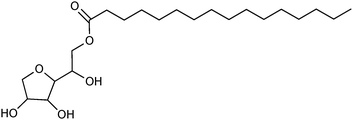 | ||
| Fig. 7 Span 60, a surfactant frequently used for preparing surfactant-coated lipases. | ||
In addition to the commercially available surfactants, special surfactants have been synthesized in research laboratories128 and used for enzyme coating. Very good results have been obtained with esters of N-D-glucono-L-glutamic acid (Fig. 8).129 In a comparison of different derivatives, the highest activity in an esterification reaction was obtained with dioleoyl N-D-glucono-L-glutamate (2C18Δ9GE).130
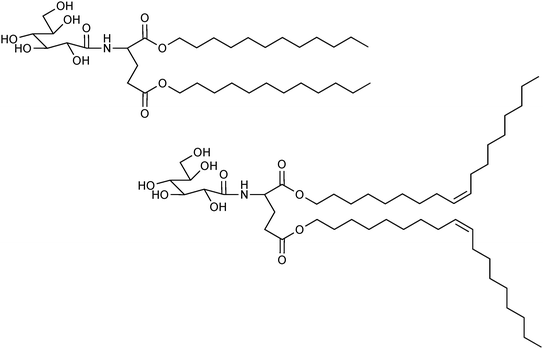 | ||
| Fig. 8 Two surfactants used for preparing surfactant-coated lipases. Above: dilauryl N-D-glucono-L-glutamate (2C12GE) and below: dioleoyl N-D-glucono-L-glutamate (2C18Δ9GE). | ||
Most surfactants used for lipase coating are non-ionic, but the use of ionic surfactants has also been reported. BCL coated with an ionic surfactant was shown to be useful in dynamic kinetic resolution of secondary alcohols, since it was compatible with the ruthenium catalyst used for the racemisation reaction.123 Lipases containing ionic surfactants can be called ion-paired lipases if they are dissolved, but if the solubility is not proven, they should rather be included in the surfactant-coated lipases.
Ionic liquids have been used to coat lipases in a similar way to surfactants. D-Pro-Me (Fig. 9) was found to be the most favourable ionic liquid for coating BCL for the catalysis of a transesterification reaction with vinyl acetate in diisopropyl ether.131
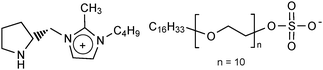 | ||
| Fig. 9 An ionic liquid used for coating lipase (abbreviated: D-Pro-Me). | ||
Conflicting reports are found in the literature on the solubility of surfactant-coated lipases in organic solvents. Lipases coated with 2C18Δ9GE were easily dissolved in isooctane, while those coated with Span 60 or dilauryl N-D-glucono-L-glutamate (2C12GE) were reported to be insoluble.122 However, it has been claimed that lipases coated with 2C12GE and several related surfactants are freely soluble in most organic solvents such as isooctane, diisopropyl ether, ethyl acetate and dimethyl sulfoxide.119,132 Good evidence of true solubilisation of single lipase molecules has been obtained using gel permeation chromatography. A Rhizopus lipase coated with 2C12GE was studied in dichloromethane and was shown to consist of enzyme molecules coated with about 150 surfactant molecules.129 The solubility of complexes obtained with sorbitan monostearate was quite low in most dry organic solvents, but increased with increasing water content.133 It is possible that the presence of salts or other components in some of the surfactant-coated lipases could have caused the apparent contradicting reports concerning solubility in organic solvents. Furthermore, reversed micelles or other structures might also have been formed when water was present.
A different way of forming surfactant-coated lipases is to solubilise the lipase in an Aerosol OT-based microemulsion, followed by drying.85 The preparations obtained expressed good catalytic activity, although lower than preparations of the same lipases on porous polypropene.
Surfactant-coated lipases not soluble in the reaction medium can be considered rather typical immobilised preparations with the surfactant as support material.85 As with other immobilisation methods, lipase powder is often used for the comparison of catalytic activity, and the surfactant-coated lipases usually express considerably higher activity.85,121
• Injection of an aqueous enzyme solution into an organic solution containing the surfactant
• Extraction of the enzyme from an aqueous solution into an organic phase containing the surfactant
• Dissolution of the enzyme from a solid phase into an already prepared microemulsion
One important advantage of microemulsions as media for enzymatic reactions is that hydrophobic substrates can be dissolved in the organic phase and hydrophilic ones in the aqueous phase. They are, therefore, suitable for reactions involving both types of substrates, such as the synthesis of sugar esters.137 Mass transfer between the phases is rapid because of the dynamics and small dimensions of the systems. In most cases, the size of the reversed micelles is suitable for one enzyme molecule.
Most studies of enzymatic reactions in microemulsions have focussed on fundamental aspects and measurements of catalytic activity, while the isolation of reaction products has seldom been carried out. Product isolation is complicated by the presence of surfactants, but in some cases this problem can be solved by breaking the microemulsion by a moderate change in temperature.138
A drawback of some microemulsion systems is that enzyme inactivation can occur, due to the presence of the surfactant and the large interface between the organic and aqueous microphases. Structural changes compared to aqueous solutions have been detected using a variety of methods, such as circular dichroism spectroscopy, fluorescence spectroscopy and NMR.136 The sensitivity to this kind of inactivation varies between the lipases139 and depends on the location of the enzyme in the microemulsion.135,136 When a Rhizopus lipase was solubilised in a microemulsion, the observed kcat values of a hydrolysis reaction decreased with increasing surfactant concentration, which was interpreted as being due to unfavourable interaction with the surfactant layer.140
Aerosol OT has been used in the majority of the studies on lipases in microemulsions,134–136 and isooctane is the most common organic solvent. However, it has been observed that microemulsions can cause inactivation, as exemplified by cutinases, which lost their catalytic activity in Aerosol OT/isooctane-based microemulsions due to unfolding in minutes or hours, depending on the type of cutinase.141 It has been shown that the addition of non-ionic surfactants, such as Spans, Tweens, short-chain PEG and alkyl glycosides can increase the activity of the Chromobacterium viscosum lipase in an Aerosol OT/isooctane-based microemulsion.142,143 Interestingly, both the catalytic activity and stability of the same lipase were increased by the presence of aprotic solvents in the Aerosol OT/isooctane-based microemulsions, dimethyl sulfoxide being the best solvent.144 The half-life was increased from 33 days without solvent to 125 days with dimethyl sulfoxide.
As an alternative to Aerosol OT, phospholipids have been evaluated in the preparation of microemulsions for lipases. Microemulsions based on different types of lecithin were shown to provide TLL with higher activity than Aerosol OT-based microemulsions.145 In a study of a Rhizopus lipase in microemulsions, higher catalytic activity was observed when a mixture of Aerosol OT and lecithin was used, than when either surfactant was used alone.146 Also, cutinase has been shown to be catalytically active in phosphatidylcholine-based microemulsions.147
In most cases hydrocarbons have been used as the organic phase in the microemulsions.134,135 However, other solvents can be used, and an interesting recent development is the use of ionic liquids as the organic phase. It has been shown that Vmax in a hydrolysis reaction catalysed by BCL was higher in an AOT/ionic liquid-based microemulsion, than in the corresponding AOT/isooctane-based system.148 The activities and stabilities of CRL, TLL and Chromobacterium viscosum lipase have been studied in microemulsions based on non-ionic surfactants (Tween 20 or Triton X-100) and the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF6).139 The longest enzyme half-lives were obtained in the microemulsions with low water content, and the Chromobacterium lipase generally exhibited better stability than CRL and TLL.139
Incorporation of nanoparticles into microemulsions was recently evaluated in a system based on Aerosol OT or CTAB with 1-hexanol as cosurfactant in isooctane. The addition of gold nanorods more than doubled the observed catalytic activity in both cases.149
2.7 Comparison of lipase preparation methods
Unfortunately, few direct comparisons of several immobilised lipase preparations have been made. It is difficult to compare the results obtained in different studies because they often differ in the reaction catalysed, the solvent, the substrate concentration, the reaction temperature, etc. In a typical report on a new lipase immobilisation method, the properties of the immobilised lipase were simply compared with that of “free lipase”, which is usually not sufficiently well defined as to allow direct comparisons. Excellent guidelines for reporting biocatalytic reactions including those using immobilised biocatalysts have been published,152 but still many reports in the literature lack important information.In one of the few comparative studies involving several immobilisation methods, an esterification reaction was catalysed by TLL prepared in different ways and used under identical reaction conditions (Table 3).85 The specific activity of unmodified TLL powder (free enzyme) was low, as expected. Freeze-drying from an appropriate buffer solution led to an increase in activity, which was further increased when a crown ether or KCl was used as additive before freeze-drying. Even higher specific activity was observed after surfactant coating or sol–gel entrapment, and the highest specific activity was obtained when the enzyme was adsorbed on a porous polypropene support. It is worth noting that the most active preparation was 400 times more active than the unmodified TLL. Similar effects were observed with CRL, RML and BCL, although the differences were somewhat smaller: the activity of the polypropene-adsorbed lipase was between 20 and 110 times that of the lowest activity. However, in a study of a Pseudomonas fluorescens lipase adsorbed on different supports, the highest activity was observed for that adsorbed on mesoporous, underivatised silica, while the activity on porous polypropene was almost 3 times lower.153 The reason for this discrepancy may be that the latter study was carried out at low enzyme loading,153 which could have caused inactivation of this particular lipase on the polypropene surface.
| Lipase preparation method | Relative specific activity (%) |
|---|---|
| a Reaction: TTL-catalysed esterification of 1-phenylethanol (50 mM) with caproic acid (100 mM) at a water activity of 0.33 in hexane. 100% relative specific activity corresponds to 6.8 μmol min−1 g protein−1. Data from Persson et al.85 | |
| Adsorption on polypropene | 100 |
| Sol–gel entrapment | 79 |
| Coating with Span 60 | 38 |
| Freeze-drying with KCl | 11 |
| Coating with Span 65 | 10 |
| Freeze-drying with crown ether | 10 |
| Coating with Aerosol OT | 2.1 |
| Freeze-drying | 1.1 |
| Unmodified enzyme | 0.25 |
It is clear that adsorption on porous hydrophobic supports is a very useful method for lipase immobilisation. A highly hydrophobic surface has been shown to be beneficial in several studies mentioned in this review, although there are contradicting observations. The hydrolytic activity of TLL was found to be higher when it was adsorbed on the underivatised silicon dioxide surface than when adsorbed on the corresponding surface derivatised with octadecyl groups.154 In the case of octadecyl-containing Sepabeads, the addition of polyethyleneimine to the preparation increased the initial reaction rate of RML and CRL when catalysing the esterification of lauric acid with octanol.155 The addition of polyethyleneimine must have made the microenvironment of the lipases more hydrophilic, and it thus appears that the highest hydrophobicity is not always the best.
Concerning pore size, diameters around 100 nm seem to be advantageous, although few direct studies have been carried out on the importance of this parameter. Most mesoporous silicas are thus likely to have somewhat too narrow pores,53 while some of the porous organic polymers used for lipase immobilisation have a considerable fraction of their pore volume in large enough pores, such as the polypropene Accurel EP-100,156 and Accurel MP-1004.157
In order to avoid mass transfer limitations, the diffusion of substrates and products should not be hindered and diffusion distances should not be too long. When enzyme molecules are present inside the particles, the particle size should be as small as possible. On the other hand, the preparation should be easy to handle, which often means that the particles should not be too small, so in reality a suitable compromise must be found. In most cases, the support particles used for lipase immobilisation are more or less spherical in shape. Alternative particle shapes include fibres. Electrospun polyacrylonitrile fibres with a diameter of around 400 nm constitute an interesting example.158 Further examples of lipase adsorption on small particles include nanoparticles of fumed silica,75 tin dioxide159 or superparamagnetic Fe3O4,160 as well as silica nanotubes.161 These lipase preparations could be of interest in special applications, especially when mass transfer limitations would occur in other types of preparations.
In this review, lipase preparations are mainly compared with respect to the specific activity of the lipase, which is highly relevant if the enzyme is to be utilised as efficiently as possible. However, it should be pointed out that for some applications a high catalytic activity per unit weight of immobilised preparation, or per unit reactor volume, may be more important.
The surfactant-based methods have great potential to provide highly active lipase preparations, because not only can the lipases be activated, but in some cases the surfactant can lead to isolated lipase molecules dissolved in the organic medium as ion pairs, soluble surfactant-coated enzymes or in reversed micelles, thus allowing high mass transfer.
For industrial applications of immobilised enzymes, all factors influencing the costs should be considered. The costs of enzyme, support and other ingredients are thus important, as well as the operational stability of the lipase.162
3. Water dependence of lipase-catalysed reactions
Some water is always present, even when using organic solvents as reaction media for lipase-catalysed reactions, and that water has a profound effect on the performance of the enzyme.163 In early studies of biocatalysis in organic solvents, it was observed that the catalytic activity of enzymes correlated well with the amount of water bound to the enzyme.164 However, it is difficult to measure and adjust the amount of water bound to the enzyme. Halling proposed the use of water activity to quantify the amount of water present in organic reaction mixtures for biocatalysis, since this parameter correlates well with the amount of water bound to the enzyme under a wide range of conditions, especially in water immiscible solvents.165,166 The water activity varies between 0 in a completely dry system and 1 in a water-saturated system, and it is equal in all phases at equilibrium. The advantage of using the water activity is illustrated by the water dependence of an enzymatic reaction in a range of solvents. The optimal water concentrations in the different solvents are different, while the optimal water activity is largely the same.165 Likewise, early observations on the effects of the support material on enzymatic activity indicated that the water absorbing capacity (called the aquaphilicity) of the support material influenced the catalytic activity of the immobilised enzyme to a large extent.167 When reactions were carried out at fixed water activity, almost identical activity profiles were obtained on different supports,168 although there may be deviations for some combinations of supports and enzymes.1693.1 Optimal water activity for lipases
It is important to have quantitative knowledge on the influence of water activity on enzyme activity since the effects are so large. Simple experiments can be used to determine this relationship, and the resulting curves showing enzyme activity as a function of water activity (water activity profiles) constitute important guidelines for practical applications. Ideally, the water activity dependence of kinetic constants (such as kcat and Km) should be determined, but in most cases only the enzyme activities observed at a fixed substrate concentration are reported in the literature, and it should be kept in mind that the profiles can be shifted by a change in substrate concentration.28There is a large variation among the enzymes concerning how the water activity in organic media influences their catalytic activity.170–172 Some lipases, such as BCL, behave in a way similar to other enzymes, with maximal activity at high water activity. However, CRL has an optimum at intermediate water activity according to most reports, while RML, Rhizopus lipases and CALB show optimal activity at low water activity.173 In the case of RML, catalytic activity has been detected down to a water activity of 0.0001.174 Reported optimal water activities for lipases are listed in Table 4.
| Lipase | Optimal water activity | Substrates, medium | Ref. |
|---|---|---|---|
| a Different values depending on the support used.b From different Rhizopus strains. | |||
| BCL | 0.69 | Vinyl acetate + 1-phenylethanol, hexane | 175 |
| 0.75 | Caproic acid + 1-phenylethanol, hexane | 77 | |
| 0.97 | Decanoic acid + dodecanol, diisopropyl ether | 28 | |
| 0.97 | Decanoic acid + dodecanol, hexane | 172 | |
| CALB | 0 | Methyl propionate + propanol, gas phase | 176 |
| CRL | 0.53 | Ethyl decanoate + hexanol, diisopropyl ether | 177 |
| 0.56 | Vinyl acetate + 1-phenylethanol, hexane | 175 | |
| 0.75 | Decanoic acid + dodecanol, diisopropyl ether | 28 | |
| 0.77–0.97a | Decanoic acid + dodecanol, hexane | 172 | |
| RML | 0.55 | Decanoic acid + dodecanol, hexane | 172 |
| Rhizopusb | 0.06 | Ethyl decanoate + hexanol, diisopropyl ether | 177 |
| 0.19 | Vinyl acetate + 1-phenylethanol, hexane | 175 | |
| 0.32–0.55a | Decanoic acid + dodecanol, hexane | 172 | |
| 0.53 | Decanoic acid + dodecanol, diisopropyl ether | 28 | |
| TLL | 0.33 | Caproic acid + 1-phenylethanol, hexane | 77 |
| 0.77–0.97a | Decanoic acid + dodecanol, hexane | 172 | |
Water can thus have both positive and negative effects on the rate of lipase-catalysed reactions. Several such effects are listed below and will be further discussed in the following sections.
• Increased active site polarity
• Increased proton conductivity
• Functions as substrate (increases hydrolysis only)
• Formation of a diffusion barrier for hydrophobic substrates
• Causes hydrolysis which competes with the desired reaction (esterification, transesterification)
3.2 Activation by water
Different amounts of water are bound to an enzyme molecule, depending on the water activity in the surroundings. Simulations of molecular dynamics have been used to model the hydration of a lipase (CALB) in the gas phase178 and in an organic solvent.179 In an organic medium with low water activity, single water molecules are bound to specific sites, while at higher water activity, networks of water molecules are formed on the enzyme surface.178 In most cases, the bound water molecules have a positive effect on enzyme activity. Water has been described as a molecular lubricant of enzymes,170 and the increased internal flexibility can be assumed to facilitate the movements necessary for catalysis.163 Simulations of molecular dynamics for CALB indeed showed that water caused increased internal flexibility.178 In addition, an increase in the amount of water in the region of the active site increases the local polarity, which stabilises polar transition states, thereby promoting enzymatic reactions having such transition states.180 Another activating effect of water could be due to facilitation of proton conductivity.163 While the positive lubricating effect can be expected to be common to most enzymes, the effects due to increased active site polarity and proton conductivity depend on the catalytic mechanism of the enzyme.3.3 Inhibition by water
As mentioned above, most bound water molecules have a positive influence on enzyme activity. However, if water molecules bind in the active site they can interfere with binding of the substrate, thereby acting as a true competitive inhibitor.181 This phenomenon has been studied in great detail for CALB-catalysed alcoholysis of methyl propionate with n-propanol in solid/gas systems,176,182 and the inhibition constant for water in terms of activity has been determined to be 0.128.176 CALB has a higher affinity for the substrates in gas phase than in organic solvents,176 but the data from the solid/gas systems are useful for predictions of the kinetics in organic solvents.183If multilayers of water molecules are present between the bulk organic solution and the active site of the enzyme, this can form a diffusion barrier for hydrophobic substrates, which slows down the overall conversion rate.184
3.4 Water as substrate
Water is a substrate in hydrolysis reactions, and it is thus natural that increasing water activity has a positive effect on such reactions, although other effects discussed above sometimes dominate. On the other hand, when esterification or transesterification are the main reactions, water molecules can interfere by deacylating the acyl-enzyme. In the esterification reaction, this is a form of product inhibition leading to a reduction in the observed reaction rate. An increase in alcohol concentration favours esterification vs. hydrolysis, and the effect of water on the kinetics can thus be observed as an increase in the apparent Km value for the alcohol.28 Furthermore, the water activity profiles were shifted towards higher water activity with increasing substrate (alcohol) concentrations, due to the competition between water and alcohol.28In the transesterification case, competing hydrolysis leads to separate products. Normally an increase in water concentration leads to an increase in the hydrolytic side reaction and a decrease in the rate of transesterification itself. The acyl donor is consumed in both the competing reactions and is thus consumed at a rate that is the sum of the two individual rates.
3.5 Water activity profiles of different types of reactions
The kinetics of lipase-catalysed esterification reactions have been studied in detail,28 and a useful model has been constructed.185 Likewise, the kinetics of alcoholysis reactions have been studied in great detail, especially in gas phase reactions.176,182 Water activity profiles of esterification and transesterification reactions are often bell-shaped because of the activating effect of water in the low water activity range and the competing hydrolytic reactions in the high water activity range.28,176 Many lipases express good catalytic activity at low water activity, and under these conditions, other nucleophiles can compete successfully with water for deacylation of the acyl-enzyme, explaining the observation that lipases are much more efficient catalysts in transesterification reactions, than other hydrolytic enzymes in corresponding reactions, such as glycosidases in transglycosylation reactions.177Intuitively, one would expect transesterification and hydrolysis reactions to have quite different dependences on the water activity. However, if the acylation step is rate determining and alcohol inhibition is of less importance, the two reactions can actually have almost identical water activity profiles.186
Water clearly has competing effects in hydrolysis reactions. Water acts as a substrate, which causes an increase in the reaction rate with increasing water activity, while the inhibiting effect of water acts in the other direction. As a result, bell-shaped activity profiles are often observed with a maximum at intermediate water activity.173 In extreme cases, the inhibiting effects dominate, and the fastest hydrolysis reaction occurs at the lowest water activity. CALB-catalysed hydrolysis of lutein esters is one such example.184 This behaviour is more likely to occur when low substrate concentrations are used, as it is then easier for water to act as a competitive inhibitor. Furthermore, the lipase used should be one with high activity at low water activity.
3.6 Effects of water on stereoselectivity
Water can influence the stereoselectivity of reactions by binding in substrate-binding pockets (stereospecificity pockets), thereby selectively interfering with the binding of one of the enantiomers of the substrate.187 In the case of CALB, molecular modelling has been combined with detailed experimental studies to show that the binding of a single water molecule was of crucial importance in determining the enantioselectivity in the reaction.1883.7 Control of water activity
Because the water activity of the organic reaction medium is of great importance in lipase catalysis, there is a need to measure and control water activity in such media, and there are a number of methods to do this.189 In small-scale reactions, the water activity can be controlled by equilibration with saturated solutions of inorganic salts.189,190 Normally, equilibration occurs via a gas phase, but the process can be speeded up by shortening the diffusion distance for water. Thus, the saturated salt solution can be contacted with the reaction mixture by pumping it through silicon tubing passing through the reactor.191 An alternative way of controlling the water activity in organic reaction media is to add pairs of salt hydrates that act as a buffer of the water activity.192 In this method, it is important to ensure that the salt used is compatible with the enzyme, since it is added directly to the reaction mixture.Good sensors for direct measurement of the water activity in a gas phase are commercially available. Such a sensor can be used to measure the water activity in the gas phase of a bioreactor, and the data can be used to adjust the water activity in the desired direction. The system works best with a gas flow through the reactor, which facilitates water activity equilibration between the phases, and the gas flow can be either dry, to decrease the water activity, or water saturated to supply more water to the reactor.193 The whole set-up should preferably be computer controlled.
4. Applications involving lipase-catalysed reactions
4.1 Ester synthesis
The majority of applications of lipases in organic media are in the preparation of esters, and a wide variety of esters has indeed been prepared.120 This first section focuses on the synthesis of esters from monovalent carboxylic acids and monovalent alcohols, while more complex examples are presented in later sections.Esters can be prepared using chemical catalysts, but lipase catalysis is preferable for some applications. For example, there is great interest in the lipase-catalysed synthesis of flavour and fragrance esters for food applications because the products can be labelled natural, which is not the case when a chemical catalyst is used. Typical esters in this group include esters of acetic, butyric and caproic acid, and alcohols such as ethanol, butanol and isopentanol, as well as more complex ones such as geraniol.194,195 Short-chain carboxylic acids are relatively polar and tend to partition to the microenvironment around the lipase and decrease the local pH, which can cause a reduction in catalytic activity. It has been observed that the activity of Novozym 435 was quite low with acrylic acid as acyl donor.196 In a study of Novozym 435-catalysed esterification reactions involving several carboxylic acids it was found that high activity was obtained with propionic and butyric acid, but low activity with acetic acid and several substituted carboxylic acids, such as malic acid and 2-chlorobutyric acid.197 It was noted that the substituted carboxylic acids had lower pKa values, which may be the reason for the rate reducing effect. Further indications that the reduced activity was caused by the acidity are the positive effects of adding bases such as triethylamine to the reaction mixtures.196,197 A practical way to avoid the problem is to use esters of the carboxylic acids as acyl donors instead.197,198 The fact that this approach works shows that steric factors are not the major problem with these acids.
Esters composed of long-chain carboxylic acids and long-chain alcohols are called wax esters. Enzymatic production is used especially for wax esters intended for use in personal care products. Almost quantitative conversion can be achieved from equimolar mixtures of reactants, provided that water is efficiently removed from the reaction mixture, which can be achieved by blowing dry air through a stirred tank reactor,199 or by using a bubble column reactor.200 In wax ester synthesis, all the reactants have a sufficiently low volatility to make selective water removal easy, but in esterification involving short-chain alcohols and/or volatile solvents, extra precautions must be taken. In the solvent-free esterification between isopropanol and myristic acid, water and isopropanol were removed by azeotropic distillation at reduced pressure,201 and dry isopropanol was added to compensate for the alcohol removed. Distillation has also been used to remove water in esterification with short-chain alcohols in hydrocarbons such as hexane, thereby increasing the esterification yields.202 The hydrocarbon solvent that evaporated was condensed and recirculated after being passed through a drying agent. The condensate can also be allowed to phase separate to remove most of the water (together with some alcohol), before returning the hydrocarbon solvent to the reactor via a drying column.
Lactic acid constitutes an interesting substrate in esterification reactions since it contains both a carboxyl group and a hydroxyl group. CALB in the form of Novozym 435 was shown to be an efficient catalyst for making esters from alcohols and lactic acid, but not when using lactic acid as a nucleophile, and lactic acid esters were, therefore, produced without interfering oligomerisation reactions.203 In other cases, where oligomerisation or polycondensation is desired to yield polyesters, this can be efficiently carried out using lipases as catalysts, as recently reviewed.204
4.2 Triacylglycerols
Lipases are excellent tools for the synthesis of triacylglycerols or the modification of existing triacylglycerols in organic media.205 Lipases express various types of specificity with respect to triacylglycerol substrates. The regiospecificity is of special importance since many lipases are specific for acyl residues in the sn-1 and sn-3 positions on the glycerol backbone. Specificities for fatty acid chain length and unsaturation must also be kept in mind, but have been utilised to a minor extent for triacylglycerol production. Stereospecificity, which is important in other applications, has been exploited in the synthesis of chiral triacylglycerols, although to a rather limited extent.206sn-1,3-specific lipases are excellent tools for the production of triacylglycerols with different fatty acid compositions in the sn-1 and sn-3 positions compared with the sn-2 position. Lipases are now also being used for fatty acid exchange reactions in triacylglycerols requiring no regiospecificity, an important example being interesterification (the exchange of fatty acids between different triacylglycerol molecules) of margarine fats (see Section 4.2.2).Many other examples of lipase-catalysed preparation of structured lipids have been presented, as reviewed by Xu.208 The fatty acid to be incorporated is normally added as a free fatty acid, and the reaction is called acidolysis (Scheme 3), but fatty acid esters209 or triacylglycerols207 can also be used as acyl donors. In order to achieve a very high proportion of the new fatty acid in the sn-1,3 positions, a high excess of this fatty acid must be used. One way to obtain even purer products is to use a two-step process in which the fatty acids in the sn-1,3 positions of the original substrate are removed, followed by isolation of the 2-monoacylglycerol, before the new fatty acid is introduced in the second step (Scheme 3).210 The first step, the conversion of a triacylglycerol to a 2-monoacylglycerol, is best carried out by alcoholysis using ethanol.211 It is important to minimise acyl migration in both the one-step acidolysis reaction and the two-step process, as this can lead to products with fatty acids in the wrong positions. Parameters of crucial importance are the choice of support used for lipase immobilisation, the solvent, and the water activity of the reaction medium.211–213 In one case, the use of ethyl esters as acyl donors was shown to give less acyl migration than free fatty acids.209
 | ||
| Scheme 3 Lipase-catalysed preparation of structured lipids. (A) One-step acidolysis. (B) Two-step process. In the first step fatty acids in the sn-1,3 positions are removed by alcoholysis (as shown above) or by hydrolysis. After isolation, the 2-monoacylglycerol is esterified with the fatty acid to be incorporated. | ||
Important structured lipids include human milk fat substitutes, which contain a large proportion of palmitic acid in the sn-2 position and oleic acid in the sn-1,3 positions.214,215 Long-chain omega-3 fatty acids have been incorporated into structured lipids because of their health-promoting properties. In most cases they are present in the sn-2 position, but structured lipids with these fatty acids in the sn-1,3 positions have also recently been produced.209
The operational stability of the lipase is of key importance for process economy, and it has been shown that lipase stability depends on the purity of the substrate oil. Acidic contaminants and lipid oxidation products are thus detrimental to stability.218,220
The enzymatic interesterification process has so far mainly been used to prepare products resembling those obtained with the chemical method, thus completely randomised in all positions. However, the lipases used are more or less sn-1,3-specific and randomisation in the sn-1,3 positions is therefore much more rapid than in the sn-2 position.216,221 Almost completely randomised products can often be obtained with an extended reaction time due to acyl migration and/or incomplete sn-1,3 specificity. It should be pointed out that the sn-1,3 specificity of lipases should be regarded as an opportunity rather than a problem, since this makes it possible to produce a range of products with a high degree of randomisation in the sn-1 and 3 positions, and a degree of randomisation in the sn-2 position that can be controlled by choosing the appropriate reaction conditions.
4.3 Enantiomer resolution
Lipases are the most frequently used biocatalysts in organic synthesis. Most applications concern the synthesis of chiral intermediates by kinetic resolution or desymmetrization reactions.222–224 In the kinetic resolution of a racemic mixture, the lipase converts one of the enantiomers into a product that can easily be separated from the unreacted isomer. In the ideal case, the enzyme converts just one of the enantiomers, but in reality the other enantiomer is often converted, but at a lower rate. The success of kinetic resolution depends critically on the specificity of the lipase, which is best described as the enantiomeric ratio, defined as the ratio of the specificity constants of the enzymatic conversion of the two enantiomers. A quantitative analysis of the influence of the enantiomeric ratio and the conversion in the reaction on the enantiomeric excess of the product and the remaining substrate has been presented by Sih and co-workers.225The traditional method of using hydrolases for kinetic resolution is to carry out hydrolysis reactions, in the case of lipases, the hydrolysis of esters. However, in organic media the lipases can also be used to catalyse esterification and transesterification reactions. Unless the equilibrium position is shifted far towards the product side, the reversibility of these reactions can cause a reduction in the enantiomeric excess.226 On the other hand, the fact that the same lipase can be used to catalyse three types of reactions (hydrolysis, esterification and transesterification) is a great advantage since this increases the chance of finding a reaction with a sufficiently high enantiomeric ratio to be practically useful (Scheme 4). Furthermore, the problems associated with reversibility can be avoided by the use of vinyl or isopropenyl esters as acyl donors, as they give unstable alcohols as side products, thereby making the reactions practically irreversible.227
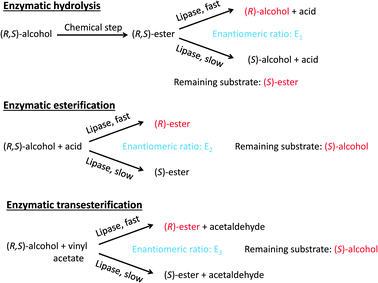 | ||
| Scheme 4 Lipase-catalysed reactions used for the resolution of the two enantiomers of a racemic alcohol. The two main products in each case are given in red (the reaction product and the remaining substrate). The lipase converts the R-enantiomer of the substrate faster than the S enantiomer in all cases, but the enantiomeric ratio (E) can be different in each case. | ||
Lipase-catalysed reactions can be scaled up to industrial scale:228 for example, the BASF process for the production of chiral amines (Scheme 5A). In this case, one enantiomer of the racemic amine is acylated using an alkyl methoxyacetate as acyl donor and an immobilised Burkholderia lipase or CALB as catalyst. Several different amines can be resolved using this process. In a few other examples, vinyl acetate or isopropenyl acetate are used as acyl donors in stereoselective acylation to yield chiral building blocks that are useful as intermediates in the production of pharmaceuticals (Scheme 5B–D). The ring opening of an oxazolin with butanol in Scheme 5E is a step in the synthesis of L-tert-leucine. All reactions shown in Scheme 5 are catalysed by immobilised lipases and are carried out in organic solvents or solvent free mixtures.
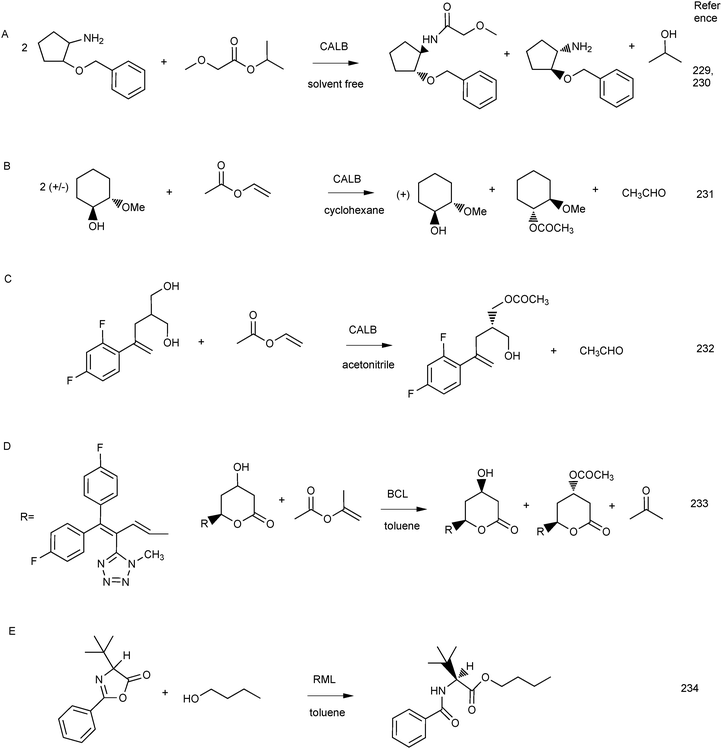 | ||
| Scheme 5 Examples of lipase-catalysed reactions used on a large scale for the production of chiral building blocks. | ||
A key issue in the practical use of enantiomer resolution is the utilisation of not only the desired enantiomer, which is the product, but also the other enantiomer, which constitutes 50% of the starting material.235 This can be achieved by batch-wise racemisation, but even better with continuous racemisation of the substrate during enzymatic conversion. The latter approach is called dynamic kinetic resolution, and can be used to convert a racemic substrate into an enantiomerically pure product. Ruthenium, rhodium and iridium complexes have been shown to be useful for the racemisation of alcohols during their conversion to esters,236,237 and a palladium catalyst can be used in the resolution of amines.228 It is of vital importance that the metal–organic catalyst does not racemise the esters and does not inactivate the lipase. Typical reaction conditions for CALB involve isopropenyl acetate as acyl donor, a ruthenium-based catalyst, and toluene as solvent.237
4.4 Fatty-acid enrichment
In analogy with the kinetic resolution of enantiomers, enzymes can also be used for the enrichment of other isomers or closely related substances, provided that they have the appropriate substrate specificity. The basic principle is that the substance to be enriched is converted considerably faster or slower than the other substances in the starting mixture. An important example is the lipase-catalysed enrichment of fatty acids. The specificity of lipases for fatty acids depends on chain length, the number and position of double bonds, as well as other structural features such as further substitution (branching, hydroxylation, etc.) of the fatty acid.The main driver for research in enzymatic fatty acid enrichment is the interest in long-chain omega-3 fatty acids, such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). These are present primarily in marine oils and have been shown to have considerable health-promoting effects, and there is thus an interest in products with higher proportions of these fatty acids (nutraceuticals) than in natural oils. Lipases can be useful tools since many of them discriminate strongly against these fatty acids compared to other fatty acids normally present. Enzymatic processing is attractive due to the mild reaction conditions, although other separation methods such as urea fractionation and distillation still dominate. Lipase-catalysed enrichment of long-chain omega-3 fatty acids was recently reviewed.238
In practical applications, the starting material for fatty acid enrichment is typically a fat or oil containing mainly triacylglycerols, which can be used directly as substrate for the lipase-catalysed reaction. The reaction mixture should preferably be liquid, and organic solvents are thus sometimes used to dissolve high-melting fats, while solvent-free mixtures are often used for low-melting substrates. The reactions are normally carried out as hydrolysis or alcoholysis reactions.
The lipase fatty acid selectivity is often expressed in terms of competitive factors, which are ratios of the specificity factors of a pair of substrates, analogous to the enantiomeric ratio for enantiomer resolution (see Section 4.3). One fatty acid is selected as the reference substrate and the specificity of the enzyme for other fatty acids is related to the specificity to the reference. Several competitive factors for EPA and DHA in the range 10–100 have been published, showing that lipase-catalysed enrichment of these fatty acids can be quite efficient.239–241 When carrying out enrichment using triacylglycerols as substrates, it is important to bear in mind that the regiospecificity of the lipase also influences the outcome.242 Depending on the distribution of fatty acids within the triacylglycerols, it may thus be advantageous to choose either an sn-1,3-specific lipase or a nonregiospecific one. In order to achieve high degrees of enrichment, it can be beneficial to carry out repeated enzymatic steps with separation steps in between. Using repeated hydrolysis of salmon oil, the content of long-chain omega-3 fatty acids was increased from 16 to 51%.243 In the enrichment of EPA and DHA from squid oil, ethanolysis of the oil without extra solvent was quite efficient, increasing the combined content of EPA and DHA from 31 to 51% in a single enzymatic step.244
Although enrichment starting directly from the natural oil can be attractive, it has been shown that higher competitive factors, and therefore more efficient enrichment, can be achieved when free fatty acids or simple fatty acid esters are used as substrates.241 The full fatty acid selectivity is only expressed when each substrate molecule contains just one fatty acid residue. It has been found that esterification provides the best enrichment for some combinations of enzymes and substrates, while hydrolysis is more selective in other cases.245,246 Almost all lipases discriminate against the long-chain omega-3 fatty acids, but lipases originating from fish intestines hydrolysed EPA and DHA from triacylglycerols faster than more saturated fatty acids.247
It has been shown that the methodology for quantitative analysis of enantiomer resolution developed by Sih and co-workers can be developed further to include multisubstrate mixtures.246 The competitive factors can easily be determined from experiments using initial rate measurements in mixtures of the substrates of interest. Based on the competitive factors, a strategy can be developed for the enrichment of a certain fatty acid with a certain purity (Fig. 10). If high purity is desired, it is beneficial to use lipases with higher specificity for the fatty acids to be removed, so that the fatty acid of interest is enriched in the remaining substrate fraction.
 | ||
| Fig. 10 Simulation of fatty acid enrichment in an equimolar mixture of three fatty acids A, B and C. The competitive factors (ratios of specificity constants) compared to the slowest reacting fatty acid (C) are equal to 10 for the fastest reacting one (A), and between 2 and 10 for the one of intermediate reactivity (B) (denoted α2 and shown next to the curves). The diagrams show the mole fraction of C (left) and B (right) as a function of the conversion of these fatty acids. The diagram to the left shows that C can be enriched to high purity and that this is reached at a low degree of conversion (which corresponds to a high recovery of the remaining substrate C) when the value of the competitive factor is high. Reprinted with permission from Kaki and Adlercreutz. Biotechnology and Bioengineering 2013, 110, 78–86. Copyright (2012) Wiley Periodicals, Inc.246 | ||
The discrimination against long-chain omega-3, compared to other fatty acids, is clearly efficient enough for the method to be practically useful. Other interesting examples of enzymatic fatty acid enrichment include erucic acid248 and gamma-linolenic acid.249 Interestingly, the fatty acid selectivity of lipases can be modified by site-directed mutagenesis.250 A lipase with increased specificity for trans fatty acids has been created, with the potential application to remove these undesired fatty acids from food lipids.251 To achieve increased fatty acid selectivity based on fatty acid chain length, Pleiss and co-workers constructed a set of mutants of C. rugosa lipase 1.252 Some of the mutations caused blocking of the active site so that fatty acids longer than a critical length (C10 or C14) were strongly discriminated against.252 This strategy has great potential in the development of more efficient lipases for the enrichment of long-chain fatty acids.
4.5 Biodiesel
The main constituent of biodiesel is alkyl esters of fatty acids, most commonly methyl esters. Biodiesel is used as fuel and has the advantage of being produced from renewable resources. Furthermore, emissions of hydrocarbons and particulate matter from biodiesel are considerably lower than from petrochemically derived diesel.253 Biodiesel is produced by the alcoholysis of triacylglycerol-rich fats or oils, using acidic, alkaline or enzymatic catalysts (Scheme 6). In industry, alkaline catalysis dominates because it is fast and cheap.253,254 Enzymatic biodiesel production has the advantage that oils with high contents of free fatty acids or water can be processed directly, while they require pretreatment prior to alkaline-catalysed processing.254 | ||
| Scheme 6 Alcoholysis of a triacylglycerol to produce esters (biodiesel). | ||
Lipase-catalysed transesterification for biodiesel production was recently reviewed.254–257 Since triacylglycerols are the normal substrates, many lipases express high activity in biodiesel production. CALB, as well as other Candida and Pseudomonas lipases, are among the most commonly used enzymes, often in the immobilised form.258 The reactions are carried out in solvent-free systems or with the addition of organic solvents.254,259 Immobilised CALB (Novozym 435) can be used for biodiesel production at very low water contents, while other lipases have been used with as much as 10% added water.258,260 The presence of relatively large amounts of water increases the formation of free fatty acids, which are undesirable in the final product.
Although biodiesel production is the major target for the alcoholysis of triacylglycerols, other applications of the same reactions are of interest. The production of ethyl esters from fish oils constitutes an important example.244,261
The alcoholysis of triacylglycerols may appear simple, but the reaction suffers from several problems, which must be solved before enzymatic processing can become efficient and cheap enough to compete with chemical processing. A few of the most important process issues are discussed below.
4.6 Phospholipid conversion
Because of their chirality, it is attractive to use natural starting materials in the preparation of specific glycerophospholipids.269 The fatty acids and/or the polar group can be replaced using chemical or enzymatic methods to obtain the desired lipid. Lipases are often able to convert phospholipids, and many sn-1,3-specific lipases express specificity for the sn-1 position of glycerophospholipids, thus constituting an attractive alternative to phospholipase A1. The most frequently used lipases are RML,270 ROL271 and other Rhizopus lipases.272 The lipase can be used to catalyse the hydrolysis of a diacyl glycerophospholipid to the corresponding 2-monoacyl lipid, or to add a fatty acid to a 2-monoacyl glycerophospholipid in an esterification or transesterification reaction.271,273,274 Lipase-catalysed incorporation of fatty acids in the sn-1 position normally gives higher yields than the corresponding incorporation in the sn-2 position, catalysed by phospholipase A2, partly because lipases can be used at lower water activity, and partly because the equilibrium position for incorporation in the sn-1 position is more favourable.275 Lipase-catalysed conversion has been carried out in toluene,276 hexane272 and in a solvent-free system.270 Enzymes are especially attractive when handling sensitive fatty acids such as the polyunsaturated ones,270,277 but the choice of lipase is critical since many lipases discriminate strongly against some of these fatty acids. In some cases, chemoenzymatic approaches have been found to be the best alternative.2784.7 Carbohydrate modification
Several different approaches have been used to make the carbohydrate more hydrophobic and thereby more soluble in relatively nonpolar solvents. One attractive approach is to convert the carbohydrate to an acetal. Acetals resulting from acetone and monosaccharides were successfully acylated in a solvent-free system, and after the enzymatic reaction, the acetal groups were removed by mild acid-catalysed hydrolysis.281 The methodology has been extended to be applicable to disaccharides. Both lactose and maltose were converted to their monoesters via triacetals, in which one of the primary hydroxyls was protected.282 A second method of making carbohydrates more hydrophobic and thus promoting their esterification is to convert them to complexes with hydrophobic boronic acids.283 Glucose was acylated in the presence of butyl or phenyl boronic acid, but among the disaccharides tested only Palatinose™ was converted. After the enzymatic conversion, the boronic acid complexes were easily hydrolysed with water. Yet another approach is to use alkyl glycosides as substrates instead of pure carbohydrates. Efficient solvent-free esterification of ethyl D-glucopyranoside was carried out using CALB as catalyst284 and, as alkyl glucoside esters are good surfactants, the ethyl group was maintained in the final product.
Direct condensation of carbohydrates and carboxylic acids is possible even without derivatisation or complex formation. Glucose was acylated with octanoic acid in acetonitrile using CALB as catalyst.285 Glucose had limited solubility in the solvent, but dissolved during the course of the reaction. The limited solubility of the carbohydrate in the reaction medium was thus sufficient to make the reaction possible, and this approach was further developed by decreasing the solvent volume to make it an adjuvant. Systems containing a small amount of a solvent such as acetone or dioxane, while the main part was the undissolved carbohydrate, have been successfully used for acylation of glucose, mannose, sorbitol and mannitol with a range of fatty acids.286 Studies of glucose acylation in 2-methyl-2-butanol catalysed by CALB have shown that the reaction rate is highly influenced by the glucose concentration in the reaction medium, and it is thus advantageous to use supersaturated glucose solutions.287 Detailed kinetic analysis of this reaction showed that the initial reaction rate decreased with increasing water concentration, as in other cases of CALB-catalysed esterification.288
Some ionic liquids are good solvents for carbohydrates and they are therefore of interest as media for carbohydrate acylation. In the acylation of glucose with fatty acid vinyl esters, the best results were obtained in mixtures of ionic liquids and t-butanol.289,290 High conversion (87%) was achieved in the RML-catalysed acylation of D-galactose with oleic acid in a mixture of ionic liquid and dimethyl sulfoxide.291 However, the ionic liquids that are the best solvents for carbohydrates often interact too strongly with enzymes, causing their inactivation. The method of using supersaturated carbohydrate solutions in ionic liquids with moderate ability to dissolve carbohydrates is a practical approach.292 The supersaturated solutions can be obtained by adding an aqueous carbohydrate solution to the ionic liquid and subsequently removing water. Ultrasound has been found to further enhance the reaction rate in the supersaturated solutions, possibly by improving the mass transfer in these viscous systems.293
As mentioned above, acylation of longer carbohydrates is considerably more difficult than the corresponding reaction with monosaccharides. Acylation of monosaccharides by CALB was successful in a t-butanol–pyridine mixture, while only limited acylation of maltose occurred, and other di- and trisaccharides were completely unreactive.294 However, acylation of maltose, leucrose, maltotriose295 and sucrose296 was achieved using TLL as catalyst, vinyl esters as acyl donors and 2-methyl-2-butanol/dimethyl sulfoxide as solvent. Interestingly, immobilisation of TLL in silica granules led to highly selective monoacylation of sucrose, while diacylation was prominent using TLL immobilised on porous polypropene.297
4.8 Increasing the lipophilicity of bioactive compounds
Many bioactive compounds of potential interest for practical use as antioxidants, nutraceuticals, etc., have poor solubility in nonpolar media. One way to increase their solubility is to modify them by attaching hydrophobic groups. Compounds with hydroxyl and/or carboxyl groups can then be converted into esters using long-chain carboxylic acids or long-chain alcohols. Since many of the compounds are quite sensitive, mild reaction conditions must be used, and lipases are therefore attractive catalysts for the esterification reactions. This topic will be discussed only briefly here, since several excellent reviews have recently been published.The use of enzymes, primarily lipases but also proteases, for the acylation of natural glycosides, with ginsenosides and cardioactive glycosides as interesting examples, has been reviewed.302 The more general use of lipases in conversions to increase the lipophilicity of target compounds has been reviewed, with emphasis on the modification of carbohydrates, amino acids and phenolics.303 Phenolic acids constitute an interesting group of antioxidants, and several studies have concerned their lipase-catalysed esterification with alcohols of different chain length.304 A typical reaction mixture contains a hydrophilic substrate (the bioactive compound) and a hydrophobic substrate (the reagent), and the same types of problems as in the acylation of carbohydrates (see previous section) thus arise. Solvents frequently used are tertiary alcohols and short-chain ketones.303,305 The use of a highly concentrated substrate solution with a small addition of dimethyl sulfoxide as solvent or adjuvant constitutes an interesting system used in the esterification of naringin with palmitic acid.306
Products with different properties can be obtained, depending on the length of the hydrophobic chain attached to the parent molecule. In the case of cardioactive glycosides, it was shown that the pharmacokinetic properties of the substances varied considerably depending on their lipophilicity.302 In the case of antioxidants it was shown that the antioxidative capacity often increased with increasing length of the added hydrophobic chain up to a certain critical length, after which the capacity decreased considerably.307 For alkyl esters of chlorogenic acid, the best performance in protecting fibroblast cells from oxidative stress was obtained with a chain length of 12 carbons,308 while in other cases 4 or 8 carbon atoms have been found to be optimal.307
There are several reports on the use of lipases to catalyse the preparation of esters containing two building blocks with interesting, often health-promoting, properties. The antioxidants rutin and vanillyl alcohol were acylated with a mixture of fatty acids originating from fish, which were rich in long-chain omega-3 fatty acids.309 Naringin was converted to naringin 6′′-ricinoleate using ricinoleic acid or, even better, castor oil, as acyl donor (Scheme 7).305L-Ascorbic acid acylated with palmitic acid is a commercial product, and the ester with cinnamic acid was recently prepared with the intention of creating an efficient antioxidant (Scheme 7).310 Vinyl cinnamate was found to be the best acyl donor and a yield of 68% was obtained. Immobilised CALB was used as catalyst in these last three reactions.
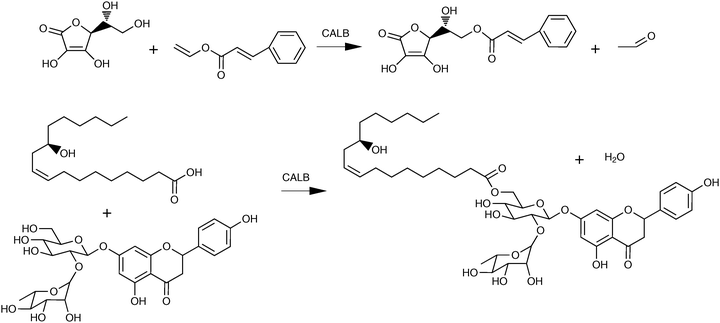 | ||
| Scheme 7 Lipase-catalysed coupling of pairs of bioactive compounds: ascorbic acid and vinyl cinnamate (upper reaction),310 ricinoleic acid and naringin (lower reaction).305 | ||
5. Concluding remarks
5.1 In which applications are lipases attractive?
Lipase-catalysed reactions have proven to be better than other approaches for the preparation of many products. The most thoroughly studied applications are those that have been realised on an industrial scale. Most industrial applications of lipases for synthesis concern chiral products, in which the stereoselectivity of enzymes in general and lipases in particular is extremely valuable. Likewise, the regioselectivity of lipases makes them the natural choice as catalysts for the production of structured lipids. Another important advantage of enzyme catalysis is the mild reaction conditions which, in many cases, lead to fewer side reactions, higher product yields and less waste. Product purification is simplified by the reduction in by-products. This has a positive impact on process economy which, in many cases, is sufficient to compensate for the cost of the enzyme. Furthermore, enzymatic methods often have considerably less environmental impact than alternative methods, which is naturally desirable. The mild reaction conditions are more important when handling sensitive substrates and products. However, lipase catalysis has also proven to be advantageous from an economic point of view when handling relatively stable compounds, such as in the interesterification of margarine fats. Enzymatic ester synthesis is another example of a process that does not require high selectivity, but is still competitive on industrial scale. Considerable effort is currently being devoted to making enzymatic production of biodiesel competitive with chemical methods.5.2 Which is the best method of lipase immobilisation?
The following are important in obtaining an immobilised lipase preparation that uses the enzyme as efficiently as possible.• No enzyme inactivation should occur during immobilisation
• No enzyme leakage should occur after immobilisation
• The lipase should be present in fully activated form
• Mass transfer limitations should be negligible
The first two are sometimes difficult to combine. Covalent coupling to a support or covalent cross-linking are the best ways to avoid enzyme leakage, but they often cause enzyme inactivation. For most applications of lipases in organic media, non-covalent immobilisation is sufficient because the lipase is not soluble in the reaction medium, but remains in the immobilised preparation. However, if the reaction mixture contains surface active compounds, there is a risk of enzyme leakage, and covalent immobilisation is recommended. In some cases, the best approach may be not to immobilise the lipase, but to use surfactants to form hydrophobic ion pairs, surfactant-coated lipases or microemulsions. However, in these cases there is a need for a more sophisticated separation step to separate the enzyme from the product mixture.
Immobilisation is of special importance where lipases are concerned because, in addition to the normal benefits of enzyme immobilisation, it can also lead to a considerable increase in lipase activity, probably due to conformational changes in the lipase molecules. Although dramatic activation effects have been demonstrated following adsorption on hydrophobic supports and treatment with surfactants, it has not been convincingly shown that maximal activation has been achieved. It is thus recommended that research be continued to develop even better immobilisation methods, allowing full lipase activation to be achieved. It may well be that the flexibility of lipases acting at the organic–aqueous interface of an emulsion is of importance in achieving maximal activity, and that this is more difficult to achieve in an immobilised preparation.
Mass transfer limitations can cause severe reductions in catalytic activity when diffusion is too slow compared to the reaction being catalysed. However, some of the porous materials evaluated as lipase supports have a particle size, porosity and surface area that make efficient conversions with negligible mass transfer limitations possible for normal lipase-catalysed reactions. To enable even faster reactions without mass transfer limitations, it may be advantageous to use even more dispersed lipase molecules, such as lipase on nanoparticles or lipase solubilised in the organic medium using surfactants.
Additional factors are important for the large-scale application of lipases. The cost of supporting materials and other costs associated with the preparation of the lipase must be considered more critically than in small-scale work. In addition, the immobilised preparation must be suitable for use in the intended reactor, which may be a stirred-tank reactor, a packed-bed reactor or some other type. The particle size and shape, as well as the resistance to shear, compression, etc., should also be taken into consideration. Furthermore, the operational stability is important since it determines how much product can be produced per unit weight of enzyme. Existing industrial applications have shown that lipases can remain active for months of continuous use which, of course, is beneficial for process economy.
Finally, it should be pointed out that each lipase has its own unique properties, and that fine-tuning of any methodology employing lipases to suit the individual enzyme will be fruitful. There are considerable differences between lipases, for example, concerning activation and inactivation during immobilisation, and activation by surfactants. Protein engineering of lipases and the further improvement of lipase preparations and reaction methodology have great potential to generate even better bioconversions in the future.
Acknowledgements
The author is grateful for financial support from The Swedish Research Council and from The Swedish Research Council Formas and for help with Fig. 1 from Pontus Lundemo.Notes and references
- L. Sarda and P. Desnuelle, Biochim. Biophys. Acta, 1958, 30, 513–521 CrossRef CAS.
- H. W. Anthonsen, A. Baptista, F. Drabloes, P. Martel, S. B. Petersen, M. Sebastiao and L. Vaz, Biotechnol. Annu. Rev., 1995, 1, 315–371 CrossRef CAS.
- R. Verger, Trends Biotechnol., 1997, 15, 32–38 CrossRef CAS.
- L. Brady, A. M. Brzozowski, Z. S. Derewenda, E. Dodson, G. Dodson, S. Tolley, J. P. Turkenburg, L. Christiansen, B. Hugejensen, L. Norskov, L. Thim and U. Menge, Nature, 1990, 343, 767–770 CrossRef CAS.
- J. D. Schrag, Y. Li, S. Wu and M. Cygler, Nature, 1991, 351, 761–764 CrossRef CAS.
- A. M. Brzozowski, U. Derewenda, Z. S. Derewenda, G. G. Dodson, D. M. Lawson, J. P. Turkenburg, F. Bjorkling, B. Hugejensen, S. A. Patkar and L. Thim, Nature, 1991, 351, 491–494 CrossRef CAS.
- A. Maraite, P. Hoyos, J. D. Carballeira, A. C. Cabrera, M. B. Ansorge-Schumacher and A. R. Alcantara, J. Mol. Catal. B: Enzym., 2013, 87, 88–98 CrossRef CAS.
- J. Uppenberg, M. T. Hansen, S. Patkar and T. A. Jones, Structure, 1994, 2, 293–308 CrossRef CAS.
- M. Martinelle, M. Holmquist and K. Hult, Biochim. Biophys. Acta, 1995, 1258, 272–276 CrossRef.
- D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, J. Mol. Biol., 1984, 179, 125–142 CrossRef CAS.
- R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608–1633 CrossRef.
- R. Sangeetha, I. Arulpandi and A. Geetha, Res. J. Microbiol., 2011, 6, 1–24 CrossRef CAS.
- A. K. Singh and M. Mukhopadhyay, Appl. Biochem. Biotechnol., 2012, 166, 486–520 CrossRef CAS.
- M. Lotti, A. Tramontano, S. Longhi, F. Fusetti, S. Brocca, E. Pizzi and L. Alberghina, Protein Eng., 1994, 7, 531–535 CrossRef CAS.
- N. Lopez, M. A. Pernas, L. M. Pastrana, A. Sanchez, F. Valero and M. L. Rua, Biotechnol. Prog., 2004, 20, 65–73 CrossRef CAS.
- E. M. Andersson, K. M. Larsson and O. Kirk, Biocatal. Biotransform., 1998, 16, 181–204 CrossRef.
- R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 62, 197–212 CrossRef CAS.
- R. C. Rodrigues and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 66, 15–32 CrossRef CAS.
- R. C. Rodrigues and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 64, 1–22 CrossRef CAS.
- M. Masomian, R. N. Z. Raja AbdRahman, A. B. Salleh and M. Basri, Process Biochem. DOI:10.1016/j.procbio.2012.11.002.
- D. Chakravorty, S. Parameswaran, V. K. Dubey and S. Patra, Appl. Biochem. Biotechnol., 2012, 167, 439–461 CrossRef CAS.
- M. S. Humble and P. Berglund, Eur. J. Org. Chem., 2011, 3391–3401 CrossRef CAS.
- C. Branneby, P. Carlqvist, K. Hult, T. Brinck and P. Berglund, J. Mol. Catal. B: Enzym., 2004, 31, 123–128 CrossRef CAS.
- P. Vongvilai, M. Linder, M. Sakulsombat, M. S. Humble, P. Berglund, T. Brinck and O. Ramstrom, Angew. Chem., Int. Ed., 2011, 50, 6592–6595 CrossRef CAS.
- M. Svedendahl, P. Carlqvist, C. Branneby, O. Allner, A. Frise, K. Hult, P. Berglund and T. Brinck, ChemBioChem, 2008, 9, 2443–2451 CrossRef CAS.
- J. L. Schmitke, C. R. Wescott and A. M. Klibanov, J. Am. Chem. Soc., 1996, 118, 3360–3365 CrossRef CAS.
- K. Ryu and J. S. Dordick, Biochemistry, 1992, 31, 2588–2598 CrossRef CAS.
- E. Wehtje and P. Adlercreutz, Biotechnol. Bioeng., 1997, 55, 798–806 CrossRef CAS.
- L. F. Garcia-Alles and V. Gotor, Biotechnol. Bioeng., 1998, 59, 684–694 CrossRef CAS.
- R. Wedberg, J. Abildskov and G. H. Peters, J. Phys. Chem. B, 2012, 116, 2575–2585 CrossRef CAS.
- P. Trodler, R. D. Schmid and J. Pleiss, BMC Struct. Biol., 2008, 8, 9 CrossRef.
- S. Rehm, P. Trodler and J. Pleiss, Protein Sci., 2010, 19, 2122–2130 CrossRef CAS.
- M. Graber, R. Irague, E. Rosenfeld, S. Lamare, L. Franson and K. Hult, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1052–1057 CrossRef CAS.
- A. S. Ghatorae, M. J. Guerra, G. Bell and P. J. Halling, Biotechnol. Bioeng., 1994, 44, 1355–1361 CrossRef CAS.
- P. Adlercreutz, in Organic synthesis with enzymes in non-aqueous media, ed. G. Carrea and S. Riva, Wiley-VCH, Weinheim, 2008, pp. 3–24 Search PubMed.
- H. J. Park, J. C. Joo, K. Park and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2012, 17, 722–728 CrossRef CAS.
- J. Abildskov, M. B. van Leeuwen, C. G. Boeriu and L. A. M. van den Broek, J. Mol. Catal. B: Enzym., 2013, 85–86, 200–213 CrossRef CAS.
- A. D. Blackwood, L. J. Curran, B. D. Moore and P. J. Halling, Biochim. Biophys. Acta, 1994, 1206, 161–165 CrossRef CAS.
- J. Partridge, P. J. Halling and B. D. Moore, J. Chem. Soc., Perkin Trans. 2, 2000, 465–471 RSC.
- J. Partridge, N. Harper, B. D. Moore and P. J. Halling, in Enzymes in Nonaqueous Solvents, ed. H. N. Vulfson, P. J. Halling and H. L. Holland, Humana Press, Totowa, New Jersey, 2001, vol. 15, pp. 227–234 Search PubMed.
- Immobilization of Enzymes and Cells, Methods Biotechnol., ed. J. M. Guisan, 2006, vol. 22 Search PubMed.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. J. Barros, E. Wehtje and P. Adlercreutz, Biotechnol. Bioeng., 1998, 59, 364–373 CrossRef CAS.
- F. Hasan, A. A. Shah and A. Hameed, Biotechnol. Adv., 2009, 27, 782–798 CrossRef CAS.
- A. B. Majumder and M. N. Gupta, Bioresour. Technol., 2010, 101, 2877–2879 CrossRef CAS.
- B. Berger and K. Faber, J. Chem. Soc., Chem. Commun., 1991, 1198–1200 RSC.
- M. M. Zheng, Y. Lu, L. Dong, P. M. Guo, Q. C. Deng, W. L. Li, Y. Q. Feng and F. H. Huang, Bioresour. Technol., 2012, 115, 141–146 CrossRef CAS.
- T. Gitlesen, M. Bauer and P. Adlercreutz, Biochim. Biophys. Acta, 1997, 1345, 188–196 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernandez-Lafuente, J. Huguet and J. M. Guisan, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS.
- R. Fernandez-Lafuente, P. Armisen, P. Sabuquillo, G. Fernandez-Lorente and J. M. Guisan, Chem. Phys. Lipids, 1998, 93, 185–197 CrossRef CAS.
- A. R. Macrae, J. Am. Oil Chem. Soc., 1983, 60, 243A–246A CrossRef.
- V. Pace, J. V. Sinisterra and A. R. Alcantara, Curr. Org. Chem., 2010, 14, 2384–2408 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS.
- H. Gustafsson, C. Thorn and K. Holmberg, Colloids Surf., B, 2011, 87, 464–471 CrossRef CAS.
- J. A. Bosley and J. C. Clayton, Biotechnol. Bioeng., 1994, 43, 934–938 CrossRef CAS.
- J. S. Clifford and R. L. Legge, Biotechnol. Bioeng., 2005, 92, 231–237 CrossRef CAS.
- Q. R. Jin, G. Q. Jia, Y. M. Zhang, Q. H. Yang and C. Li, Langmuir, 2011, 27, 12016–12024 CrossRef CAS.
- Z. Zhou, A. Inayat, W. Schwieger and M. Hartmann, Microporous Mesoporous Mater., 2012, 154, 133–141 CrossRef CAS.
- M. Shakeri and K. Kawakami, Catal. Commun., 2008, 10, 165–168 CrossRef CAS.
- Y. M. Zhang, J. Li, D. F. Han, H. D. Zhang, P. Liu and C. Li, Biochem. Biophys. Res. Commun., 2008, 365, 609–613 CrossRef CAS.
- R. M. Blanco, P. Terreros, M. Fernandez-Perez, C. Otero and G. Diaz-Gonzalez, J. Mol. Catal. B: Enzym., 2004, 30, 83–93 CrossRef CAS.
- Y. M. Zhang, L. F. Zhao, J. Li, H. D. Zhang, L. Y. Zheng, S. G. Cao and C. Li, Biochem. Biophys. Res. Commun., 2008, 372, 650–655 CrossRef CAS.
- N. G. Graebin, A. B. Martins, A. S. G. Lorenzoni, C. Garcia-Galan, R. Fernandez-Lafuente, M. A. Z. Ayub and R. C. Rodrigues, Biotechnol. Prog., 2012, 28, 406–412 CrossRef CAS.
- M. D. Truppo and G. Hughes, Org. Process Res. Dev., 2011, 15, 1033–1035 CrossRef CAS.
- A. E. V. Hagstrom, M. Nordblad and P. Adlercretz, Biotechnol. Bioeng., 2009, 102, 693–699 CrossRef.
- Y. Mei, L. Miller, W. Gao and R. A. Gross, Biomacromolecules, 2003, 4, 70–74 CrossRef CAS.
- K. Mondal, P. Mehta, B. R. Mehta, D. Varandani and M. N. Gupta, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1080–1086 CrossRef CAS.
- M. Filice, M. Marciello, L. Betancor, A. V. Carrascosa, J. M. Guisan and G. Fernandez-Lorente, Biotechnol. Prog., 2011, 27, 961–968 CrossRef CAS.
- J. Partridge, P. J. Halling and B. D. Moore, Chem. Commun., 1998, 841–842 RSC.
- A. B. Majumder, S. Shah and M. N. Gupta, Chem. Cent. J., 2007, 1, 10 CrossRef.
- M. Kreiner, B. D. Moore and M. C. Parker, Chem. Commun., 2001, 1096–1097 RSC.
- M. Kreiner, J. F. A. Fernandes, N. O'Farrell, P. J. Halling and M.-C. Parker, J. Mol. Catal. B: Enzym., 2005, 33, 65–72 CrossRef CAS.
- J. J. Zheng, L. Xu, Y. Liu, X. Y. Zhang and Y. J. Yan, Bioresour. Technol., 2012, 110, 224–231 CrossRef CAS.
- S. Shah, A. Sharma and M. N. Gupta, Biocatal. Biotransform., 2008, 26, 266–271 CrossRef CAS.
- J. C. Cruz, P. H. Pfromm, J. M. Tomich and M. E. Rezac, Colloids Surf., B, 2010, 79, 97–104 CrossRef CAS.
- J. A. Laszlo, M. Jackson and R. M. Blanco, J. Mol. Catal. B: Enzym., 2011, 69, 60–65 CrossRef CAS.
- M. Persson, E. Wehtje and P. Adlercreutz, ChemBioChem, 2002, 3, 101–106 CrossRef.
- E. Wehtje, P. Adlercreutz and B. Mattiasson, Biotechnol. Bioeng., 1993, 41, 171–178 CrossRef CAS.
- S. Shah, K. Solanki and M. N. Gupta, Chem. Cent. J., 2007, 1 CrossRef CAS , I:30.
- A. Salis, I. Svensson, M. Monduzzi, V. Solinas and P. Adlercreutz, Biochim. Biophys. Acta, 2003, 1646, 145–151 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Angew. Chem., Int. Ed. Engl., 1995, 34, 301–303 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Biotechnol. Bioeng., 1996, 49, 527–534 CrossRef CAS.
- D. Weiser, Z. Boros, G. Hornyanszky, A. Toth and L. Poppe, Process Biochem., 2012, 47, 428–434 CrossRef CAS.
- M. T. Reetz, P. Tielmann, W. Wiesenhofer, W. Konen and A. Zonta, Adv. Synth. Catal., 2003, 345, 717–728 CrossRef CAS.
- M. Persson, I. Mladenoska, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 2002, 31, 833–841 CrossRef CAS.
- K. Kawakami, T. Urakawa, Y. Oda and Y. Iwai, J. Chem. Technol. Biotechnol., 2009, 84, 1412–1417 CrossRef CAS.
- D. Pirozzi, E. Fanelli, A. Aronne, P. Pernice and A. Mingione, J. Mol. Catal. B: Enzym., 2009, 59, 116–120 CrossRef CAS.
- S. Dech, V. Wruk, C. P. Fik and J. C. Tiller, Polymer, 2012, 53, 701–707 CrossRef CAS.
- S. Sakai, K. Antoku, T. Yamaguchi and K. Kawakami, J. Biosci. Bioeng., 2008, 105, 687–689 CrossRef CAS.
- K. Gawlitza, C. Z. Wu, R. Georgieva, D. Y. Wang, M. B. Ansorge-Schumacher and R. von Klitzing, Phys. Chem. Chem. Phys., 2012, 14, 9594–9600 RSC.
- Z. Knezevic, N. Milosavic, D. Bezbradica, Z. Jakovljevic and R. Prodanovic, Biochem. Eng. J., 2006, 30, 269–278 CrossRef CAS.
- C. Pizarro, M. C. Branes, A. Markovits, G. Fernandez-Lorente, J. M. Guisan, R. Chamy and L. Wilson, J. Mol. Catal. B: Enzym., 2012, 78, 111–118 CrossRef CAS.
- J. M. Guisan, Enzyme Microb. Technol., 1988, 10, 375–382 CrossRef CAS.
- R. C. Rodrigues, C. A. Godoy, M. Filice, J. M. Bolivar, A. Palau-Ors, J. M. Garcia-Vargas, O. Romero, L. Wilson, M. A. Z. Ayub, R. Fernandez-Lafuente and J. M. Guisan, Process Biochem., 2009, 44, 641–646 CrossRef CAS.
- J. Ge, D. A. Lu, J. Wang and Z. Liu, Biomacromolecules, 2009, 10, 1612–1618 CrossRef CAS.
- D. D. Xu, L. Tonggu, X. P. Bao, D. N. Lu and Z. Liu, Soft Matter, 2012, 8, 2036–2042 RSC.
- K. Solanki and M. N. Gupta, Bioorg. Med. Chem. Lett., 2011, 21, 2934–2936 CrossRef CAS.
- Y. Inada, K. Takahashi, T. Yoshimoto, A. Ajima, A. Matsushima and Y. Saito, Trends Biotechnol., 1986, 4, 190–194 CrossRef CAS.
- S. A. Khan, P. J. Halling, J. A. Bosley, A. H. Clark, A. D. Peilow, E. G. Pelan and D. W. Rowlands, Enzyme Microb. Technol., 1992, 14, 96–100 CrossRef CAS.
- K. Nakashima, J. Okada, T. Maruyama, N. Kamiya and M. Goto, Sci. Technol. Adv. Mater., 2006, 7, 692–698 CrossRef CAS.
- D. R. Walt and V. I. Agayn, TrAC, Trends Anal. Chem., 1994, 13, 425–430 CrossRef CAS.
- N. L. St. Clair and M. A. Naiva, J. Am. Chem. Soc., 1992, 114, 7314–7316 CrossRef CAS.
- A. L. Margolin, Trends Biotechnol., 1996, 14, 223–230 CrossRef CAS.
- J. J. Roy and T. E. Abraham, Chem. Rev., 2004, 104, 3705–3721 CrossRef CAS.
- A. M. Collins, C. Maslin and R. J. Davies, Org. Process Res. Dev., 1998, 2, 400–406 CrossRef CAS.
- N. Khalaf, C. P. Govardhan, J. L. Lalonde, R. A. Persichetti, Y. F. Wang and A. L. Margolin, J. Am. Chem. Soc., 1996, 118, 5494–5495 CrossRef CAS.
- Z. J. Dijkstra, H. Weyten, L. Willems and J. T. F. Keurentjes, J. Mol. Catal. B: Enzym., 2006, 39, 112–116 CrossRef CAS.
- L. Q. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS.
- P. Lopez-Serrano, L. Cao, F. van Rantwijk and R. A. Sheldon, Biotechnol. Lett., 2002, 24, 1379–1383 CrossRef CAS.
- B. Devi, Z. Guo and X. B. Xu, J. Am. Oil Chem. Soc., 2009, 86, 637–642 CrossRef.
- F. Kartal, M. H. A. Janssen, F. Hollmann, R. A. Sheldon and A. Kilinc, J. Mol. Catal. B: Enzym., 2011, 71, 85–89 CrossRef CAS.
- A. B. Majumder, K. Mondal, T. P. Singh and M. N. Gupta, Biocatal. Biotransform., 2008, 26, 235–242 CrossRef CAS.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS.
- J. Cruz, O. Barbosa, R. C. Rodrigues, R. Fernandez-Lafuente, R. Torres and C. Ortiz, J. Mol. Catal. B: Enzym., 2012, 80, 7–14 CrossRef CAS.
- D. H. Altreuter, J. S. Dordick and D. S. Clark, Enzyme Microb. Technol., 2002, 31, 10–19 CrossRef CAS.
- J. C. Wu, P. S. Chua, Y. Chow, R. J. Li, K. Carpenter and H. W. Yu, J. Chem. Technol. Biotechnol., 2006, 81, 1003–1008 CrossRef CAS.
- S. S. Adkins, H. R. Hobbs, K. Benaissi, K. P. Johnston, M. Poliakoff and N. R. Thomas, J. Phys. Chem. B, 2008, 112, 4760–4769 CrossRef CAS.
- Y. Okahata and T. Mori, Trends Biotechnol., 1997, 15, 50–54 CrossRef CAS.
- A. R. M. Yahya, W. A. Anderson and M. Moo-Young, Enzyme Microb. Technol., 1998, 23, 438–450 CrossRef CAS.
- N. Kamiya, M. Goto and F. Nakashio, Biotechnol. Prog., 1995, 11, 270–275 CrossRef CAS.
- N. Kamiya, E. Murakami, M. Goto and F. Nakashio, J. Ferment. Bioeng., 1996, 82, 37–41 CrossRef CAS.
- H. Kim, Y. K. Choi, J. Lee, E. Lee, J. Park and M. J. Kim, Angew. Chem., Int. Ed., 2011, 50, 10944–10948 CrossRef CAS.
- S. Basheer, M. Nakajima and U. Cogan, J. Am. Oil Chem. Soc., 1996, 73, 1475–1479 CrossRef CAS.
- S. Y. Huang, H. L. Chang and M. Goto, Enzyme Microb. Technol., 1998, 22, 552–557 CrossRef CAS.
- A. Thakar and D. Madamwar, Process Biochem., 2005, 40, 3263–3266 CrossRef CAS.
- H. J. Hsieh, G. R. Nair and W. T. Wu, J. Agric. Food Chem., 2006, 54, 5777–5781 CrossRef CAS.
- M. Goto, M. Matsumoto, K. Kondo and F. Nakashio, J. Chem. Eng. Jpn., 1987, 20, 157–164 CrossRef CAS.
- Y. Okahata and K. Ijiro, Bull. Chem. Soc. Jpn., 1992, 65, 2411–2420 CrossRef CAS.
- M. Goto, H. Kameyama, M. Goto, M. Miyata and F. Nakashio, J. Chem. Eng. Jpn., 1993, 26, 109–111 CrossRef CAS.
- Y. Abe, T. Hirakawa, S. Nakajima, N. Okano, S. Hayase, M. Kawatsura, Y. Hirose and T. Itoh, Adv. Synth. Catal., 2008, 350, 1954–1958 CrossRef CAS.
- Y. Okahata, Y. Fujimoto and K. Ijiro, J. Org. Chem., 1995, 60, 2244–2250 CrossRef CAS.
- S. Basheer, K. Mogi and M. Nakajima, Biotechnol. Bioeng., 1995, 45, 187–195 CrossRef CAS.
- H. Stamatis, A. Xenakis and F. N. Kolisis, Biotechnol. Adv., 1999, 17, 293–318 CrossRef CAS.
- C. M. L. Carvalho and J. M. S. Cabral, Biochimie, 2000, 82, 1063–1085 CrossRef CAS.
- M. A. Biasutti, E. B. Abuin, J. J. Silber, N. M. Correa and E. A. Lissi, Adv. Colloid Interface Sci., 2008, 136, 1–24 CrossRef CAS.
- S. Z. B. A. Puat, M. Taib and H. Suhaimi, J. Sustainability Sci. Manage., 2010, 5, 87–93 CAS.
- K. M. Larsson, P. Adlercreutz and B. Mattiasson, Biotechnol. Bioeng., 1990, 36, 135–141 CrossRef CAS.
- I. V. Pavlidis, D. Gournis, G. K. Papadopoulos and H. Stamatis, J. Mol. Catal. B: Enzym., 2009, 60, 50–56 CrossRef CAS.
- E. D. Brown, R. Y. Yada and A. G. Marangoni, Biochim. Biophys. Acta, 1993, 1161, 66–72 CrossRef CAS.
- T. Ternstrom, A. Svendsen, M. Akke and P. Adlercreutz, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1748, 74–83 CrossRef.
- Y. Yamada, R. Kuboi and I. Komasawa, Biotechnol. Prog., 1993, 9, 468–472 CrossRef CAS.
- N. M. R. Talukder, M. M. Zaman, Y. Hayashi, J. C. Wu, T. Kawanishi, C. Ogino and N. Shimizu, J. Chem. Technol. Biotechnol., 2004, 79, 273–276 CrossRef.
- M. Moniruzzaman, Y. Hayashi, M. M. R. Talukder, E. Saito and T. Kawanishi, Biochem. Eng. J., 2006, 30, 237–244 CrossRef CAS.
- M. Svensson, G. D. Rees, B. H. Robinson and G. R. Stephenson, Colloids Surf., B, 1996, 8, 101–111 CrossRef CAS.
- K. Nagayama, S. Matsu-ura, T. Doi and M. Imai, J. Mol. Catal. B: Enzym., 1998, 4, 25–32 CrossRef CAS.
- A. M. C. Pintosousa, J. M. S. Cabral and M. R. Airesbarros, Biocatalysis, 1994, 9, 169–179 CrossRef CAS.
- M. Moniruzzaman, N. Kamiya, K. Nakashima and M. Goto, Green Chem., 2008, 10, 497–500 RSC.
- S. Maiti, M. Ghosh and P. K. Das, Chem. Commun., 2011, 47, 9864–9866 RSC.
- M. Zoumpanioti, H. Stamatis and A. Xenakis, Biotechnol. Adv., 2010, 28, 395–406 CrossRef CAS.
- R. Dave and D. Madamwar, Process Biochem., 2008, 43, 70–75 CrossRef CAS.
- L. Gardossi, P. B. Poulsen, A. Ballesteros, K. Hult, V. K. Svedas, D. Vasic-Racki, G. Carrea, A. Magnusson, A. Schmid, R. Wohlgemuth and P. J. Halling, Trends Biotechnol., 2010, 28, 171–180 CrossRef CAS.
- A. Salis, M. S. Bhattacharyya, M. Monduzzi and V. Solinas, J. Mol. Catal. B: Enzym., 2009, 57, 262–269 CrossRef CAS.
- A. W. Sonesson, T. H. Callisen, H. Brismar and U. M. Elofsson, Colloids Surf., B, 2008, 61, 208–215 CrossRef CAS.
- J. M. Guisan, P. Sabuquillo, R. Fernandez-Lafuente, G. Fernandez-Lorente, C. Mateo, P. J. Halling, D. Kennedy, E. Miyata and D. Re, J. Mol. Catal. B: Enzym., 2001, 11, 817–824 CrossRef CAS.
- R. J. Barros, E. Wehtje, F. A. P. Garcia and P. Adlercreutz, Biocatal. Biotransform., 1998, 16, 67–85 CrossRef CAS.
- A. Salis, E. Sanjust, V. Solinas and M. Monduzzi, J. Mol. Catal. B: Enzym., 2003, 24–25, 75–82 CrossRef CAS.
- S. Sakai, Y. P. Liu, T. Yamaguchi, R. Watanabe, M. Kawabe and K. Kawakami, Biotechnol. Lett., 2010, 32, 1059–1062 CrossRef CAS.
- M. Guncheva, M. Dimitrov and D. Zhiryakova, Catal. Commun., 2011, 16, 205–209 CrossRef CAS.
- K. Solanki and M. N. Gupta, New J. Chem., 2011, 35, 2551–2556 RSC.
- W. Bai, Y.-J. Yang, X. Tao, J.-F. Chen and T.-W. Tan, J. Mol. Catal. B: Enzym., 2012, 76, 82–88 CrossRef CAS.
- E. Severac, O. Galy, F. Turon, C. A. Pantel, J. S. Condoret, P. Monsan and A. Marty, Enzyme Microb. Technol., 2011, 48, 61–70 CrossRef CAS.
- P. J. Halling, Philos. Trans. R. Soc., London, Ser. B, 2004, 359, 1287–1297 CrossRef CAS.
- A. Zaks and A. Klibanov, J. Biol. Chem., 1988, 263, 8017–8021 CAS.
- P. J. Halling, Enzyme Microb. Technol., 1994, 16, 178–206 CrossRef CAS.
- P. J. Halling, Biochim. Biophys. Acta, 1990, 1040, 225–228 CrossRef CAS.
- M. Reslow, P. Adlercreutz and B. Mattiasson, Eur. J. Biochem., 1988, 172, 573–578 CrossRef CAS.
- D. K. Oladepo, P. J. Halling and V. F. Larsen, Biocatalysis, 1994, 8, 283–287 CrossRef CAS.
- P. Adlercreutz, Eur. J. Biochem., 1991, 199, 609–614 CrossRef CAS.
- A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 3194–3201 CAS.
- T. Hansson, M. Andersson, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 2001, 29, 527–534 CrossRef CAS.
- R. H. Valivety, P. J. Halling, A. D. Peilow and A. R. Macrae, Biochim. Biophys. Acta, 1992, 1122, 143–146 CrossRef CAS.
- M. Nordblad and P. Adlercreutz, Biocatal. Biotransform. Search PubMed , in press.
- R. H. Valivety, P. J. Halling and A. R. Macrae, FEBS Lett., 1992, 301, 258–260 CrossRef CAS.
- X. L. Xia, C. Wang, B. Yang, Y. H. Wang and X. N. Wang, Appl. Biochem. Biotechnol., 2009, 159, 759–767 CrossRef CAS.
- M. P. Bousquet-Dubouch, M. Graber, N. Sousa, S. Lamare and M. D. Legoy, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1550, 90–99 CrossRef CAS.
- L. Ma, M. Persson and P. Adlercreutz, Enzyme Microb. Technol., 2002, 31, 1024–1029 CrossRef CAS.
- R. J. F. Branco, M. Graber, V. Denis and J. Pleiss, ChemBioChem, 2009, 10, 2913–2919 CrossRef CAS.
- P. Trodler and J. Pleiss, BMC Struct. Biol., 2009, 9, 38 CrossRef.
- D. S. Clark, Philos. Trans. R. Soc., London, Ser. B, 2004, 359, 1299–1307 CrossRef CAS.
- R. H. Valivety, P. J. Halling and A. R. Macrae, Biotechnol. Lett., 1993, 15, 1133–1138 CrossRef CAS.
- M. Graber, M. P. Bousquet-Dubouch, S. Lamare and M. D. Legoy, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1648, 24–32 CrossRef CAS.
- M. Graber, V. Leonard, Z. Marton, C. Cusatis and S. Lamare, J. Mol. Catal. B: Enzym., 2008, 52–53, 121–127 CrossRef CAS.
- J. M. Mora-Pale, S. Perez-Munguia, J. C. Gonzalez-Mejia, J. S. Dordick and E. Barzana, Biotechnol. Bioeng., 2007, 98, 535–542 CrossRef CAS.
- A. E. M. Janssen, B. J. Sjursnes, A. V. Vakurov and P. Halling, Enzyme Microb. Technol., 1999, 24, 463–470 CrossRef CAS.
- E. Wehtje and P. Adlercreutz, Biotechnol. Lett., 1997, 19, 537–540 CrossRef CAS.
- V. Leonard-Nevers, Z. Marton, S. Lamare, K. Hult and M. Graber, J. Mol. Catal. B: Enzym., 2009, 59, 90–95 CrossRef CAS.
- V. Leonard, L. Fransson, S. Lamare, K. Hult and M. Graber, ChemBioChem, 2007, 8, 662–667 CrossRef CAS.
- G. Bell, P. J. Halling, M. Lindsay, B. D. Moore, D. A. Robb, R. Ulijn and R. Valivety, in Enzymes in Nonaqueous Solvents, ed. E. N. Vulfson, P. J. Halling and H. L. Holland, Humana Press, Totowa, New Jersey, 2001, vol. 15, pp. 105–126 Search PubMed.
- H. Goderis, G. Ampe, M. Feyton, B. Fouwe, W. Guffens, S. Van Cauwenbergh and P. Tobback, Biotechnol. Bioeng., 1987, 30, 258–266 CrossRef CAS.
- E. Wehtje, I. Svensson, P. Adlercreutz and B. Mattiasson, Biotechnol. Tech., 1994, 7, 873–878 CrossRef CAS.
- P. J. Halling, Biotechnol. Tech., 1992, 6, 271–276 CrossRef CAS.
- A. E. V. Petersson, P. Adlercreutz and B. Mattiasson, Biotechnol. Bioeng., 2007, 97, 235–241 CrossRef CAS.
- A. Larios, H. S. Garcia, R. M. Oliart and G. Valerio-Alfaro, Appl. Microbiol. Biotechnol., 2004, 65, 373–376 CrossRef CAS.
- Z. Jin, J. Ntwali, S.-Y. Han, S.-P. Zheng and Y. Lin, J. Biotechnol., 2012, 159, 108–114 CrossRef CAS.
- M. Nordblad and P. Adlercreutz, J. Biotechnol., 2008, 133, 127–133 CrossRef CAS.
- F. Hollmann, P. Grzebyk, V. Heinrichs, K. Doderer and O. Thum, J. Mol. Catal. B: Enzym., 2009, 57, 257–261 CrossRef CAS.
- M. Nordblad and P. Adlercreutz, Biotechnol. Bioeng., 2008, 99, 1518–1524 CrossRef CAS.
- A. E. V. Petersson, L. M. Gustafsson, M. Nordblad, P. Borjesson, B. Mattiasson and P. Adlercreutz, Green Chem., 2005, 7, 837–843 RSC.
- L. Hilterhaus, O. Thum and A. Liese, Org. Process Res. Dev., 2008, 12, 618–625 CrossRef CAS.
- G. A. Hills, A. R. Macrae and R. R. Poulina, Patent application, EP 0383405 B1, 1990 Search PubMed.
- S. Bloomer, P. Adlercreutz and B. Mattiasson, Enzyme Microb. Technol., 1992, 14, 546–552 CrossRef CAS.
- M. From, P. Adlercreutz and B. Mattiasson, Biotechnol. Lett., 1997, 19, 315–317 CrossRef CAS.
- Y. Yu, D. Wu, C. Liu, Z. Zhao, Y. Yang and Q. Li, Process Biochem., 2012, 47, 1027–1036 CrossRef CAS.
- Enzymes in lipid modification, ed. U. T. Bornscheuer, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- I. C. Chandler, P. T. Quinlan and G. P. McNeill, J. Am. Oil Chem. Soc., 1998, 75, 1513–1518 CrossRef CAS.
- A. R. Macrae, Biochem. Soc. Trans., 1989, 17, 1146–1148 CAS.
- X. B. Xu, Eur. J. Lipid Sci. Technol., 2000, 102, 287–303 CrossRef CAS.
- B. Chen, H. Zhang, L.-Z. Cheong, T. Tan and X. Xu, Food Bioprocess Technol., 2012, 5, 541–547 CrossRef CAS.
- M. M. Soumanou, U. T. Bornscheuer and R. D. Schmid, J. Am. Oil Chem. Soc., 1998, 75, 703–710 CrossRef CAS.
- A. Millqvist, P. Adlercreutz and B. Mattiasson, Enzyme Microb. Technol., 1994, 16, 1042–1047 CrossRef CAS.
- A. M. Fureby, C. Virto, P. Adlercreutz and B. Mattiasson, Biocatal. Biotransform., 1996, 14, 89–111 CrossRef CAS.
- M. M. Soumanou, U. T. Bornscheuer, U. Schmid and R. D. Schmid, Biocatal. Biotransform., 1999, 16, 443–459 CrossRef CAS.
- U. Schmid, U. T. Bornscheuer, M. M. Soumanou, G. P. McNeill and R. D. Schmid, Biotechnol. Bioeng., 1999, 64, 678–684 CrossRef CAS.
- A. D. M. Sorensen, X. B. Xu, L. Zhang, J. B. Kristensen and C. Jacobsen, J. Am. Oil Chem. Soc., 2010, 87, 185–194 CrossRef.
- H. Zhang, X. B. Xu, J. Nilsson, H. L. Mu, J. Adler-Nissen and C. E. Hoy, J. Am. Oil Chem. Soc., 2001, 78, 57–64 CrossRef CAS.
- M. W. Christensen, L. Andersen, T. L. Husum and O. Kirk, Eur. J. Lipid Sci. Technol., 2003, 105, 318–321 CrossRef CAS.
- H. C. Holm and D. Cowan, Eur. J. Lipid Sci. Technol., 2008, 110, 679–691 CrossRef CAS.
- J. Svensson and P. Adlercreutz, Eur. J. Lipid Sci. Technol., 2008, 110, 1007–1013 CrossRef CAS.
- N. M. Osorio, M. M. R. da Fonseca and S. Ferreira-Dias, Eur. J. Lipid Sci. Technol., 2006, 108, 545–553 CrossRef CAS.
- J. Svensson and P. Adlercreutz, Eur. J. Lipid Sci. Technol., 2011, 113, 1258–1265 CrossRef CAS.
- F. Theil, Chem. Rev., 1995, 95, 2203–2227 CrossRef CAS.
- Hydrolases in Organic Synthesis, ed. U. T. Bornscheuer and R. J. Kazlauskas, 2nd edn, Wiley-VCH, 2005 Search PubMed.
- E. Garcia-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110–PR180 CrossRef CAS.
- C.-S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
- C.-S. Chen, S.-H. Wu, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1987, 109, 2812–2817 CrossRef CAS.
- U. Hanefeld, Org. Biomol. Chem., 2003, 1, 2405–2415 CAS.
- Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, Weinheim, Germany, 2nd edn, 2006 Search PubMed.
- F. Balkenhohl, K. Ditrich, B. Hauer and W. Ladner, J. Prakt. Chem./Chem.- Ztg., 1997, 339, 381–384 CrossRef CAS.
- F. Balkenhohl, K. Ditrich and C. Nubling, Patent application, US 6,214,608, 2001 Search PubMed.
- P. Stead, H. Marley, M. Mahmoudian, G. Webb, D. Noble, Y. T. Ip, E. Piga, T. Rossi, S. Roberts and M. J. Dawson, Tetrahedron: Asymmetry, 1996, 7, 2247–2250 CrossRef CAS.
- A. Zaks and D. R. Dodds, Drug Discovery Today, 1997, 2, 513–531 CrossRef CAS.
- R. N. Patel, C. M. McNamee and L. J. Szarka, Appl. Microbiol. Biotechnol., 1992, 38, 56–60 CrossRef CAS.
- N. J. Turner, J. R. Winterman, R. McCague, J. S. Parratt and S. J. C. Taylor, Tetrahedron Lett., 1995, 36, 1113–1116 CrossRef CAS.
- Y. Chikusa, Y. Hirayama, M. Ikunaka, T. Inoue, S. Kamiyama, M. Moriwaki, Y. Nishimoto, F. Nomoto, K. Ogawa, T. Ohno, K. Otsuka, A. K. Sakota, N. Shirasaka, A. Uzura and K. Uzura, Org. Process Res. Dev., 2003, 7, 289–296 CrossRef CAS.
- B. A. Persson, A. L. E. Larsson, M. Le Ray and J. E. Backvall, J. Am. Chem. Soc., 1999, 121, 1645–1650 CrossRef CAS.
- B. Martin-Matute, M. Edin, K. Bogar and J. E. Backvall, Angew. Chem., Int. Ed., 2004, 43, 6535–6539 CrossRef CAS.
- J. A. Kralovec, S. Zhang, W. Zhang and C. J. Barrow, Food Chem., 2012, 131, 639–644 CrossRef CAS.
- K. D. Mukherjee, I. Kiewitt and M. J. Hills, Appl. Microbiol. Biotechnol., 1993, 40, 489–493 CrossRef CAS.
- I. Jachmanian, E. Schulte and K. D. Mukherjee, Appl. Microbiol. Biotechnol., 1996, 44, 563–567 CrossRef CAS.
- A.-M. Lyberg and P. Adlercreutz, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 343–350 CrossRef CAS.
- B. Mbatia, P. Adlercreutz, F. Mulaa and B. Mattiasson, Eur. J. Lipid Sci. Technol., 2010, 112, 977–984 CrossRef CAS.
- D. Kahveci and X.-B. Xu, Food Chem., 2011, 129, 1552–1558 CrossRef CAS.
- A.-M. Lyberg and P. Adlercreutz, Eur. J. Lipid Sci. Technol., 2008, 110, 317–324 CrossRef CAS.
- B. Mbatia, B. Mattiasson, F. Mulaa and P. Adlercreutz, Eur. J. Lipid Sci. Technol., 2011, 113, 717–723 CrossRef CAS.
- S. S. Kaki and P. Adlercreutz, Biotechnol. Bioeng., 2013, 110, 78–86 CrossRef CAS.
- A. Halldorsson, B. Kristinsson and G. G. Haraldsson, Eur. J. Lipid Sci. Technol., 2004, 106, 79–87 CrossRef CAS.
- G. P. McNeill and P. E. Sonnet, J. Am. Oil Chem. Soc., 1995, 72, 213–218 CrossRef CAS.
- M. Rahmatullah, V. K. S. Shukla and K. D. Mukherjee, J. Am. Oil Chem. Soc., 1994, 71, 563–567 CrossRef CAS.
- R. Kourist, H. Brundiek and U. T. Bornscheuer, Eur. J. Lipid Sci. Technol., 2009, 112, 64–74 CrossRef.
- H. B. Brundiek, A. S. Evitt, R. Kourist and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2012, 51, 412–414 CrossRef CAS.
- J. Schmitt, S. Brocca, R. D. Schmid and J. Pleiss, Protein Eng., 2002, 15, 595–601 CrossRef CAS.
- Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 89, 1–16 CrossRef CAS.
- L. Fjerbaek, K. V. Christensen and B. Norddahl, Biotechnol. Bioeng., 2009, 102, 1298–1315 CrossRef CAS.
- W. Du, W. Li, T. Sun, X. Chen and D. H. Liu, Appl. Microbiol. Biotechnol., 2008, 79, 331–337 CrossRef CAS.
- A. Bajaj, P. Lohan, P. N. Jha and R. Mehrotra, J. Mol. Catal. B: Enzym., 2009, 62, 9–14 CrossRef.
- A. Gog, M. Roman, M. Tosa, C. Paizs and F. D. Irimie, Renewable Energy, 2012, 39, 10–16 CrossRef CAS.
- T. W. Tan, J. K. Lu, K. L. Nie, L. Deng and F. Wang, Biotechnol. Adv., 2010, 28, 628–634 CrossRef CAS.
- M. M. Soumanou and U. T. Bornscheuer, Enzyme Microb. Technol., 2003, 33, 97–103 CrossRef CAS.
- L. Deng, X. B. Xu, G. G. Haraldsson, T. W. Tan and F. Wang, J. Am. Oil Chem. Soc., 2005, 82, 341–347 CrossRef CAS.
- Y. Shimada, Y. Watanabe, A. Sugihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 133–142 CrossRef CAS.
- M. Lee, J. Lee, D. Lee, J. Cho, S. Kim and C. Park, Enzyme Microb. Technol., 2011, 49, 402–406 CrossRef CAS.
- E. Hernandez-Martin and C. Otero, Bioresour. Technol., 2008, 99, 277–286 CrossRef CAS.
- Y. Xu, M. Nordblad, P. M. Nielsen, J. Brask and J. M. Woodley, J. Mol. Catal. B: Enzym., 2011, 72, 213–219 CrossRef CAS.
- Y. Huang, H. Zheng and Y. J. Yan, Appl. Biochem. Biotechnol., 2010, 160, 504–515 CrossRef CAS.
- W. Du, Y. Y. Xu, D. H. Liu and Z. B. Li, J. Mol. Catal. B: Enzym., 2005, 37, 68–71 CrossRef CAS.
- Y. D. Wang, X. Y. Shen, Z. L. Li, X. Li, F. Wang, X. A. Nie and J. C. Jiang, J. Mol. Catal. B: Enzym., 2010, 67, 45–51 CrossRef CAS.
- M. J. Ko, H. J. Park, S. Y. Hong and Y. J. Yoo, Bioprocess Biosyst. Eng., 2012, 35, 69–75 CrossRef CAS.
- F. Paltauf and A. Hermetter, Prog. Lipid Res., 1994, 33, 239–328 CrossRef CAS.
- G. G. Haraldsson and A. Thorarensen, J. Am. Oil Chem. Soc., 1999, 76, 1143–1149 CrossRef CAS.
- D. Adlercreutz and E. Wehtje, J. Am. Oil Chem. Soc., 2004, 81, 553–557 CrossRef CAS.
- F. Hara and T. Nakashima, J. Am. Oil Chem. Soc., 1996, 73, 657–659 CrossRef CAS.
- P. Adlercreutz, A. M. Lyberg and D. Adlercreutz, Eur. J. Lipid Sci. Technol., 2003, 105, 638–645 CrossRef CAS.
- S. S. Kaki and P. Adlercreutz, Chem. Phys. Lipids, 2011, 164, 246–250 CrossRef CAS.
- D. Adlercreutz, H. Budde and E. Wehtje, Biotechnol. Bioeng., 2002, 78, 403–411 CrossRef CAS.
- I. Svensson, P. Adlercreutz and B. Mattiasson, Appl. Microbiol. Biotechnol., 1990, 33, 255–258 CrossRef CAS.
- M. Hosokawa, K. Takahashi, N. Miyazaki, K. Okamura and M. Hatano, J. Am. Oil Chem. Soc., 1995, 72, 421–425 CrossRef CAS.
- A. M. Lyberg, D. Adlercreutz and P. Adlercreutz, Eur. J. Lipid Sci. Technol., 2005, 107, 279–290 CrossRef CAS.
- A. M. Gumel, M. S. M. Annuar, T. Heidelberg and Y. Chisti, Process Biochem., 2011, 46, 2079–2090 CrossRef CAS.
- L. Q. Cao, U. T. Bornscheuer and R. D. Schmid, Biocatal. Biotransform., 1998, 16, 249–257 CrossRef CAS.
- G. Fregapane, D. B. Sarney and E. N. Vulfson, Enzyme Microb. Technol., 1991, 13, 796–800 CrossRef CAS.
- D. B. Sarney, H. Kapeller, G. Fregapane and E. N. Vulfson, J. Am. Oil Chem. Soc., 1994, 71, 711–714 CrossRef CAS.
- G. B. Oguntimein, H. Erdmann and R. D. Schmid, Biotechnol. Lett., 1993, 15, 175–180 CrossRef CAS.
- F. Bjorkling, S. E. Godtfredsen and O. Kirk, J. Chem. Soc., Chem. Commun., 1989, 934–935 RSC.
- G. Ljunger, P. Adlercreutz and B. Mattiasson, Biotechnol. Lett., 1994, 16, 1167–1172 CrossRef CAS.
- L. Cao, A. Fischer, U. T. Bornscheuer and R. D. Schmid, Biocatal. Biotransform., 1997, 14, 269–283 CrossRef CAS.
- M. V. Flores, K. Naraghi, J. M. Engasser and P. J. Halling, Biotechnol. Bioeng., 2002, 78, 814–820 CAS.
- M. V. Flores and P. J. Halling, Biotechnol. Bioeng., 2002, 78, 794–800 CAS.
- F. Ganske and U. T. Bornscheuer, J. Mol. Catal. B: Enzym., 2005, 36, 40–42 CrossRef CAS.
- F. Ganske and U. T. Bornscheuer, Org. Lett., 2005, 7, 3097–3098 CrossRef CAS.
- E. Abdulmalek, H. S. M. Saupi, B. A. Tejo, M. Basri, A. B. Salleh, R. N. Z. R. Abd Rahman and M. B. Abdul Rahman, J. Mol. Catal. B: Enzym., 2012, 76, 37–43 CrossRef CAS.
- S. H. Lee, D. T. Dang, S. H. Ha, W.-J. Chang and Y.-M. Koo, Biotechnol. Bioeng., 2007, 99, 1–8 CrossRef.
- S. H. Lee, H. M. Nguyen, Y.-M. Koo and S. H. Ha, Process Biochem., 2008, 43, 1009–1012 CrossRef CAS.
- P. Degn and W. Zimmermann, Biotechnol. Bioeng., 2001, 74, 483–491 CrossRef CAS.
- M. Ferrer, M. A. Cruces, F. J. Plou, M. Bernabe and A. Ballesteros, Tetrahedron, 2000, 56, 4053–4061 CrossRef CAS.
- M. Ferrer, F. J. Plou, G. Fuentes, M. A. Cruces, L. Andersen, O. Kirk, M. Christensen and A. Ballesteros, Biocatal. Biotransform., 2002, 20, 63–71 CrossRef CAS.
- F. J. Plou, M. A. Cruces, M. Ferrer, G. Fuentes, E. Pastor, M. Bernabe, M. Christensen, F. Comelles, J. L. Parra and A. Ballesteros, J. Biotechnol., 2002, 96, 55–66 CrossRef CAS.
- A. Alissandratos and P. J. Halling, Bioresour. Technol., 2012, 115, 41–47 CrossRef CAS.
- A. Alissandratos, N. Baudendistel, S. L. Flitsch, B. Hauer and P. J. Halling, BMC Biotechnol., 2010, 10, 82 CrossRef CAS.
- A. Alissandratos, N. Baudendistel, B. Hauer, K. Baldenius, S. Flitsch and P. Halling, Chem. Commun., 2011, 47, 683–685 RSC.
- S. Chakraborty, B. Sahoo, I. Teraoka, L. M. Miller and R. A. Gross, Macromolecules, 2005, 38, 61–68 CrossRef CAS.
- S. Riva, J. Mol. Catal. B: Enzym., 2002, 19, 43–54 CrossRef.
- P. Villeneuve, Biotechnol. Adv., 2007, 25, 515–536 CrossRef CAS.
- M. C. Figueroa-Espinoza and P. Villeneuve, J. Agric. Food Chem., 2005, 53, 2779–2787 CrossRef CAS.
- V. M. Almeida, C. R. C. Branco, S. A. Assis, I. J. C. Vieira, R. Braz and A. Branco, Chem. Cent. J., 2012, 6, 41 CrossRef CAS.
- S. H. Youn, H. J. Kim, T. H. Kim and C. S. Shin, J. Mol. Catal. B: Enzym., 2007, 46, 26–31 CrossRef CAS.
- M. Laguerre, C. Bayrasy, J. Lecomte, B. Chabi, E. A. Decker, C. Wrutniak-Cabello, G. Cabello and P. Villeneuve, Biochimie, 2013, 95, 20–26 CrossRef CAS.
- M. Laguerre, C. Wrutniak-Cabello, B. Chabi, L. J. L. Giraldo, J. Lecomte, P. Villeneuve and G. Cabello, J. Pharm. Pharmacol., 2011, 63, 531–540 CrossRef CAS.
- B. Mbatia, S. S. Kaki, B. Mattiasson, F. Mulaa and P. Adlercreutz, J. Agric. Food Chem., 2011, 59, 7021–7027 CrossRef CAS.
- Y.-H. Yang, T. Raku, E. Song, S.-H. Park, D. Yoo, H.-Y. Park, B.-G. Kim, H.-J. Kim, S.-H. Lee, H.-S. Kim and Y. Tokiwa, Biotechnol. Bioprocess Eng., 2012, 17, 50–54 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |