 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilised enzymes in biorenewables production
Maurice C. R. Franssen*a, Peter Steunenberga, Elinor L. Scottb, Han Zuilhofac and Johan P. M. Sanders*b
aLaboratory of Organic Chemistry, Wageningen University, Dreijenplein 8, 6703 HB Wageningen, The Netherlands. E-mail: Maurice.Franssen@wur.nl
bBiobased Commodity Chemicals, Wageningen University, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: Johan.Sanders@wur.nl
cDepartment of Chemical and Materials Engineering, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 21st March 2013
Abstract
Oils, fats, carbohydrates, lignin, and amino acids are all important raw materials for the production of biorenewables. These compounds already play an important role in everyday life in the form of wood, fabrics, starch, paper and rubber. Enzymatic reactions do, in principle, allow the transformation of these raw materials into biorenewables under mild and sustainable conditions. There are a few examples of processes using immobilised enzymes that are already applied on an industrial scale, such as the production of High-Fructose Corn Syrup, but these are still rather rare. Fortunately, there is a rapid expansion in the research efforts that try to improve this, driven by a combination of economic and ecological reasons. This review focusses on those efforts, by looking at attempts to use fatty acids, carbohydrates, proteins and lignin (and their building blocks), as substrates in the synthesis of biorenewables using immobilised enzymes. Therefore, many examples (390 references) from the recent literature are discussed, in which we look both at the specific reactions as well as to the methods of immobilisation of the enzymes, as the latter are shown to be a crucial factor with respect to stability and reuse. The applications of the renewables produced in this way range from building blocks for the pharmaceutical and polymer industry, transport fuels, to additives for the food industry. A critical evaluation of the relevant factors that need to be improved for large-scale use of these examples is presented in the outlook of this review.
 Maurice C. R. Franssen | Maurice Franssen is Associate Professor Enzymes and Surfaces at the Laboratory of Organic Chemistry of Wageningen University. His main interests are the application of surface-bound enzymes and biocatalytic processes in organic chemistry. He obtained a PhD in Organic Chemistry from Wageningen University. In 1987 he was appointed as assistant professor at the Laboratory of Organic Chemistry and since 1999 he is an Associate Professor. In 1988 he won the Royal/Dutch Shell Award and in 1989 he was a visiting scientist at the Laboratory of prof. A. M. Klibanov (MIT). He is a member of the Permanent Steering Committee of the Biotrans symposium series and the Editorial Board of J. Mol. Catal. B: Enzymatic. |
 Peter Steunenberg | Peter Steunenberg is Post-Doctoral researcher at the Laboratory of Organic Chemistry of Wageningen University. His research interests focus on synthetic organic chemistry, green chemistry and biocatalysis. He obtained a PhD in Organic Chemistry from École Polytechnique Féderale de Lausanne for researching methods to synthesise C-disaccharides. Afterwards he did postdoctoral work at the Karolinska Institutet and worked as a scientist for Kiadis Pharma and Axon Medchem. Subsequently he returned to academia and was employed as a postdoctoral researcher at the University of Twente and in 2011 he started his postdoctoral work at Wageningen University. |
 Elinor L. Scott | Elinor Scott is Assistant Professor at Wageningen University of the Biobased Commodity Chemicals group. Since 1997 she has been concerned with the development of economically and ecologically attractive conversion routes and processes of biomass to industrial chemicals. She obtained her PhD in Chemistry at Heriot-Watt University. Following positions as Post-Doctoral researcher in the field of polymer chemistry at the University of Strathclyde (1994–1995) and Heriot-Watt University (1995–1996), she moved to the Netherlands where she worked as an industrial Post-Doctoral scientist working on composite materials at DSM and later as a scientist at ATO looking into biomass conversions. |
 Han Zuilhof | Han Zuilhof is Chair of Organic Chemistry at the Laboratory of Organic Chemistry of Wageningen University. His interests focus on surface-bound organic chemistry, and the use of detailed (bio-)organic chemistry knowledge in a wide variety of phenomena and devices. He obtained both an MSc in Chemistry and an MA in Philosophy, before deciding that chemistry was the more social of the two disciplines. After obtaining a PhD in chemistry (Leiden University, 1994; highest honors) he did postdoctoral work at the University of Rochester, NY, and at Columbia University. Subsequently he joined the faculty at Wageningen University, and is a Professor of Organic Chemistry since 2007. He is also a Distinguished Adjunct Professor of Chemical Engineering at the King Abdulaziz University in Jeddah, Saudi Arabia, is the founder of Surfix, a company specializing in surface modifications, and serves on the Editorial Advisory Board of Langmuir and Applied Surface Science. |
 Johan P. M. Sanders | Johan Sanders is Full Professor Biobased Commodity Chemicals at Wageningen University. His work focuses on reducing the CO2 production in a cost-effective way using chemistry, fermentation and biorefineries at a large as well as at small scale to enable optimal application of all plant components. He holds a PhD in Molecular Biology from the University of Amsterdam for researching the physical map of yeast mitochondrial DNA. From 1977 to 1993 he worked at Gist Brocades, starting the Genetic Engineering group and working on various projects in the field of enzyme research; he became Associate Director of Food Research. From 1993 to 2001 Sanders worked at AVEBE as R&D Director focusing on the enzymatic and genetic modification of starch. |
1. Introduction
Mankind is used to apply large amounts of plant material as food. We are also accustomed to apply biomass for some non-food products, such as wood for construction purposes or for paper production, cotton for clothes, and rubber for the production of car tyres.1 In the next few decades we will also need to apply biomass for applications that are up to now produced from fossil resources, like oil, natural gas and coal. There are several long-term drivers that urge us in this direction, such as the depletion of oil reserves, climate change, the dependency on a limited number of countries that have fossil reserves, and the fast economic growth of e.g. the BRIC countries. Several forms of renewable energy sources other than biomass are currently being further developed, such as photovoltaic cells, hydropower, tidal energy and wind energy. Biomass can contribute here by substituting natural gas, coal and oil for the large-scale application as energy production and transportation fuels, but it should be emphasised that biomass is the only source that can substitute oil for the production of chemicals and materials.2From an economic point of view the substitution of fossil-based chemicals by biomass sources would be attractive since different components of biomass already contain much of the molecular functionalities that are now introduced in oil-derived base chemicals at the cost of a lot of energy and capital costs.3 A major part of the capital costs arises from the need for heat exchange under industrial conditions of high temperature, high pressures, corrosive conditions or the need to contain dangerous chemicals or intermediates.4
We can benefit from enzymes in the conversion from functionalised natural molecules into the functionalised chemicals or intermediates that are nowadays produced from oil,5–9 when we use these enzymes at relatively low temperatures as compared to the conditions of the petrochemical industry. Functionalised chemicals will have a higher added value than chemicals with no or only a few functional moieties. In contrast, transportation fuels, which come at a larger volume than the chemicals in the market, have a lower added value per ton of product, leaving less economic room for the application of enzymes. This puts pressure on e.g. the use of lipases for the transesterification of oils to biodiesel: they can only contribute to a competitive process when they are very cheap.
Enzymes generally have some important properties that can give them an economic advantage. Many enzymes catalyse reactions with a very high substrate specificity. Therefore even in complex mixtures dissolved in water at high and low concentrations, one single component is converted into its product. Enzymes are active at modest temperatures, mainly in the range of 20–80 °C, although certain enzymes can operate under more extreme conditions between 0 and 130 °C.10
This combination of specificity and low temperature demand imply that capital costs can be significantly lower per unit of product manufactured, even though the concentration of the substrates and products is much lower than usual in the petrochemical industry. This lower capital cost is also a consequence of the fact that much less energy is produced in the enzymatic conversions. The production of chemicals from biomass therefore can be much more energy efficient than in the petrochemical processes. Another potential advantage of enzymatic reactions is that no or much less salts are created as waste streams because no or significantly less acids and bases are required for the reactions to proceed. Finally, the structure of enzymes can relatively easily be modified by enzyme engineering technologies towards their desired reaction conditions such as pH and temperature, but also in their specific activity, substrate specificity or their sensitivity towards inhibitors.
On the other hand enzymes also have some disadvantages as compared to petrochemical catalysts. The volumetric conversion rates of enzymatic conversions are lower because of the low concentration of the reactants. Generally the costs of enzyme usage are high because enzymes are not very stable or can be inactivated by inhibitors or reduced in their activity because of the product formed by their own activities. Some enzymes also need cofactors that should be regenerated by another reaction.11 For other enzymes the prosthetic group undergoes turnover-mediated alterations leading to enzyme inactivation.12
The immobilisation of enzymes might reduce some disadvantages of the use of enzymes, mainly the cost factor because immobilised enzymes can be easily reused. Lowering the costs of enzymatic conversions will open routes for the production of biobased chemicals but also of transportation fuels. If enzymatic reactions would run at larger volumes, their price would go down because a larger sales volume of enzymes tolerates a larger effort to lower the cost of production of the enzymes by genetic and fermentation technologies.
In this review, we will discuss the techniques that have been applied for immobilisation of enzymes used in the production of biorenewables (Section 2), followed by a detailed overview of the applications themselves, grouped by the type of compound: fatty acids (Section 3), carbohydrates (4), amino acids (5), lignin and its building blocks (6) and polymers (7). The review will be concluded with an outlook (Section 8) and some closing remarks (Section 9). It should be borne in mind that whenever a KM or a Vmax value is mentioned this refers to apparent kinetic constants because immobilised enzymes do not obey Michaelis–Menten kinetics due to mass transfer limitations.
2. Immobilisation techniques
Immobilised enzymes are enzymes that are physically confined or localised, with retention of its catalytic activity, so that they can be used repeatedly and continuously.13 Research and application of immobilised enzymes started in the late 1960s. The books of Zaborsky14 and Royer15 served as a source of inspiration for many academic and industrial scientists. Basically, all enzyme immobilisation techniques still can be classified in the same way as done in those days:1. binding to a solid support
a. by adsorption (by hydrogen bonds or hydrophobic interaction)
b. by ionic binding (e.g. to ion exchange resins)
c. by covalent binding (e.g. to epoxy groups)
2. cross-linking (e.g. by glutaraldehyde)
3. entrapment
a. in gels (e.g. calcium alginate)
b. in membrane reactors (e.g. hollow fibre reactors)
c. in reversed micelles, microemulsions, etc.
As discussed elsewhere in this themed issue, the application of immobilised enzymes is widespread nowadays, thanks to the commercial availability of stable immobilised enzyme preparations (e.g. Novozym 435, CLEAs), the development of novel supports (e.g. microporous or nanostructured materials), highly active commercially available supports (Sepabeads©, Immobeads) and novel techniques (sol–gels, CLEAs).16 Almost all techniques mentioned above have been used in the enzymatic preparation of biorenewables. Some of these techniques will be highlighted below, before discussing their application in detail in separate sections.
2.1 Binding to a solid support
A robust enzyme that is used commercially in China for biodiesel production is the lipase from Candida sp. 99–125. Its immobilisation by adsorption onto a textile membrane is facile,29 yielding a preparation that is claimed to be cheaper than Novozym 435, but it is not clear to us if it is indeed this form that is used in the factory.
Other very useful modern supports for enzyme immobilisation by adsorption are microporous beads made of polyethylene or polypropylene, marketed under the name Accurel®. Lipases are once more the enzymes that are most often immobilised onto this material, and not only for the advantages of lipases mentioned earlier but also because they possess hydrophobic patches that bind well to these hydrophobic carriers. E.g., Burkholderia cepacia lipase immobilised on Accurel 1282 was used for biodiesel production,30 while CalB adsorbed on Accurel MP 1000 was used for various esterification and amidation reactions, including biodiesel production.31 This lipase preparation was claimed to be cheaper than Novozym 435 while having similar stability.
Immobilisation involves a matrix, a treatment, or both, and since costs play a crucial role especially in the production of bulk chemicals, it is important to look for cheap materials and methods. For this reason, Malaysian researchers have proposed mica as a cheap and useful adsorption matrix for enzymes. Mica is a very cheap and porous silicate that is used on a large scale for industrial applications, like the ceramics and tile industry.32 It can be modified by alkoxysilanes,33 and an aminopropyl derivative has been successfully used for the adsorption of Candida rugosa lipase (CRL). The authors claim that there is covalent binding, but this is unlikely because no coupling reagent was used in their experiments. The derivative was applied for the preparation of propyl laurate and lactose esters.34 The activities of the enzyme increased 2.4-fold over the free enzyme; CRL adsorbed to mica and subsequently cross-linked showed a 2.6 times increase in activity. Porous alumina has also been mentioned as a cheap and useful support for enzyme adsorption. Reshmi et al. studied Bacillus subtilis α-amylase, adsorbed to alumina, for the hydrolysis of starch to maltose and glucose.35 XRD analysis showed that the enzyme was only adsorbed to the surface, while IR spectra proved that mainly OH-groups of the alumina are involved in binding. The immobilised enzyme is much more stable at higher pH than free enzyme. A higher calcination temperature of the alumina also led to increased stability of the adsorbed enzyme. Novozymes has immobilised Thermomyces lanuginosa lipase on silica by adsorption and subsequent granulation, leading to a very stable preparation that contains lipase that is both adsorbed and entrapped.36 It is marketed under the name Lipozyme TL IM and used for biodiesel production.
Magnetic particles are useful because of their ease of separation. Tran et al.37 used hybrid magnetic nanoparticles made of Fe3O4 modified with silicates containing long alkyl chains for the adsorption of the lipase of Burkholderia sp. C20. This preparation was successfully applied for transforming the oil of the microalga Chlorella vulgaris into biodiesel. For a very recent overview of the applications of magnetic nanoparticles for enzyme immobilisation, the reader is referred to Netto et al.38
An interesting new approach to enzyme immobilisation was recently published by a Korean group.39 The gene of L-glutamate α-decarboxylase was fused to the cellulose binding domain of Trichoderma harzianum endoglucanase II, and bound to the resulting enzyme to microcrystalline cellulose (Avicel). The enzyme still had 60% of the initial activity after being reused 10 times. See for applications of this enzyme further down in this review.
An interesting development was made by Zheng et al.,48 who made a mixed-mode support based on silica possessing both charged groups and hydrophobic chains. Candida rugosa lipase was immobilised on octyl and sulfonic acid co-bonded silica (OSS), a kind of silica bead functionalized with octyl and thiol moieties that after oxidation with hydrogen peroxide yielded sulfonic acid groups. These mixed-mode silica particles49 would, in principle, allow any enzyme to accommodate itself inside the pores, orienting its charged or hydrophobic parts to the corresponding patches of the support. It has been shown that lipases, especially the hydrophobic areas surrounding the active center, prefer to absorb on hydrophobic surfaces,50 leaving the active site fully exposed, thereby allowing a high activity.51–53 CRL was immobilised in this way and used to couple phytosterols to fatty acids.
A potentially cheap matrix is a monolith made by polymerisation of organic or inorganic (silica) materials. This matrix is highly porous, so there are hardly any mass transfer problems. For example, lignin peroxidase was coupled to methacrylate based monolithic supports containing epoxy groups.57
In principle, membranes can also be regarded as porous materials, onto which enzymes can be immobilised by adsorption or covalent bonds.58 This is especially interesting if the enzymatic reaction can be combined with downstream processing, i.e. the product diffuses through the membrane and is in this way separated from both the enzyme and the substrate. It can, however, also be used to remove unwanted byproducts. Sen et al.59 immobilised β-galactosidase on a polyamide–poly(ethersulphone) composite film membrane that was modified with polyethylenimine, using glutaraldehyde as the coupling reagent. The system was used for converting lactose into galactooligosaccharides (GOS) with simultaneous separation of the GOS and the enzyme from the low-molecular weight byproducts caused by hydrolysis of the lactose. The yield of the resulting product was higher for this membrane system than for a batch system. Unfortunately, possible effects on the average MW were not checked. Edwards et al.60 used manganese peroxidase and laccase adsorbed to a poly(ethersulphone) membrane to degrade in situ any insoluble phenolic foulant that would block the pores of the membrane.
Covalent immobilisation to solid supports by glutaraldehyde is also the technique that was used by an Italian group to immobilise laccase61 and horseradish peroxidase (HRP)62 for lignin functionalisation. However, in order to protect the enzyme against denaturation by high-molecular weight polymers and microorganisms, the enzyme-containing particles were covered by alternating layers of oppositely charged polymers. In this so-called layer-by-layer (LbL) technique, the enzyme particle was first covered by poly(allylamine hydrochloride), then poly(styrene sulphonate) followed by poly(allylamine hydrochloride) again. The layers are sufficiently porous for low-MW substrates and products to diffuse to and from the enzyme. In the case of entrapped HRP, there are even indications that some of the immobilised enzymes have direct contact with the insoluble lignin polymer. Enzyme stability was indeed improved by the LbL technique, but thus immobilised preparations were only compared with soluble enzymes, not with immobilised enzymes that were not covered by polyelectrolyte layers. When covalently bound tyrosinase was covered with polyelectrolyte layers, 13% loss of activity was observed, with only marginal improvement of stability.63 In a paper on the immobilisation of lignin peroxidase and manganese peroxidase,64 the enzymes themselves were used as the negatively charged polyelectrolyte in the LbL technique.
2.2 Cross-linking
As mentioned before, cross-linking of enzymes was already known in the 1970s but was never popular because of enzyme inactivation by the cross-linker, and bad packing properties of the resulting particles. This picture changed upon the introduction of cross-linked enzyme crystals (CLECs).65,66 Although CLECs have been applied for the conversion of biobased compounds,67,68 large scale applications in industry are not likely because they need expensive and cumbersome crystallisation. The real breakthrough came when cross-linked enzyme aggregates (CLEAs) entered the field,69,70 because CLEAs involve simple precipitation of the enzyme by e.g. ammonium sulphate or t-butanol instead of crystallisation. Both methods are followed by cross-linking by e.g. glutaraldehyde, but because the enzyme is ‘solidified’ inactivation is much less severe than in solution. The method for making CLEAs and their advantages is reviewed by Schoevaart et al.71 Quite remarkably, CLEAs have hardly been used up to now for making biobased materials. Gaur et al.72 compared three different techniques for the immobilisation of Aspergillus oryzae β-galactosidase, namely adsorption on Celite®, covalent coupling to chitosan, and CLEAs. The covalently coupled enzyme preparation appeared to be advantageous in terms of yields when making GOS, while the CLEA was superior in lactose hydrolysis. The fact that the immobilisation matrix affects the synthesis/hydrolysis ratio in transfer reactions was already noticed in the first paper on CLEAs.69 Jones and Vasudevan73 have used CLEAs of Trichoderma reesei cellulase for the hydrolysis of cellulose to glucose. The highest enzyme activity was obtained when acetone was used as the precipitant. Significantly higher yields were obtained when the cellulose was pretreated with an ionic liquid in order to enhance its solubilisation.2.3 Entrapment
Entrapment in matrices like calcium alginate is still a common practice because of its ease and low cost,74 but it can only be used for high-molecular weight enzymes (above ∼100 kDa) because of the large pore size of the material. A solution was offered by Tee and Kaletunç, who combined entrapment with covalent binding to alginate.75 The carboxyl groups of the alginate were activated with EDC and sulfo-NHS and subsequently reacted with amino groups of α-amylase. A modern alternative to alginate is LentiKats®, lens-shaped particles mainly composed of poly(vinyl alcohol).76 Just like alginate, this matrix is only suitable for large enzymes (>50 kDa), as otherwise the enzyme will leak out. It has also been used as a matrix for aspartase for the conversion of fumaric acid to L-aspartic acid.46 Since this reaction is reversible it could, in principle, also be used to make biobased fumaric acid.Enzymes can also be entrapped in magnetic nanoparticles by making them in situ, as shown by Bellino and Regazzoni.77 In their elegant approach, ferrous chloride was mixed with urea, urease and α-amylase in an oxygen-saturated solution. Urease-mediated hydrolysis of urea leads to an increase in the pH with concomitant oxidation of Fe2+ and formation of insoluble ferric (hydr)oxide particles, in which the α-amylase (and the urease) is entrapped. The amylase was used for starch hydrolysis. It is just as active as the free enzyme and does not lose activity after five consecutive reactions.
Entrapment in membrane reactors is also well known and has interesting advantages when combined with downstream processing. For instance, Das et al.78 used the β-galactosidase from Bacillus circulans to make GOS from lactose in a membrane reactor in which the membrane serves as a barrier for the enzyme and the produced GOS, but is permeable for the produced monosaccharides glucose and galactose. This enables facile isolation of the product but also diminishes the inhibitory effect of the monosaccharides on the enzyme. Constant removal of these compounds resulted in a 33% higher yield of GOS compared to a batch system. On the other hand, in a method for the production of cellobiose,79 a membrane reactor was used to separate the enzyme and substrate from the product. The ultrafiltration membrane now acts as a barrier for the enzyme and the substrate (sugar beet pulp), while the permeate stream only consists of the desired product cellobiose. Cellulases are usually mixtures of enzymes and although the authors removed β-glucosidase activity from the Trichoderma reesei enzyme preparation, they only studied cellulose hydrolysis, ignoring the hydrolysis of pectin and hemicellulose. In this way 94% of the cellulose present in sugar beet pulp was converted with a cellobiose yield of 79%.
López et al. used an ultrafiltration membrane together with a CSTR for the manganese peroxidase-mediated decoloration of azo dyes.80 A membrane reactor was also useful in the production of biodiesel, where it was applied to mitigate product inhibition by glycerol, and to keep methanol levels low. In this study, however, the enzyme was immobilised onto an acrylic resin (Novozym 435).81
It is also possible to entrap enzymes in hollow polyelectrolyte microspheres, such as prepared by the layer-by-layer technique. For this, eight layers of oppositely charged polyelectrolyte macromolecules were physisorbed to the surface of manganese carbonate beads, after which the MnCO3 was dissolved in a strong acid and the enzyme was allowed to penetrate inside the microspheres at pH 5. At this pH, the permeability of the microcapsules increases dramatically, allowing enzyme molecules to penetrate into the spheres. Laccase immobilised in this way was used for the functionalisation of lignin.61
Entrapment by cross-linking of acrylic monomers has up to now not been very successful, because the monomers often inactivate the enzyme, e.g. by irreversible Michael addition of lysine or cysteine side chains to the α,β-unsaturated system of the monomers. In a recent patent,82 however, CalB was successfully entrapped in photochemically cross-linked methacrylated derivatives of chitosan and PEG, in the presence of sucrose. The resulting enzyme preparation was used for the esterification of sucrose in an organic solvent (t-amyl alcohol). The matrix is claimed to be highly resistant to organic solvents and elevated temperatures, permeable to substrate and product while retaining the enzyme. Sucrose acts as a (weakly binding) substrate for the lipase so this may be the reason for the resistance of the enzyme against the polymerisation conditions. It should be borne in mind, however, that sugars and sugar alcohols are in general good stabilising agents for enzymes, and that CalB is an extremely robust enzyme.
Entrapment in sol–gels is a very useful addition to the toolbox of applied biocatalysis. This method, pioneered by Reetz,51 involves a two-step process: in the first step alkoxysilanes (with or without [substituted] alkyl groups) are mixed in water and react to form a silica sol. Enzyme is added during or after sol formation. In the second step, the sol is destabilised by the addition of fluoride ions, and the whole mixture gellifies. The gel containing the entrapped enzyme can be cut or grinded. When the gel is subsequently dried and shrunk it is called a xerogel, when the solvent is removed under supercritical conditions the gel does not shrink and the result is called an aerogel. Both gels have very high porosity and surface area while having small pores. Enzymes having hydrophobic patches are often activated when entrapped in hydrophobic sol–gels, which is remarkable because usually enzymes become less active upon immobilisation due to diffusional limitations. Beneficial conformational changes may play a role here, but enhanced partitioning of hydrophobic substrates will undoubtedly be important too. Within the scope of this review, two lignin peroxidases were successfully immobilised in a xerogel made of Si(OMe)4/nPrSi(OMe)3.83,84
Different enzymes can be co-immobilised on one matrix, for making enzyme cascades, removing deleterious side products or ensuring slow production of substrate. The latter reason was the background for the research by Qiu et al.,85 who co-immobilised glucose oxidase with lignin peroxidase. The peroxidase needs H2O2 as a substrate, as its name implies, but very high concentrations lead to inactivation. Slow and continuous production of H2O2 by the co-immobilised glucose oxidase prevented this problem. A disadvantage of such a system is, however, that the two enzymes usually have different inactivation rates, which makes the kinetic regime difficult. A solution to this is not co-immobilise, but to have the two enzymes on separate beads in the reactor, so that the fast inactivating enzyme can be replenished independently from the other.
3. Fatty acids, biodiesel and beyond
Fats and oils are water-insoluble, hydrophobic substances in plants and animals that consist of one equivalent of glycerol and three equivalents of fatty acids, therefore usually named triglycerides. In this part of the review the focus is on the production of renewables from oils and fats or the products that are formed after hydrolysis of these oils and fats: fatty acids and glycerol. Oils and fats are one of the most important and abundant renewable raw materials for the chemical industry. After a short introduction to biodiesel (3.1.), recent developments for the production of biodiesel based on edible and non-edible oils and fats will be highlighted (3.2.). Afterwards the production of functionalised chemicals starting from renewable resources based on triglycerides etc. will be discussed (3.3.).3.1 Introduction to biodiesel
Due to the depletion of the world petroleum reserves, combined with the destructive impact of environmental pollution by the rapidly increasing exhaust emission, there is an urgent need for suitable alternative fuels for use in combustion engines. The feasibility to use biodiesel for this purpose was already shown in 1900 at the Paris World exposition, where the French Otto Company showed that it was possible to run an engine on peanut oil. In the same time period, Rudolf Diesel also tested his engine on the suitability of using vegetable oil.86,87 Biodiesel combines several advantageous features: it has several environmental benefits, it has better lubricating properties than petroleum diesel, and a much higher cetane value. Additionally it contains little sulphur and generates less dust pollution.88–90 However, the main problem for large-scale introduction is that the overall production costs for biodiesel are currently twice that of fossil-derived diesel, which is mainly based on the expense made on raw materials.91 Biodiesel can be used in its neat form or as a blend with conventional diesel fuel in diesel engines. Several excellent reviews about the production of biodiesel starting from plant oils have already been published.24,92–102 The focus of the first part of this section is to give an overview of the latest developments regarding the production of biodiesel from fats and oils, i.e. transesterification catalysed by immobilised lipases. In the second part, reactions with immobilised enzymes for the preparation of other fatty acid derivatives will be described.3.2 Biodiesel production
The chemical description of biodiesel is monoalkyl esters of long chain fatty acids, derived from renewable feedstocks like vegetable oils and animal fats. Animal tallow, grease, fish oil, and lard have been used in biodiesel production. Nevertheless, the most common triglyceride source is plant oil. Main sources are vegetable oil, soybean oil, sunflower oil, palm oil and rapeseed oil.103–107 Different countries use different sources of vegetable oil, depending on the climate. Soybean oil is used mostly in the United States, rapeseed and sunflower oil in Europe and palm or coconut oil in Asia. Other sources of triglycerides are microalgae or waste oil from factories and restaurants. The main drawback of the use of waste oils is the high content of free fatty acid and water, which causes saponification and hydrolysis.108The conventional method for producing biodiesel usually involves the acid or base-catalysed formation of fatty acid alkyl esters by transesterification of oils of the corresponding fatty acid to short chain alcohols (see Scheme 1). Alternative routes for the production of biodiesel are pyrolysis, transesterification under supercritical conditions, and the formation of micro-emulsions.109–111 These methods are typically costly and yield low-quality biodiesel.
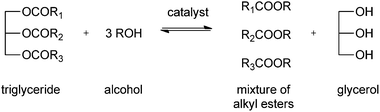 | ||
| Scheme 1 Formation of biodiesel: transesterification of triglycerides. | ||
Enzymatic reactions have become an alternative for the production of biodiesel through the so-called alcoholysis process, a transesterification catalysed by lipases. The transesterification at the same time reduces the viscosity characteristic of triacylglycerides (TAG). This enhances the physical properties and improves the engine performance. In this process the alcohol (typically methanol) plays a dual role: on the one hand, high alcohol concentrations will drive the reaction to products, given the reversible nature of the reaction.112 On the other hand high concentrations of methanol cause problems, since it is a known enzyme inhibitor.113,114 Finding the optimum alcohol concentration is thus typically an important and enzyme-dependent step in the optimisation of the reaction. The transesterification reaction also generates glycerol,115 which can also affect the reaction efficiency (see below). In a later section of this review useful applications of glycerol for the production of renewables will be discussed. The use of immobilised lipases or whole cells lowers the overall production costs and also minimizes downstream processing problems. Whereas free enzymes have lower production costs, immobilisation of the enzymes improves the stability, reusability and allows higher reaction temperatures.25,36,116,117 Plant oils and especially fatty acids offer many more possibilities for applications other than biodiesel alone. Petroleum-based production of products like coatings, paints, lubricants and surfactants have in many cases already been replaced by a fatty acids-based production.118–120
An example is the production of Fatty Acid Ethyl Esters (FAEE), one of the main components of biodiesel, which have better lubrication properties compared to Fatty Acid Methyl Esters (FAME), the standard when it comes to the use as fuel. In addition, ethanol is preferred due to its low costs and abundant availability.121 Compared to conventional diesel fuel, Fatty Acid Alkyl Esters FAEE have higher cetane numbers, pour points, cloud points, flash points, a higher viscosity, a higher oxygen but lower sulphur content and heat of combustion.122 FAEE can also be used as a replacement of fossil-based products as a green solvent123 and as a drilling fluid applicable to high temperatures.124 The cost of the lipase is the main drawback for the industrial applications of enzyme-catalysed processes for biodiesel production.
Bornscheuer et al.30 reported the use of Burkholderia cepacia lipase in its immobilised form on an Accurel 1282 carrier for the formation of biodiesel from Jatropha curcas oil. Jatropha curcas is a drought-resistant perennial, easily cultivated on marginal or poor soil. Jatropha is also known as the wonder plant, because its seeds contain around 37% of oil. The oil can even be used without being refined, it gives a clear, smoke-free flame and is tested for use in simple diesel engines.95Jatropha curcas oil is a plant oil that is not suitable for human nutrition, and therefore an ideal starting material for biodiesel production. Jatropha curcas seeds can be obtained from several locations in Benin, and the oil can be extracted in high yields from these seeds. By using a genetically modified and short chain alcohol-tolerant lipase from Pseudomonas cepacia,125 the highest conversion of Jatropha oil (93%) was obtained using only a low enzyme loading (3% w/w) after a reaction time of 16 h. This lipase from Pseudomonas cepacia was the only lipase (of the ones tested, when immobilised on an acrylic support) that showed a high activity for the formation of the methyl ester of the corresponding fatty acid, as well as the formation of the ethyl and butyl ester of the fatty acid. However, high reusability – no observable loss of enzyme activity during 160 h – and conversion was only observed for the transformation into the ethyl ester. For alcoholysis in methanol or n-butanol, after 10 cycles more than 60% of the enzyme activity was lost.
Li et al. have shown that biodiesel can also be prepared from Pistacia chinensis bge seed oil (PCO).126Pistacia chinensis is a woody plant from central and western China that can withstand harsh conditions and poor quality soils. The content of oil in the seeds is above 30% and contains up to 82% of unsaturated fatty acids.127 An enzyme that catalyses the transesterification is the lipase from Rhizopus oryzae (ROL), which induces methanolysis even with low amounts or no water present in oil.128 Therefore this enzyme is considered as a potential candidate for solvent-free reaction processes. For these transesterification reactions ROL was immobilised on microporous (MI-ROL) and anion exchange (AI-ROL) resins, because they have an enhancing effect on the transesterification process.129,130 Optimum conditions for the transesterification and the effect of water content were determined to be 20% (w/w) water relative to oil, with MI-ROL or AI-ROL as the catalyst at 37 °C, in which after 60 h 81.8% conversion was reached for AI-ROL and 91.8% for MI-ROL. On the other hand, AI-ROL showed a higher stability, as there was no significant loss in the yield of biodiesel after 6 cycles, whereas for the MI-ROL, after 4 cycles the yield dropped to 60%. This process has significant potential, because the biodiesel obtained from PCO has properties close to biodiesel obtained from soybean oil and meets the American Society for Testing Materials (ASTM) specifications: the density is 0.88 g cm−3 (ASTM: 0.86–0.90), kinematic viscosity 4.15 mm2 s−1 (3.5–5.0), flashpoint 102 (>100) and cetane number 49 (47). However, the calorific value of PCO is still somewhat lower (39.8 MJ kg−1) than observed for standard diesel fuel (44.3 MJ kg−1), which can be attributed to the presence of more heteroatoms per kg fuel.
One of the main reasons for the high manufacturing costs of biodiesel is the high cost of virgin vegetable oil. Therefore waste cooking oil, a much less expensive starting material than vegetable oil, is a promising alternative. An example of the transesterification of fatty acids from waste oil has been reported by Sonare and Rathod.42 Lipozyme RM IM showed the highest activity for this reaction at a mild reaction temperature of 45 °C, with an optimum loading of 4–6% (w/w) relative to oil. Tertiary butanol was used as a solvent here. It is a moderately hydrophilic solvent that can solubilise oil, methanol and glycerol, but it is not a substrate for lipases,131 and it enhances the activity of the enzyme compared to a solvent-free system, thereby effecting a higher conversion. The solvent was used because a solvent-free system gives a maximum conversion of 22% and deactivates the enzyme. This compensates for the additional costs of the solvent. Under optimised conditions, with a 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol-to-oil ratio a maximum conversion of 67% of waste frying oil was reached. The reusability of the Lipozyme RM IM was also tested and the enzyme remained active after 6 cycles. The conversion of used sunflower oil into FAME was also compared to refined sunflower oil, which gave a higher conversion of 74%. This was mainly caused by the higher water content of frying oil, because water inhibits the transesterification reaction.
:1 methanol-to-oil ratio a maximum conversion of 67% of waste frying oil was reached. The reusability of the Lipozyme RM IM was also tested and the enzyme remained active after 6 cycles. The conversion of used sunflower oil into FAME was also compared to refined sunflower oil, which gave a higher conversion of 74%. This was mainly caused by the higher water content of frying oil, because water inhibits the transesterification reaction.
A more recent example of the use of waste oil has been described by Balasubramaniam et al.132 Waste cooking oil, obtained from e.g. restaurants and canteens and mainly containing sunflower oil, was used as a substrate for biodiesel production. A 1/3 molar ratio of oil/solvent was used for all reactions. The whole-cell biocatalyst of Rhizopus oryzae was compared to its pure lipase enzyme, both immobilised on alginate beads by entrapment. The use of the whole cell was investigated as a possibly more cost-effective method, because whole-cell biocatalysts are cheaper to produce than the purified enzyme. However, the purified enzyme showed both a higher catalytic activity and a wider catalytic ability, being able to accept several solvents as acyl acceptor. The somewhat poorer performance of the whole cells can probably be explained by the inability of the acyl acceptors and oil to effectively reach the active site. The highest conversion, 93% for the free enzyme and 84% for the whole cell biocatalyst, was obtained for the formation of the methyl ester, at a reaction temperature of 30 °C for 24 h. Comparable conversions were obtained for ethanol, n-propanol and iso-propanol with the free enzyme, but with the whole cell biocatalyst a high conversion was only obtained with methanol. Besides vegetable oils also animal waste oils such as slaughterhouse lipid waste (beef, goat and chicken tallow) can be used to produce biodiesel.54
Microalgae are another source of oil that has attracted attention over the last decade.133 Microalgae have a high photosynthesis efficiency, biomass productivity and growth rate compared to other energy crops.134–137 Additionally, oil-rich microalgae have a lipid content that can reach 50–60% of the total dry weight.138,139 Extracting oil from microalgae for transesterification therefore has a high potential for biodiesel production, and many methods have been described.140 Tran et al.37 have shown that microalgal oil from Chlorella vulgaris ESP-31 can be transformed into biodiesel by immobilised Burkholderia lipase.141 The lipase was immobilised by absorption onto a hybrid nanostructured material (Fe3O4–SiO2) with a long chain alkyl group.142 The oil was isolated from the microalgae by sonication (to disrupt the cell walls in the most effective way)113,114 and extraction with hexane. The extracted oil was used for transesterification experiments directly, by the addition of methanol, and produced biodiesel (FAME) efficiently (up to 97 wt% of the oil). An important problem here is that the extracted microalgae oil has a high water content. This influences the conversion performance: although lipases need some water (0–30 wt%), in the presence of high content of water in the reaction mixture, the fatty acid esters are hydrolysed to fatty acids and glycerol. The reactivity of the enzyme decreases by the formation of a shell, consisting of glycerol, fatty acids and FAME, around the enzyme that blocks the active site. In addition, the concentration of methanol is important. This immobilised enzyme can, in principle, function under high methanol-to-oil molar ratios, which enables the direct conversion of the ultrasound-extracted wet microalgae biomass to FAME, without the need of dewatering or further oil extraction. This is advantageous to drive the transesterification reaction.94 To minimize the inhibiting effects of high methanol concentrations, methanol was added stepwise.112 Initially, the conversion increased when the ratio was increased from 6 to 12, but a subsequent increase of this ratio to 24 decreased the conversion again. Further increase to ratios of 37, 62 and 123 also led to decreased conversion. The immobilised enzyme was recovered and checked for reusability, which showed that the enzyme remains active for 288 hours and six cycles, but that the yield drops to 65%. All together the process showed potential for the commercial production of biodiesel from microalgae, if the reusability of the immobilised enzyme can be improved.
A side product obtained from biodiesel production is glycerol. When biodiesel is formed through the transesterification of vegetable oils with methanol or other short-chain alcohols, large amounts of this glycerol are liberated. It is known that glycerol decreases enzyme activity,143,144 but on the other hand increases the stability.145 During the biodiesel production, glycerol forms a hydrophilic phase with FAME, creating a film layer around the immobilised enzymes. Membrane bioreactors that in situ separate the glycerol during the biodiesel production offer an alternative. Another advantage of the use of membrane reactors is that the enzymes can be used in a continuous process. Ko et al.81 reported the continuous production of biodiesel by such a membrane bioreactor system, while isolating the glycerol at the same time. The reactor system consists of two parts, in which the first part contained the transesterification reaction and the second part a membrane unit for glycerol removal. Due to this hydrophilic 10 kDa molecular weight cut-off (MWCO) membrane in the continuous reactor, the hydrophobic and hydrophilic phases were separated. The glycerol concentration increased on the hydrophilic methanol side of the membrane and the biodiesel conversion continued on the hydrophobic side. By removing the glycerol steadily its negative side effects on reaction reversibility and enzyme inactivation were minimized, resulting in a 95% conversion. This novel continuous membrane bioreactor provides a highly promising approach for industrial applications.
3.3 Non-fuel usages of fats and oil products
Fatty acid/fatty ester conversion from lipid-rich waste is a process that is usually carried out at high temperature and high pressure in places that generate large amounts of lipid rich waste such as slaughterhouses.146 This takes place to overcome problems like clogging and mass transfer of substrates adsorbed to microbial biomass surfaces in anaerobic digesters. Intrinsically selective enzymatic hydrolysis takes place at lower temperatures and leads to products with high purity. This should lead to a cost-effective procedure for hydrolysis.147,148 An acidic lipase from Pseudomonas gessardi was covalently immobilised using ethylenediamine and glutaraldehyde on surface-modified mesoporous activated carbon (SMMAC), derived from rice husk.149 The slaughterhouse lipid waste was then hydrolysed using these SMMAC-immobilised enzymes to give a nearly quantitative conversion into the product of hydrolysis. Also the reusability of these catalysts was very high, after 18 cycles the conversion was still 100%, and only after 50 cycles the conversion dropped to 64%.Epoxidation of the C–C double bond in fatty acids leads to the formation of useful building blocks for the production of polyurethanes,150 cross-linkers in powder coatings,151 paints,152 lubricants,153,154 PVC-plasticizers,155 stabilizers156 and surfactants.157 Currently these epoxidised oils and fatty acids are produced at a scale of 200000 tons p.a.,158via the Prilezhaev epoxidation, in which a peracid is used for oxygen transfer to the double bond.159 The peracid is formed usually from hydrogen peroxide and an organic acid, with the help of a strong mineral acid or ion exchange resin. This epoxidation reaction can also be carried out enzymatically, offering a green alternative for the production of fatty epoxides.
Warwel and Klaas25 reported in 1995 a method for converting carboxylic acids into percarboxylic acids catalysed by immobilised Candida antarctica lipase B (Novozym 435). As a result, the unsaturated fatty acid is first converted into an unsaturated peroxy acid that afterwards epoxidises itself (see Scheme 2). For internal unsaturated acids the epoxy acids are obtained in a yield of 70–90%, whereas ω-unsaturated carboxylic acids give the epoxy acid in 10% lower yield. Lipases are normally deactivated in non-polar solvents, like toluene, if these solvents contain large amounts of water. However, the use of 60% H2O2, which forms water during the process, did not hamper reusing the enzyme 15 times for the epoxidation of oleic acid, to produce 200 gram peroxy acid per gram of Novozym 435. To improve the reusability and to overcome problems related with solvents, like generation of extra waste and the need for additional purification steps, Törnvall et al.160 developed a solvent-free method for the epoxidation of oleic acid and its methyl ester. For a successful application of this process catalyst stability is crucial, however the reusability of Novozym 435 for the solvent-free epoxidation of oleic acid dropped drastically after 5 runs. The main advantage of the solvent-free reaction conditions are the simple recovery of substrate and enzyme. Furthermore the epoxidation was carried out with quantitative conversion of oleic acid. In 2007 the same group reported the chemo-enzymatic epoxidation of rapeseed (RME) and tall oil methyl esters (TOME) under the same solvent-free conditions. Quite similar results were obtained for the two types of corresponding fatty acids, tall oil fatty acids (TOFA) and rapeseed oil fatty acids, especially in terms of reaction speed, energy requirements and final conversion. The initial productivity during RME epoxidation was 196 g l−1 h−1, and conversion of 90% was achieved within only 6 h at 40 °C. At the same H2O2 addition rate, the reaction with TOME reached 82% conversion of the double bonds within 12 h at 40 °C. For the production of fatty acids from rapeseed and tall oil derivatives a life cycle analysis was also carried out. This analysis shows that when energy and greenhouse gases were taken into account, TOFA/TOME showed a 5% lower greenhouse gas emission value than RME (and 20% lower than fossil fuel) and TOME also requires a 60% lower energy input than RME. Another benefit of TOFA/TOME is that it will not cause competition of arable land, being a renewable waste stream from the paper and pulp industry.
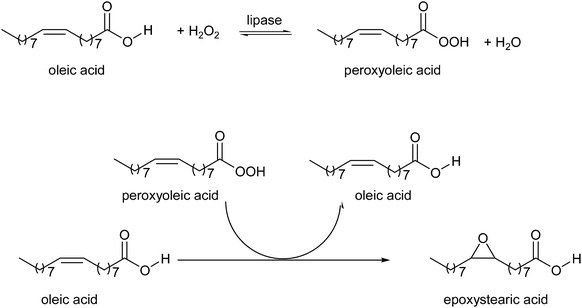 | ||
| Scheme 2 Chemo-enzymatic epoxidation of oleic acid, by Warwel et al.25 | ||
Later studies by Törnvall et al. to improve the stability of the enzyme, again in solution, showed that temperature and dosage of hydrogen peroxide are essential for the stability of Novozym 435.161,162 The enzyme activity decreased when increased H2O2 concentrations were applied for the chemo-enzymatic epoxidation of oleic acid. H2O2 has been reported to oxidise surface residues of the enzyme and thereby affect the enzyme activity. Desorption of the enzyme from the carrier caused by degradation of the carrier by H2O2 also takes place. The reaction temperature has an important role as well, no deactivation was observed after 48 h at 20 °C with a H2O2 concentration of 6–12 M, while at 60 °C under the same conditions the enzyme lost activity rapidly.
The main drawback of the use of enzymes in these conversions is still, however, the production cost of the enzymes, and cheaper and more stable enzyme preparations are needed to make biocatalysis an attractive alternative for the production of these chemicals. The added cost of the enzyme can, in principle, be reduced in two ways: by reducing the cost of the enzyme preparation or by improving its activity and stability. In 2011 a more cost-effective route for esterifications and amidations was developed by immobilising Candida antarctica B (CALB) on different resins.31 Immobilisation of CALB on Accurel MP 1000 and Lewatit resins provided a cost-effective biocatalyst preparation for industrial applications such as the synthesis of low cost commodities. While the enzyme stability on MP 1000 was similar to that of Novozym 435, the cost for MP 1000 was significantly lower. The so-developed immobilised enzyme could be used in a normal batch stirred tank reactor as well as in a packed bed reactor. From an industrial perspective this enzyme preparation is very promising, because the unit cost is one quarter of the available preparations. It was also shown that amidation reactions could be carried out quite cost-efficiently. The drawback of this system is mechanical disintegration of the immobilised enzymes at high agitation speed, causing shear stress on the particles.163 The choice of the right reactor is key to the success of the upscaling of a biocatalytic process, success of scaling up a biocatalytic process, although the enzymatic efficiency for this process needs to be further optimized to be economically feasible,164 as also discussed above for the continuous membrane reactor.80 However, in the latter case80 initial experiments provided low conversion (40%) and low enzyme stability (27% loss of activity). Under optimized conditions, a continuous operation with conversions higher than 85% and minimal enzymatic deactivation was feasible for 18 days.
Fatty amides and their derivatives have attracted much attention over the last decades. This is due to their biological applications165 and industrial applications such as surfactants, lubricants, cosmetics, detergents and anti-blocking agents in the plastic processing industry.166,167 These fatty amides can be prepared by reacting fatty acids or their corresponding esters with amine-containing compounds. The amidation can be catalysed by enzymes. Al-Mulla et al.43 reported the enzymatic synthesis of fatty amides from palm olein and urea, instead of ammonia, using Novozym 435 or Lipozyme as a catalyst. In this way triglycerides are converted to fatty amides, glycerol and ammonia (see Scheme 3). The highest yield of fatty amides (96%) was obtained by using Lipozyme, at a reaction temperature of 40 °C and reaction time of 36 h, with hexane as the appropriate solvent. This method to produce fatty amides offers, compared to other methods to produce biorenewables, several advantages like renewable and abundant amount of starting material, technical simplicity, high conversion and easy isolation of the enzyme.
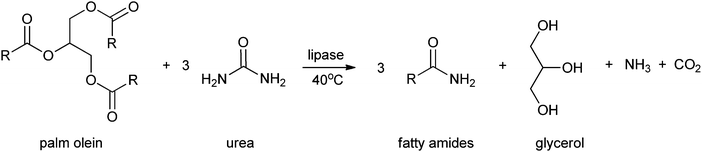 | ||
| Scheme 3 Formation of fatty amides by Al-Mulla et al.43 | ||
Alkanolamides of fatty acids were prepared in a similar manner by Fernandez-Perez and Otero.26 Fatty acid alkanolamides are surfactants that are applied in cosmetics because they show high skin tolerance and good biological degradability.166,168 A series of fatty acids was transformed into alkanolamides by transamidation by ethanolamine catalysed by Novozym 435. A good reusability was obtained, the enzyme stays completely active, but it was only verified after one round of reactions. The conversion obtained varied from 90% for palmitic acid to 94% for capric acid.
Wax esters can also be obtained from a lipase-catalysed esterification. Wax esters are long-chain esters derived from fatty acids and alcohols, both having chain lengths of twelve carbons or more169 which can be isolated from animal and plant materials. Wax esters are applied in lubricants, pharmaceutical, cosmetic and plasticiser industries.170,171 Jojoba oil and spermaceti oil are the main sources of unsaturated wax esters, but since the sperm whale is an endangered species and the growth of jojoba plants is restricted to desert environment, the esters are scarce and too expensive. There has been an ongoing search to find substitutes for jojoba oil and spermaceti oil already for many years. Salis et al.44 synthesised wax esters from heavy fractions of sheep milk fat and cetyl alcohol in hexane catalysed by Lipozyme RM IM or Novozym 435. Hadzir et al. synthesised the wax esters by alcoholysis of triolein and oleyl alcohol.172 However, the procedures are limited to laboratory scale and too expensive to scale up. As a cheap and scalable alternative Deng et al. proposed the synthesis of wax esters of oleic acid and cetyl alcohol (see Scheme 4).173 The lipase from Candida sp. 99–125 was immobilised and the esterification reaction was carried out under solvent-free conditions. The reactor capacity could be scaled up to 1 L, and optimised reaction conditions appeared to be acid/alcohol 1:0.9 with 2.5% (by mass) of immobilised enzyme to obtain the wax esters in 95% yield. The immobilised lipase could be reused 4 times, after which a noticeable decrease in activity was observed. The physical and chemical properties of these wax esters from oleic acid and cetyl alcohol are similar to jojoba oil and could be used as a substitute. A similar route towards wax esters was developed by Guncheva and Zhiryakova.174
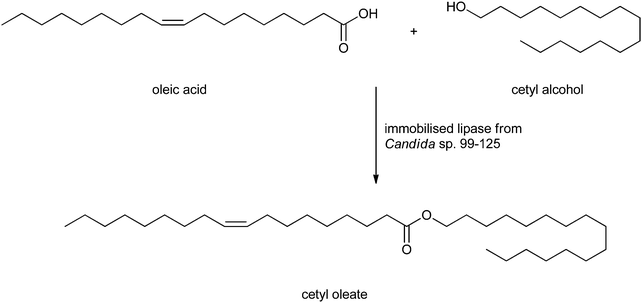 | ||
| Scheme 4 Formation of a wax ester by Deng et al.173 | ||
An interesting and somewhat sophisticated application of immobilised enzymes for the production of renewables is the synthesis of phytosterol esters. Zheng et al. immobilised Candida rugosa lipase on mixed-mode silica particles via hydrophobic and cation-exchange interaction, in order to catalyse the esterification of phytosterols to free fatty acids (see Scheme 5).48 Phytosterols are steroid compounds similar to cholesterol, which occur in plants and vary only in carbon side chains and/or presence or absence of a double bond. Their fatty acid esters recently gained importance because they can be applied in the food and nutraceutical industry to reduce cholesterol-related health problems.175,176 Several lipases have been tested for the esterification of phytosterols, Candida rugosa is the most used and gives the best results.177,178 The esterification of phytosterol with several fatty acids (palmitic acid, oleic acid, linolenic acid) could be carried out under mild conditions, with a conversion of 90–95%. The octyl and sulfonic acid co-bonded silica (OSS) particles also showed improved thermal stability compared to the free lipase and reusability, giving 80% yield after 7 cycles. Transesterification of the fatty acid methyl ester or triglycerides with phytosterol catalysed by the OSS-immobilised enzyme yielded the phytosterol esters with a conversion of 80–90%. A review about the enzymatic synthesis of these designer lipids was written by Devi et al. in 2008.179
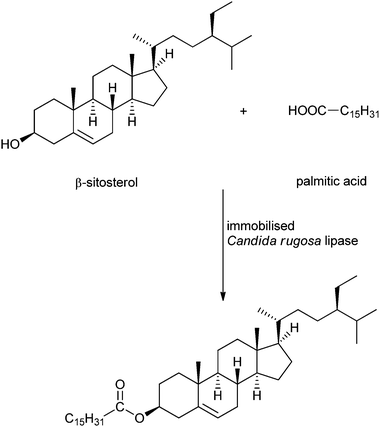 | ||
| Scheme 5 Example of a phytosterol ester synthesis as reported by Zheng et al.48 | ||
A special class of fatty acids are omega-3 fats, long chain poly-unsaturated fats containing methylene separated double bonds. Omega-3 fats are ingredients used in dietary supplements, healthy food and pharmaceutical products. Kralovec et al. have described recently a nice and comprehensive review about the enzymatic concentration of these omega-3 fatty acids from fish oil.180
The plant-oil methanolysis-based production of renewable esters or amides yields one equivalent of glycerol as a by-product.181 Therefore the conversion of glycerol to useful material has also gained increasing attention.182 A possible application of glycerol could be the lipase-catalysed production of glycerol carbonate, starting from glycerol and dimethyl carbonate, as shown by Kim et al.28 (Scheme 6). Glycerol carbonate can be used in coatings, in detergents and as a source of polymeric materials.183–185 Under optimised conditions Novozym 435 catalysed the formation of glycerol carbonate from glycerol and dimethyl carbonate. The reaction was carried out in THF, at 60 °C for 28 h with equimolar amounts of glycerol to dimethyl carbonate to obtain the product in a quantitative yield. Molecular sieves were added to scavenge the formed methanol, which otherwise would have deactivated the catalyst.
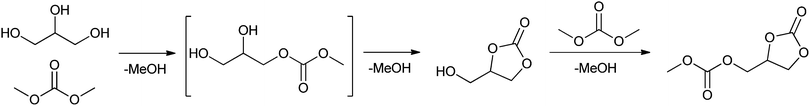 | ||
| Scheme 6 Lipase-catalysed synthesis of glycerol carbonate.28 | ||
Another interesting application of glycerol is the production of heat-sensitive mono- and diacylglycerols (MAG and DAG).186,187 Enzyme technology offers the opportunity to produce on a large scale MAGs and DAGs of polyunsaturated fatty acids. These unsaturated MAG or DAG products, especially when the MAG could be produced selectively, can be used as food additives and biocompatible emulsifiers, and can find their application in functional foods, pharmaceutical products, and cosmetics.188 Damstrup et al. and others developed an efficient and continuous process for glycerolysis with an immobilised Novozym 435-packed reactor.188–192 Glycerol and sunflower oil were dissolved in a binary tert-butanol:tert-pentanol medium. After 20 min an equilibrium was reached with a MAG content of 50–55 wt%. The immobilised enzyme showed high stability and retained its activity for 92 days, leading to a very high enzyme capacity of 200 L pure MAG produced per kg enzyme. This packed bed reactor set up with simple dry enzyme packing of the column, no mass transfer limitations, high capacity and long-lasting activity of the enzyme displays high potential for scale-up and industrial applications.
Finally, Liebminger et al. have shown that glycerol can be oxidised by TEMPO/laccase.193 The laccase, from Trametes hirsuta, was immobilised on silanised alumina pellets and the oxidation was carried out in a sodium acetate buffer (pH 4.5). Glycerol was oxidised by TEMPO, which is then regenerated by the laccase.194 The oxidation products of glycerol (see Scheme 7) are all of practical value. First, glyceraldehyde is formed,195 and subsequently glyceric acid, an important intermediate in organic synthesis, and in particular in the synthesis of serine.195 Upon further oxidation, tartronic acid193 – an ingredient in cosmetics and pharmaceuticals – is formed, and finally mesoxalic acid, which is a valuable organic synthon.196 Conditions need to be tuned to selectively produce and isolate one of the materials. Depending on the conditions, especially the concentration of TEMPO, 70% conversion to glyceraldehyde could be obtained. By varying the conditions other ratios of product could be obtained with an overall conversion of 55–70%.
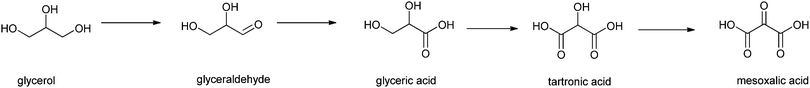 | ||
| Scheme 7 Glycerol oxidation by TEMPO in the presence of laccase.193 | ||
4. Carbohydrates
Sugars and polysaccharides, especially cellulose and hemicellulose, are abundantly present in nature, and therefore can be seen as practically inexhaustible resources for the renewable production of functional chemicals and energy. This section is devoted to the formation of mono- and disaccharides from polysaccharides, and their subsequent conversion to useful compounds (4.1) and the enzymatic degradation of cellulose (4.2).4.1 Production of carbohydrate-based renewables for pharma and food applications
One of the most important enzymatic processes is the isomerisation of glucose into fructose by glucose isomerase (GI), because of the unique dietary properties of fructose. The benefits of fructose are that it is sweeter than sucrose and that it is metabolised without the need of insulin. This isomerisation process has been studied in detail over the last decades, and can be catalysed by free or immobilised GI and even by GI-containing cells197–200 at 50–60 °C, pH 7–8 and 40–50% dry substance matter.201 The isomerisation of glucose into fructose is used for the production of high fructose corn syrup, a process that is industrially applied by immobilising GI in a packed bed reactor.202 The immobilisation of GI has been carried out on almost all possible carrier materials.203–207 A carrier that has only recently drawn attention is a composite called GAMM,208 which is prepared by an inverse suspension polymerisation of glycidyl methacrylate (G), allyl glycidyl ether (A), N,N′-methylene-bis-(acrylamide) (M) and acrylamide (M).209 This support was compared to Eupergit C 250L, benzyl DEAE cellulose, TEAE cellulose and DEAE cellulose, and GI immobilised on GAMM indeed showed better operational stability, thermal stability and storage stability. Immobilised GI has a broader application range of pH values than free enzyme, it retained 92% of its maximum activity at pH = 6.0 and 88% of its maximum activity at pH = 8.5 while free GI only retained 60% of its maximum activity at pH = 6.0 and 70% of its maximum activity at pH = 8.5. Immobilisation of enzymes onto GAMM thus appears to be a promising approach.Instead of D-fructose, its epimer D-psicose – which has a similar sweetness intensity – can also be produced from glucose via fructose, by an immobilised enzyme (see Scheme 8). In contrast to D-fructose, D-psicose is hardly metabolised and therefore yields an almost zero-calorie intake, making it is an effective ingredient of diet foods. Another benefit is that D-psicose does not cause any side effects common for artificial sweeteners, like diarrhoea when taken in large amounts.210 Hong has developed a method to produce D-psicose using an inexpensive substrate such as glucose or fructose by immobilising psicose-epimerase.211 The psicose epimerase, derived from Agrobacterium tumefaciens, was immobilised on sodium alginate, a natural colloidal polysaccharide from the cell wall of algae, and packed onto a column. If two sequential columns are used with glucose isomerase and psicose epimerase, respectively, glucose can be transformed via fructose into psicose. A limitation of this methodology is that both conversions have to be improved, with the optimum result so far being a conversion of glucose to psicose of 18%, while the fructose to psicose conversion yields 47%. Lim et al. have shown that a 2-fold increase of conversion, from 28% to 62%, could be obtained by immobilising this psicose epimerase on Duolite A 658 beads, when the conversion was carried out in the presence of borate in the buffer solution.212
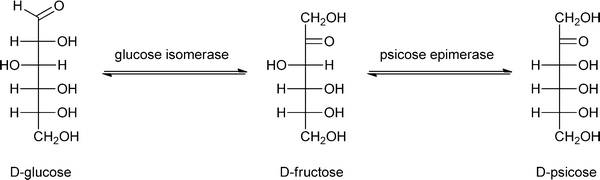 | ||
| Scheme 8 Two-step conversion of glucose into the low-caloric sugar psicose. | ||
Another main target are sugar esters, which are prepared on a scale of several thousands of tons per year by acid-catalysed esterification of sorbitans and fatty acids (sorbitan esters:Span or Tween). They are widely used as non-ionic biosurfactants or chemical emulsifiers,213 for the cosmetics, health-care, pharmaceuticals and food industries.214,215 They have the advantage over other surfactants in that they are renewable, tasteless, odourless and non-irritant. Recently, Neta et al.216 optimized the enzymatic preparation of fructose esters, to yield fructose ester in 88.4%, an improvement of 5–10% compared to the method described by Sabader et al.217
We have studied the esterification of sorbitol with decanoic acid by Chromobacterium viscosum lipase,218 while Adnani et al.219 reported an efficient route towards xylitol esters.
Instead of making use of commercial or standard enzyme formulations like Novozym 435 or Lipozyme RM IM, Kahn et al.82 reported the application of a new enzyme-catalysed process to make so-called sugar-6-esters. The enzyme is immobilised/encapsulated within a biopolymer scaffold based on chitosan and polyethylene glycol (PEG), reinforced by treatment with methacrylic anhydride. This appeared remarkably useful for the production of biodegradable surfactants like fatty acid esters of suitable sugars, as well as for the production of intermediates leading to useful sugar derivatives.220 An example is the production of 6-O-acyl-sucrose (Scheme 9), a key intermediate in the route towards the high-intensity sweetener 4,1′,6′-trichloro-4,1′,6′-trideoxygalactosucrose (sucralose). In this particular method the substrate as well as the enzyme are encapsulated in each other's vicinity, creating a reactive environment. This esterification between sucrose and a fatty acid is selective for the 6-position of the sucrose, and yielded the desired product in >80%. Distinct from many other methods, the described method makes use of t-amyl alcohol, a solvent that is easy to recycle, and thus has many advantages over alternatives like DMSO. Other advantages are that the activity of the enzyme is retained for a longer period of time and at higher temperatures than the native enzyme. In this way a more environmentally acceptable biotechnological process (green chemistry) is developed for the production of sugar-6-esters.
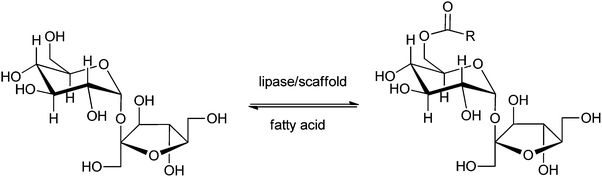 | ||
| Scheme 9 Lipase-catalysed synthesis of 6-O-acyl-sucrose fatty acid esters.82 | ||
Also in this field several attempts have been made to use low-cost supports to immobilise enzymes. For example, mica was used by Zaidan et al. as a low-cost carrier for Candida rugosa lipase (CRL), for the production of lactose esters.34 Mica is a very cheap silicate that is used on a large scale for industrial applications, like the ceramics and tile industry.32 Previously, Zaidan et al. already showed that mica can be used to immobilise CRL for the production of propyl laurate.33 CRL was immobilised onto amino-activated mica via covalent binding (amino-CRL) and by crosslinking (nanoporous mica NER-CRL). The activities of the enzyme increased 2.5-fold over the free enzyme. As an example for the formation of sugar fatty acids, optimum conditions (85% conversion) for the coupling of lactose with capric acid in the presence of NER-CRL were found to be a molar ratio of 2:1 at 55 °C for 48 h. Under these conditions immobilised amino-CRL and NER-CRL both showed a high stability. However, after 6 cycles the conversion started to decrease, and after 10 cycles the conversion dropped below 40% for amino-CRL, while the same occurred for NER-CRL after 13 cycles.
Sugar esters of aromatic carboxylic acids are used in tumour treatment.221 Given the desired use thereof, Croitoru et al. recently described the enzyme-catalysed formation of several aromatic esters of sugar alcohols.27 The sugar alcohols used are xylitol, arabitol, mannitol and sorbitol, while the aromatic carboxylic acids are based on hydroxycinnamic acids. Their esters show interesting biological activities.222 Dihydro-p-coumaric esters were formed upon catalysis by Novozym 435 in tert-butanol at 60 °C for 3 days. Under these conditions, high conversions of the sugar were obtained: 80% for mannitol, 93% for sorbitol, 94% for xylitol, and even 98% for arabitol (see Scheme 10). Analysis of the aromatic esters showed the predominant formation of monoesters and diesters, and even small amounts of triesters were formed. These new esters can find application as surfactants and anti-oxidants, but an increased selectivity in the directed formation of mono- and diesters is required for practical scaling-up.
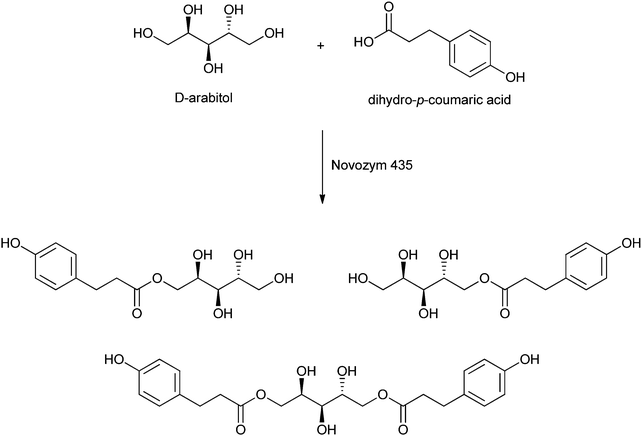 | ||
| Scheme 10 Example of the lipase-catalysed formation of esters of dihydro-p-coumaric acid with a sugar alcohol as reported by Croitoru et al.27 | ||
The group of Riva used the laccase from Trametes pubescens, covalently immobilised on Eupergit, for the oxidation of several glycosides, using TEMPO as the mediator.223 The obtained glycosides can be applied as therapeutic agents against human gout and other inflammations.224 The C6–OH's of the sugars were selectively oxidised to the carboxylic acids. The substrates were thiocolchicoside, colchicoside, amygdalin and asiaticoside (see Scheme 11). Isolated yields were 24–77%, depending on the substrate. Remarkably, the sulfur atom of thiocolchicoside was not touched at all by this oxidative system.
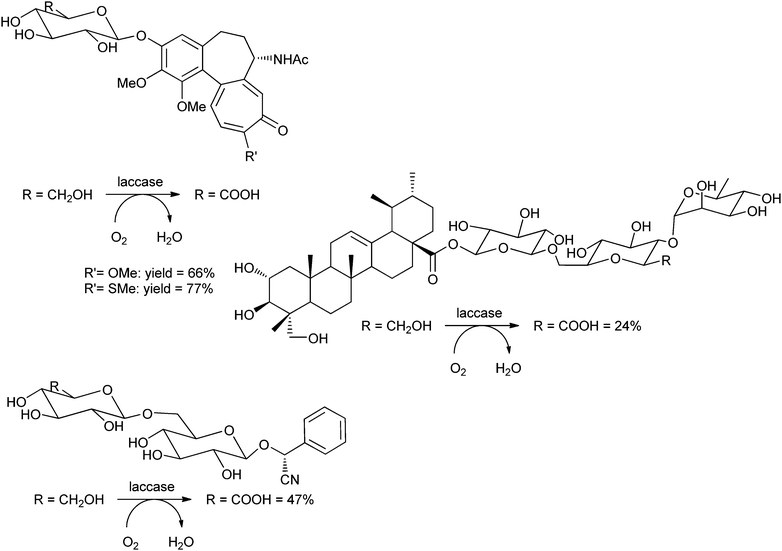 | ||
| Scheme 11 Laccase-TEMPO-mediated oxidation of natural glycosides.223 | ||
Another category of interest is formed by esters of isosorbide, which can be used as flavours, emulsifiers, lubricants and additives in food, cosmetics and pharmaceutical products.225 El Boulifi et al. reported the lipase-catalysed synthesis of isosorbide monoricinoleate (Scheme 12).226 Isosorbide (1,4:3,6-dianhydro-D-glucitol) is a byproduct obtained by selective dehydratation of sorbitol.227 The mono-esterification of isosorbide was described before,228,229 but these processes needed further optimisation in order to be applied for the production of renewables. The fatty acid used is ricinoleic acid, derived from castor oil. Under optimised reaction conditions (determined by the Response Surface Method:230,231 6.3 wt% catalyst Novozym 435 at 63 °C with a reaction time of 2 h), the monoester was obtained in a yield of 93% and the diester in a low yield of only 1.7%.
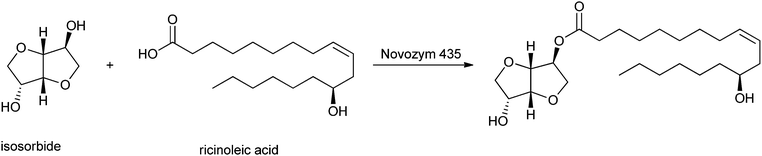 | ||
| Scheme 12 Lipase-mediated esterification of isosorbide to ricinoleic acid. | ||
Galactooligosaccharides (GOS) received a large interest in recent years. GOS are functional food elements that improve the intestinal microflora.232,233 They are produced by enzymatic oligomerisation of galactose, often using lactose as the source of galactose entities.234,235 Lactose is obtained in large amounts as a side product in the dairy industry via low-temperature crystallisation of milk, to obtain low-allergenic lactose-free milk.236 Iwasaki et al. were one of the first to study the production of GOS from lactose.232 By using a β-galactosidase from Aspergillus oryzae it was shown that by increasing the lactose concentration an increase in GOS production was also observed. The purity of GOS in this process was affected by free enzymes, monosaccharides and unreacted lactose that remain in the final product, hampering its purification. This could be partially solved by using immobilised enzymes, but it was finally Engel et al., who showed that the amendment of a membrane separation technique, together with enzyme immobilisation improved the purity and production process significantly.237,238 Das et al. and Sen et al. showed that GOS can be produced from lactose with a membrane-immobilised enzyme.78,239 The required enzyme, a β-galactosidase, was immobilised on a poly(ethersulphone) ultrafiltration membrane using polyethyleneimine,240 an extremely branched cationic chain polymer,241 for use in a rotating disk membrane reactor (RDMBR). The β-galactosidase enzyme synthesises oligosaccharides through a transgalactosylation reaction.72 By using a membrane reactor, GOS can be separated from other low molecular weight sugars, which thus enriches the purity of the GOS. The conversion of lactose into GOS using the RDMBR was 67% and the product purity was 80%. Compared to other methods to produce GOS from lactose, this methodology by far gives the highest yield and purity.
Carbohydrates can also be converted into their corresponding keto-sugars by an immobilised pyranose oxidase together with catalase to keep the concentration of the produced hydrogen peroxide low.242 Keto sugars are interesting synthons that can be converted into pyrrolidine- and piperidine-aminosugars, which are used as antibiotics and glycosidase inhibitors. In 2008 Ludwig et al. had already reported on the production of keto-aldoses by the trienzyme system pyranose 2-oxidase (P2O), laccase and catalase.243 The three enzymes were covalently immobilised on acrylic carriers containing epoxide groups. After immobilisation a higher operational stability was observed, and it was shown that the enzymes are more stable against thermodynamic inactivation. To improve the P2O–catalase system laccase was introduced, which boosted the specific productivity of P2O to a value of 34.2 g kU−1 h−1. Laccase provides an alternative electron acceptor, quinones, which show a higher catalytic efficiency than oxygen for the oxidation carried out by P2O. By the use of this regeneration system of H2O2, the dosage of H2O2 is optimised and does not inhibit the laccase.244 Another method for the production of keto-sugars was described by Karmali.242 The oxidation of carbohydrates into keto-sugars by enzymes is slow and time consuming under normal temperature and pressure. However, in this case a solution of a carbohydrate such as glucose in phosphate buffer was brought under high pressure in the presence of immobilised pyranose oxidase and catalase by compressed air in a batch reactor at room temperature. The enzyme was immobilised by covalent bonding through glutaraldehyde on a poly(ethersulphone) membrane disk. The reaction time of total conversion was reduced by applying high pressure for several hours. When conversion was complete, the membrane containing the immobilised enzymes was removed and stored, so it could be reused several times for this conversion over a period of months. The solution containing the keto sugar was evaporated and afterwards freeze-dried to give the pure keto-sugar.
4.2 Cellulose hydrolysis
Cellulose is the most abundant renewable polysaccharide with a high potential for degradation to functional compounds, e.g. bioethanol.245,246 The polymer is composed of β-1,4-glucosidic bonds. The ability to form hydrogen bonds between adjacent cellulose chains, in combination with increased hydrophobic interactions and van der Waals forces, leads to a parallel alignment and to a crystalline structure of fibers of great strength and low accessibility.247 Hydrolysis leads to the disaccharide cellobiose and ultimately to glucose (see Fig. 1).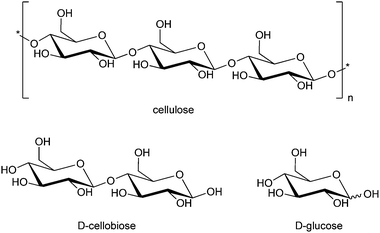 | ||
| Fig. 1 Structure of cellulose and its hydrolysis products cellobiose and glucose. | ||
Jones and Vasudevan have shown that microcrystalline cellulose can be hydrolysed by CLEAs of Trichoderma reesei cellulase.73 The immobilised cellulase is used in conjunction with a low concentration (2% or 4%) of 1-ethyl-3-methylimidazolium diethyl phosphate (EMIM-DEP), an ionic liquid that increased the initial reaction rate 2.7 times. In addition, the immobilised cellulase could be reused 5 times. After 8 h, the yields achieved with 2 and 4% (v/v) EMIM-DEP were similar: 1.05 and 0.95 g glucose per g cellulose, respectively.
Shen and Xia248 have shown that by a two-step process cellulosic material derived from corn cob can be hydrolysed effectively into glucose by an immobilised cellulase. In a second step the cellulosic hydrolysate is converted into lactic acid by immobilised cells of Lactobacillus delbrueckii. Lactic acid is used in food, pharmaceutical, leather, textile and chemical industries and to make polylactic acid (PLA). The enzymatic hydrolysis of cellulosic materials in the first step depends on the synergism of the three components of the cellulase: endo-β-glucanase, exo-β-glucanase, and cellobiase.249,250 Because cellulase of Trichoderma reesei shows a low activity for the conversion of cellobiose to glucose,251,252 an immobilised cellobiase is added. By the synergism of cellulase and the immobilised cellobiase, cellobiose formed was quickly converted into glucose and the hydrolysis yield went up from 65% for cellulose alone to 83% for cellulase/cellobiase. In a second step immobilised cells of L. delbruecki were used and cellulosic hydrolysate was converted into lactic acid with a conversion efficiency of 95%. The formed lactic acid can be converted into PLA by enzymatic polymerisation. The cellulose can also be partly hydrolysed in order to obtain short oligomers or cellobiose.
As described before, the most used cellulase from T. reesei is poor in cellobiase activity. Therefore for an increased saccharification yield, the activity of cellobiase in the cellulase system has to be improved.253 Shen and Xia proposed the improvement of the cellobiase activity by immobilisation of spores from Aspergillus niger ZU-07 into calcium alginate gels.254 A cellulosic residue of corn cob, ground to particles of 3 mm diameter, was pretreated with dilute acid, and afterwards hydrolysed with cellulase of T. reesei, giving the desired glucose in a yield of 69%. A significant amount of cellobiose was found in the hydrolysate, and by addition of the immobilised spores the yield could be improved to 84%. These immobilised spores are quite stable, with a half-life of 38 h and a yield of enzymatic hydrolysis of cellobiose staying over 97% over 10 runs.
Cellobiose production from biomass catalysed by immobilised enzymes has been reported by Kohl et al.79 Cellobiose can be applied in the production of food, animal feed, chemicals, cosmetics and pharmaceutical industries. It can be produced from cellulosic feedstock by chemical acidolysis or enzymatic hydrolysis. Many cellulose containing materials are suitable substrates for the process, in particular microcrystalline cellulose. The enzymatic method leads in general to higher yields and has an advantage over the chemical methodology that it does not require toxic reagents nor creates waste disposal problems. The sugar beet biomass used in this case was derived from the root tissue of Beta vulgaris, of which the dry tissue contains 80% sucrose, while the beet pulp contains 7% pectin, 7% cellulose and 7% hemicellulose. Sugar beet pulp was first treated with 1% H2SO4, and then the solid phase was used for the cellobiose production. The pre-treated sugar beet pulp was then mixed with a cellulase preparation of T. reesei (12% w/w) and an acetate buffer was added to obtain a dry mass content of 16.5% w/w. The so-obtained mixture was incubated for 48 h at 50 °C, afterwards followed by a separation step of the liquid phase. The liquid phase contained cellobiose and glucose. The latter is subsequently converted into gluconic acid by glucose oxidase and removed from the mixture by ion-exchange chromatography (see Scheme 13). The glucose oxidase used in this process was immobilised on glass beads. Glucose oxidase is capable of oxidising glucose in the presence of oxygen into D-gluconolactone. Catalase is added to remove the formed hydrogen peroxide that otherwise could have oxidized the cellobiose. Gluconic acid is formed by hydrolysis of the D-gluconolactone. The obtained solution can be concentrated or the cellobiose can be crystallised, to obtain a product with a purity of more than 99%. The cellobiose preparation can also be used directly as food ingredient, animal feed additive, cosmetics ingredient or anti-inflammatory pharmaceutical preparation. Alternative uses are low-caloric ingredient in food, building block for polymers or an active pharmaceutical ingredient in therapeutic applications. Altogether 94% of the cellulose present was converted into cellobiose, which was obtained in an overall yield of 79%.
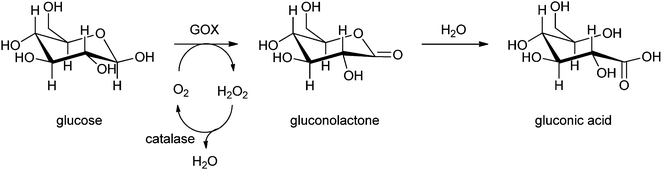 | ||
| Scheme 13 Glucose oxidase (GOX)-mediated conversion of glucose into gluconolactone and gluconic acid.79 | ||
4.3 Starch hydrolysis
After cellulose, starch is the most abundant carbohydrate available from agricultural raw materials. The estimated world production of starch amounts to 58 million tons, extracted from maize (46 million), wheat (4.6 million), and potatoes (3.5 million), with the remainder coming from rice and cassava roots (tapioca).255 The structure and properties of starch are strongly dependent on its botanical source. Starch is usually found as a heterogeneous mixture of two polymers, amylose and amylopectin. Both have a backbone of α-1,4-linked D-glucose units, but amylopectin has additional α-1,6-glucosidic branches (see Fig. 2). For the complete hydrolysis of starch to glucose two enzymes are needed, namely α-amylase (active on α-1,4-glucosidic bonds) and γ-amylase (also called glucoamylase, active on both α-1,4- and α-1,6-glucosidic bonds). | ||
| Fig. 2 Partial structure of the starch constituents amylose and amylopectin, and their hydrolysis products maltose and glucose. | ||
The immobilisation of amylases for starch hydrolysis is a very active research area. Numerous papers are published yearly but the only comprehensive reviews available were published more than 30 years ago.256,257 These extensive studies have led to highly efficient reuse of the immobilised enzymes (up to 50x), which is quite different from the other fields covered in this review, and might function as an example of the potential of the use of immobilised enzymes. However, to the best of our knowledge, industry does not use immobilised amylases because the enzymes have become cheap enough over the years to dispose of them after one-time use. In this section on immobilised enzymes for starch hydrolysis, we confine ourselves to the most significant recent papers due to space restrictions.
A highly porous form of silica was made by Bellino et al., and used for the adsorption of Bacillus α-amylase.258 A sol–gel solution was made in the usual way and mixed with Pluronics F127, i.e. (EO)106(PO)70(EO)106, where EO and PO represent ethylene oxide and propylene oxide blocks, respectively. The material was deposited on a glass slide by dip-coating. After drying the organic template was removed by calcination, yielding a highly porous material. Amylase was adsorbed to it and appeared to be slightly more active on the hydrolysis of potato starch than free enzyme. The immobilised enzyme had 60% remaining activity after 7 reuses, but it could not be ruled out that this reduction was due to some enzyme leakage. Unfortunately, the authors did not report how high the activity of the immobilised enzyme was compared to that of free enzyme. In the research by Singh and Ahmad, silver nanoparticles were included into a sol–gel, which was made on a cellulose template.259 The template was removed by calcination and the result was a highly porous silica network containing antimicrobial silver nanoparticles. Diastase amylase was adsorbed onto it and the resulting immobilised enzyme, when used for the hydrolysis of soluble starch, had a slightly higher Vmax than the free enzyme (6.35 mg ml−1 min−1vs. 4.29 mg ml−1 min−1) and lower KM (3.46 mg ml−1vs. 4.16 mg ml−1). The enzyme was recycled six times and still had 74% of its original activity. The use of α-amylase adsorbed to porous alumina has been mentioned already in Section 2.1.35
A novel adsorption material was devised by Kara et al.260 These authors copolymerised ethylene glycol di-methacrylate with N-vinyl imidazole and loaded the resulting hydrogel beads with Cu2+ ions. Aspergillus oryzae α-amylase was adsorbed to the metal ions of this polymer. The KM value increases from 15.8 to 22.5 mM but Vmax increases too, from 214 × 103 to 234 × 103 U mg−1. The enzyme can be liberated by the addition of EDTA and the enzyme hardly showed any deactivation during ten successive adsorption–desorption cycles. Chang and Juang adsorbed α-amylase and glucoamylase onto a support composed of chitosan and activated clay that was cross-linked with glutaraldehyde.261 For α-amylase, the KM value for soluble starch doubled upon immobilisation but Vmax was more or less unchanged; for glucoamylase there was not much change in the KM value but a 15% decrease in Vmax. The thermostability was improved, as well as the stability and turnover: both enzymes could be reused no less than 50 times with only 19% loss of activity in the case of glucoamylase and even no measurable activity decrease in the case of α-amylase.
α-Amylase has been adsorbed to magnetic nanoparticles, with high activity (94%) and good activity (almost 80%) after 8 reuses.262 After 6 h, starch conversion by free enzyme halted at 73% conversion whereas the immobilised enzyme had converted 83% and went on to 92% in the next 2 hours. When magnetic nanoparticles were added in the copolymerisation of 2-hydroxyethyl methacrylate (HEMA) and N-methacryloyl-(L)-phenylalanine (MAPA), a hydrophobic magnetic nanoparticle was formed that appeared to be a good adsorbent for Bacillus licheniformis α-amylase.263 A very high loading of 705 mg of enzyme per gram of nanoparticle was observed. Remarkably, the KM value decreased by a factor 2.2 upon adsorption but unfortunately the Vmax value also decreased by a factor of four. Enzyme reuse was not tested, but the magnetic carrier could be reused ten times without significant loss in affinity for the enzyme. Very specific affinity for enzymes can be obtained by molecular imprinted polymers (MIPs). Lee et al. included α-amylase in poly(ethylene-co-vinyl alcohol) droplets containing magnetic nanoparticles.264 The solvent was removed and the enzyme washed out yielding a MIP containing a cavity that specifically recognises α-amylase. Newly bound amylase had 94% of the activity of free enzyme and the thus bound enzyme could be reused 29 times with 86% remaining activity. Remarkably, the enzyme molecules that were involved in the imprinting remained active even though they were dissolved in 100% DMSO, an organic solvent that is known to ‘turn enzymes inside out’. The preparation containing the imprinted protein was even more stable upon reuse (83% remaining activity after 50 reuses).
In another study, α-amylase was covalently bound to magnetic particles that were covered by a copolymer of styrene and maleic anhydride.265 Polyethylene glycol was added to improve the contact between the magnetite core and the polymeric shell. The optimum temperature of the enzyme shifted from 50 to 80 °C and the temperature stability also increased strongly. The KM value of the enzyme did not change upon immobilisation. The enzyme was reused 10 times with only 19% activity loss. Mukherjee et al. used cyanamide for the covalent coupling of Bacillus alcalophilus α-amylase to magnetic particles, and optimised the binding procedure using factorial design and the Response Surface Method.266 About 70% of the enzyme was bound, showing a remarkable twenty-six fold increase in specific activity due to both a decrease in KM and an increase in Vmax. The enzyme had about 70% of remaining activity after 10 reuses.
Shewale and Pandit have bound B. licheniformis α-amylase to superporous cellulose beads that were subsequently activated with epichlorohydrin, ethylene diamine and glutaraldehyde.267 Although the immobilised enzyme has only 9% of its original activity, it retains full activity after eight batches of hydrolysis. Interestingly, the oligosaccharide product profile of the immobilised enzyme was different (mostly tri- and pentasaccharides) from the free enzyme (mostly tri- and hexasaccharides).
Hasirci et al. covalently bound pork pancreas α-amylase to poly(dimer acid-co-alkyl polyamine) particles that were activated by a carbodiimide, ethylene diamine or hexamethylene diamine.268 The latter preparation gave the best results, with hardly any loss in activity after one month of storage or 40 times reuse in batch reactions. However the Vmax for that enzyme preparation was decreased fivefold while the KM was slightly increased (2.51 to 3.71 g l−1). Another covalent immobilisation method was used by Park et al.,269 who coupled α-amylase and glucoamylase to glutaraldehyde-activated aminopropylated silica, and to glutaraldehyde-activated polyethyleneimine deposited on DEAE–cellulose particles. The latter preparation was entrapped in calcium alginate beads. There was not much improvement in pH or temperature stability, but the enzymes, when immobilised together, were reused ten times with only 10% deactivation. The KM values were halved but the Vmax values also decreased, by 60 and 84% for the two immobilised enzyme preparations. Glutaraldehyde-activated aminopropylated silica (in the form of montmorillonite) was also used for covalent coupling of B. subtilis α-amylase and Aspergillus niger glucoamylase.270 The immobilised amylase was completely stable during 84 h of continuous operation, the glucoamylase during at least 96 h. Porc pancreas α-amylase has been immobilised by mixing it with a monomer containing acrylate and epoxide groups, followed by photo-crosslinking.271 The method of immobilisation is likely covalent binding to the epoxide groups. The activity of the immobilised enzyme was 78% and it retained 73% of its original activity after 35 reuses. The rather complex structure of the monomer is, however, a disadvantage of this method. El-Ghaffar and Hashem coupled B. subtilis α-amylase to chitosan that was modified with amino acids, using glutaraldehyde as the coupling/cross-linking reagent, with 97% remaining activity when L-glutamic acid was used.272 After 25 uses the remaining activity of this preparation was still 79%. Glutaraldehyde-activated polyaniline is another popular support for covalent enzyme immobilisation. Diastase α-amylase, bound in this way, had about 80% of remaining activity after 10 reaction cycles.273 The enzyme was stable under storage conditions for 5 months, however the KM value for soluble potato starch was doubled and the Vmax value halved.
Giordano et al. converted amylase-liquefied starch directly to bioethanol using co-immobilised glucoamylase and baker's yeast, a so-called simultaneous saccharification and fermentation (SSF) approach.274 The enzyme was covalently coupled to glutaraldehyde-activated aminopropylated macroporous silica; this preparation was subsequently entrapped into a pectin gel together with yeast cells. A packed bed system yielded 65.4 g l−1 of ethanol (5.9 g l−1 h−1), corresponding to 97% conversion, in 226 h of continuous operation.
An original method of enzyme immobilisation was reported by Wang et al.275 These authors used the ability of the enzyme transglutaminase to couple the ε-amino groups of lysine in proteins to the γ-amido group of glutamine in other proteins. In their system, the glutamines were derived from casein that was cross-linked to chitosan and deposited on a silica support; the amino groups were from B. licheniformis α-amylase and the coupling enzyme was transglutaminase from Streptomyces mobaraensis. Amylase binding was quantitative; no binding was detected in the absence of transglutaminase. The enzyme preparation was 20× reused, having 70% remaining activity. Enzyme activity or kinetic constants were not reported.
As mentioned before, cross-linking of enzyme usually does not give very good results but in the hands of Nwagu et al., raw starch digesting amylase from Aspergillus carbonarius that was adsorbed to sepa beads (poly(methacrylate)) and subsequently cross-linked using glutaraldehyde showed 86% activity yield and virtually no loss of activity after ten reuses.276 The immobilised enzyme preparation showed an increased KM value for raw potato starch and a decreased Vmax value. Alternatively, the enzyme that was covalently bound to polyglutaraldehyde-activated sepa beads gave 97% immobilisation yields and 92% remaining activity after 10 reuses. The KM value of this preparation slightly decreased while the Vmax value increased by 23%, making this a very interesting approach for enzyme immobilisation.
Kumar et al. combined enzyme purification with immobilisation.277 The α-amylase of A. oryzae binds strongly to alginate so entrapment into calcium alginate beads was an elegant way to purify the enzyme. The enzyme could be released again by dissolving the beads in 0.5 M NaCl and 0.2 M CaCl2. As an immobilisation method this approach is less useful, because the entrapped enzyme has less than 10% of its original activity. Another study came to the conclusion that activity of the calcium alginate-entrapped B. subtilis α-amylase could be enhanced by adding equal amounts of silicate to the alginate solution.278 However, the efficiency of the enzyme to hydrolyse soluble starch was about two-third of the free enzyme, due to diffusion limitation. Ninety percent of its initial activity remained after 20 batch reactions, compared to 70% without the silicate. The successful entrapment of α-amylase in iron (hydr)oxide particles has already been discussed in Section 2.3.77
5. Proteins and amino acids as starting materials for functionalised chemicals
In recent years, with the need to find biobased raw materials for the production of chemicals and materials, bio-chemical conversions of biomass, derived either from dedicated crops or from rest-streams from agro-food industries, have been investigated. While there has been prominent and well established research on the conversion of carbohydrates, fatty acids and lignin towards a wide variety of industrial products using (bio)catalysis (as discussed in Sections 2–4 above), much less attention has been given to the use of proteins and amino acids. Conventionally, proteins from animal and plant sources have been primarily used, and transformed, with the aim of producing improved quality food. However, given the incentives across the globe to increase the production and use of biofuels, large volumes of rest streams will be generated that contain significant amounts of protein (20–40% of dry mass). More tangibly, if guidelines to replace 10% of the global transportation fuels were to originate from a biobased source this would result in the generation of ca. 100 million tons of protein. Amino acids, resulting from the hydrolysis of proteins, have functional similarities to a number of current industrial chemicals such as small aliphatic (di)amines and carboxylic acids. By reactions, such as deamination or decarboxylation, these can result in the formation of the desired chemicals. For example, alanine can undergo decarboxylation to form ethylamine, a high-volume product used in the dye and agro-chemical industry. Up to now, such types of reactions using enzymes have been explored mainly within the context of nutrition, or product quality, where the amino acid content has been measured using biosensors based on immobilised enzymatic conversion. We will give some examples of those systems, but the main focus will be on those few publications in which the main goal is the transformation of amino acids into building blocks for other chemicals. Non-essential amino acids, or surplus essential amino acids, should be considered as raw materials for the production of chemicals and reactions with immobilised enzymes could lead to their efficient conversion routes.5.1 Proteases and protein hydrolysis
Free amino acids do not occur in high concentrations in nature, so protein hydrolysis is a way to obtain them in larger amounts. This can be done catalytically with acid or base,279 or using enzymes (proteases). Research on the immobilisation and use of proteases has mainly focused on the food and drink industry. In the dairy industry research was prompted to allow reuse of the enzyme, greater control over the milk clotting process and allow a continuous coagulation operation. In the case of pepsin, covalent attachment to ethylene-maleic anhydride copolymer (EMA)280 and aminoalkylated porous glass281 has been achieved, as has physical adsorption onto alumina.282 Hydrolysis of proteins and peptides present in wine has also been widely studied. Proteins in wine can precipitate with time resulting in cloudy wine, which cannot be marketed. Fine clay has been used to clarify wine by adsorbing proteins, however, it also removes some of the taste and colour components.283 To overcome this, proteases could be used to produce soluble peptides and amino acids via the immobilisation of acid proteases on Eupergit® and aminopropyl porous glass (APG).284 Isolation and hydrolysis of plant proteins for human consumption, as an alternative to meat, has also been examined. For example, a soybean drink can be prepared from water extracted protein treated with an immobilised protease.285 Hydrolysed soybean protein with a high amino acid content using proteases immobilised on shrimp chitin by coupling with formaldehyde and cross-linking with glutaraldehyde has also been described.286If the aim is to obtain amino acids from rest protein sources to produce chemicals, a high degree of hydrolysis of these proteins will be required. Chemical hydrolysis under acidic conditions can result in complete hydrolysis. However, during this process some of the amino acids are decomposed and large amounts of salts are produced. Thus, after removal of non-essential acids for use as chemical raw materials, the nutritional quality of the remaining fraction will be compromised. Hydrolysis under basic conditions is generally slower than acidic hydrolysis and also leads to racemisation of amino acids.277 This leads to problems with potential conversions of amino acids. If a subsequent reaction is carried out using a specific enzyme, such as an L-amino acid decarboxylase, then the overall conversion will be reduced. This will affect the raw materials costs and thus increase costs of conversion. The use of immobilised proteases could circumvent amino acid degradation and salt formation. However, complete hydrolysis of proteins to produce amino acids as bulk chemical feedstocks is not yet cost-effective using the current methods.
5.2 An overview of enzymatic conversions of amino acids
Most conversions involving immobilised enzymes utilising amino acid transformations have centred on biosensors for food quality and medical needs. Unfortunately, few studies are devoted to the applications of amino acid-converting enzymes for biobased synthesis.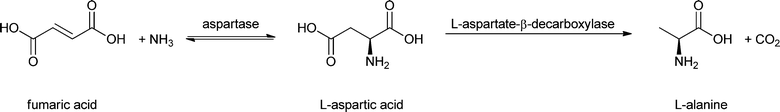 | ||
| Scheme 14 Application of aspartase and L-aspartate β-decarboxylase. | ||
Phenylalanine ammonia lyase (PAL) has been widely studied for the deamination of phenylalanine to cinnamic acid as well as the reverse reaction for industrial phenylalanine production.289 PAL can be found in yeasts, such as Rhodotorula glutinis, which was shown to use L-phenylalanine as a substrate leading to an accumulation of trans-cinnamic acid.290 Yeast cells with PAL activity have also been immobilised with glutaraldehyde-crosslinked alginate for the synthesis of phenylalanine (esters).291,292 From the methods tested it was found that the activity in the forward reaction was 17.2 units mg−1 dry free cells and ca. 85% of the activity was retained after immobilisation by entrapment in alginate or crosslinked egg white.292
L-Glutamic acid can undergo decarboxylation using the E. coli GAD immobilised by entrapment in calcium alginate or covalent binding to Eupergit® 250 C to form γ-aminobutyric acid (GABA), a compound that was proposed to be a useful intermediate for the synthesis of biobased chemicals.74 Operational stability, when reactions were carried out in a fed batch reactor, was found to decrease by only 30% over a 24 h period at 30 °C. This, allied with the high activity of the enzyme, led to high volumetric productivity (35 kg m−3 h−1). It was estimated that the cost of using immobilised GAD, would be ca. € 5 per ton GABA produced using the reactor conditions chosen. Later the same authors reported the cyclisation and alkylation of GABA to N-alkyl pyrrolidones which have a variety of industrial applications.293 When the petrochemical route to N-alkyl pyrrolidones was compared to the biobased route via GABA, the biobased route showed less environmental impact294 and the techno-economic feasibility was also positive.6 In another study, to improve GABA production, high expression levels of GAD (gadA) in E. coli were achieved and the crude enzyme was immobilised by entrapment in alginate and carrageenan gel beads.295 Using a batch mode process, ca. 85% of the relative activity of the immobilised enzyme was retained after 5 cycles (each cycle is 8 h at 37 °C) after the tenth consecutive batch 50% of the relative activity remained. In other efforts to improve productivity of GABA and reduce costs of production, GAD has been fused to the cellulose binding site (CBD) to form GAD-CBD with an activity of ca. 25 U mg−1, expressed in E. coli and immobilised on Avicel as a low-cost matrix.296 A similar retention in relative activity to that described above was shown. However in this case each batch cycle was 0.5 h at 37 °C.
Decarboxylation of L-aspartic acid by E. coliL-aspartate α-decarboxylase (ADC) results in the formation of β-alanine, which has been proposed as an intermediate for the synthesis of acrylonitrile and acrylamide, which are monomers in the polymer industry.12 Covalent immobilisation of ADC on a variety of epoxy resins (Sepabeads) was carried out but it was found that operational stability was still very poor: the total turnover number (TTN) of the enzyme increased from 2390 to only 3730 upon immobilisation. This deactivation was attributed to irreversible transamination of the pyruvoyl catalytic site of the enzyme during the catalytic cycle. It was calculated from the activity that the cost of the immobilised enzyme was very high (ca. €2200 kmol−1 product) compared to the proposed product acrylamide (ca. €220 kmol−1) and therefore currently such a route would not be feasible.297
Arginase hydrolyses the guanidine group in L-arginine, leading to L-ornithine and urea (see Scheme 15). Ornithine can be further converted to 1,4-diaminobutane (see the next paragraph) and urea is a very important fertiliser. The arginase from Bacillus subtilis has been studied in detail.9 Although immobilisation of the enzyme on epoxy activated polymethacrylate beads (Sepabeads®) was facile, the native enzyme was found to be robust under the operating conditions (TTN >109) and did not merit the additional costs associated with the immobilisation method for potential application.
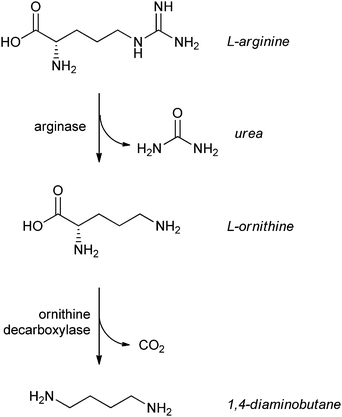 | ||
| Scheme 15 Two-step conversion of L-arginine to 1,4-diaminobutane and urea. | ||
Trypanosoma brucei ornithine decarboxylase (ODC) catalyses the transformation of L-ornithine to 1,4-butanediamine (DAB). DAB, a co-monomer in nylon-4,6, is currently produced by the addition of HCN to acrylonitrile to form succinonitrile, which is subsequently reduced to DAB. ODC was shown to be stabilised and activated in the presence of dithiothreitol and that immobilisation on epoxy resins (Sepabeads®) improved the operational stability (TTN increased from 6.90 × 104 to 1.95 × 105).7 Although this was encouraging, again the costs of the immobilised enzyme per unit products were found to be prohibitive in the current situation.297
Phenylalanine, an essential amino acid used in the food, drink and feed industries, can be produced from cinnamic acid as described earlier. An alternative methodology involves the reaction of phenylpyruvate and L-aspartate in the presence of L-aspartate aminotransferase (see Scheme 16). The immobilisation of the enzyme at low loadings (to prevent diffusion limitations) on Eupergit® C and MANA-agarose and entrapment in LentiKats has been carried out and the productivity and stability of the system studied.47 Using MANA-agarose, low yields (4.7%), residual activity (55%) and initial rates (0.07 mmol L−1 h−1) were obtained. However, in both the case of Eupergit® C and LentiKats, comparable initial rates (0.8 and 0.84 mmol L−1 h−1) and good yields (ca. 70%) were found when compared to the native enzyme (0.74 mmol L−1 h−1 and 68%). This was coupled with an improvement in the stability of the enzyme. Over a 150 h period, the native enzyme showed a decrease in activity of 40% while the immobilised enzyme reported a loss of ca. 10% of activity. Other authors have reported the immobilisation of this enzyme in κ-carrageenan and gelatine and its application in the synthesis of phenylalanine.299 The enzyme in κ-carrageenan showed a retention in enzyme activity of 75.6%, but has a low mechanical strength and high solidifying point (around 50 °C), which leads to unstable gels. In contrast, the gelatin-based carrier exhibited great mechanical strength (694 g cm−3) but the lowest aspartate transaminase activity recoveries (51.9%). A mixed gel containing 87.5% κ-carrageenan and 12.5% gelatin appeared to be the perfect compromise, giving 94% remaining enzyme activity after immobilisation and over 80% after 14 reuses.
 | ||
| Scheme 16 Preparation of L-phenylalanine using asparate aminotransferase. | ||
The use of transaminases has been explored for the synthesis of chemicals for the pharmaceutical and agrochemical industry, e.g. in improving the synthesis of the pharmaceutical compounds (S)-aminotetralin (a pharmaceutically active ingredient), using calcium alginate immobilised aminotransferase,300 and Sitagliptin (an oral antihyperglycemic drug) using the transaminase immobilised on Sepabeads® EXE 12.301 The application of covalently bound transaminase (on VA-Biosynth, a commercially available epoxy-activated carrier) in the synthesis of the herbicide phosphinothricin302 has also been reported. In general the ability to prepare enantiomerically pure amines and amino alcohols as chiral auxiliaries for fine and pharmaceutical chemistry is of interest. In the synthesis of the chiral amino alcohol, (2S,3R)-2-amino-1,3,4-butanetriol, the use of alanine aminotransferase was performed initially with whole cells,303 but later an immobilised enzyme reactor (based on 3-aminopropyltriethoxysilane-treated silica) was also shown to allow the conversion.304 The specific ω-transaminases required can also be immobilised on catechol–chitosan iron oxide nanoparticle composites, by reaction of the catechol with the enzyme.305
 | ||
| Scheme 17 Preparation of 7-aminocephalosporanic acid from cephalosporin C using amino acid oxidase and amidase. | ||
Frost has also investigated the use of L-lysine to synthesise caprolactam, the precursor of nylon-6, by chemical means.308 In an alternative approach the use of L-lysine α-oxidase from Trichoderma viride, immobilised on an epoxy-activated solid support (Sepabeads EC-EP), has been explored. Similar to the reaction depicted in Scheme 17, this resulted in the spontaneous oxidative decarboxylation of the intermediate 6-amino-2-oxocaproic acid in the reaction medium and the formation of 5-aminovaleric acid, another potential nylon monomer.309 Using 5 U of immobilised enzyme, 0.88 g of L-lysine was transformed into 0.67 g (95%) of 5-aminovaleric acid in 3 days.
 | ||
| Scheme 18 Use of haloperoxidase to perform oxidative decarboxylation of amino acids. | ||
5.3 From amino acids to chemical intermediates and products
A review in 2007 describes the potential to convert amino acids into a wide variety of different industrial chemicals by various (bio)catalytic approaches.312 As the concept to use enzymes to convert amino acids to useful chemicals and intermediates develops, research to develop high throughput screening for enzymes with amino acid (decarboxylase) activity is also now being carried out.313 The advances with the use of immobilised enzymes when it comes to biobased production of chemicals from amino acids are depicted in Fig. 3. | ||
| Fig. 3 An overview of the present and potential future role of immobilised enzymes in the conversion of proteins and amino acids in the production of chemicals. | ||
The starting material for these routes can be any protein that is available in sufficiently large amounts in agro rest streams,314 but then all twenty amino acids in the hydrolysate have to be separated which will be cumbersome. The use of cyanophycin as a protein source would circumvent this problem. Cyanophycin is a non-ribosomal peptide containing only two amino acids, a poly(L-aspartic acid) backbone with L-arginine side chains (see Fig. 4).315 Accumulation of the peptide occurs within the cell which, when separated, can be extracted and precipitated. It has been shown that cyanophycin can be prepared from agro rest streams when these are fed to Saccharomyces cerevisiae containing the cyanophycin synthase gene.277,316 This would be a good source of arginine and aspartic acid for the conversions described above.
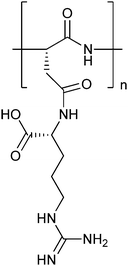 | ||
| Fig. 4 Structure of cyanophycin. | ||
It has been described earlier how PAL can used in the conversion of L-phenylalanine to cinnamic acid. It is also known that decarboxylation of cinnamic acid results in the formation of styrene, either chemically317 or biochemically.318,319 Combining the concept of deamination of L-phenylalanine to cinnamic acid followed by subsequent decarboxylation then it may be possible to devise routes to biobased styrene (see Scheme 19). Other authors have also investigated this approach.320
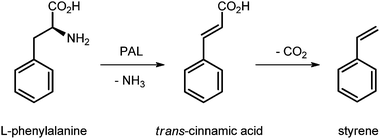 | ||
| Scheme 19 The application of PAL in the synthesis of styrene. | ||
All of the above conversions rely on the use of individual, isolated amino acids. However if the source of the amino acids is from hydrolysed rest proteins, the amino acids will exist as a heterogeneous aqueous mixture. In order to effect isolation techniques, immobilised enzymes can also play a role. Difficulties arise in the complete separation of all amino acids by electrodialysis, due to the similar isoelectric points of the amino acids within each group of acidic, basic and neutral amino acids and obtained by electrodialysis. Specific enzymatic modification of a particular amino acid within each group, by decarboxylation for example, will lead to a change in charge and allow separation. If the choice of reaction leads to the formation of a particular chemical (intermediate), product formation and separation can be coupled. Examples in the literature describe the use of alginate entrapped decarboxylases which perform a specific modification of an amino acid to a potential chemical intermediate where the change in charge will facilitate separation.8,321
In summary, immobilised enzymes can lead to opportunities in the synthesis of fine and pharmaceutical chemicals and biobased industrial chemicals from amino acids obtained from rest proteins (as opposed to the use of fossil resources), as well as the possibilities to couple synthesis with product separation in complex aqueous media.
6. Lignin and its building blocks
6.1 Introduction
Lignin is one of the most abundant polymers in nature. It is a recalcitrant, three dimensional, high-MW biopolymer, composed of hydroxy- and methoxy-phenylpropanoids, interlinked by ether and carbon–carbon bonds. Up to 35% of plants and trees may consist of lignin, which makes it very attractive to further develop its potential for the production of aromatics.322 Yet, because of its very limited solubility, inertness and heterogeneity, it is regarded as the most difficult biomass fraction to be valorised. In nature several enzymes are involved in its degradation:323,324–lignin peroxidase (LiP)
–manganese peroxidase (MnP)325
–versatile peroxidase (VP)
–laccase
Lignin peroxidase is a heme-containing enzyme that is able to break C–C and ether bonds, using hydrogen peroxide as the oxidant. Manganese peroxidase is also a heme enzyme, catalysing the oxidation of Mn2+ ions by hydrogen peroxide. The resulting Mn3+ acts as a mediator that is able to oxidise a variety of lignin building blocks. The recently discovered Versatile Peroxidase is a kind of hybrid between LiP and MnP, having a heme prosthetic group and binding sites for both Mn2+ and small phenolic and nonphenolic substrates. Laccase is a totally different enzyme, having four Cu2+ ions in the active site that are able to oxidise four phenolic substrates while reducing molecular oxygen to water. The action of all four enzymes ultimately results in the formation of radicals, which may oligomerise/polymerise, or lead to oxygenated products. The reader is referred to the excellent review of Wong323 for information on the occurrence and reaction mechanisms of these enzymes.
Another enzyme that has often been used for the conversion of lignin building blocks and related aromatics is tyrosinase. We will therefore also discuss applications of this enzyme. In contrast to laccase, tyrosinase has only two copper ions. It is involved in o-hydroxylation reactions of phenols and the subsequent oxidation of the resulting catechols to o-quinones, which may polymerise to e.g. the natural pigment melanin.326 Reactions with immobilised horseradish peroxidase (HRP) are not included in this review: this enzyme is often bound to surfaces and immobilised biomolecules, using the oxidation of ABTS as a ‘reporter’. There are also many applications for sensors327 and in bioremediation;328 the reader is referred to the cited reviews for further information.
It does not seem to make sense to use immobilised enzymes when converting a highly insoluble substrate like lignin. It should, however, be borne in mind that, in addition to direct interaction with lignin, many of the enzymes mentioned above can also act by the use of mediators, i.e. they oxidise a low-MW substrate into a reactive species (a radical or a high-valent metal ion like Mn3+). The resulting reactive species can then diffuse to the lignin and induces bond cleavages, removing the need for direct interaction between the enzyme and the (sterically bulky) lignin substrate.
The next section is devoted to conversion of lignin using immobilised enzymes. Subsequent sections deal with the application of immobilised forms of the enzymes mentioned above for the conversion of lignin constituents, model compounds thereof and a few other aromatic natural compounds. It should be stressed that the large majority of all published papers on immobilised forms of these enzymes deal with waste water treatment or biosensors for pollutants. In many cases the products are insoluble polymers, or low-MW compounds of which the chemical structures have not been determined by the authors. Nevertheless, the paragraphs on LiP and MnP are comprehensive and therefore also contain immobilisation techniques for these purposes. Because of its recent discovery, there is only one paper on immobilised VP: the enzyme has been adsorbed to a graphite electrode and used for the electrochemical detection of veratryl alcohol, methoxy-p-hydroquinone and 2,6-dimethoxyphenol.329
Finally, in the paragraph on laccase and tyrosinase, only synthetic applications with immobilised enzymes are taken into account, i.e. reactions with well-defined and isolated products, made from compounds that (in principle) can be biobased.
6.2 Conversion of lignin by immobilised enzymes
Only a few research groups have studied the reaction of lignin with an immobilised enzyme. In one of the reports, laccase was covalently linked to alumina beads that were treated with γ-aminopropyltriethoxysilane and glutaraldehyde. Subsequently, oppositely charged polymer layers were deposited on the immobilised enzyme preparation by the LbL technique (cLbL). In addition, laccase was entrapped in LbL microspheres (mLbL). Both enzyme preparations were used for the oxidation of softwood milled wood lignin and kraft residual lignin.61 The stability of the enzyme was strongly increased upon immobilisation: cLbL treatment yielded a remaining activity of 84% after 10 reactions of 12 h each, mLbL 68%, while free enzyme had only 22% remaining activity. The oxidation of lignin was carried out both with and without external mediators. In the absence of mediators, it is assumed that small, soluble, lignin fragments are able to access the immobilised enzyme and act as mediators. In all cases, yield of oxidised, solubilised lignin is 77–94% with the immobilised enzymes, compared to 45–68% with free enzyme. Remarkably, the immobilised laccases follow a different reaction pathway than the free enzyme. Free laccase not only depolymerises, but also induces crosslinks, probably because of direct contact of the enzyme with the lignin. The immobilised enzymes, however, only showed depolymerisation, with cLbL-laccase mainly performing side chain oxidations, while mLbL-laccase favours alkyl-phenyl ether bond cleavage. The different product ratios in the immobilised enzymes are hard to explain considering that the reaction is done by the same mediators. Perhaps there are pores in the polyelectrolyte layers, thereby allowing some direct contact between the enzyme and the lignin. This has been suggested by the same research group for the reaction of LbL-immobilised HRP with lignin.62 In this reaction, both polymerisation and depolymerisation was found with the immobilised enzyme. Laccases and HRP were also co-immobilised using the LbL-technique.330 In the depolymerisation of wheat straw milled lignin, the co-immobilised enzyme preparation gave a higher amount of depolymerisation and more soluble oligomers than the separately immobilised enzymes; the background of this phenomenon is not clear.Recently a patent appeared on the depolymerisation of lignin by LiP, MnP, or VP adsorbed on magnetic nanoparticles.331 The products were soluble aromatics like coniferyl, sinapyl, and coumaryl alcohols or derivatives thereof. Enzyme activities were increased with respect to soluble enzymes, but no product yields or stability studies were given.
Areskogh and Henriksson332 studied the polymerisation of lignosulphonate with immobilised laccase. Lignosulphonates are generated during the ‘sulphite pulping process’, in which the native lignin is solubilised by treating it with (bi)sulphite, leading to the introduction of charged sulphonic acid groups on the lignin. The resulting biopolymer is an amphiphilic compound, but the dispersing and plasticising ability is not optimal. It can be improved by cross-linking using the laccase from Trametes villosa. The enzyme was covalently immobilised on either controlled-pore silica or aminopropylated alumina, after treatment with glutaraldehyde. The silica beads gave much a higher immobilisation yields (89%) than the alumina beads (20%), but in the end the alumina beads worked better because of extensive adsorption of resulting lignosulphonate to the highly porous matrix of the silica beads. The average MW of the lignosulphonate increased from 50000 to 100–150000 because of the enzymatic reaction.
6.3 Reactions of lignin peroxidase with lignin building blocks
There are only few reports about immobilised LiP, and in all of them the oxidation of the lignin building block veratryl alcohol (VA) to veratraldehyde is the only well-characterised reaction (see Scheme 20). In fact, this reaction is the standard assay for LiP.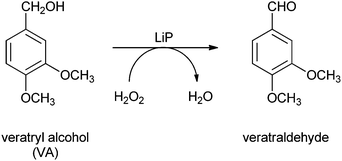 | ||
| Scheme 20 Lignin peroxidase-mediated conversion of veratryl alcohol (VA) to veratraldehyde. | ||
In the first report on the immobilisation of LiP from Phanerochaete chrysosporium, the enzyme was immobilised by covalent coupling of native enzyme to glutaraldehyde-activated glass beads and to epoxy-activated acrylic beads (Eupergit© C), and by coupling of NaIO4-oxidised LiP to hydrazide-derivatised agarose.333 The latter method relates to immobilisation of glycoproteins. The immobilised enzymes were put in a flow injection analysis (FIA) column. The glutaraldehyde-glass method gave the highest immobilisation yield, but the activity was low due to diffusion limitation and the apparent KM for H2O2 was 3 mM, compared to 44 μM for the soluble enzyme. Immobilisation protected the enzyme against inactivation (bleaching) by hydrogen peroxide: at 100 μM H2O2 concentration, a constant signal was detected in FIA. The system can be used for monitoring lignin degradation.
Presnell et al.334 used three classical methods for the immobilisation of LiP to aminopropyl-functionalised controlled-pore glass beads. In the first method carboxylate groups of the enzyme were coupled to the amino beads using EDC. In the second method the amino groups of the beads were reacted with succinic acid anhydride, yielding carboxylate-functionalised glass beads. Coupling to the enzyme was performed with EDC again. The third method involved reacting the aldehyde groups derived from periodate oxidation of the sugars of this glycoprotein to the amino beads. For all three enzyme preparations Vmax for VA conversion was lower than for free enzyme, probably because of conformational changes. Mass transfer limitation does not play a role because KM for VA was lower except for the carbohydrate-based method. The KM for H2O2 was decreased for all three enzyme preparations. The enzymes were applied for the degradation of anthracene and pulp bleach plant waste from a paper mill, using VA as a mediator.
Another study on the covalent immobilisation of LiP from Phanerochaete chrysosporium involved three similar methods: coupling to CNBr-activated Sepharose, to N-hydroxysuccinimide-activated Affigel 15, and to the amine groups of Affigel 102 using EDC.335 The CNBr-Sepharose method gave by far the best results, although the activity towards VA dropped to only 35%. No effect on pH profile and vulnerability towards organic solvents was observed, and there was a small increase in temperature stability. The stability towards low pH, however, was strongly increased, which is important because the activity of the enzyme is highest at low pH.
Podgornik and Podgornik coupled two LiP isozymes from P. chrysosporium to methacrylate based monolithic supports containing epoxy groups.57 The advantage of the monolith is that there is hardly any diffusion limitation. However, enzyme activity and stability was low because of product inhibition. Furthermore, immobilisation led to a fourfold increase of KM while Vmax decreased by a factor 20.
Qiu et al. co-immobilised LiP and glucose oxidase to nanoporous gold.85 The second enzyme produces H2O2 upon oxidising glucose, which in this way is formed in a slow and continuous way, thereby minimising inactivation of LiP by H2O2. Although the enzyme is only physisorbed, the interaction between the enzyme and the support is very strong because the enzyme does not detach in citrate buffer, 0.5 M NaCl or 10% (w/v) polyethylene glycol. Immobilisation strongly increased LiP stability at 45 °C as well as stability under storage and turnover conditions. The enzyme still had 65% of its original activity after 7 reuses, but any loss of activity upon immobilisation was not reported.
Asgher et al. entrapped LiP from P. chrysosporium in a Si(OMe)4/nPrSi(OMe)3 xerogel.83 The immobilised enzyme appeared to be more active than the soluble enzyme, judged by the conversion of VA. The background is a decrease of KM (from 13 to 10.6 mg ml−1) and an increase of Vmax (from 11.8 to 16.7 μmol min−1). Entrapping the enzyme makes it much more stable at temperatures above 40 °C and above pH 4. The authors isolated a new LiP from Trametes versicolor, a common white rot fungus that is well known for its laccase, and immobilised it in the same way.84 Also for this enzyme the activity at basic pH and temperature above 60 °C was enhanced by this immobilisation approach: KM and Vmax were 70 and 56 μM and 588 and 417 U mg−1 for the free and immobilised enzyme.
The LiP and the MnP from P. chrysosporium have also been entrapped between polyelectrolytes using the LbL technique, in which the enzyme was used as the negatively charged polyelectrolyte for the outer layers.63 Both flat surfaces and melamine-formaldehyde colloidal microparticles were used. Enzyme activity, measured by VA oxidation, was decreased tenfold by the immobilisation, but stability was claimed to be increased.
6.4 Reactions of manganese peroxidase with lignin building blocks
MnP has been immobilised by adsorption to mesoporous silica FSM-16 with different pore sizes.55 As discussed earlier in this review, immobilisation strongly increased its stability. The same kind of support was used by Takahashi et al. for the immobilisation of a genetically optimised MnP.336 The optimisation and the immobilisation together accounted for a 50-fold increase of stability against 10 mM H2O2. The MnP from Anthracophyllum discolor has been immobilised on a natural nanoclay by adsorption,337 leading to an enhanced storage stability. Neither of these studies have reported kinetic constants for the immobilised enzymes.MnP from Lentinula edodes was covalently immobilised on an azlactone-functional copolymer.338 The reaction involves nucleophilic ring opening of the azlactone by amino groups of the enzyme. The coupling efficiency was only about 50% because of the low fraction of available lysines in this enzyme. In addition, the enzymatic activity decreased threefold by this treatment, so this seems not the method of choice for MnP. Better results were obtained by the same group using an amine support and 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) or EDC as coupling reagents.339 Now the coupling efficiency was 95% for EEDQ as coupling agent (and 46% for EDC), without loss of activity. Activity was tested by the degradation of 4-(4-hydroxy-3-methoxy-phenyl)-2-butanone (vanillylacetone). Upon immobilisation, the KM for Mn2+ increased from 91 to 440 μM while Vmax decreased from 7.3 to 6.1 mM mg−1 min−1. The stability of the immobilised enzyme was not very different from the free enzyme, except that storage stability was enhanced (t1/2 was increased threefold) and the enzyme was slightly more stable towards H2O2. This immobilised MnP system was used in a two-stage bioreactor for the oxidation of 2,4-dichlorophenol and 2,4,6-trichlorophenol.340 Mn3+ was generated in the first reactor as an oxalate complex, while the oxidation took place in the second one.
Just like for LiP, co-immobilisation of MnP with glucose oxidase proved to be beneficial to minimise H2O2 inactivation.341 The support was aminopropylated controlled-pore glass, treated with glutaraldehyde, used for the covalent binding of the MnP from Phlebia radiata. However, the observed positive effects were not large and immobilisation occurred at the expense of activity and substrate affinity.
Mielgo et al. used glutaraldehyde-functionalised agarose for the covalent immobilisation of MnPs from P. chrysosporium and Bjerkandera sp. BOS55.342 The resulting imine bonds were reduced using sodium borohydride. Best results were obtained with supports having a high density of functional groups using low ionic strength during the immobilisation procedure. The latter showed that adsorption of the enzyme to the support precedes the final covalent bonding. Activity assays were done using Mn2+ with malonate as the ligand for the formed Mn3+, and 2,6-dimethoxyphenol as the reporter for the formation of Mn3+. The product is the oxidised dimer called cerulignone (see Scheme 21). The MnP of P. chrysosporium was slightly stabilised by immobilisation, the one from Bjerkandera more strongly, especially at 50 °C and during storage (50% remaining activity after 12 months compared to 5% for the soluble enzyme, at 4 °C). Concerning the kinetic data upon immobilisation, Vmax was halved for P. chrysosporium MnP while there was not much difference for Bjerkandera MnP; the KM's for H2O2 increased by about 40%.
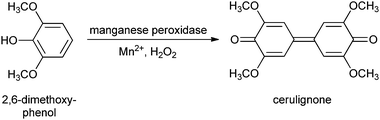 | ||
| Scheme 21 Manganese peroxidase mediated conversion of 2,6-dimethoxyphenol to cerulignone. | ||
The same research group used the Bjerkandera MnP for the continuous decolourisation of azo dyes from waste water.80 The enzyme was freely soluble in a CSTR but was kept inside the reactor by a poly(ethersulphone) ultrafiltration membrane that is permeable for the dye products but not for the enzyme. There was only minimal decrease in enzyme activity during continuous operation over 18 days. The enzymatic membrane reactor attained a decolorization percentage of 98% at a dye loading rate of 0.1 g l h−1, which equals 20 mg dye per MnP unit consumed. In another paper, MnP covalently bound to glutaraldehyde-functionalised agarose was compared to free enzyme in a membrane reactor, used for the degradation of phenol.343 The fact that the agarose-bound enzyme was hardly stabilised compared to the free enzyme, and the high costs of the immobilisation matrix, led the authors to the conclusion that soluble enzyme in a membrane reactor was the most cost-effective system for this reaction.
Edwards et al. immobilised MnP and laccase from Trametes versicolor on a poly(ethersulphone) ultrafiltration membrane used for wastewater treatment from a petrochemical plant.60 The membrane acts as a filter but becomes fouled by organic components. The adsorbed enzymes were used to remove phenolic foulants in situ by oxidation. In the presence of the enzymes over 30% sustained improvement in membrane flux was achieved compared with the controls. The authors assume that the phenolics are turned into soluble compounds that no longer adsorb to the membrane, but evidence from our group has shown that poly(ethersulphone) membranes are in fact easily covalently modified by phenolics using laccases, thereby creating an antifouling layer.344,345 The approach of Edwards et al. was successful when the effluent contained only phenolics, but in ‘real’ industrial effluents fouling still occurred, most likely because of nonphenolic hydrophobic organic compounds.
6.5 Synthetic reactions of laccase and tyrosinase
Many papers have been written on the application of immobilised laccases and tyrosinases for waste water treatment and biosensors.346–348 Few also include well-defined organic reactions,349 and in most of those cases the starting material is a man-made pollutant (e.g. 2,4,6-trichlorophenol)350 or a clearly fossil-derived substrate (e.g. ABTS;351 the cited reference is important for mechanistic laccase studies). Given the focus of the current review on the production of biorenewables some recent preparative transformations are given below.The research on the degradation of organic pollutants with these enzymes has resulted in several useful preparative reactions. p-Cresol (1a, see Scheme 22), which can be considered as a lignin model, is easily oxidised by tyrosinase from the common mushroom Agaricus bisporus (in this paper called polyphenol oxidase) to the corresponding catechol 2.352 The enzyme was either adsorbed to a nylon membrane and subsequently cross-linked with glutaraldehyde, or adsorbed to a poly(ethersulphone) membrane. The nylon-immobilised enzyme showed about the same activity as soluble enzyme; both only gave the catechol product 2a. The enzyme immobilised on the poly(ethersulphone) membrane, however, gave only the o-quinone 3a, albeit at a much lower rate than free or nylon-immobilised enzyme. The authors think the change in enzyme selectivity is caused by the hydrophilic (water, nylon) versus hydrophobic (poly(ethersulphone)) environment of the enzyme. In a subsequent more detailed study, tyrosinases from A. bisporus and Neurospora crassa were either covalently linked by glutaraldehyde to a nylon membrane, or adsorbed to a poly(ethersulphone) membrane as mentioned before.353 This study showed that substrate flux was important: a high flux (i.e., a short contact time between the enzyme and the substrate/product) gave the highest yield of catechols with nylon-immobilised enzyme. The productivity of the enzyme (expressed in amounts of phenol converted per unit of enzyme per hour) increased from 0.011 to 2.60 upon immobilisation. Most likely this is caused by enzyme inhibition by the o-quinones and/or by the polymeric products that are spontaneously formed in water. When enzyme adsorbed to glass beads was used for the oxidation in organic solvents, only the o-quinones were observed at productivities of 0.96 μmol phenol converted U−1 h−1. In this system the quinones apparently do not polymerise or adhere to the enzyme, and therefore do not lead to inhibition. Other substrates studied in this paper are p-methoxyphenol (1b), p-chlorophenol (1c) and m-cresol (4), which behave analogously.
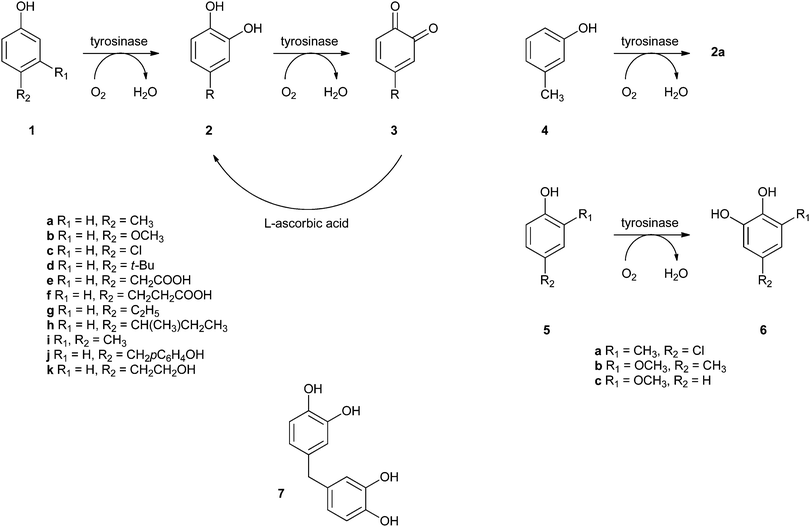 | ||
| Scheme 22 Oxidation of phenols by tyrosinase. Formation of the o-quinone could be reversed by adding ascorbic acid. | ||
Marín-Zamora et al. adsorbed Agaricus bisporus tyrosinase to chiral supports, to study the effect of the support on the stereoselectivity of the enzyme in the o-hydroxylation and/or o-quinone formation of chiral aromatic amino acids.354 The support was UV-crosslinked, fully cinnamoylated sorbitol or glycerol, adsorbed onto porous glass beads. Vmax and KM values were obtained for the conversion of the D- and L enantiomers of tyrosine, dopa, α-methyldopa, adrenaline and noradrenaline with laccase adsorbed on chiral (sorbitol-based) and achiral (glycerol-based) supports. No differences in stereoselectivity were found for the two enzyme preparations. The KM values for the L-enantiomers were always lower than for the D-enantiomers. In a subsequent paper the authors used the tyrosinase adsorbed to the sorbitol-based support for the preparation of o-diphenols (2) from a number of phenols (see Scheme 22).355 Product yields (based on GC) were >95% for the hydroxylation of 1a and the phenolic acids 1e and 1f, and about 88% for p-methoxyphenol (1b) and p-t-butylphenol (1d). The reaction was carried out in the presence of L-ascorbic acid in order to reduce any undesired overoxidation product (o-quinones 3). Suppression of quinone production may also be achieved by complexation of the formed catechols to boronic acid derivatives.
An even larger array of phenols (1a, 1c–k, 4, 5) was hydroxylated by the Crestini group, applying their LbL immobilisation technique to tyrosinase from A. bisporus that was covalently coupled to Eupergit® C250L.63 Although about 70% of the protein in solution was bound to Eupergit, the yield in terms of enzyme activity was only 30–40%. When three alternating layers of poly(allylamine) and poly(styrene sulphonate) were layered on top of the Eupergit-tyrosinase conjugate another 13% of activity was lost (compared to the unlayered enzyme). The storage stability of the enzyme improved by 10–20% by the LbL technique and also the reusability improved to some extent. The reactions were done in the presence of 1.5 equivalent of L-ascorbic acid to avoid formation of o-quinones (3). Yields were 92–99% for the p-substituted phenols, 48–89% for the m- and o-substituted phenols and 77–84% for the phenolic acids 1e and 1f and alcohol 1k. The bisphenol 1j, a building block of the well-known polymer Bakelite®, gave in addition to the monohydroxylated compound 2j (32%) also the dihydroxylated product 7 (66%).
Rhus vernicifera laccase was immobilised in various ways for the conversion of the lignin model isoeugenol (see Scheme 23).356 The enzyme was adsorbed to zirconium oxide particles or chitosan beads, covalently linked to glutaraldehyde-modified chitosan beads, and entrapped in κ-carrageenan, calcium alginate, chitosan and agar. The highest conversion (80%) was found for the zirconium oxide-adsorbed and the agar-entrapped enzyme. The product mixture mainly contained compound 8 (∼54%) followed by 9 (∼29%) and 10 (∼15%), with some traces of oligomers and polymers.
 | ||
| Scheme 23 Conversion of isoeugenol by laccase. | ||
Trametes villosa laccase, adsorbed to mesoporous molecular sieve SBA-15, was used for the dimerisation of trans-resveratrol 11a and some derivatives (see Scheme 24).56 The dimerisation proceeded with a 50% higher rate for the more polar glucoside 11c. The reaction medium was 80% n-butanol in buffer, which is remarkable considering the low stability of laccases towards organic solvents. Nevertheless, the enzyme could be reused up to four times with only 22% loss of activity. Unfortunately, isolated yields of the products were not given.
 | ||
| Scheme 24 Dimerisation of resveratrol (7a) and two derivatives by laccase. | ||
Roy and Abraham studied the dimerisation of pyrogallol to purpurogallin (see Scheme 25) catalysed by Trametes versicolor laccase cross-linked enzyme crystals.67 The CLECs were coated with β-cyclodextrin as a surfactant, to make the enzyme preparation more powder-like and easier to handle. The reaction was carried out in a packed-bed reactor and gave up to 76% of product, depending on residence time. Purpurogallin is a natural product found in galls of white oak; it is an antioxidant and an inhibitor of the epidermal growth factor receptor and of protein tyrosine kinase activity.
 | ||
| Scheme 25 Conversion of pyrogallol to purpurogallin by cross-linked enzyme crystals of laccase. | ||
A CLEC from Trametes versicolor laccase (and a Celite-adsorbed preparation) was also used for the in situ oxidation of catechin, followed by coupling of the resulting o-quinone to polyallylamine (see Scheme 26).68 Compounds like catechin are powerful anti-oxidants, but they suffer from a relatively low water solubility and a limited stability towards light. Conjugation to a polymer is thought to improve its bioavailability. The Vmax value in 70% methanol for the CLEC laccase is higher than for the free enzyme (50.0 vs. 31.3 mmol mg−1 min−1), but the KM value is also higher (29.5 vs. 11.8 mM); for the Celite-absorbed enzyme there were hardly any differences in kinetics. The temperature stability is increased for both immobilised enzyme preparations. The intermediate o-quinone is an enzyme inhibitor and immobilisation apparently protects against this effect, because free enzyme was almost totally inactivated after one reaction cycle, whereas the immobilised enzymes hardly show any inactivation after three consecutive batch reactions.
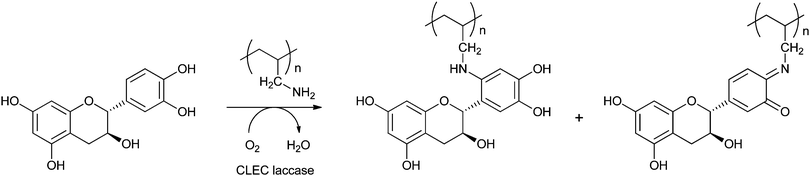 | ||
| Scheme 26 Laccase-catalysed conjugation of catechin with poly(allylamine). | ||
This type of reaction was exploited in more detail by Wellington et al.357 The authors enzymatically oxidised four different p-hydroquinones (13, see Scheme 27) in situ, and the resulting corresponding benzoquinones were reacted with six different primary amines to give 2,5-diaminated benzoquinones (14). The catalyst was the commercially available laccase from Myceliophthora thermophilia immobilised on an inert carrier (Denilite® II Base, Novozymes). The yields of this one-pot, three-step reaction ranged from 5% to 58%; the highest yields were obtained with the aromatic amines.
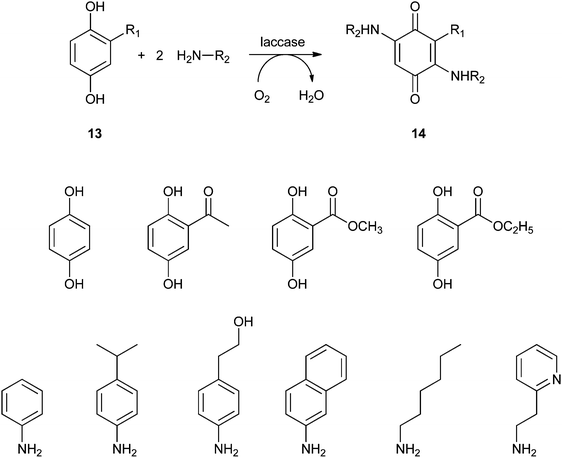 | ||
| Scheme 27 Laccase-mediated oxidation and amination of p-hydroquinones. The used p-hydroquinones are depicted on the second row, the amines on the third row. | ||
The dimerisation of tea epicatechin and its derivatives (epigallocatechin, epigallocatechin gallate) to theaflavins by tea tyrosinase (polyphenol oxidase) was investigated by Sharma et al. (see Scheme 28).358 The enzyme was covalently immobilised to carbonyl diimidazole-activated cellulose and had 84% of its original activity. The Vmax of the immobilised enzyme was 60× higher than of the free enzyme but this was attributed to the removal of inhibitors in the tea extract during the immobilisation procedure. The enzyme was reused 14 times without significant reduction in product yield. The yield (according to HPLC) was about 85%, but the reaction should be stopped in time to avoid overoxidation.
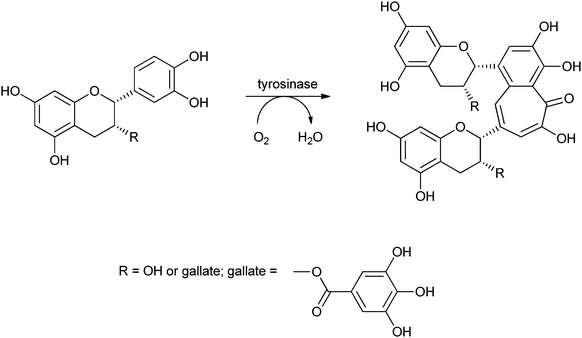 | ||
| Scheme 28 The dimerisation of tea epicatechins to theaflavins by tyrosinase. | ||
Brandi et al. performed a thorough study on four different ways to immobilise Trametes villosa laccase: covalent attachment to Eupergit© C and glutaraldehyde-terminated active carbon, and entrapment into copper and calcium alginate gels.359 The authors evaluated the performance of the different preparations by the efficiency of the oxidation of p-anisyl alcohol to p-anisaldehyde, using four different mediators (see Scheme 29). The immobilisation yield and final enzyme activity was highest for the entrapped enzymes and lowest for the active carbon-bound enzyme. The best combination in terms of product yield (85%) was laccase entrapped in copper alginate, with 1-hydroxybenzotriazole as the mediator. This preparation gave also the best residual activity after 3× reuse.
 | ||
| Scheme 29 Laccase-mediated oxidation of p-anisyl alcohol to p-anisaldehyde. | ||
Cinnabarinic acid is a phenoxazinone dye found in the basidiomycete mushroom Pycnoporus cinnabarinus (see Scheme 30). Osiadacz et al. made this compound and its analogue actinocin from two naturally occurring 3-hydroxyanthranilic acids using Trametes versicolor laccase entrapped in a polyacrylamide gel.360 The phenoxazinone ring system is the central core of actinomycins and the authors study the use the enzyme for the chemoenzymatic synthesis of actinomycin analogues. The yield of actinocin with immobilised enzyme in aqueous buffer was higher (74%) than in 60% acetonitrile (53%), and also than with free enzyme (53%).
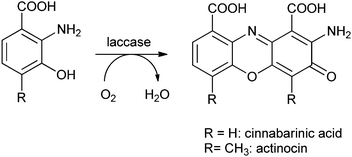 | ||
| Scheme 30 Laccase-mediated oxidation of 3-hydroxy anthranilic acids to cinnabarinic acid and actinocin. | ||
Wan et al.361 elucidated the structure of the reaction product of 2,6-dimethoxyphenol with Rhus laccase and found it to be the symmetrical dimer (see Scheme 31). Both free and immobilised laccase were used, but no details were given about the immobilisation method, the organic solvent used, or the yield of the reaction product.
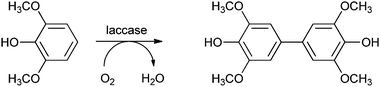 | ||
| Scheme 31 Dimerisation of 2,6-dimethoxyphenol by laccase. | ||
Finally, Fontana et al.362 used laccases for the efficient semisynthesis of antitumor agents. Although not based on lignin-like materials, the efficiency of this reaction is worth mentioning. Immobilised laccases from various sources were used for the in situ oxidation of catharantin. The product was nonenzymatically coupled to vindoline and the resulting adduct was reduced with NaBH4, giving anhydrovinblastine, in 56% overall isolated yield over three steps. The laccases were covalently coupled to Eupergit© or Sepabeads©. Water addition to anhydrovinblastine would give vinblastine, an important alkaloid drug for the treatment of carcinoma.
7. Polymers
The discovery that lipases are able to perform (trans)esterification reactions raised the question whether they are also able to catalyse polymerisation reactions. This indeed appeared to be the case and many polycondensation reactions of diacids/esters with dialcohols, or hydroxyacids/esters alone have been reported, often catalysed by Candida antarctica lipase B in its immobilised form Novozym 435. Also enzymatic ring-opening polymerisations (ROP) of lactones have been reported. Mw values of up to 75000 have been attained. The field has recently been reviewed by Gross et al.363 According to these authors, advantages of the enzymatic methods are their biobased origin, mild reaction conditions, no need for toxic metal catalysts and high regio- and stereoselectivity. Biobased building blocks for polyesters or polyamides that are currently available are succinic acid, citric acid, lactic acid (see below), 3-hydroxypropionic acid, 3-hydroxybutyric acid, furan-2,5-dicarboxylic acid, glutamic acid, lysine, and the esters thereof, and the dialcohols ethylene glycol, 1,3-propanediol, 1,2-propanediol, glycerol, isosorbide (see below), sugars and sugar alcohols like sorbitol. Compounds that are expected to be marketed as biobased compounds soon include adipic acid, glucaric acid, itaconic acid, glycolic acid, levulinic acid, 1,4-butanediol and caprolactam.364 The large majority of these compounds are obtained by fermentation.364The polymer of lactic acid has gained interest because of its mechanical strength, biodegradability, transparency and biocompatibility.365,366 Polylactic acid (PLA) has been applied as an environmentally friendly alternative to plastics derived from fossil resources. Except as plastic, it also has been used for the production of chirurgical sutures or clips, prosthetic devices and pharmaceutical carriers.367,368 The polymerisation of lactic acid usually takes place by ring-opening polymerisation of its cyclic dimer (called lactide). Metal oxides are used as catalysts but also lipases have been used a few times. Novozym 435 is the only immobilised enzyme that has been applied for PLA synthesis; this topic has very recently been reviewed by Idris and Bukhari.369
The following paragraphs are devoted to a few reports that appeared after the cited reviews. Habeych et al. studied the CALB-mediated esterification of the di-anhydrohexitols isomannide, isosorbide and isoidide with succinic acid in toluene (see Scheme 32).370 The cyclic diols arise from double dehydration of reduced D-glucose, D-mannose and L-idose. The esterification results in linear and cyclic ester oligomers. The conversion is highest for isomannide and decreases in the order isomannide – isosorbide – isoidide. The maximum conversions under optimized conditions were 88% and 94% for succinic acid and isomannide, respectively. MALDI-TOF detected products at 24 h were a mixture of cyclic (35%) and linear ester oligomers (65%). Cyclic ester oligomers were the most abundant products during the first 8 h of reaction (33–49%), where the simplest cyclic species of the series (one isomannide molecule di-esterified to one succinic acid molecule) was the most predominant cyclic product (23–40%).
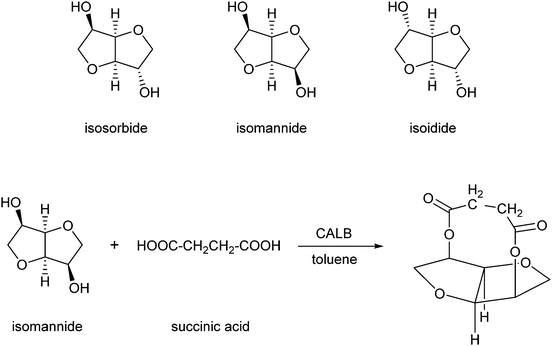 | ||
| Scheme 32 Structures of isomannide, isosorbide and isoidide, and the dilactone product produced by enzyme-mediated condensation of one molecule of isomannide with one molecule of succinic acid. | ||
Li and Li adsorbed a thermostable esterase from the archaeon Archaeoglobus fulgidus to the hydrophobic macroporous resin Octadecyl-Sepabeads EC-OD and used it for the ROP of ε-caprolactone.371 The substrate was fully converted but yielded polymer with low average molecular weight (∼940 at 80 °C, PDI = 1.34, 95 h reaction time). After 5 reuses the immobilised enzyme only had 36% of its original activity, probably due to the loss of adsorbed enzyme from the support during each consecutive reaction.
Tanaka et al. coupled α,ω-diols and aromatic diols with dimethyl mercaptosuccinate to give thiolated polyesters in 67–90% with Mw ranging from 17 000 to 37000 and Mw/Mn of 1.6–1.8 (see Scheme 33).372 The enzyme used was once more Novozym 435. The best results (lowest Mw/Mn) were obtained at 70 °C; above 80 °C thioester formation was observed resulting in chloroform-insoluble material. The polymers were cross-linked by air in a mixture of chloroform and DMSO to form gels or films that have potential for use as an absorbent for haloalkanes. Interestingly, the cross-linking could be reversed by the addition of tributylphosphine and the resulting polymer could be converted into cyclic monomers and oligomers by reaction with CALB in dilute toluene.
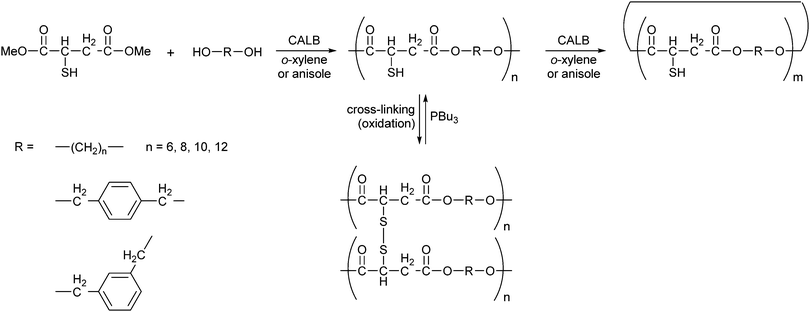 | ||
| Scheme 33 Formation of polymers of mercaptosuccinic acid and α,ω-diols by CALB. The polymer can be reversibly oxidised or converted into small cyclic oligomers by another treatment with CALB. | ||
8. Outlook
In this review, many chemical reactions have been described that are catalysed by immobilised enzymes. The enzymatic reactions show their advantage over chemically catalysed reactions because of their specificity, use of aqueous conditions, and mild conditions that do not demand a high expenditure for process equipment. Several industrially produced enzymes are used in their soluble form, because of the nature of their activities such as the insoluble substrates in laundry applications, but also in animal feed, in beverages as well as in the second generation ethanol production. Thermostable α-amylase is used in its soluble form in order to enable its starch-solubilising activity at high temperatures, even though there are numerous reports on the immobilisation of the enzyme. Chymosine is used in its soluble form not to be entrapped into the curd of the cheese production. Most of these applications are in food or in consumer goods that can afford higher costs than for the production of bulk chemicals and transportation fuels. According to BCCresearch, the turnover of all industrial enzymes was around 4 billion US $ in 2011 and is expected to become 6 billion US $ in 2016.373 However, we should mention that there are only four large-scale processes that involve immobilised enzymes: the production of high-fructose corn syrups by glucose isomerase, the conversion of acrylonitrile to acrylamide by nitrile hydratase immobilised in permeabilised cells and the lipase-mediated production of biodiesel and food-grade fats and oils, although no exact volumes of the latter two applications can be found. Immobilised lipases are used in the food industry for the modification of fat products for margarine and bakery products.374 Transesterification of the various fatty acids from different origins enables tailor-made specification of the resulting fat mixtures to suit a particular food. In the case of margarin the melting point, shelf life, spreadability and nutritional properties can be modified.375 The technology enables the use of low cost lipid sources that on their own do not fulfil the high specifications for use in margarin. In this way e.g. sunflower oil, palm stearin as well as palm olein have been used as raw material inputs. A variety of lipases have been described for this aim, some have specific properties that favour their use in industrial processes. Ronne et al.376 describe the advantages of using immobilised 1,3-specific lipases that give more specific reactions allowing the conservation of essential polyunsaturated fatty acids, most often found in the sn-2 position, thereby leading to less side products and higher product yields.This outlook discusses several factors that will influence the further development of the use of immobilised enzymes for the production of biorenewables, such as the costs of immobilised enzymes (8.1), novel enzymes and conditions (8.2), co-immobilisation of multiple enzymes (8.3), advanced uses of enzyme specificity (8.4), and further improvements in immobilisation methods (8.5).
8.1 Costs of immobilised enzymes
Glucose isomerase is an intracellular enzyme, therefore not trivial to isolate, making immobilisation useful. Advantage is taken of the high stability of the enzyme as was optimised by protein engineering.377 Tufvesson et al. indicates that for an effective industrial process a productivity of around 10 tons of product per kg of immobilised enzyme is required.378 This normally requires considerable stability. For example, immobilised glucose isomerase as used to produce high-fructose corn syrup has an operational lifetime of about one year and productivities are typically around 15 tons of syrup dry substance per kg immobilized enzyme.201 Depending on the number of recycles of the enzyme (up to 200 is usually required) the cost contribution to the produced product varies between a few hundred euro per kg (for pharmaceuticals) down to a few cents per kg (for bulk chemicals), but is normally in the range of 10–0.1 € kg−1.379,380 Lammens et al.74 calculate the cost for the alginate-immobilised glutamate decarboxylase to be around 5€ per ton of γ-aminobutyric acid (GABA) produced. Another fine example is the case of the cell-immobilised nitrile hydratase, in which with only little amount of enzyme conversions of almost 100% could be obtained at very high product concentrations of 400 g l−1 of acrylamide.381A kg of enzyme can be manufactured at a cost of 100€ provided that genetic optimisation as well as a fermentation production process has been performed. Pollard correctly indicated that there is no need to purify the enzyme, as the crudest possible form of the enzyme, acceptable to maintain product quality, should be used for industrial purposes.382 To the best of our knowledge, industry does not use immobilised amylases because due to genetic engineering the enzymes have become cheap and stable enough to dispose of them after one-time use.
Protein engineering has been used to increase the (thermal) stability of glucose isomerase, but also the apparent affinity for the glucose substrate can be improved, judged from the ten times lowered KM value.383 These examples show that both enzyme costs and costs of immobilisation play an important role in the economic viability of a process.
8.2 Novel enzymes and reaction conditions
Since cost price will always be a critical factor, strategies should be developed to lower the cost price of immobilised enzymes and/or increase the value of what can be obtained by using these enzymes. Selection of novel enzymes with high specific activities and/or robustness to extreme conditions such as temperature, pH, concentration of products or substrates by enzyme mining is an essential novel technology apart from the protein engineering, which was already mentioned.384These tools might not only help in lowering the catalytic conversion costs, but at the same time under well-chosen conditions, they might contribute to the overall economy of the production process of (bulk) products, because an elegant recovery can be obtained by e.g. the properties of the product at a certain pH or temperature. High temperatures might allow specific products to be recovered in their gaseous form while at certain pH conditions, products might be recovered and purified because of their insolubility. Other benefits that might result from immobilisation of enzymes are highlighted here below.
8.3 Co-immobilisation of multiple enzymes
Betancor and Luckarift state that there are three primary reasons for the utilisation of co-immobilised enzymes in tandem reactions: to enhance the efficiency of one of the enzymes by the in situ generation of its substrate, to simplify a process that is conventionally carried out in several steps and/or to eliminate undesired by-products of an enzymatic reaction.385 As such, co-immobilisation provides benefits that span numerous biotechnological applications, from biosensing of molecules to cofactor recycling and to combination of multiple biocatalysts for the synthesis of valuable products.Co-immobilisation actually allows turning a negative aspect into positive use. In several reactions mentioned in this review hydrogen peroxide is formed, e.g. in the oxidation of alcohols to carboxylic groups under mild conditions by oxygen using the TEMPO laccase system, the oxidative amination of a variety of aromatic compounds,357 or the formation of gluconic acid out of glucose.85 While this hydrogen peroxide – which is in principle detrimental to a wide variety of enzymes – can be removed by catalase activity, a better use of hydrogen peroxide can be obtained by using its oxidative power in another reaction, e.g. the immobilised enzymatic production of fatty epoxides from rape seed and tall oil derivatives.161 A report of the use of vanadium chloroperoxidases for the decarboxylation of amino acids resulting in the formation of nitriles (e.g. 3-cyanopropanoic acid from glutamic acid) and H2O2 has recently been reported.311 Here the selective transformation of glutamic acid to 3-cyanopropanoic acid using hydrogen peroxide, was described. Analogously, in the synthesis of succinonitrile,386 a precursor to diaminobutane, and acrylonitrile,387 the conversion of L-glutamic acid by oxidative decarboxylation to the intermediate cyanopropanoic acid was identified. This approach was highly successful, but did, however, require the use of hypochlorite, which is disadvantageous in view of its environmental impact and techno-economic feasibility.6,294 The use of haloperoxidase enzymes using the hydrogen peroxide to perform this reaction may provide an alternative method to carry out the conversion to cyanopropanoic acid.311 If the respective enzymes can be immobilised on either side of a membrane system, this would allow the migration of the hydrogen peroxide while the membrane forms a barrier for the substrates and the products (see Fig. 5). In this way, immobilisation could be the answer for not mixing up all different reagents.
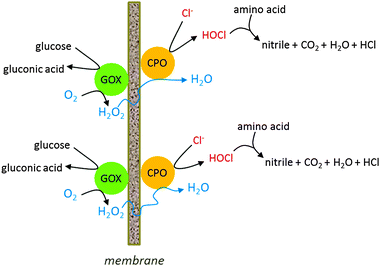 | ||
| Fig. 5 Possible method for co-immobilisation of enzymes with concomitant separation of products. GOX = glucose oxidase, CPO = chloroperoxidase. | ||
Finally, Lopez-Gallego and Schmidt-Dannert describe the co-immobilisation of enzymes to enable the coupling of two reactions,388 not only as described here above for hydrogen peroxide, but also for the regeneration of ATP and for NADH utilised by the other enzyme.
8.4 Advanced uses of enzyme specificity
The isolation of individual amino acids from complex mixtures requires attention. For example, difficulties arise in the complete separation of all amino acid by electrodialysis, attributed to the similar isoelectric points of the amino acids within each group of acidic, basic and neutral amino acids. Electrodialysis will chiefly lead to these three groups being available. It has been proposed that specific enzymatic modification of a particular amino acid within each group, by decarboxylation for example, will lead to a change in charge and allow separation. If the choice of reaction leads to the formation of a particular chemical (intermediate), then the product formation and separation can be coupled. An example of this is the use of calcium alginate-immobilised lysine decarboxylase, which specifically reacts with L-lysine in the basic amino acid stream (potentially obtained after electrodialysis).8 The resultant 1,5 pentanediamine, an industrial chemical in the polymer industry, has a charge difference compared to the L-arginine and therefore separation can in a subsequent electrodialysis step can be achieved. A similar strategy can be applied to the acidic amino acid stream where immobilised GAD is used to make GABA, which can be separated from L-aspartic acid.3218.5 Further improvements in immobilisation methods
As mentioned above, the properties of the enzymes, such as specific activity and stability under operational conditions are of major importance for application in renewable products manufacture. Immobilisation often leads to increased stability and enables reuse. While very many materials and methods for enzyme immobilisation have been described, only few of them pay attention to costs. Commodity chemicals typically have very low added value, leaving very little space for catalysts costs. Therefore, cheap carriers/reagents and very high reusability are key factors in the use of immobilised enzyme in biobased commodity chemistry. For non-aqueous systems such as biodiesel, covalent coupling is not required, with corresponding reduction of costs. Cheap neutral supports like Accurel®,30,31 porous alumina35 and silica55 are advantageous here, but ionic supports have also proven their added value (e.g. Lipozyme RM IM),42 as have magnetic nanoparticles.38,331 For reactions in aqueous media, adsorption to ion exchange resins is important. E.g., glucose isomerase is immobilised on ion exchange resins that can be recycled as the support once inactivated enzymes have been dissociated from the ion exchange resin.389 Ion exchange resins could become suitable for other enzymes if the binding to the resin is enabled by the introduction of a binding site engineered into the enzyme molecule by protein engineering. However, regeneration of the ion exchange resin when all enzyme is inactivated produces salt. This is not a problem at present but there will be environmental pressure in the future. Entrapment in alginate is probably the most cost-effective immobilisation method for enzymes with high molecular weight;74,300 co-immobilisation of silica particles seems also useful to increase the stability.275 Alternatively, LentiKats seem to be useful and cost-effective entrapment vehicles.47 Entrapping enzymes in situ in magnetic nanoparticles is an elegant new method.77 Immobilisation in membrane systems as has been described for several reactor configurations by Jochems et al.58 but suffers, while elegant, from high capital costs. When combined with downstream processing, however, the system has strong potential to be cost effective.78–81 For the industrial production of acrylamide, an immobilised system is applied that keeps the hydratase within the microbial cells in which this enzyme was accumulated. Obviously, this is a very cost-effective method to keep the enzyme within the reactor.Regarding future techniques for enzyme immobilisation, specific binding of enzymes to molecularly imprinted polymers (MIP's)262 could become important. Cross-linking and covalent attachment seem less likely techniques in this field, because of costs and risk of inactivation of the enzyme. Cross-linking by chemical (CLEA's)73 and enzymatic (transglutaminase)275 methods hold the greatest promise.
It is remarkable that, while industrially 100–200 reuses of immobilised enzymes are needed to make their use economically feasible, only few published studies really focus on this large number of reuses. They can be found in the areas where competition is apparently high, like starch hydrolysis259,262 and β-lactam antibiotic synthesis.307 Yet, it is a fair statement that a significantly improved reusability is imperative for the switch from petrol-based to enzymatic biobased routes.
9. Closing remarks
Up to now, only a few immobilised enzymes are used in large-scale industrial processes. Glucose isomerase, lipase and nitrile hydratase are good examples to show the current state of the art. These examples also point to the boundary conditions for further development of the numerous enzyme uses described in this review, to the large-scale production of chemicals and fuels from biomass resources. If we assume that only 10% of the bulk chemical market of today, being about 330 Mton per year will be manufactured using immobilised enzymes in the next few decades at a cost of 100 US $ per ton, this would mean a turnover of 3.3 billion US $. In addition to this, the use of immobilised enzymes in the biodiesel industry will likely also grow, which will thus add significant potential to the total of the enzyme market, which is around 4 billion US $ today.373 The scale of these numbers indicates both the potential and necessity of further developments in the use of immobilised enzymes in biobased chemistry.References
- A. Raschka and M. Carus, Stoffliche Nutzung von Biomasse, Basisdaten fur Deutschland, Europa und die Welt, Nova Institut, Hurth, 2012, Forderkernzeichen 371093109.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 Search PubMed.
- J. P. M. Sanders, E. L. Scott, R. A. Weusthuis and H. Mooibroek, Macromol. Biosci., 2007, 7, 105–117 CrossRef CAS.
- J. P. M. Sanders, J. H. Clark, G. J. Harmsen, H. J. Heeres, J. J. Heijnen, S. R. A. Kersten, W. P. M. Swaaij and J. A. Moulijn, Chem. Eng. Process., 2012, 51, 117–136 Search PubMed.
- M. Kobayashi, T. Nagasawa and H. Yamada, Trends Biotechnol., 1992, 10, 402–408 CrossRef CAS.
- T. M. Lammens, S. Gangarapu, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biofuels, Bioprod. Biorefin., 2012, 6, 177–187 Search PubMed.
- P. M. Könst, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2011, 3, 1167–1174 Search PubMed.
- Y. Teng, E. L. Scott, A. N. T. van Zeeland and J. P. M. Sanders, Green Chem., 2011, 13, 624–630 RSC.
- P. M. Könst, P. M. C. C. D. Turras, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Adv. Synth. Catal., 2010, 352, 1493–1502 CrossRef.
- M. J. Liszka, M. E. Clark, E. Schneider and D. S. Clark, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 77–102 Search PubMed.
- W. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS.
- P. M. Könst, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2009, 11, 1646–1652 RSC.
- I. Chibata, Pure Appl. Chem., 1978, 50, 667–675 CAS.
- O. R. Zaborsky, Immobilized Enzymes, CRC Press, Cleveland, Ohio, 1973 Search PubMed.
- G. P. Royer, Immobilized Enzymes, Antigens, Antibodies, and Peptides, Marcel Dekker, New York, 1975, vol. 1 Search PubMed.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- K. Faber, Biotransformations in Organic Chemistry, Springer, Heidelberg, 6th edn, 2011 Search PubMed.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656–2665 CrossRef CAS.
- S. N. Ahmed, R. J. Kazlauskas, A. H. Morinville, P. Grochulski, J. D. Schrag and M. Cygler, Biocatalysis, 1994, 9, 209–225 Search PubMed.
- M. C. R. Franssen, H. Jongejan, H. Kooijman, A. L. Spek, N. L. F. L. Camacho Mondril, P. M. A. C. Boavido dos Santos and Ae. de Groot, Tetrahedron: Asymmetry, 1996, 7, 497–510 CrossRef CAS.
- E. M. Anderson, K. M. Larsson and O. Kirk, Biocatal. Biotransform., 1998, 16, 181–204 CrossRef CAS.
- L. Fjerback, K. V. Christensen and B. Nohrdahl, Biotechnol. Bioeng., 2009, 102, 1298–1315 CrossRef CAS.
- M. M. Rahman Talukder, J. C. Wu, S. K. Lau, L. C. Cui, G. Shimin and A. Lim, Energy Fuels, 2009, 23, 1–4 CrossRef.
- T. Tan, J. Lu, K. Nie, L. Deng and F. Wang, Biotechnol. Adv., 2010, 28, 628–634 CrossRef CAS.
- S. Warwel and M. Rüsch gen. Klaas, J. Mol. Catal. B: Enzym., 1995, 1, 29–35 CrossRef CAS.
- M. Fernandez-Perez and C. Otero, Enzyme Microb. Technol., 2001, 28, 527–536 Search PubMed.
- R. Croitoru, F. Fitigau, L. A. M. van den Broek, A. E. Frissen, C. M. Davidescu, C. G. Boeriu and F. Peter, Process Biochem., 2012, 47, 1894–1902 Search PubMed.
- S. C. Kim, Y. H. Kim, H. L. Hyuk, D. Y. Yoon and B. K. Song, J. Mol. Catal. B: Enzym., 2007, 49, 75–78 CrossRef CAS.
- J. Lu, K. Nie, F. Xie, F. Wang and T. Tan, Process Biochem., 2007, 42, 1367–1370 Search PubMed.
- M. M. Soumanou, S. T. Djenontin, F. P. Tchobo, D. C. K. Sohounhloue and U. T. Bornscheuer, Lipid Technol., 2012, 24, 158–160 Search PubMed.
- P. Tufvesson, U. Törnvall, J. Carvalho, A. J. Karlsson and R. Hatti-Kaul, J. Mol. Catal. B: Enzym., 2011, 68, 200–205 Search PubMed.
- S. Miao, Z. Liu, H. Ma, B. Han, J. Du, Z. Sun and Z. Miao, Microporous Mesoporous Mater., 2005, 83, 277–282 Search PubMed.
- U. H. Zaidan, M. B. A. Rahman, M. Basri, S. S. Othman, R. N. Z. R. A. Rahman and A. B. Salleh, Appl. Clay Sci., 2010, 47, 276–282 Search PubMed.
- U. H. Zaidan, M. B. A. Rahman, S. S. Othman, M. Basri, E. Abdulmalek, R. N. Z. R. A. Rahman and A. B. Salleh, Food Chem., 2012, 131, 199–205 Search PubMed.
- R. Reshmi, G. Sanjay and S. Sugunan, Catal. Commun., 2006, 7, 460–465 CrossRef CAS.
- M. W. Christensen, L. Andersen, T. L. Husum and O. Kirk, Eur. J. Lipid Sci. Technol., 2003, 105, 318–321 Search PubMed.
- D.-T. Tran, K.-L. Yeh, C.-L. Chen and J.-S. Chang, Bioresour. Technol., 2012, 108, 119–127 Search PubMed.
- C. G. C. M. Netto, H. E. Toma and L. H. Andrade, J. Mol. Catal. B: Enzym., 2013, 85–86, 71–92 Search PubMed.
- H. Park, J. Ahn, J. Lee, H. Lee, C. Kim, J.-K. Jung, H. Lee and E. G. Lee, Int. J. Mol. Sci., 2012, 13, 358–368 Search PubMed.
- M. Y. Arica, H. Soydogan and G. Bayramoglu, Bioprocess Biosyst. Eng., 2010, 33, 227–236 Search PubMed.
- S. L. Gao, Y. L. Wang, T. Wang, G. S. Luo and Y. Y. Dai, Bioresour. Technol., 2009, 101, 7231–7238 Search PubMed.
- N. R. Sonare and V. K. Rathod, J. Mol. Catal. B: Enzym., 2010, 66, 142–147 Search PubMed.
- E. A. J. Al-Mulla, W. M. Z. W. Yunus, N. A. B. Ibrahim and M. Z. A. Rahman, J. Oleo Sci., 2010, 59, 59–64 Search PubMed.
- A. Salis, V. Solinas and M. Monduzzi, J. Mol. Catal. B: Enzym., 2003, 21, 167–174 Search PubMed.
- R. A. Kahn, D. Perin, E. Murano and M. Bergamin, WO2012/053017, 26 April 2012.
- M. Cárdenas-Fernández, C. López, G. Álvaro and J. López-Santín, Bioprocess Biosyst. Eng., 2012, 35, 1437–1444 Search PubMed.
- M. Cárdenas-Fernández, C. López, G. Álvaro and J. López-Santín, Biochem. Eng. J., 2012, 63, 15–21 Search PubMed.
- M.-M. Zheng, Y. Lu, L. Dong, P.-M. Guo, Q.-C. Deng, W.-L. Li, Y.-Q. Feng and F.-H. Huang, Bioresour. Technol., 2012, 115, 141–146 Search PubMed.
- X. T. Peng, Z. G. Shi and Y. Q. Feng, Food Anal. Methods, 2010, 4, 381–388 Search PubMed.
- Y. Kang, J. He, X. Guo, X. Guo and Z. Song, Ind. Eng. Chem. Res., 2007, 46, 4474–4479 CrossRef CAS.
- M. T. Reetz, Adv. Mater., 1997, 9, 943–954 CrossRef CAS.
- R. Fernandez-Lafuente, P. Amisen, P. Sabuquillo, G. Fernandez-Lorente and J. M. Guisan, Chem. Phys. Lipids, 1998, 93, 185–197 CrossRef CAS.
- C. Matteo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- K. Ramani, R. Boopathy, A. B. Mandal and G. Sekaran, Catal. Commun., 2011, 14, 82–88 Search PubMed.
- T. Sasaki, T. Kajino, B. Li and H. Sugiyama, Appl. Environ. Microbiol., 2001, 67, 2208–2212 CrossRef.
- H. Zhang, E. Xun, J. Wang, G. Chen, T. Cheng, Z. Wang, T. Ji and L. Wang, Int. J. Mol. Sci., 2012, 13, 5998–6008 Search PubMed.
- H. Podgornik and A. Podgornik, Enzyme Microb. Technol., 2002, 31, 855–861 Search PubMed.
- P. Jochems, Y. Satyawali, L. Diels and W. Dejonghe, Green Chem., 2011, 13, 1609–1623 RSC.
- D. Sen, A. Sarkar, S. Das, R. Chowdhurry and C. Bhattacharjee, Ind. Eng. Chem. Res., 2012, 51, 10671–10681 Search PubMed.
- W. Edwards, W. D. Leukes and J. J. Bezuidenhout, Desalination, 2002, 149, 275–278 Search PubMed.
- C. Crestini, R. Perazzini and R. Saladino, Appl. Catal., A, 2010, 372, 115–123 Search PubMed.
- R. Perazzini, R. Saladino, M. Guazzaroni and C. Crestini, Bioorg. Med. Chem., 2011, 19, 440–447 Search PubMed.
- M. Guazzaroni, C. Crestini and R. Saladino, Bioorg. Med. Chem., 2012, 20, 157–166 Search PubMed.
- D. S. Patel, R. K. Aithal, G. Krishna, Y. M. Lvov, M. Tien and D. Kuila, Colloids Surf., B, 2005, 43, 13–19 Search PubMed.
- N. L. St. Clair and M. A. Navia, J. Am. Chem. Soc., 1992, 114, 7314–7316 CrossRef CAS.
- J. J. Roy and T. E. Abraham, Chem. Rev., 2004, 104, 3705–3722 CrossRef CAS.
- J. J. Roy and T. E. Abraham, J. Chem. Technol. Biotechnol., 2006, 81, 1836–1839 Search PubMed.
- P. Gogoi, S. Hazarika, N. N. Dutta and P. G. Rao, Chem. Eng. J., 2010, 163, 86–92 Search PubMed.
- L. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS.
- L. Cao, L. van Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS.
- R. Schoevaart, M. W. Wolbers, M. Golubovic, M. Ottens, A. P. G. Kieboom, F. van Rantwijk, L. A. M. van der Wielen and R. A. Sheldon, Biotechnol. Bioeng., 2004, 87, 754–762 CrossRef CAS.
- R. Gaur, H. Pant, R. Jain and S. K. Khare, Food Chem., 2006, 97, 426–430 Search PubMed.
- P. O. Jones and P. T. Vasudevan, Biotechnol. Lett., 2010, 32, 103–106 CrossRef CAS.
- T. M. Lammens, D. de Biase, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2009, 11, 1562–1567 RSC.
- B. L. Tee and G. Kaletunç, Biotechnol. Prog., 2009, 25, 436–445 Search PubMed.
- M. Schlieker and K.-D. Vorlop, in Methods in Biotechnology: Immobilisation of Enzymes and Cells, ed. J. M. Guisan, Humana Press Inc., Totowa, NJ, 2nd edn, 2006 Search PubMed.
- M. G. Bellino and A. E. Regazzoni, Appl. Catal., A, 2011, 408, 73–77 Search PubMed.
- R. Das, D. Sen, A. Sarkar, S. Bhattacharyya and C. Bhattacharjee, Ind. Eng. Chem. Res., 2011, 50, 806–810 Search PubMed.
- A. Kohl, T. Bruck, J. Gerlach, M. Zavrel, L. Rocher and M. Kraus, WO2012/001102, 2012.
- C. López, M. T. Moreira, G. Feijoo and J. M. Lema, Biotechnol. Prog., 2004, 20, 74–81 Search PubMed.
- M. J. Ko, H. J. Park, S. Y. Hong and Y. J. Yoo, Bioprocess Biosyst. Eng., 2012, 35, 69–75 Search PubMed.
- R. A. Kahn, D. Perin, E. Murano and M. Bergamin, WO2012/053017, 26 April 2012.
- M. Asgher, M. J. Asad, H. N. Bhatti and R. L. Legge, World J. Microbiol. Biotechnol., 2007, 23, 525–531 Search PubMed.
- M. Asgher, H. M. N. Iqbal and M. Irshad, BMC Biotechnol., 2012, 12, 46–53 Search PubMed.
- H. Qiu, Y. Li, G. Ji, G. Zhou, X. Huang, Y. Qu and P. Gao, Bioresour. Technol., 2009, 100, 3837–3842 CrossRef CAS.
- The Biodiesel Handbook, ed. G. Knothe, J. V. Gerpen and J. Krahl, AOCS Press, Illinois, 2005, pp. 4–16 Search PubMed.
- E. G. Shay, Biomass Bioenergy, 1993, 4, 227–242 CrossRef CAS.
- J. M. Marchetti, V. U. Miguel and A. F. Errazu, Renewable Sustainable Energy Rev., 2007, 11, 1300–1311 CrossRef CAS.
- O. Kose, M. Tuter and A. H. Aksoy, Bioresour. Technol., 2002, 83, 125–129 CrossRef CAS.
- Y. Shimada, Y. Watanabe, A. Sughihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 133–142 CrossRef CAS.
- L. Wright, Biomass Bioenergy, 2006, 30, 706–714 Search PubMed.
- D. Ganesan, A. Rajendran and V. Thangavelu, Rev. Environ. Sci. Bio/Technol., 2009, 8, 367–394 Search PubMed.
- B. Zhang, Y. Weng, H. Xu and Z. Mao, Appl. Microbiol. Biotechnol., 2012, 93, 61–70 Search PubMed.
- R. Jothiramalingam and M. K. Wang, Ind. Eng. Chem. Res., 2009, 48, 6162–6172 CrossRef CAS.
- P. T. Palligarni and B. Fu, Waste Biomass Valorization, 2010, 1, 47–63 Search PubMed.
- S. K. Padhi and R. K. Singh, J. Chem. Pharm. Res., 2011, 3, 39–49 Search PubMed.
- W. Parawira, Crit. Rev. Biotechnol., 2009, 29, 82–93 Search PubMed.
- A. Bajaj, P. Lohan, P. N. Jha and R. Mehrotra, J. Mol. Catal. B: Enzym., 2010, 62, 9–14 CrossRef CAS.
- H. Taher, S. Al-Zuhair, A. H. Al-Marzouqi, Y. Haik and M. M. Farid, Enzyme Res., 2011 Search PubMed , 468292.
- S. Uthoff, D. Bröker and A. Steinbüchel, Microb. Biotechnol., 2009, 2, 551–565 Search PubMed.
- M. G. Kulkarni and A. K. Dalai, Ind. Eng. Chem. Res., 2006, 45, 2901–2913 CrossRef CAS.
- S. P. Singh and D. Singh, Renewable Sustainable Energy Rev., 2010, 14, 200–216 CrossRef CAS.
- A. Demirbas, Energy Convers. Manage., 2002, 43, 2349–2356 CrossRef CAS.
- W. Cao, H. Han and J. Zhang, Fuel, 2005, 44, 256–262 Search PubMed.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. M. Tost, J. Santemaria and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS.
- S. Mekhilef, S. Siga and R. Saidur, Renewable Sustainable Energy Rev., 2010, 14, 217–232 CrossRef CAS.
- K. G. Georgogianni, A. K. Katsoulidis, P. J. Pomonis, G. Manos and M. G. Kontominas, Fuel Process. Technol., 2007, 90, 1016–1022 Search PubMed.
- A. J. Kinney and T. E. Clemente, Fuel Process. Technol., 2005, 86, 1137–1147 CrossRef CAS.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- Z. Helwani, M. R. Othman, N. Aziz, W. J. N. Fernando and J. Kim, Fuel Process. Technol., 2009, 90, 1502–1514 CrossRef CAS.
- A. Demirbas, Prog. Energy Combust. Sci., 2007, 33, 1638–1643 Search PubMed.
- Y. Shimada, Y. Watenabe, T. Samukawa, A. Sugihara, H. Noda, H. Fukuda and Y. Tominaga, J. Am. Oil Chem. Soc., 1999, 76, 445–450 Search PubMed.
- K. H. Wiltshire, M. Boersma, A. Moeller and H. Buhtz, Aquat. Ecol., 2000, 34, 119–126 CrossRef CAS.
- F. Pernet and R. Tremblay, Lipids, 2003, 38, 1191–1195 Search PubMed.
- H. E. Hoydonckx, D. E. de Vos, S. A. Chavan and P. A. Jacobs, Top. Catal., 2004, 27, 83–96 CrossRef CAS.
- W. Du, W. Li, T. Sun, X. Chen and C. Lui, Appl. Microbiol. Biotechnol., 2008, 79, 331–337 CrossRef CAS.
- P. M. Nielsen, J. Brask and L. Fjerbaek, Eur. J. Lipid Sci. Technol., 2008, 110, 662–700 Search PubMed.
- S. Nakamura, Oleochemicals, 1997, 8, 866–874 Search PubMed.
- T. Watanabe, Foods Food Ingredients J. Jpn., 1999, 180, 18–25 Search PubMed.
- U. Biermann, W. Friedt, S. Lang, W. Luhs, G. Machmuller, J. O. Metzger, M. Rüsch gen. Klaas, H. J. Schaffer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS.
- S. Al-Zuhair, Biofuels, Bioprod. Biorefin., 2007, 1, 57–66 Search PubMed.
- G. Knothe, Prog. Energy Combust. Sci., 2010, 36, 364–373 CrossRef CAS.
- J. Hu, Z. Du, Z. Tang and E. Min, Ind. Eng. Chem. Res., 2004, 43, 7928–7931 CrossRef CAS.
- Z. Ilham and S. Zaka, Bioresour. Technol., 2009, 100, 1793–1796 Search PubMed.
- J. Yang, D. Guo and Y. Yan, J. Mol. Catal. B: Enzym., 2007, 45, 91–96 CrossRef CAS.
- X. Li, X.-Y. He, Z.-L. Li, Y.-D. Wang, C.-Y. Wang, H. Shi and F. Wang, Fuel, 2012, 92, 89–93 CrossRef CAS.
- X. Chang, X. A. Nie, W. D. Dai and J. Zhou, Mod. Chem. Ind., 2008, 28, 117–119 Search PubMed.
- M. Kaieda, T. Samukawa, T. Matsumoto, K. Ban, A. Kondo, Y. Shimada, H. Noda, F. Nomoto, K. Ohtsuka, E. Izumoto and H. Fukuda, J. Biosci. Bioeng., 1999, 88, 627–631 CrossRef CAS.
- N. Dizge, C. Aydiner, D. Y. Imer, M. Bayramoglu, A. Tanriseven and B. Keskinler, Bioresour. Technol., 2009, 100, 1983–1991 CrossRef CAS.
- Y. Wang, H. Wu and M. H. Zong, Bioresour. Technol., 2008, 99, 7232–7237 Search PubMed.
- D. Royon, M. Daz, G. Ellenrieder and S. Loccatelli, Bioresour. Technol., 2007, 98, 648–653 CrossRef CAS.
- B. Balasubramaniam, A. S. Perumal, J. Jayaraman, J. Mani and P. Ramanujam, Integr. Waste Manage., 2012, 32, 1539–1547 Search PubMed.
- Y. Christi, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS.
- X. L. Miao and Q. Y. Wu, Bioresour. Technol., 2006, 97, 841–846 CrossRef CAS.
- L. Gouveia and A. C. Oliviera, J. Ind. Microbiol. Biotechnol., 2009, 36, 269–274 Search PubMed.
- C. Posten and G. Schaub, J. Biotechnol., 2009, 142, 64–69 CrossRef CAS.
- T. M. Mata, A. A. Martins and N. S. Caetano, Renewable Sustainable Energy Rev., 2011, 14, 841–846 Search PubMed.
- K. L. Yeh, J. S. Chang and W. M. Chen, Eng. Life Sci., 2010, 10, 201–208 Search PubMed.
- K. L. Yeh and J. S. Chang, Biotechnol. J., 2011, 6, 1–9 Search PubMed.
- L. Y. Lee, C. Yoo, S. Y. Jun, C. Y. Ahn and H. M. Oh, Bioresour. Technol., 2010, 101, S75–S77 CrossRef CAS.
- F. Rosenau, J. Tommassen and K.-E. Jaeger, ChemBioChem, 2004, 5, 152–161 Search PubMed.
- D.-T. Tran, C.-L. Chen and J.-S. Chang, J. Biotechnol., 2012, 158, 112–119 Search PubMed.
- K. Belafi-Bako, F. Kovacs, L. Gubicza and J. Hancsok, Biocatal. Biotransform., 2002, 20, 437–439 Search PubMed.
- V. Dossat, D. Combes and A. Marty, Enzyme Microb. Technol., 1999, 25, 194–200 CrossRef CAS.
- Y. Watenabe, P. Pinsirodom, T. Nagao, A. Yamaguchi, T. Kobayasji, Y. Nishida, Y. Takagi and Y. Shimada, J. Mol. Catal. B: Enzym., 2007, 44, 99–105 CrossRef CAS.
- D. G. Crine, X. Paloumet, L. Bjornsson, M. M. Alves and B. Mattiasson, Renewable Energy, 2007, 32, 965–975 Search PubMed.
- Y. Kimura, A. Tanaka, K. Sonomota, T. Nihira and S. Fukui, Eur. J. Appl. Microbiol. Biotechnol., 1983, 17, 107–112 Search PubMed.
- W. M. Linfield, R. A. Baruaskas, L. Sivieri, S. Serota and R. W. Stevenson, J. Am. Oil Chem. Soc., 1984, 61, 191–195 Search PubMed.
- K. Ramani, L. J. Kennedy, C. Vidya, R. Boopathy and G. Sekaran, J. Mol. Catal. B: Enzym., 2010, 62, 59–66 Search PubMed.
- G. Lligades, J. C. Rondes, M. Galia, U. Bierman and J. O. Metzger, J. Polym. Sci., Polym. Chem. Ed., 2000, 44, 2206–2224 Search PubMed.
- G. J. H. Buisman, Surf. Coat. Int., 1999, 82, 127–130 Search PubMed.
- S. Ahmad, S. M. Ashraf and A. Hasnat, Paintindia, 2002, 52, 47–52 Search PubMed.
- A. Adhvaryu and S. Erhan, Ind. Crops Prod., 2002, 15, 247–254 CrossRef CAS.
- B. K. Sharma, K. M. Doll and S. Z. Erhan, Green Chem., 2007, 9, 469–474 RSC.
- T. Benaniba, N. Belhaneche-Bensemra and G. Gelhard, Polym. Degrad. Stab., 2003, 82, 245–249 CrossRef CAS.
- R. Joseph, K. N. Madhusofhanan, R. Alex, S. Varghese, K. E. George and B. Kuriakose, Plast., Rubber Compos., 2004, 33, 217–222 Search PubMed.
- S. Warwel, F. Bruse, C. Demes, M. Kunz and M. Rüsch gen. Klaas, Chemosphere, 2001, 43, 39–48 CrossRef CAS.
- M. Rüsch gen. Klaas and S. Warwel, Ind. Crops Prod., 1999, 9, 125–132 CrossRef CAS.
- D. Swern, J. Am. Chem. Soc., 1947, 69, 1692–1698 CrossRef CAS.
- C. Orellana-Coca, U. Törnvall, D. Adlercreutz, B. Mattiasson and R. Hatti-Kaul, Biocatal. Biotransform., 2005, 23, 431–437 Search PubMed.
- U. Törnvall, C. Orellana-Coca, R. Hatti-Kaul and P. Adlercreutz, Enzyme Microb. Technol., 2007, 40, 447–451 CrossRef CAS.
- U. Törnvall, P. Börjesson, L. M. Tufvesson and R. Hatti-Kaul, Ind. Biotechnol., 2009, 5, 184–192 Search PubMed.
- X. Xu, Eur. J. Lipid Sci. Technol., 2003, 105, 289–304 Search PubMed.
- A. E. V. Hagström, U. Törnvall, M. Nordblad, R. Hatti-Kaul and J. M. Woodley, Biotechnol. Prog., 2011, 27, 67–76 Search PubMed.
- S. Omura, M. Katagiri, J. Awaya, T. Furukawa, I. Umezewa, N. Oi, M. Mizoguchi, B. Aoki and M. Shindo, Antimicrob. Agents Chemother., 1974, 6, 207–215 Search PubMed.
- H. Maag, J. Am. Oil Chem. Soc., 1985, 61, 259–267 Search PubMed.
- T. M. Kuo, H. Kim and C. T. Hou, Curr. Microbiol., 2001, 43, 198–203 Search PubMed.
- J. Garison, Deterg. Age, 1968, 27–29 Search PubMed.
- J. P. Chen and J. B. Wang, Enzyme Microb. Technol., 1997, 20, 615–622 Search PubMed.
- E. R. Gunawan, M. Basri, M. B. A. Rahman, A. B. Salleh and R. N. Z. A. Rahman, J. Oleo Sci., 2004, 53, 471–477 Search PubMed.
- R. Kahlscheuer, T. Stöveken, H. Lauftmann, U. Malkus, R. Reichelt and A. Steinbüchel, Appl. Environ. Microbiol., 2006, 72, 1373–1379 Search PubMed.
- N. M. Hadzir, M. Basri, M. B. A. Rahman, C. N. A. Razak, R. N. Z. A. Rahman and A. B. Salleh, J. Chem. Technol. Biotechnol., 2001, 76, 511–515 Search PubMed.
- L. Deng, X. Wang, K. Nie, F. Wang, J. Lui, P. Wang and T. Tan, Chin. J. Chem. Eng., 2011, 19, 978–982 Search PubMed.
- M. H. Guncheva and D. Zhiryakova, Biotechnol. Lett., 2008, 30, 509–512 Search PubMed.
- I. Demonty, R. T. Ras, H. C. M. van der Knaap and G. S. M. J. E. Duchateau, J. Nutr., 2009, 139, 271–284 CAS.
- Q. C. Deng, P. Zhang, Q. D. Huang, F. Wei, M. M. Zheng, X. Yu, Q. Zhou and C. Zheng, Eur. J. Lipid Sci. Technol., 2011, 113, 441–449 Search PubMed.
- N. Weber, P. Weitkamp and K. D. Mukherjee, Bioresour. Technol., 2001, 49, 67–71 Search PubMed.
- G. Torello, C. F. Torres, F. J. Senorans, R. M. Blanco and G. Reglero, J. Chem. Technol. Biotechnol., 2009, 84, 745–750 Search PubMed.
- B. L. A. P. Devi, H. Zhang, M. L. Damstrup, Z. Guo, L. Zhang, B.-M. Lue and X. Xu, Ol., Corps Gras, Lipides, 2008, 15, 189–195 Search PubMed.
- J. A. Kralovec, S. Zhang, W. Zhang and C. J. Barrow, Food Chem., 2012, 131, 639–644 Search PubMed.
- S. Claude, M. Heming and K. Hill, Lipid Tech. Newsl., 2000, 6, 105–109 Search PubMed.
- R. Garcia, M. Beson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- D. Randall and R. De Vos, Eur. Pat., EP 419114, 1991 Search PubMed.
- M. Weuthen and U. Hees, Ger. Pat., DE 4335947, 1995 Search PubMed.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529–534 RSC.
- W. Kaewthong and A.-H. Kittikun, Enzyme Microb. Technol., 2004, 35, 218–222 Search PubMed.
- U. T. Bornscheuer, Enzyme Microb. Technol., 1995, 17, 578–586 CrossRef CAS.
- M. L. Damstrup, S. Z. Kiil, A. D. Jensen, F. V. Sparsø and X. Xu, J. Agric. Food Chem., 2007, 55, 7786–7792 Search PubMed.
- M. L. Damstrup, T. Jensen, F. V. Sparsø, S. Z. Kiil, A. D. Jensen and X. Xu, J. Am. Oil Chem. Soc., 2005, 82, 559–564 CrossRef CAS.
- M. L. Damstrup, T. Jensen, F. V. Sparsø, S. Z. Kiil, A. D. Jensen and X. Xu, J. Am. Oil Chem. Soc., 2006, 83, 27–33 Search PubMed.
- W. Kaewthong, S. Sirisanseeyakul, P. Prasertsan and A. H-Kittikun, Proc. Biochem., 2005, 43, 122–130 Search PubMed.
- U. T. Bornscheuer, Enzyme Microb. Technol., 1995, 17, 578–586 CrossRef CAS.
- S. Liebminger, M. Siebenhofer and G. Guebitz, Bioresour. Technol., 2009, 100, 4541–4545 Search PubMed.
- I. W. C. E. Arends, Y. X. Li and R. Ausan, Tetrahedron Lett., 2006, 62, 6659–6665 Search PubMed.
- A. Behr, J. Eiltingh and K. Irawadi, J. Org. Chem., 2001, 66, 8650–8653 CrossRef CAS.
- P. Fordham, M. Bessom and P. Gallezot, Catal. Lett., 1997, 46, 195–199 CrossRef CAS.
- A. M. Dehkordi, M. S. Tehrany and I. Safari, Ind. Eng. Chem. Res., 2009, 48, 3271–3278 Search PubMed.
- N. M. Faqir and M. M. Attarakih, Biotechnol. Bioeng., 2002, 77, 163–173 Search PubMed.
- A. Toumi and S. Engell, Chem. Eng. Sci., 2004, 59, 3777–3792 Search PubMed.
- K. M. Vilonen, A. Vuolanto, J. Jokela, M. S. A. Leisola and A. O. I. Krause, Biotechnol. Prog., 2004, 20, 1555–1560 CrossRef CAS.
- A. Boisen, T. B. Christensen, W. Fu, Y. Y. Gorbanev, T. S. Hansen, J. S. Jensen, S. K. Klitgaard, S. Pedersen, A. Riisager, T. Ståhlberg and J. M. Woodley, Chem. Eng. Res. Des., 2009, 8, 1318–1327 Search PubMed.
- H. Tumturk, G. Demirel, H. Altinok, S. Aksoy and N. Hasirci, J. Food Biochem., 2008, 32, 234–246 CrossRef CAS.
- W. P. Chen and A. W. Anderson, Appl. Environ. Microbiol., 1979, 38, 1111–1119 Search PubMed.
- M. Rhimi, E. B. Messaud, M. A. Borgi, K. B. Khadra and S. Bejar, Enzyme Microb. Technol., 2007, 40, 1531–1537 CrossRef CAS.
- G. A. Kovalenko, L. V. Perminova, T. G. Terent'eva, L. I. Sapunova, A. G. Lobanok, T. V. Chuenko, N. A. Rudina and E. I. Chernyak, Appl. Biochem. Microbiol., 2008, 44, 174–181 Search PubMed.
- L. V. Perminova, G. A. Kovalenko, N. A. Rudina, L. I. Sapunova, I. O. Tamkovic and A. G. Lobanok, Appl. Biochem. Microbiol., 2009, 45, 389–394 Search PubMed.
- E. Katchalski-Katzir and D. M. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef.
- H. Yu, Y. Guo, D. Wu, W. Zhan and G. Lu, J. Mol. Catal. B: Enzym., 2011, 72, 73–76 Search PubMed.
- D. L. Wu, Q. L. Zhao, Y. L. Guo, Y. Wang, Y. S. Wang, W. C. Zhan and G. Z. Lu, Chin. J. Catal., 2010, 31, 586–590 Search PubMed.
- W. Mu, W. Zhang, Y. Feng, B. Jiang and L. Zhou, Appl. Microbiol. Biotechnol., 2012, 94, 1461–1467 Search PubMed.
- Y. H. Hong, WO040708, 2011.
- B.-C. Lim, H.-J. Kim and D.-K. Oh, Proc. Biochem., 2009, 44, 822–828 Search PubMed.
- R. D. Schmidt and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608–1633 CrossRef.
- S. Nakamura, Oleochemicals, 1997, 8, 866–874 Search PubMed.
- T. Watanabe, Foods Food Ingredients J. Jpn., 1999, 180, 18–25 Search PubMed.
- N. S. Neta, A. M. Peres, J. A. Teixeira and L. R. Rodrigues, New Biotechnol., 2011, 28, 349–355 Search PubMed.
- S. Sabader, M. Habulin and Z. Knez, J. Food Eng., 2006, 77, 880–886 Search PubMed.
- R. J. Barros, P. G. V. Moura-Pinto, M. C. R. Franssen, A. E. M. Janssen and Ae. de Groot, Bioorg. Med. Chem., 1994, 2, 707–713 Search PubMed.
- A. Adnani, M. Basri, E. A. Malek, A. B. Salleh, M. B. Abdul Rahman, N. Chaibakhsh and R. N. R. A. Rahman, Ind. Crops Prod., 2010, 31, 350–356 Search PubMed.
- T. Polat and R. J. Linhardt, J. Surfactants Deterg., 2001, 4, 415–421 Search PubMed.
- A. Bernhaus, M. Fritzer-Szekeres, M. Grusch, P. Saiko, G. Krupitza, S. Venkateswarlu, G. Trimurtulu, W. Jaeger and T. Szekeres, Cancer Lett., 2009, 274, 299–304 Search PubMed.
- Y. H. P. Zhang, M. E. Himmel and J. R. Mielenz, Biotechnol. Adv., 2006, 24, 452–481 CrossRef.
- L. Barrato, A. Candido, M. Marzorati, F. Sagui, S. Riva and B. Danieli, J. Mol. Catal. B: Enzym., 2006, 39, 3–8 Search PubMed.
- J. I. Clark and T. N. Margulis, Life Sci., 1980, 26, 833–836 Search PubMed.
- J. H. Chen and C. T. Ho, J. Agric. Food Chem., 1997, 45, 2374–2378 CrossRef CAS.
- N. El Boulifi, J. Aricil and M. Martinez, Bioresour. Technol., 2010, 101, 8520–8525 Search PubMed.
- G. Fleche and M. Huchette, Starch/Staerke, 1986, 38, 26–30 Search PubMed.
- A. Aracil, M. Martinez and N. E. Boulifi, Spanish patent P200801042, 2008.
- C. Ceccuti, Z. Mouloungui and A. Gaset, Bioresour. Technol., 1998, 66, 63–67 CrossRef CAS.
- P. J. Halling, Enzyme Microb. Technol., 1994, 16, 178–206 CrossRef CAS.
- G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for Experimenters: Design, Innovation, and Discovery, John Wiley and Sons, New York, 2nd edn, 1978, ch. 12, pp. 489–530 Search PubMed.
- K. Iwasaki, M. Nakajima and S. Nakao, Proc. Biochem., 1996, 31, 69–76 Search PubMed.
- N. Albayrak and S. T. Yang, Biotechnol. Bioeng., 2002, 77, 8–19 Search PubMed.
- K. Esakkiyappan, S. Shatabhisa and S. S. Rekha, Trends Carbohydr. Res., 2011, 3, 1–16 Search PubMed.
- D. P. M. Torres, M. dP. F. Gonçalves, J. A. Teixeira and L. R. Rodrigues, Compr. Rev. Food Sci. Food Saf., 2010, 9, 471–482 Search PubMed.
- J. R. Bourne, M. Hegglin and J. E. Prenosil, Biotechnol. Bioeng., 1983, 25, 1625–1639 Search PubMed.
- L. Engel, P. Schneider, M. Ebrahimi and P. Czermak, Open Food Sci. J., 2007, 1, 17–23 Search PubMed.
- L. Engel, M. Ebrahimi and P. Czermak, Desalination, 2008, 224, 46–51 Search PubMed.
- D. Sen, A. Sarkar, S. Das, R. Chowdhurry and C. Bhattacharjee, Ind. Eng. Chem. Res., 2012, 51, 10671–10681 Search PubMed.
- F. H. Isgrove, R. J. H. Williams, G. W. Niven and A. T. Andrews, Enzyme Microb. Technol., 2001, 28, 225232 Search PubMed.
- J. Zemek, L. Kuniak, P. Gemeiner, J. Zamocky and S. Kucar, Enzyme Microb. Technol., 1982, 4, 233238 Search PubMed.
- A. Karmali, EP2210953, 2009.
- P. Sukyai, T. Rezic, C. Lorenz, K. Mueangtoom, W. Lorenz, D. Haltrich and R. Ludwig, J. Biotechnol., 2008, 135, 281–290 CrossRef CAS.
- R. Branden, B. G. Malmstrom and T. Vanngard, Eur. J. Biochem., 1971, 18, 238–241 Search PubMed.
- P. Chandrakant and V. S. Bisaria, Crit. Rev. Biotechnol., 1998, 18, 295–331 Search PubMed.
- M. Dashtban, H. Schraft and W. Qin, Int. J. Biol. Sci., 2009, 6, 578–595 Search PubMed.
- Y. H. P. Zhang, M. E. Himmel and J. R. Mielenz, Biotechnol. Adv., 2006, 24, 452–481 CrossRef.
- X. Shen and L. Xia, Appl. Biochem. Biotechnol., 2006, 133, 251–262 Search PubMed.
- J. S. Tolan and B. Foody, Adv. Biochem. Eng./Biotechnol., 1999, 65, 41–67 Search PubMed.
- B. Nidetzky, S. Steiner, M. Hayn and M. Claeyssens, Biochem. J., 1994, 298, 705–710 CAS.
- B. C. Stockton, D. J. Mitchell and K. Gromann, Biotechnol. Lett., 1991, 13, 57–62 Search PubMed.
- L. Xia and X. Shen, Bioresour. Technol., 2004, 91, 259–262 Search PubMed.
- H. Esterbauer, W. Steiner and I. Labudova, Bioresour. Technol., 1991, 36, 51–65 Search PubMed.
- X. Shen and L. Xia, Process Biotechnol., 2004, 39, 1363–1367 Search PubMed.
- B. Kamm, P. R. Gruber and M. Kamm, Biorefineries – Industrial processes and products, Wiley, Weinheim, 2006, ch. 2, pp. 61–90 Search PubMed.
- N. F. Olson and T. Richardson, J. Food Sci., 1974, 39, 653–659 Search PubMed.
- P. J. Reilly, in Immobilized Enzymes for Food Processing, ed. W. H. Pitcher Jr., CRC Press, Boca Raton, Florida, USA, 1980, pp. 113–151 Search PubMed.
- M. G. Bellino, A. E. Regazzoni and G. J. A. A. Soler-Illia, ACS Appl. Mater. Interfaces, 2010, 2, 360–365 Search PubMed.
- V. Singh and S. Ahmad, Cellulose, 2012, 19, 1759–1769 Search PubMed.
- A. Kara, B. Osman, H. Yavuz, N. Beşirli and A. Denizli, React. Funct. Polym., 2005, 62, 61–68 Search PubMed.
- M.-Y. Chang and R.-S. Juang, Enzyme Microb. Technol., 2005, 36, 75–82 Search PubMed.
- M. J. Khan, Q. Husain and A. Azam, Biotechnol. Bioprocess. Eng., 2012, 17, 377–384 Search PubMed.
- D. A. Uygun, N. Öztürk, S. Akgöl and A. Denizli, J. Appl. Polym. Sci., 2012, 123, 2574–2581 Search PubMed.
- M.-H. Lee, J. L. Thomas, Y.-C. Chen, H.-Y. Wang and H.-Y. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 916–921 Search PubMed.
- G.-M. Qiu, B.-K. Zhu and Y.-Y. Xu, J. Appl. Polym. Sci., 2005, 95, 328–335 Search PubMed.
- A. K. Mukherjee, T. S. Kumar, S. K. Rai and J. K. Roy, Biotechnol. Bioprocess Eng., 2010, 15, 984–992 Search PubMed.
- S. D. Shewale and A. B. Pandit, Carbohydr. Res., 2007, 342, 997–1008 Search PubMed.
- N. Hasirci, S. Aksoy and H. Tumturk, React. Funct. Polym., 2006, 66, 1546–1551 CrossRef CAS.
- D. Park, S. Haam, K. Jang, I.-S. Ahn and W.-S. Kim, Proc. Biochem., 2005, 40, 53–61 Search PubMed.
- G. Sanjay and S. Sugunan, J. Porous Mater., 2008, 15, 359–367 Search PubMed.
- M. V. Kahraman, G. Bayramoğlu, N. Kayaman-Apohan and A. Güngör, React. Funct. Polym., 2007, 67, 97–103 Search PubMed.
- M. A. A. El-Ghaffar and M. S. Hashem, J. Appl. Polym. Sci., 2009, 112, 805–814 Search PubMed.
- P. C. Ashly, M. J. Joseph and P. V. Mohanan, Food Chem., 2011, 127, 1808–1813 Search PubMed.
- R. L. C. Giordano, J. Trovati and W. Schmidell, Appl. Biochem. Biotechnol., 2008, 147, 47–61 Search PubMed.
- H.-W. Wang, I. H. Kim, C.-S. Park and J.-H. Lee, Korean J. Chem. Eng., 2008, 25, 801–803 Search PubMed.
- T. N. Nwagu, H. Aoyagi, B. N. Okolo and S. Yoshida, J. Mol. Catal. B: Enzym., 2012, 78, 1–8 Search PubMed.
- R. S. S. Kumar, K. S. Vishwanath, S. A. Singh and A. G. A. Rao, Proc. Biochem., 2006, 41, 2282–2288 Search PubMed.
- Z. Konsoula and M. Liakopoulou-Kyriakides, Proc. Biochem., 2006, 41, 343–349 Search PubMed.
- P. M. Könst, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, J. Biobased Mater. Bioenergy, 2011, 5, 102–108 Search PubMed.
- L. Goldstein, Biochim. Biophys. Acta, 1973, 327, 132–137 Search PubMed.
- M. Cheryan, P. J. van Wyk, T. Richardson and N. F. Olsen, Biotechnol. Bioeng., 1976, 18, 273–279 Search PubMed.
- M. J. Taylor, M. Cheryan, T. Richardson and N. F. Olsen, Biotechnol. Bioeng., 1977, 19, 683–700 Search PubMed.
- J. C. Høj, D. B. Tattersall, K. Adams, K. F. Pocok, Y. Hayasakka, R. Van Heeswijck and E. J. Waters, Proceeding of the ASEV 50th Anniversary Annual Meeting, Seattle, WA 149–154, 2000.
- K. Liburdi, I. Benucci and M. Esti, Food Biotechnol., 2010, 24, 282–292 Search PubMed.
- L. Bin and L. Bin, CN 102138588 A 20110803, 2011.
- H. Bai, S.-H. Ge and L.-X. Zhang, Int. J. Food Sci. Technol., 1999, 34, 95–99 Search PubMed.
- T. Takamatsu and T. Tosa, Bioprocess Technol., 1993, 16, 25–35 Search PubMed.
- T. Takamatsu, T. Tosa and I. Chibata, J. Chem. Eng. Jpn., 1986, 19, 31–36 Search PubMed.
- A. Ben-Bassat, F. S. Sariaslani, L. L. Huang, R. Patnaik and D. J. Lowe, WO20050260724 A1, 2005.
- K. Ogata, K. Uchiyama and H. Yamada, Agric. Biol. Chem., 1966, 30, 311–312 Search PubMed.
- A. I. El-Batal, Acta Microbiol. Pol., 2002, 51, 153–169 Search PubMed.
- G. B. D'Cunha, V. Satyanarayna and P. M. Nair, Enzyme Microb. Technol., 1996, 19, 421–427 Search PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2010, 12, 1430–1436 RSC.
- T. M. Lammens, J. Potting, J. P. M. Sanders and I. J. M. de Boer, Environ. Sci. Technol., 2011, 45, 8521–8528 Search PubMed.
- Q. Wang, Y. Xin, F. Zhang, Z. Feng, J. Fu, L. Luo and Z. Yin, World J. Microbiol. Biotechnol., 2011, 27, 693–700 Search PubMed.
- H. Park, J. Ahn, J. Lee, H. Lee, C. Kim, J.-K. Jung, H. Lee and E. G. Lee, Int. J. Mol. Sci., 2012, 13, 358–369 Search PubMed.
- P. M. Könst, Production of nitrogen containing chemicals from cyanophycin, PhD thesis, Wageningen University, 2011 Search PubMed.
- C. Zhang, C. Liu, X. Yue, J. Sun and N. Chang, Farming Zhuanli Shenqing, CN 102010859 A 20110412, 2011 Search PubMed.
- Y. Hu, T. Tang, W. Yang and H. Zhou, Process Biochem., 2009, 44, 142–145 Search PubMed.
- A. R. Martin-Garcia, D. R. Shonnard and S. Pannuri, AIChE Annual Meeting, Conference Proceeding, San Francisco, United States, Nov 12–17, 2006.
- M. D. Truppo, H. Strotman and G. Hughes, ChemCatChem, 2012, 4, 1071–1074 Search PubMed.
- A. Schulz, P. Taggeselle, D. Tripier and K. Bartsch, Appl. Environ. Microbiol., 1990, 56, 1–6 Search PubMed.
- C. U. Ingram, M. Bommer, M. E. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. Y. Lye, Biotechnol. Bioeng., 2007, 96, 559–569 CrossRef CAS.
- S. Matosevic and G. J. Lye, J. Biotechnol., 2011, 155, 320–329 Search PubMed.
- K. Ni, X. Zhao, L. Zhao, H. Wang, Y. Ren and D. Wei, PLoS One, 2012, 7, e41101 Search PubMed.
- J. W. Kim, S. K. Shin, H. Y. Hwang, S. Y. Choi and J. C. Yun, PCT Int. Appl., WO2003031610 A1 20030417, 2003 Search PubMed.
- X. Zhang, S. Cao and Z. Mo, Zhongguo Yiyao Gongye Zazhi, 2007, 37, 846–848 Search PubMed.
- J. W. Frost, Synthesis of Caprolactam from Lysine, WO2005123669, 2005 Search PubMed.
- A. Pukin, C. G. Boeriu, E. L. Scott, J. P. M. Sanders and M. C. R. Franssen, J. Mol. Catal. B: Enzym., 2010, 65, 58–62 Search PubMed.
- S. Aguila, R. Vazquez-Duhalt, C. Covarrubias, G. Pecchi and J. B. Alderete, J. Mol. Catal. B: Enzym., 2011, 70, 81–87 Search PubMed.
- A. But, J. E. L. Le Notre, E. L. Scott, R. Wever and J. P. M. Sanders, ChemSusChem, 2012, 5, 1199–1202 Search PubMed.
- E. L. Scott, F. Peter and J. P. M. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS.
- R. Medici, P. Dominguez de Maria, L. G. Otten and A. J. J. Straathof, Adv. Synth. Catal., 2011, 353, 2369–2376 Search PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biomass Bioenergy, 2012, 44, 168–181 Search PubMed.
- K. M. Frey, F. B. Oppermann-Sanio, H. Schmidt and A. Steinbüchel, Appl. Environ. Microbiol., 2002, 68, 3377–3384 CrossRef CAS.
- Y. Elbahloul, K. Frey, J. P. M. Sanders and A. Steinbüchel, Appl. Environ. Microbiol., 2005, 71, 7759–7767 CrossRef CAS.
- W. Dahlig, Przem. Chem., 1955, 11, 518–520 Search PubMed.
- W. J. Middelhoven and M. D. Sollewijn Gelpke, Antonie van Leeuwenhoek, 1995, 67, 217–219 Search PubMed.
- M. Takemoto and K. Achiwa, Chem. Pharm. Bull., 2001, 49, 639–641 Search PubMed.
- F. S. Sariaslani, Annu. Rev. Microbiol., 2007, 61, 51–69 Search PubMed.
- Y. Teng, E. L. Scott and J. P. M. Sanders, in preparation.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- D. W. S. Wong, Appl. Biochem. Biotechnol., 2009, 157, 174–209 CrossRef.
- A. T. Martínez, F. J. Ruiz-Dueñas, M. J. Martínez, J. C. del Río and A. Gutiérrez, Curr. Opin. Biotechnol., 2009, 20, 348–357 Search PubMed.
- M. Hofrichter, Enzyme Microb. Technol., 2002, 30, 454–466 CrossRef.
- E. Chiacchierini, D. Restuccia and G. Vinci, Food Sci. Technol. Int., 2004, 10, 373–382 Search PubMed.
- H. Li, S. Liu, Z. Dai, J. Bao and X. Yang, Sensors, 2009, 9, 8547–8561 CrossRef CAS.
- Q. Husain and R. Ulber, Crit. Rev. Environ. Sci. Technol., 2011, 41, 770–804 Search PubMed.
- F.-D. Munteanu, F. J. Ruiz-Dueñas, A. T. Martínez and A. Cavaco-Paulo, J. Electroanal. Chem., 2005, 580, 35–40 Search PubMed.
- C. Crestini, F. Melone and R. Saladino, Bioorg. Med. Chem., 2011, 19, 5071–5078 Search PubMed.
- S. C. Corgie, P. Kahawong, E. Giannelis and L. P. Walker, WO2012122437, 2012.
- D. Areskogh and G. Henriksson, Proc. Biochem., 2011, 46, 1071–1075 Search PubMed.
- M. S. Fawer, J. Stierli, S. Cliffe and A. Fiechter, Biochim. Biophys. Acta, 1991, 1076, 15–22 Search PubMed.
- T. L. Presnell, H. E. Swaisgood, T. W. Joyce and H. Chang, J. Biotechnol., 1994, 35, 77–85 Search PubMed.
- M. Asther and J.-C. Meunier, Appl. Biochem. Biotechnol., 1993, 38, 57–67 Search PubMed.
- H. Takahashi and C. Miyazaki-Imamura, e-J. Surf. Sci. Nanotechnol., 2005, 3, 207–212 Search PubMed.
- F. Acevedo, L. Pizzul, M. dP. Castillo, M. E. González, M. Cea, L. Gianfreda and M. C. Diez, Chemosphere, 2010, 80, 271–278 Search PubMed.
- A. C. Grabski, P. L. Coleman, G. J. Drtina and R. R. Burgess, Appl. Biochem. Biotechnol., 1995, 55, 55–73 Search PubMed.
- A. C. Grabski, J. K. Rasmussen, P. L. Coleman and R. R. Burgess, Appl. Biochem. Biotechnol., 1996, 60, 1–17 Search PubMed.
- A. C. Grabski, H. J. Grimek and R. R. Burgess, Biotechnol. Bioeng., 1998, 60, 204–215 Search PubMed.
- B. Van Aken, P. Ledent, H. Naveau and S. N. Agathos, Biotechnol. Lett., 2000, 22, 641–646 Search PubMed.
- I. Mielgo, C. Palma, J. M. Guisan, R. Fernandez-Lafuente, M. T. Moreira, G. Feijoo and J. M. Lema, Enzyme Microb. Technol., 2003, 32, 769–775 Search PubMed.
- C. López, M. T. Moreira, G. Feijoo and J. M. Lema, Biocatal. Biotransform., 2011, 29, 344–353 Search PubMed.
- N. Nady, K. Schroën, M. C. R. Franssen, B. van Lagen, S. Murali and R. M. Boom, ACS Appl. Mater. Interfaces, 2011, 3, 801–810 Search PubMed.
- N. Nady, K. Schroën, M. C. R. Franssen, M. S. Mohy Eldin, H. Zuilhof and R. M. Boom, J. Membr. Sci., 2012, 394–395, 69–79 Search PubMed.
- See for a review on laccase: N. Durán, M. A. Rosa, A. D'Annibale and L. Gianfreda, Enzyme Microb. Technol., 2002, 31, 907–931 Search PubMed.
- For a review on tyrosinase: L. Cordi, R. C. Minussi, R. S. Freire and N. Durán, Afr. J. Biotechnol., 2007, 6, 1255–1259 Search PubMed.
- M. Fernández-Fernández, M. Á. Sanromán and D. Moldes, Biotechnol. Adv., 2012 DOI:10.1016/j.biotechadv.2012.02.013.
- S. G. Burton, Catal. Today, 1994, 22, 459–487 CrossRef CAS.
- A. A. Leontievsky, N. M. Myasoedova, B. P. Baskunov, L. A. Golovleva, C. Bucke and C. S. Evans, Appl. Microbiol. Biotechnol., 2001, 57, 85–91 Search PubMed.
- M. Solís-Oba, V. M. Ugalde-Saldívar, I. González and G. Viniegra-González, J. Electroanal. Chem., 2005, 579, 59–66 Search PubMed.
- A. Boshoff, W. Edwards, W. D. Leukes, P. D. Rose and S. G. Burton, Desalination, 1998, 115, 307–312 CrossRef CAS.
- S. G. Burton, A. Boshoff, W. Edwards and P. D. Rose, J. Mol. Catal. B: Enzym., 1998, 5, 411–416 Search PubMed.
- M. E. Marín-Zamora, F. Rojas-Melgarejo, F. García-Cánovas and P. A. García-Ruiz, J. Agric. Food Chem., 2007, 55, 4569–4575 Search PubMed.
- M. E. Marín-Zamora, F. Rojas-Melgarejo, F. García-Cánovas and P. A. García-Ruiz, J. Biotechnol., 2009, 139, 163–168 CrossRef CAS.
- Y.-Y. Wan, R. Lub, K. Akiyama, K. Okamoto, T. Honda, Y.-M. Du, T. Yoshida, T. Miyakoshi, C. J. Knill and J. F. Kennedy, Int. J. Biol. Macromol., 2010, 47, 76–81 Search PubMed.
- K. W. Wellington, P. Steenkamp and D. Brady, Bioorg. Med. Chem., 2010, 18, 1406–1414 CrossRef CAS.
- K. Sharma, S. S. Bari and H. P. Singh, J. Mol. Catal. B: Enzym., 2009, 56, 253–258 Search PubMed.
- P. Brandi, A. D'Annibale, C. Galli, P. Gentili and A. S. N. Pontes, J. Mol. Catal. B: Enzym., 2006, 41, 61–69 Search PubMed.
- J. Osiadacz, A. J. H. Al-Adhami, D. Bajraszewska, P. Fischer and W. Peczyñska-Czoch, J. Biotechnol., 1999, 72, 141–149 CrossRef CAS.
- Y. Y. Wan, Y. M. Du and T. Miyakoshi, Chin. Chem. Lett., 2008, 19, 333–336 Search PubMed.
- G. Fontana, E. Baldelli, S. Riva and B. Danieli, WO2009153025, 2009.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS , and papers cited therein.
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, Bio-based Chemicals, Value Added Products from Biorefineries, Report on behalf of IEA Bioenergy Task 42, downloadable at http://www.iea-bioenergy.task42-biorefineries.com/publications/reports, 2012.
- M. Ajioka, K. Enomoto, K. Suzuki and A. Yamaguchi, J. Environ. Polym. Degrad., 1995, 3, 225–234 Search PubMed.
- J. Tuominen, J. Kylma, J. Kapanen, O. Venelampi, M. Itavaari and J. Seppala, Biomacromolecules, 2002, 3, 445–455 Search PubMed.
- E. T. H. Vink, K. R. Rabago, D. A. Glassner and P. R. Gruber, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef CAS.
- K. D. Newman and M. W. McBurney, Biomaterials, 2004, 25, 5763–5771 Search PubMed.
- A. Idris and A. Bukhari, Biotechnol. Adv., 2012, 30, 550–563 Search PubMed.
- D. I. Habeych, P. B. Juhl, J. Pleiss, D. Vanegas, G. Eggink and C. G. Boeriu, J. Mol. Catal. B: Enzym., 2011, 71, 1–9 CrossRef CAS.
- G. Li and Q. Li, Biotechnol. Bioprocess Eng., 2011, 16, 1201–1207 Search PubMed.
- A. Tanaka, M. Kohri, T. Takiguchi, M. Kato and S. Matsumura, Polym. Degrad. Stab., 2012, 97, 1415–1422 Search PubMed.
- http://www.bccresearch.com/report/enzymes-industrial-applications-markets-bio030g.html?tab = highlight&highlightKeyword = enzyme + market accessed 21 November 2012.
- J. W. Won, K. M. Park, S. J. Choi and P.-S. Chang, Eur. Food Res. Technol., 2012, 235, 647–657 Search PubMed.
- L. O. Ming, H. M. Ghazali and C. C. Let, Food Chem., 1999, 64, 83–88 Search PubMed.
- T. H. Ronne, L. S. Pedersen and X. Xu, J. Am. Oil Chem. Soc., 2005, 82, 737–743 Search PubMed.
- W. J. Quax, N. T. Mrabet, R. G. M. Luiten, P. W. Schuurhuizen, P. Stanssens and I. Lasters, Nat. Biotechnol., 1991, 9, 738–742 Search PubMed.
- P. Tufvesson, W. Fu, J. S. Jensen and J. M. Woodley, Food Bioprod. Process., 2010, 88, 3–11 Search PubMed.
- R. Schoevaart, personal communication.
- D. J. Rozzell, Bioorg. Med. Chem., 1999, 7, 2253–2261 CrossRef CAS.
- B.-Y. Kim and H.-H. Hyun, Biotechnol. Bioprocess Eng., 2002, 7, 194–200 Search PubMed.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS.
- J. Jenkins, J. Janin, F. Rey, M. Chiadmi, H. Van Tilbeurgh, I. Lasters, M. De Maeyer, D. Ven Belle and S. J. Wodak, Biochemistry, 1992, 31, 5449–5458 CrossRef CAS.
- M. Ferrer, O. Golyshina, A. Beloqui and P. N. Golyshin, Curr. Opin. Microbiol., 2007, 10, 207–214 CrossRef CAS.
- L. Betancor and H. Luckarift, Biotechnol. Genet. Eng. Rev., 2010, 27, 95–114 Search PubMed.
- T. M. Lammens, J. Le Nôtre, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, ChemSusChem, 2011, 4, 785–791 CrossRef CAS.
- J. Le Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, Green Chem., 2011, 13, 807–809 RSC.
- F. Lopez-Gallego and C. Schmidt-Dannert, Curr. Opin. Chem. Biol., 2010, 14, 174–183 CrossRef CAS.
- M. Suekane, Z. Allg. Mikrobiol., 1982, 22, 565–576 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |