 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and post-polymerisation modifications of aliphatic poly(carbonate)s prepared by ring-opening polymerisation
Sarah
Tempelaar
a,
Laetitia
Mespouille
b,
Olivier
Coulembier
b,
Philippe
Dubois
b and
Andrew P.
Dove
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 4107
bCenter of Innovation and Research in Materials and Polymers (CIRMAP), Laboratory of Polymeric and Composite Materials, University of Mons, Place du Parc 23, 7000 Mons, Belgium. E-mail: philippe.dubois@umons.ac.be; Fax: +32 (0)65 373484; Tel: +32 (0)65 373480
First published on 15th November 2012
Abstract
Owing to their low toxicity, biocompatibility and biodegradability, aliphatic poly(carbonate)s have been widely studied as materials for biomedical application. Furthermore, the synthetic versatility of the six-membered cyclic carbonates for the realization of functional degradable polymers by ring-opening polymerisation has driven wider interest in this area. In this review, the synthesis and ring-opening polymerisation of functional cyclic carbonates that have been reported in the literature in the past decade are discussed. Finally, the post-polymerisation modification methods that have been applied to the resulting homopolymers and copolymers and the application of the materials are also discussed.
 Sarah Tempelaar | Sarah Tempelaar graduated from Utrecht University (The Netherlands) with a MSc in chemistry in 2008. During her studies she worked for six months in the laboratory of Prof. Dr Iztok Turel to study the interaction of metal ions with quinolone compounds and completed a research thesis under the supervision of Prof. Dr R. (Bert) J. M. Klein Gebbink and Prof. Dr Gerard van Koten on the synthesis of propargylamines via the aminoarenethiolato copper(I)-catalysed Mannich reaction. She then went to the University of Warwick (UK) to undertake a PhD in 2009 where she studied the synthesis and post-polymerisation functionalisations of aliphatic poly(carbonate)s under the supervision of Dr Andrew P. Dove. Sarah completed her PhD in 2012. |
 Laetitia Mespouille | Laetitia Mespouille received her MSc in chemistry in 2003 from the University of Mons (UMONS) in Belgium. She then obtained a PhD in 2007 from the same University under the supervision of Professor Ph. Dubois where she studied the preparation of controlled polymer conetworks by ATRP. During this period she performed a short stay in the laboratory of Dr James Hedrick at IBM Research Center (San Jose, CA) and in the laboratory of Professor Curt Franck (Chemical engineering department) from Stanford University. She then started a post-doctoral stay at IBM Almaden Research Center supervised by Dr J. L. Hedrick, focused on the preparation of functional cyclic carbonates and their metal-free ring-opening polymerization. She then returned to Belgium where she was appointed as an Assistant Professor at the University of Mons in 2008. |
 Olivier Coulembier | Olivier Coulembier received his PhD degree from University of Mons-Hainaut (Belgium) in 2005 and joined the University of Stanford (United States) for his postdoctoral research in 2006. He is currently Research Associate by Belgian F.N.R.S. in the Laboratory of Polymeric and Composite Materials (LPCM) of Professor Philippe Dubois at University of Mons. His main activities are focused on the (non)organometallic ring-opening polymerization of cyclic monomers. He has (co)authored 56 scientific papers in international journals, 2 patents and 2 book chapters. |
 Philippe Dubois | Philippe Dubois, PhD in sciences, is full professor at University of Mons (UMONS, Belgium) and honorary senior research associate of the Belgian FNRS. He is currently director of the Center of Innovation and Research in Materials and Polymers (CIRMAP, ca. 150 persons). Prof. Dubois is Scientific Director at Materia Nova ASBL Research Center in Mons and Past-President of the Belgian Royal Chemical Society. His expertise covers organic chemistry, macromolecular chemistry, catalysis in polymer materials, melt (reactive) processing/engineering of nanocomposite and nanohybrid materials, incl. biodegradable and biosourced polymers. He has (co)authored more than 450 peer-reviewed scientific publications (h index: 57) and is inventor of 57 patents. Ph. Dubois is elected titular member of the Royal Academy of Sciences, Belgium. |
 Andrew P. Dove | Andrew P. Dove is an Associate Professor of Chemistry at the University of Warwick (UK). He graduated from the University of York with an MChem (Hons) degree in Chemistry in 1999 before studying for his PhD under the supervision of Prof. Vernon C. Gibson at Imperial College London, graduating in 2003. Andrew then moved to Stanford University, California and subsequently IBM Almaden research centre to undertake periods of postdoctoral research before returning to the UK in 2005. His current research is focused at the interface of chemistry, engineering and cell biology on the synthesis and application of degradable biomaterials. He has (co)authored over 40 scientific papers, patents and book chapters. |
Introduction
Alongside aliphatic poly(ester)s, aliphatic poly(carbonate)s are important biodegradable materials that have received considerable attention.1–3 These materials are excellent candidates for pharmaceutical applications as a consequence of their low toxicity, biocompatibility, and biodegradability. Copolymers of carbonates such as trimethylene carbonate (TMC) with other cyclic monomers such as lactide and glycolide have already found application as sutures and in other biomedical fields such as controlled drug delivery on account of the favourable materials properties that they bestow into the copolymers.4,5 In addition, materials prepared from poly(trimethylene carbonate) (PTMC) are known to degrade in vivo by surface erosion in contrast to the bulk degradation behaviour shown by poly(ester)s. Furthermore, poly(ester) degradation products can lead to increased levels of acidity, which are not present during poly(carbonate) degradation, that may be hazardous to loaded drugs or living tissues.6–9Poly(carbonate) synthesis is commonly realised either by the copolymerisation of epoxides with carbon dioxide or by the ring-opening polymerisation (ROP) of cyclic carbonate monomers.1,3,10–13 The synthesis of poly(carbonate)s via CO2/epoxide copolymerisation is a potentially useful method of utilising CO2; however, the formation of five-membered cyclic carbonate by-products, the presence of ether linkages, and the use of air-sensitive coordination compounds make its application synthetically challenging. The ROP of cyclic carbonates has received significantly more attention and consequently provides an attractive and versatile methodology for the synthesis of functionalised poly(carbonate)s, despite the requirement to synthesise many of the monomers, and has been studied by many groups.1,3,14 The ROP of cyclic carbonates can be realised using cationic, anionic, coordination-insertion, organocatalytic, and enzymatic methods and has resulted in high levels of control over polymer molecular weight, dispersity and end-group fidelity.15–24
For their use in biomedical applications, such as targeted drug delivery and tissue engineering, it is advantageous to be able to tailor the poly(carbonate) structure to enable specific interactions with cells or organs. The properties of poly(carbonate)s derived from TMC can be tuned by copolymerisation and blending with other biodegradable polymers (i.e. poly(lactide), poly(glycolide), poly(caprolactone)) and the introduction of functional end-groups.25–32 Precise control over the physical properties or the addition of biologically active molecules to the poly(carbonate) structure however, is best achieved by post-derivatisation of pendant functionalities in the polymer backbone. Such pendant functionalities may be introduced via the ROP of cyclic carbonate monomers bearing the desired functionality however, not all functional groups are compatible with the ring-opening polymerisation process and in some cases the added functionality limits the polymerisation efficiency.33 As a result, further modification of the polymer backbone following polymerisation is often required. While many further functionalisations of the pendant moieties in the poly(carbonate) backbone have been reported, modifications commonly involve many steps and typically only partial functionalisation is achieved. In recent years, however, post-polymerisation modifications of functional poly(carbonate)s using more efficient chemistries have been reported. In addition, many copolymerisations of functional cyclic carbonate monomers with simple cyclic esters (i.e. lactide or ε-caprolactone) have been reported to introduce functionality in the parent poly(ester) backbone in order to overcome the challenging nature of functional cyclic ester synthesis.34–36
Herein the synthesis and polymerisation of cyclic carbonates reported in the literature including post-polymerisation modifications of the resulting homo- and co-polymers are discussed with a focus on progress made in the area in the past decade.
Synthetic routes to functional poly(carbonate)s
Synthesis of cyclic carbonates can be achieved through a number of routes, commonly involving multiple steps using toxic phosgene derivatives for carbonate formation from a 1,3-diol (Fig. 1).1,3 Alternative methods that do not involve the use of phosgene include the reaction of oxetane with CO2, a two-step procedure using a pentafluorophenyl ester intermediate and the use of 1,1′-carbonyldiimidazole (CDI).37,38 This section of this review article will focus on the specific methods for monomer synthesis centred on the most common feedstocks.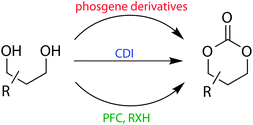 | ||
| Fig. 1 Representative scheme for the synthesis of functional six-membered cyclic carbonates from 1,3-diols using phosgene derivatives, 1,1′-carbonyldiimidazole (CDI) or bis(pentafluorophenyl)carbonate (PFC) with an alcohol or amine (RXH). | ||
Functional cyclic carbonate monomers derived from 2,2-bishydroxy(methyl)propionic acid (bis-MPA)
2,2-Bishydroxy(methyl)propionic acid (bis-MPA), also widely used in the preparation of biocompatible dendrimers,39,40 and hydrogel materials,41 was first used for the synthesis of cyclic carbonates by Bisht and coworkers in 1999.42 The reported cyclic carbonate could be prepared by direct functionalisation of the acid group in bis-MPA under either acidic or basic conditions (Scheme 1A), before cyclisation to the carbonate using phosgene or a phosgene derivative such as ethyl chloroformate, di- or triphosgene.42,43 Other possible synthetic pathways from the bis-MPA scaffold to provide functional cyclic carbonates have since been reported. Alternative pathways (Scheme 1, route B and C) are more suitable for the synthesis of monomers that have more sensitive substituents. In one example (Scheme 1, route B), cyclic carbonate synthesis involves protection of the acid functionality by conversion to a benzyl ester followed by the synthesis of benzyl functional cyclic carbonate, 5-methyl-5-benzyloxycarbonyl-1,3-dioxan-2-one (MBC, 15). Subsequent deprotection of the acid functionality followed by activation of the free carboxylic acid, typically by the use of dicyclohexylcarbodiimide (DCC) or by conversion to the acyl chloride, allow reaction with an alcohol or an amine.44 In a further alternative (Scheme 1, route C), the 1,3-diol is acetonide-protected before functionalisation of the carboxylic acid is carried out. Subsequent removal of the acetonide protecting group and formation of the functional cyclic carbonate are realised in the last step. Finally, the use of bis(pentafluorophenyl)carbonate, as demonstrated recently by Hedrick and coworkers, provides an excellent alternative to the use of phosgene derivatives and multi-step procedures (Scheme 1, route D).37 Here, the synthesis of a pentafluorophenyl-functionalised cyclic carbonate, 5-methyl-5-pentafluorophenyloxycarbonyl-1,3-dioxan-2-one, from bis-MPA is achieved in one step using bis(pentafluorophenyl)carbonate in the presence of caesium fluoride which followed by reaction of the pentafluorophenylester with amines or alcohols yields the desired monomers.37 In summary, 2,2-bishydroxy(methyl)propionic acid (bis-MPA) is an excellent choice as biocompatible and versatile scaffold for the preparation of cyclic carbonate monomers with pendant ester and amide groups.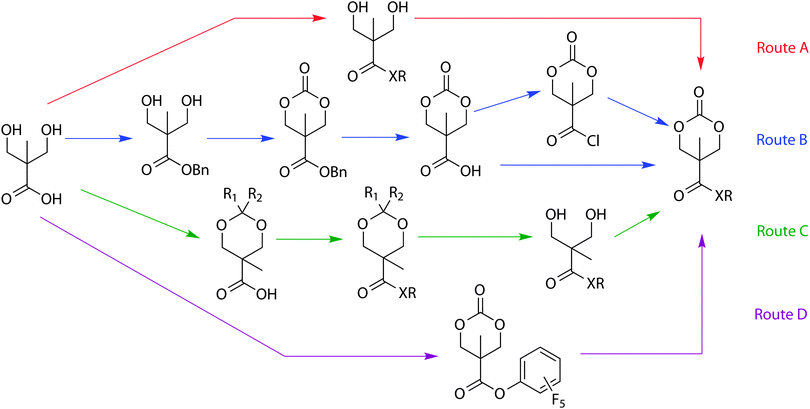 | ||
| Scheme 1 Synthesis of functional cyclic carbonates derived from 2,2-bishydroxy(methyl)propionic acid (bis-MPA). | ||
Functional cyclic carbonates derived from pentaerythritol, glycerol and trimethylolalkanes
The preparation of functional cyclic carbonates has also been reported in many cases using 1,3-diol containing compounds that have three or four hydroxyl groups in their molecular structure. Common precursors are pentaerythritol, glycerol and trimethylolalkanes which, apart from benzyl protection of the hydroxyl functionality (or functionalities) can also be used in the synthesis of other functional cyclic carbonates (Scheme 2).45–50 A number of synthetic pathways are described in the literature that use glycerol or trimethylolalkanes as a scaffold to provide cyclic carbonates with a pendant functionality. In one example (Scheme 2, route A), two of the available hydroxyl groups are protected with acetonide or benzylidene groups before further reaction of the residual alcohol with an acid chloride or via DCC-coupling with a carboxylic acid-functional molecule. The deprotection of the diol and conversion to the cyclic carbonate yields the desired monomers. Alternatively, the precursor has been reacted with a dialkyl carbonate, mediated by immobilised Candida antarctica lipase B, to yield a hydroxyl-functional cyclic carbonate intermediate (Scheme 2, route B). The hydroxyl group was then further functionalised using the abovementioned chemistries. In a report by Hatti-Kaul and coworkers, the dialkyl carbonates were also used in the functionalisation of the hydroxyl group (Scheme 2, route B).51 In a third pathway, under basic conditions, a dialkyl carbonate is used to form an oxetane by transesterification and subsequent decarboxylation. The oxetane can then be ring-opened by HX to give a 1,3-diol that can be converted to the corresponding halogen-containing carbonate (Scheme 2, route C).37,38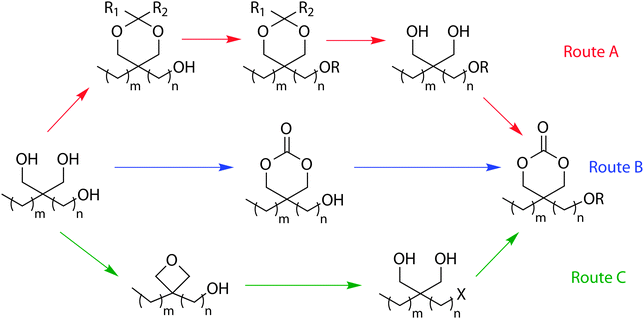 | ||
| Scheme 2 Synthesis of functional cyclic carbonates derived from glycerol and trimethylolalkanes. | ||
Synthesis of functional monomers derived from pentaerythritol is typically realised by partial functionalisation/protection of two of the hydroxyl functionalities leaving a 1,3-diol that can be cyclised using a phosgene derivative (Scheme 3).50 In conclusion, glycerol and trimethylolalkanes are good alternatives to bis-MPA in the synthesis of functional cyclic carbonates with ether and halogen substituents, whilst pentaerythritol provides an excellent opportunity to introduce two hydroxyl groups per monomer unit into the poly(carbonate) backbone.
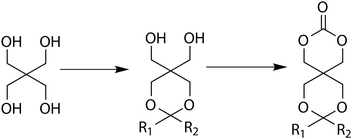 | ||
| Scheme 3 Synthesis of functional cyclic carbonates derived from pentaerythritol. | ||
Functional cyclic carbonates derived from amino acids or sugars
Amino acids and sugars are natural products that are indispensable for life processes, hence poly(amino acid)s and poly(saccharide)s or sugar-based polymers are highly attractive biocompatible materials. In view of the biocompatibility of the resulting polymers and their degradation products, cyclic carbonates derived from amino acids and sugars have been reported by Sanda et al. and by Gross and coworkers, respectively.52–54 The synthesis of cyclic carbonate monomers derived from L-serine and L-threonine was realised by protection of these amino acids via conversion of the amino groups to a carbamic acid benzyl esters (Z) or carbamic acid tert-butyl esters (Boc) followed by reduction and carbonate formation with triphosgene (Scheme 4).52 In an alternative report, the serine-derived cyclic carbonate was prepared from the alcohol analogue of serine, serinol, instead.55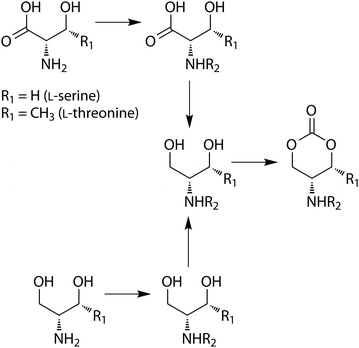 | ||
| Scheme 4 Preparation of amino-acid derived cyclic carbonate monomers. | ||
Sugar-derived cyclic carbonate monomers have been prepared from the furanose forms of D-xylose and D-glucose.53,54,56 Synthesis from D-xylofuranose involves the acetonide protection of the two hydroxyl groups to form a 1,3-diol, which is commercially available (Scheme 5A). This 1,3-diol was converted to the cyclic carbonate using ethyl chloroformate. Monomer synthesis from D-glucofuranose involved protection (acetonide and benzyl ester) of three of the hydroxyl groups to give a 1,2-diol which was converted to a 1,3-diol (Scheme 5B) through a multi-step procedure. Carbonate formation was realised using a phosgene-derivative yielding the D-glucose-derived cyclic carbonate.
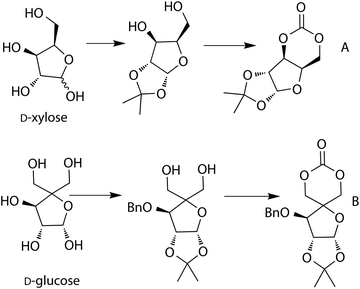 | ||
| Scheme 5 Synthesis of cyclic carbonates from (A) D-xylofuranose and (B) D-glucofuranose. | ||
Synthesis of functional cyclic carbonates from other resources
Examples of other reported cyclic carbonate syntheses include those derived from commercially available diols, oxetanes, a malonic acid diethyl ester derivative, dihydroxy acetone and N-methyl diethanolamine (Scheme 6).57–68 Direct synthesis of six-membered cyclic carbonates from oxetanes using CO2 has been reported.61 Alternatively ring-opening of 3-methyl-3-oxetanemethanol with HBr has also been reported and resulted in a bromo-functional diol. The bromide was then replaced with a thiol by reaction with NaSH, which was subsequently reacted with an alkene to generate the desired functional 1,3-diol. This diol was converted to the functional cyclic carbonate monomer by reaction with ethyl chloroformate.64 A monomer prepared from dihydroxy acetone, a glycolytic metabolite, is converted to the dimethoxy acetal before ring-closure mediated with ethyl chloroformate.62,63 In a final example, a functional malonic acid diethyl ester was functionalised with an alkyl halide in a first step followed by the reduction of the ethylesters to alcohols that enabled subsequent ring closure with diphosgene.65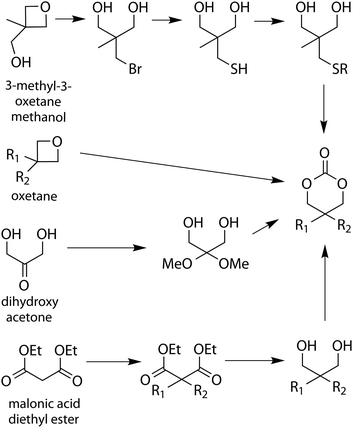 | ||
| Scheme 6 Synthesis of functional six-membered cyclic carbonates derived from oxetanes, malonic acid diethyl ester and dihydroxy acetone. | ||
Ring-opening polymerisation of functional cyclic carbonates
Cyclic carbonates with alkyl-containing functionalities
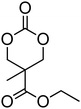
The introduction of a pendant functional group on a poly(carbonate) is desirable, but homopolymerisation of functional cyclic carbonates is not always necessary. Indeed, for some applications it is even advantageous to have a polymer backbone in which only a percentage of monomer units have a pendant functionality.66,67 Furthermore, in cases in which the polymerisability of a functional monomer is not optimal, copolymerisation often enables a solution to be achieved.33 In the preparation of functional poly(carbonate)s through copolymerisation, trimethylene carbonate (TMC) is often used as co-monomer, however other simple alkyl functional monomers have also been reported.44,68 Hedrick and coworkers have recently reported copolymerisations of functional monomers with a simple ethyl functional monomer derived from bis-MPA, 5-methyl-5-ethyloxycarbonyl-1,3-dioxan-2-one (MEC, 7), while Zhuo and coworkers report the use of dimethyl trimethylene carbonate, DTC, 1 (Table 1).37,69–78 Only two examples of MEC homopolymerisations have been reported in the literature.37,80 Using the sparteine/1-(3,5-bis(trifluoromethyl)-phenyl)-3-cyclohexyl-2-thiourea (TU) catalyst system in methylene chloride at room temperature, the polymerisation ([M]/[I] = 108) yielded a polymer with a molecular weight of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1 and a dispersity of 1.27, however application of a hexafluoroalcohol as a cocatalyst in place of the thiourea under comparable conditions resulted in polymers with decreased dispersity (1.13) and a lower molecular weight (3200 g mol−1; ([M]/[I] = 88). In contrast, homopolymerisation of DTC has been reported using a number of catalysts.79–84 The cationic ROP of DTC was reported in methylene chloride at room temperature using methyl trifluoromethane sulfonate (TfOMe) (<30% monomer conversion; Mn = 1800–2100 g mol−1; ĐM = 1.30–1.38) or trifluoromethane sulfonic acid (<50% monomer conversion; Mn = 3000–3500 g mol−1; ĐM = 1.10–1.11) or in nitrobenzene at 120 °C using methyl iodide (50% monomer conversion; Mn = 1100 g mol−1; ĐM = 1.52). Homopolymerisation of DTC has also been reported, catalysed by tin octanoate under bulk conditions at 120 °C, and resulted in polymers with molecular weights ranging from 2900 to 8300 g mol−1 (ĐM = 1.52–1.58) or using rare earth catalysts at temperatures between 0 and 45 °C (Mn = 116000–171000 g mol−1; ĐM = 2.28–2.79). Poly(DTC) is a hydrophobic, semi-crystalline material with reported melting points of 90–93 °C78 and 122 °C82 which consequently leads to decreased degradation rates of the homopolymer compared to poly(TMC) as well as for some of its copolymers.85,79–83
100 g mol−1 and a dispersity of 1.27, however application of a hexafluoroalcohol as a cocatalyst in place of the thiourea under comparable conditions resulted in polymers with decreased dispersity (1.13) and a lower molecular weight (3200 g mol−1; ([M]/[I] = 88). In contrast, homopolymerisation of DTC has been reported using a number of catalysts.79–84 The cationic ROP of DTC was reported in methylene chloride at room temperature using methyl trifluoromethane sulfonate (TfOMe) (<30% monomer conversion; Mn = 1800–2100 g mol−1; ĐM = 1.30–1.38) or trifluoromethane sulfonic acid (<50% monomer conversion; Mn = 3000–3500 g mol−1; ĐM = 1.10–1.11) or in nitrobenzene at 120 °C using methyl iodide (50% monomer conversion; Mn = 1100 g mol−1; ĐM = 1.52). Homopolymerisation of DTC has also been reported, catalysed by tin octanoate under bulk conditions at 120 °C, and resulted in polymers with molecular weights ranging from 2900 to 8300 g mol−1 (ĐM = 1.52–1.58) or using rare earth catalysts at temperatures between 0 and 45 °C (Mn = 116000–171000 g mol−1; ĐM = 2.28–2.79). Poly(DTC) is a hydrophobic, semi-crystalline material with reported melting points of 90–93 °C78 and 122 °C82 which consequently leads to decreased degradation rates of the homopolymer compared to poly(TMC) as well as for some of its copolymers.85,79–83
| Monomer | Number | Ref. | Monomer | Number | Ref. |
|---|---|---|---|---|---|
|
1 (DTC) | 75–85 |
|
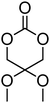
9 | 87 |
|
2 | 81 |
|
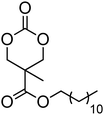
10 | 37 and 74 |

|
3 | 61 |
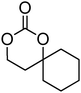
|
11 | 49, 79, 85, 90 and 91 |
|
4 | 62, 63 and 86 |
|
12 | 81 |
|
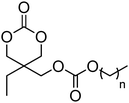
5 (n = 0) | 51 |
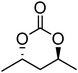
|
13 | 68 |
| 6 (n = 1) | |||||
|
7 (MEC) | 37, 69–74 and 80 |

|
14 | 68 |
|
8 | 70 |
Many other alkyl-functional monomers have been reported (Table 1; 1–14), however there are few reports on the properties of their resultant homopolymers.37,51,61–63,70,74,81,86,87 Homopolymerisation of 2 in bulk using TfOMe as a catalyst (60–100 °C; Mn = 3500–5000 g mol−1; ĐM = 1.67–2.58) or in chloro- or nitrobenzene at 120 °C initiated by methyl iodide (Mn = 600–800 g mol−1; ĐM = 1.40–1.47)81 has been reported whilst one example of the homopolymerisation of 10 using the organic catalyst 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in combination with the TU cocatalyst is reported in dichloromethane at room temperature ([M]/[I] = 99; Mn = 6600 g mol−1; ĐM = 1.24).37 Ring-opening of 4 using a range of metal-based and organic catalysts has recently received a lot of attention. Reported polymerisations were either carried out in bulk at elevated temperature (130 °C), or at temperatures between 60 and 110 °C in toluene.76,86 ROP of 4 in bulk resulted in a polymer with Mn = 56400 g mol−1 (ĐM = 1.44), whilst good results were obtained for both metal-based and the organic catalyst 4-dimethylaminopyridine (DMAP) in the solution polymerisation of 4 ([M]/[I] = 100; Mn = 6800–17000 g mol−1; ĐM = 1.18–1.25) as well. Poly(4) is a rigid, brittle, amorphous material with a glass transition temperature at 39–45 °C and a degradation temperature (225 °C) that is slightly lower than PTMC (230 °C).62,86,88,89
Other alkyl functional monomers that have a group at the alpha position on the carbonate ring are 1,3-dioxaspiro[5.5]undecan-2-one, 1281 and α-methyl trimethylene carbonate,49,79,85,90,9111. Endo and coworkers reported the cationic homopolymerisation of 11 in 2001, however only very low monomer conversions were observed and the resulting polymer was not isolated.79 Higher monomer conversions have since been reported using rare earth initiators, aluminium triflate, β-diiminate zinc complexes and organic bases.90,91 Good control was obtained using Al(OTf)3 at 110 °C and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) and 1,4,7-triazabicyclodecene (TBD) resulting in poly(carbonate)s with molecular weights between 5700 and 10000 g mol−1 and low dispersities. The substitution pattern of the carbonate ring can lead to two different propagating alcohols upon ring-opening which, in turn means that different microstructures can be expected depending on the regioselectivity of the catalysts applied in the ROP. Guillaume and coworkers investigated the regioselectivity of the resulting polymers using a range of catalytic systems.90 Polymers with a high degree of regioregularity were obtained using β-diiminate zinc complexes, whereas organocatalysts showed poor regioselectivity. It was rationalised that this difference in regioselectivity was caused by the bulky nature of the β-diiminate ligand present in the zinc-catalysed reactions, but not in those catalysed by aluminium triflate or organic bases. The thermal properties of the poly(11) homopolymers reveal amorphous polymers with a glass transition temperature (Tg) between −18 and −10 °C, which is very similar to that of PTMC (−15 °C).71 In addition, 11 was copolymerised with ε-caprolactone and lactide to form highly hydrophobic copolymers and was used in the synthesis of poly(butylene succinate-co-11) by a combination of ROP and condensation polymerisation.49,91
Cyclic carbonates with aryl-containing functionalities
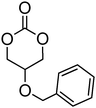
To date, many aryl-functional monomers and their respective polymers have been reported. Most commonly, those containing a benzyl group have been described. Monomers such as 5-methyl-5-benzyloxycarbonyl-1,3-dioxan-2-one, MBC 15 (Table 2) are readily polymerised and, moreover, the benzyl functionality can often be removed easily to yield hydrophilic materials or intermediates in the synthesis of carbonate monomers with more sensitive functionalities. For these reasons, benzyl-functional monomers have been used extensively in the preparation of functional aliphatic poly(carbonates) and poly(ester carbonate)s in recent years. MBC (15) and 5-benzyloxy-trimethylene carbonate (17), in particular have received much attention.42,71,75,78,86,91–102 Homopolymerisation of MBC was first reported by Bisht and coworkers in 1999 using enzymatic methods.42 Polymerisation was studied using seven different commercially available lipases at 80 °C in bulk among which lipase AK (from Pseudomonas fluorescens) gave the highest monomer conversion (97%) and molecular weight (Mn) 6100 g mol−1 with no decarboxylation observed. Molecular weights remained low due to competitive initiation from water however higher molecular weights could be obtained when the lipase was dried extensively. Increasing molecular weight also led to significant broadening of the dispersities (ĐM ≤ 7.5). In order to control the incorporation of benzyl groups in the poly(carbonate) structures, copolymerisation with TMC was conducted under the same conditions and led to statistical copolymers being isolated.92 Organocatalytic ring-opening of MBC performed at room temperature with DBU as organocatalyst has resulted in well-defined homopolymers with controlled molecular weights (Mn = 4500–11400 g mol−1) and low dispersities (ĐM = 1.18–1.19).99 The resulting poly(MBC) homopolymers are amorphous sticky polymers with a glass transition temperature (Tg) between 3 and 8 °C.92,99 MBC has furthermore been reported in copolymerisations with lactide using tin- and zinc-based catalysts, mostly at elevated temperatures,94–96 and in copolymerisations with MEC or lactide (LA) using organic catalysts (DBU or TU-amine) at room temperature.71,98 In the latter case, reactivity ratios in the copolymerisation of lactide and MBC were found to be 1.1 for lactide and 0.072 for MBC. Micelles obtained from block copolymers prepared by the statistical copolymerisation of MBC with lactide, initiated from a poly(ethylene oxide), PEO, macroinitiator, demonstrated that the introduction of carbonate moieties did not only enhance solubilisation and drug loading of the hydrophobic drug bicalutamide, but also resulted in significant reductions in the value of critical micelle concentration (CMC) compared to micelles without MBC incorporated.96
| Monomer | Number | Ref. |
|---|---|---|

|
15 (MBC) (R = H) | 42, 71 and 92–99 |
| 16 (R = NO2) | ||
|
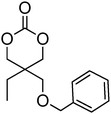
17 | 49, 75, 78, 86 and 100–102 |
|
18 | 49 |
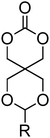
|
19 (R = Ph) | 50 and 103–105 |
| 20 (R = pOMePh) | ||
| 21 (R = 2,4,6-OMe3Ph) | ||
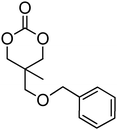
|
22 | 61 |

|
23 (R = H) | 79 |
| 24 (R = OMe) | ||
| 25 (R = NO2) | ||
|
26 (R = H) | 79 |
| 27 (R = OMe) | ||
| 28 (R = NO2) |
Another frequently reported benzyl functional monomer is 5-benzyloxy-trimethylene carbonate (17). This monomer has been polymerised under bulk conditions at elevated temperatures using aluminium- and tin-based catalysts, a range of organic catalysts, porcine pancreas lipase immobilised on silica particles and under reduced pressure without added catalyst.75,78,86,101,102 Reported poly(17) homopolymers displayed medium to large dispersities (ĐM = 1.41–1.92) and polymer molecular weights (Mn = 8800–22400 g mol−1) that are much lower than those expected for polymerisations with [M]/[I] = 1000. The resultant materials are rubbery at room temperature with Tg = 0–6 °C and no observed melting point.100,101 Reported copolymerisations include random and block copolymers with DTC, lactide and PEO.75,100,102 Another example is the combined ROP and polycondensation reaction of 17 or 18 with poly(butylene succinate) macromers.49
Other reported aryl-functional monomers, 19, 22, 23 and 26 have been studied to a lesser extent.50,61,79,103,104 ROP of benzyl-functional monomer 9-phenyl-2,4,8,10-tetraoxaspiro-[5,5]-undecan-3-one (19) was investigated using diethyl zinc as initiator at 130 °C under bulk conditions for its homopolymerisation and its copolymerisation with both lactide and ε-caprolactone.50,104 The resulting poly(19) homopolymers displayed a Tg = 124 °C, which is high compared to other poly(carbonate) homopolymers. Cationic ROP of 23 and 26 to form the respective homopolymers was reported at room temperature using a range of catalysts, however polymerisations were plagued by side reactions including backbiting and decarboxylation leading to poor control and low molecular weight products.79 Suppression of decarboxylation was achieved using MeI as initiator, but increased reaction times and temperatures (120 °C) were required for the polymerisation to proceed.
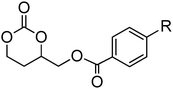
Beyond simple benzyl-functional monomers, a number of monomers with methoxy- and nitro-substituted phenyl groups have been reported (Table 2).79,105–107 The cationic ROP of 24, 25, 27 and 28 was reported by Endo and coworkers.79 All four monomers were polymerised using methyl iodide at 120 °C to yield the respective homopolymers. Homopolymers were obtained with Mn = 10000 g mol−1 (ĐM = 1.22) for poly(24), Mn = 3400 g mol−1 (ĐM = 1.88) for poly(25), Mn = 4400 g mol−1 (ĐM = 1.58) for poly(27) and Mn = 7100 g mol−1 (ĐM = 1.39) for poly(28). Polymerisation was also successfully conducted at room temperature in dichloromethane in case of 24 and 25 using TfOMe or TfOH as catalysts yielding polymers with Mn = 1300–3500 g mol−1 (ĐM = 1.13–1.24) and Mn = 3000 g mol−1 (ĐM = 1.16), respectively. Nitrobenzyl-functional monomer 5-methyl-5-(2-nitro-benzoxycarbonyl)-1,3-dioxan-2-one (16), methoxy functional monomers mono-2,4,6-trimethoxybenzylidene-pentaerythritol carbonate (21) and mono-4-methoxybenzylidene-pentaerythritol carbonate (20) have all been used in the synthesis of PEO block copolymers prepared by the homopolymerisation of the monomers or copolymerisation with lactide from methoxy-PEO macroinitiators.105 Polymerisations were performed using tin octanoate as catalyst under bulk conditions at 110–120 °C (16) or using zinc bis[bis(trimethylsilyl)amide] in methylene chloride at 50 °C (20 and 21).105–107
Cyclic carbonates with alkene- and alkyne-containing functionalities
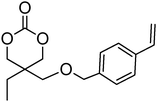
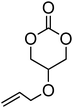
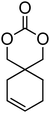
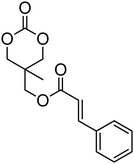
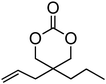
Alkene and alkyne functional groups participate in many organic reactions, including highly efficient and orthogonal reactions, as a consequence of their (double) unsaturation such as Michael addition, radical thylation, Huisgen 1,3-dipolar cycloaddition, epoxidation120 and UV-cross-linking.124,125 The introduction of such functionalities to the poly(carbonate) backbone leaves the resulting polymers open for further functionalisation.108–112 Notably, there have been several reports of the synthesis and subsequent modification of (pendant) alkenes and alkyne groups in poly(ester)s.113,114The introduction of pendant unsaturated groups in poly(carbonate)s prepared by the ring-opening of cyclic carbonate monomers has received increased attention in the past few years. The ROP of allyl ester-functional cyclic carbonate 5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one (MAC, 29), for example, has been described in the preparation of a range of (co)polymers by Storey, Jing, Zhuo, Dove, Cui and their respective coworkers (Table 3).76,115–122 Reported homopolymerisations of MAC were either performed without added catalyst at elevated temperature (at 115 °C; Mn = 13700 g mol−1; ĐM = 1.8), or in toluene at 95 °C using tin(II)-based poly(lactide) macroinitiator ([M]/[I] = 40; Mn = 9200 g mol−1; ĐM = 2.5) and at 110 °C catalyzed by diethyl zinc ([M]/[I] = 200; Mn = 19000 g mol−1; ĐM = 1.6).116,120 However, in all cases polymerisations showed relatively poor control and resulted in branched polymers in some cases. More recently Dove and coworkers demonstrated that the ROP of MAC using efficient organocatalysts led to a high level of control over the resulting polymer such that the application of the sparteine/TU catalyst system resulted in the isolation of PMAC with Mn up to ca. 13000 g mol−1 ([M]/[I] = 100) and low dispersities (ĐM = 1.12).121 Notably in this study, the polymer Tg was reported to be −26.4 °C, lower than that previously reported (Tg = −12 °C),116,120 attributed to an absence of chain branching as a consequence of very low levels of transesterification. The ROP of MAC has also been reported in the synthesis of 3-armed star polymers initiated from triethanolamine using a O,N,N,O-tetradentate salan lutetium alkyl complex.122 The resulting polymers displayed low dispersities (ĐM < 1.10) and molecular weights close to the predicted values from the monomer-to-initiator ratio. Furthermore, the polymerisation proceeded well with low catalyst loadings (compared to initiator) thus demonstrating its ‘immortal’ nature. Other studies involving MAC have mostly focused on its copolymerization with lactide,116 including the formation of amphiphilic block copolymers composed of PMAC-co-PLA and poly(ethylene oxide) to form micelles118 and polycations from poly(ethylenimine)-grafted polycarbonate.119 While ROP of the related allyl-functional monomer 5-allyloxy-1,3-dioxan-2-one (30) has been reported, only the synthesis of 5-(2-propenyl)-5-propyl-1,3-dioxan-2-one (33) is known.45,46,65 Homopolymerisation of 30 was reported by Parzuchowski et al.45 and He et al.46 Polymerisations were carried out in ethyl acetate at 65 °C using DMAP as catalyst or under bulk conditions at 120 °C using immobilised porcine pancreas lipase on silica or tin octanoate as catalyst. No evidence of decarboxylation was observed during the polymerisation and resulted in a semi-crystalline poly(30) that was characterized by a melting temperature around room temperature (Tm = 24 °C) and a low glass transition temperature (Tg = −40 °C). In an effort to improve the thermal properties of the poly(carbonate)s, while taking advantage of this lateral reactive groups, Gross and coworkers prepared the cyclohexene functional monomer 2,2-[2-pentene-1,5-diyl]-trimethylene carbonate (31).60,68,123 It was hypothesised that the introduction of the cyclohexene substituent would yield poly(carbonate)s with a more rigid backbone and, as a consequence of this rigidity, a higher glass transition temperature. Indeed, polymers obtained by the metal-mediated (Al, Sn, Zn) ROP of 31 in bulk at 120 °C exhibited a Tg = 30 °C ([M]/[I] = 200; Mn = 6000–255000 g mol−1; ĐM = 1.3–3.4) which is significantly higher than those reported for allyl-functional poly(carbonate)s and TMC. In contrast, the homopolymer obtained from ROP of cinnamate-functional 5-methyl-5-cinnamoyloxymethyl-1,3-dioxan-2-one (32) with a molecular weight of 6000 g mol−1 (ĐM = 1.87) had a Tg of 42 °C.124 Both its homopolymerisation and copolymerisation with L-lactide were performed in bulk at 120 °C using diethyl zinc as catalyst.
| Monomer | Number | Ref. | Monomer | Number | Ref. |
|---|---|---|---|---|---|
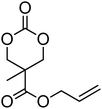
|
29 (MAC) | 76 and 115–122 |
|
36 | 48 |
|
30 | 45 and 46 |

|
37 | 128 |
|
31 | 60, 123 and 68 |
|
38 | 64 |
|
32 | 124 |
|
39 | 129 |
|
33 | 65 |
|
40 | 129 |
|
34 | 47, 126 and 127 |

|
41 (MPC) | 44 and 130–134 |
|
35 | 47 |
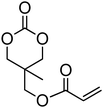
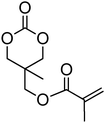
Acrylate-functional monomer 34 and methacrylate-functional monomer 35 were reported by Zhong and coworkers in copolymerisations with ε-caprolactone and lactide (incorporations lower than 33%) initiated from PEO to result in amphiphilic block copolymers.47,126,127 The monomers were polymerised in toluene using tin octanoate at 110 °C or in methylene chloride at room temperature using bis[bis(trimethylsilyl)amido] zinc as a catalyst. Styrene-functional 5-ethyl-5-[(4-vinylphenyl)methoxymethyl]-1,3-dioxan-2-one (36), reported by Miyagawa et al., was polymerised by anionic ROP in THF at 0 °C in the presence of potassium tert-butoxide (t-BuOK).48 The resulting poly(36) homopolymer was characterised by a Mn = 14000 g mol−1 (ĐM = 1.44) without polymerization of the pendant styrene groups.
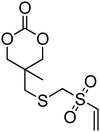
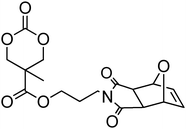
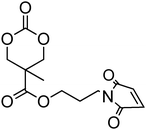
Cyclic carbonate, 5,5-(bicyclo[2.2.1]hept-2-en-5,5-ylidene)-1,3-dioxan-2-one (37), with a pendant norbornene functionality was polymerised in bulk with a range of amine catalysts at 100–140 °C among which DBU proved to be the most active.128 The resulting poly(37)s displayed high glass transition temperatures (Tg = 108 °C) as a consequence of the backbone rigidity resulting from the norbornene functionality. Copolymerisation of a vinyl sulfone-functional carbonate 38 was reported with TMC, caprolactone or lactide in toluene at 110 °C, using isopropanol as initiator and tin octanoate as catalyst has been reported.64 The pendant vinyl sulfone remained intact during the polymerisation allowing subsequent modification of the polymer surface with thiol derivatives.64 Onbulak et al. recently reported the synthesis and polymerisation of cyclic carbonate 39 which has a furan-protected maleimide as a pendant functionality.129 The possibility to expose the maleimide functionality after polymerisation was demonstrated by the synthesis of 40via deprotection in toluene at 110 °C. Homopolymerisation of 39 was carried out at room temperature using DBU as catalyst and benzyl alcohol as initiator. As polymerisations became severely retarded after 60% monomer conversion, polymerisations were stopped after 50% monomer conversion to avoid transesterification reactions resulting in poly(39)s with low dispersities (ĐM = 1.1–1.2). In order to overcome poor polymerisability of 39, its copolymerisation with LA was carried out, yielding well-defined copolymers with pendant furan-protected maleimides. Surprisingly, the only propargyl functional cyclic carbonate reported is 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC, 41) and to date this monomer has only been copolymerised with lactide or trimethylene carbonate.44,130–134 Copolymerisations were carried out in toluene or in bulk at 100–110 °C using diethyl zinc as catalyst/initiator130–134 or in methylene chloride at room temperature using DBU/TU co-catalysts44 resulting in copolymers with composition in MPC in agreement with the feed and ranging from 7 to 20%. In the latter study, it was found that MPC was incorporated preferentially to TMC at the beginning of the polymerisation.
Cyclic carbonates with halogen- and azide-containing functionalities
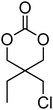
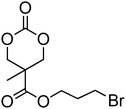
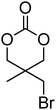
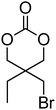
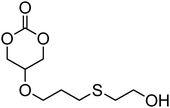
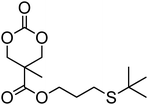
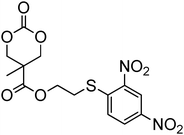
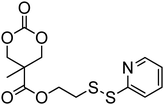
Cyclic carbonates carrying halogen groups are an attractive class of monomers as it allows subsequent nucleophilic substitution and introduction of novel functionalities even after polymerisation. Mindemark et al. recently reported the synthesis and polymerisation of four halide-containing monomers, 42–45 (Table 4).38 The resulting homopolymers of these chloro- and bromo-functional monomers, obtained by ROP at elevated temperature (110 °C) in bulk using tin octanoate as a catalyst, yielded chloride-functional polymers with Mn = 16000–38500 g mol−1 and ĐM = 1.46–1.81 and bromide functional polymers with Mn = 8500–31900 g mol−1 and ĐM = 1.41–1.63, respectively. All polymers were semi-crystalline in nature with small variations of the measured melting points and glass transitions (P42: Tg = 15 °C, Tm = 105 °C; P43: Tg = 10–13 °C, Tm = 131–152 °C; P44: Tg = 18–23 °C, Tm = 100 °C; P45: Tg = 15–18 °C, Tm = 121–153 °C) However, a significant difference in solubility was observed between halide-containing polymers with a pendant methyl and an ethyl substituent. Indeed, whereas poly(43) and poly(45) are insoluble in most common organic solvents, poly(42) and poly(44) were found to be readily soluble in methylene chloride and tetrahydrofuran. Interestingly, copolymerisation of these monomers with TMC also resulted in semicrystalline poly(carbonate)s. A similar cyclic carbonate monomer with two pendant bromide functionalities, 2,2-bis(bromomethyl)trimethylene carbonate (46), has also been reported.57,58,135 Its homopolymerisation and its copolymerisation with ε-caprolactone were reported in bulk at 120 °C using tin octanoate as catalyst and the resulting polymers were subsequently modified with NaN3 and used for the preparation of poly(carbonate) nanoparticles by the Huisgen 1,3-dipolar cycloaddition. The synthesis and polymerisation of halogen-functional cyclic carbonates derived from bis-MPA were also reported by Hedrick and coworkers (Table 4).37,44,69,72,138,139 In a recent study, the pentafluorophenol pathway was exploited to form the cyclic carbonate 53 intermediate as outlined previously.
| Monomer | Number | Ref. | Monomer | Number | Ref. |
|---|---|---|---|---|---|
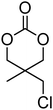
|
42 | 38 |
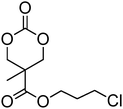
|
48 | 37, 44, 69, 72 and 139 |
|
43 | 38 |
|
49 | 37, 72 and 138 |
|
44 | 38 |

|
50 | 72 |
|
45 | 38 |
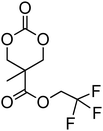
|
51 | 44 |

|
46 | 57 and 58 |
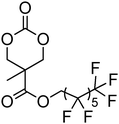
|
52 | 37 |
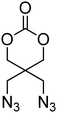
|
47 | 57, 58, 136 and 137 |

|
53 | 37 |
The synthesis of halogen-containing poly(48) ([M]/[I] = 55; Mn = 12200 g mol−1; ĐM = 1.17), poly(49) ([M]/[I] = 52; Mn = 11700 g mol−1; ĐM = 1.11), poly(50) ([M]/[I] = 51; Mn = 10500 g mol−1; ĐM = 1.22) and poly(51) ([M]/[I] = 54; Mn = 11500 g mol−1; ĐM = 1.26) was achieved via organocatalytic ROP at room temperature using the TU/DBU catalyst system.37,72 Distribution of the pendant halogen functionalities in the poly(carbonate) backbone was achieved under these conditions in copolymerisations with MEC (with 48, 49, 50) or TMC (with 52) and resulted in polymers with controlled molecular weights and low dispersities.37,44,69,72,138,139
Cyclic carbonates with lateral nitrogen-containing functionalities: amino-, amido, carbamate and urea functional cyclic carbonates
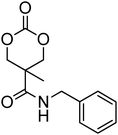
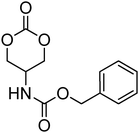
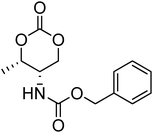
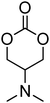
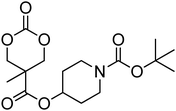
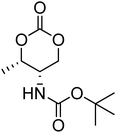
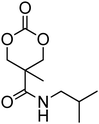
Nitrogen-containing cyclic carbonates include those with pendant amides and pendant amino, amido and urea functionalities.37,52,55,59,87,140,141 Cyclic carbonates with pendant amides are those with pendant carbamic acid benzyl ester (Z) groups and carbamic acid tert-butyl ester (Boc) functional groups, which are used to protect functional groups that have primary amines during the polymerisation. Endo and coworkers reported the anionic ROP of 55, 56 and 59 (Table 5) which was carried out in THF at temperatures between −78 and −30 °C using a range of initiators.52 Molecular weights of the resulting poly(55) homopolymers ranged from 5600 g mol−1 to 20000 g mol−1 with moderate dispersities being observed (ĐM = 1.20–1.46). Interestingly, poly(56) (Mn = 13000–24000 g mol−1) and poly(59) (Mn = 5700–21000 g mol−1) showed inverse optical rotations compared to the respective cyclic carbonates and lower dispersities (ĐM = 1.18–1.24 and 1.11–1.20, respectively) than poly(55). Additionally, ROP of 56 and 59 showed a preference (9:1) for ring-opening at the least-hindered acyl bond. The bulk copolymerisation of 55 with lactide has also been reported at 120 °C using diethylzinc as catalyst and yielded poly(55) with comparable molecular weight and dispersity (Mn = 24100 g mol−1; ĐM = 1.51).55 While the polymerisation of the Boc-protected-amine cyclic carbonate, 54, has not been reported, the organocatalytic ROP of related monomer 57 has been reported with TMC.37,44 The copolymerisation was performed using the TU/DBU catalytic system at room temperature in methylene chloride and resulted in copolymers of 57 and TMC with low dispersities (ĐM = 1.07). Synthesis of poly(58) homopolymers with low degrees of polymerisation (DP = 8–22) and copolymers of 58 with other cyclic carbonate monomers was realised using the TU/DBU catalyst system in CH2Cl2 at room temperature.87,140 Homopolymers obtained from the organocatalytic ROP of 58 exhibited low dispersities (ĐM = 1.11–1.16) for molecular weights Mn ranging from 3800 to 10000 g mol−1 and displayed excellent cellular uptake with longer oligomers being observed to mediate cell adhesion.140 Furthermore, these homopolymers were also applied as components of non-covalent, degradable hydrogels.87
| Monomer | Number | Ref. | Monomer | Number | Ref. |
|---|---|---|---|---|---|
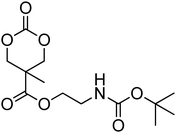
|
54 | 37 and 44 |
|
61 | 44 |
|
55 | 52 and 55 |

|
62 | 37 |
|
56 | 52 |
|
63 | 141 |
|
57 | 44 |

|
64 | 59 |

|
58 | 87 and 140 |

|
65 | 71, 73 and 142 |
|
59 | 52 |
|
66 | 71, 73 and 142 |
|
60 | 44 |
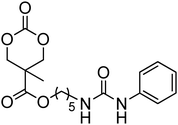
Although the synthesis of amido-functional cyclic carbonates 60 and 61 has been reported (Table 5),44 no report was found describing their polymerisation. No other reports of the ROP of this type of amido-functional cyclic carbonate monomers were found, possibly as polymerisation could be complicated by the presence of the secondary amine. In contrast, homopolymerisation of 62, with a tertiary amide pendant group, has been reported by Sanders et al. using the TU/DBU catalyst system at room temperature to yield poly(62) with Mn = 10500 g mol−1 and ĐM = 1.32 ([M]/[I] = 63).37 The homopolymerisations of the tertiary amine-containing 16-membered 6,14-dimethyl-1,3,9,11-tetraoxa-6,14-diaza-cyclohexadecane-2,10-dione (64), which contains two carbonate groups and two tertiary amines, was reported by Wang et al. in 2010.59 ROP was attempted using both enzyme- and metal-based catalyst systems. Polymerisations performed in bulk at elevated temperatures (120 °C) using tin octanoate resulted in decarboxylation of the monomer whilst polymerisations performed in toluene at temperatures between 70 and 100 °C did not result in any polymerisation being observed. In contrast, the enzymatic ROP of 64 in toluene using Novozym-435 resulted in the formation of poly(64) both at 70 °C and at room temperature and displayed living characteristics. Poly(64) with a molecular weight of 8000 g mol−1 and a ĐM = 1.34 was obtained by the enzymatic process and were amorphous materials presenting excellent biocompatibility and fast degradability. Likewise, homopolymers obtained from the enzymatic ROP (60 °C; Mn = 4900 g mol−1, ĐM = 1.40) of six-membered dimethylamine-functional cyclic carbonate, 2-dimethylaminotrimethylene carbonate (63), showed low toxicity and good degradability.141 Finally, polymerisation of urea-functional monomers 65 and 66 were reported using the TU/(−)-sparteine catalyst system at room temperature in CH2Cl2. Poly(66) was obtained with Mn = 10500 g mol−1 ([M]/[I] = 32; ĐM = 1.14) and displayed a Tg at approximately 15 °C.142 The synthesis of block copolymers with a PEO block, a poly(TMC) and a poly(66) or poly(65) block have also been reported, as well as a block copolymer with a PEO block and a poly(MEC-co-65) sequence, was reported using the same mild conditions. These block copolymers were used for the formation of micelles that displayed low CMC's and increased stability of drug-loaded micelles due to the hydrogen-bonding interactions between the urea groups.71,73 Strong H-bonding interaction arising from the urea-groups was also highlighted in semi-interpenetrating networks by a decrease of the overall degree of swelling.142,143
Cyclic carbonates with protected sugar functionalities
Beyond their degradable and biocompatible properties, carbohydrates display specific interactions with proteins (lectins) making them very desirable for targeted drug delivery systems.144 Introduction of these groups on a poly(carbonate) backbone presents an attractive, non-toxic and biodegradable method to targeted drug delivery. As the hydroxyl groups in sugar-functional cyclic carbonates are incompatible with the ring-opening process, the hydroxyl groups require protection prior to the polymerisation of the cyclic carbonate. Gross and coworkers first prepared a cyclic carbonate monomer directly from xylose saccharide, 1,2-O-isopropylidene-D-xylofuranose-3,5-cyclic carbonate (71, Table 6).54,145,146 This work was extended with the synthesis and polymerisation of glucose based 1,2-O-isopropylidene-3-benzyloxy-pentofuranose-4,4′-cyclic carbonate (70).53 Monomers were homopolymerised as well as copolymerised with L-lactide under bulk conditions at elevated temperatures (120–180 °C) or in dioxane at 25–60 °C. The resulting poly(70) and poly(71) homopolymers were obtained with number-average molecular weights of 7900 g mol−1 (ĐM = 1.47),145 and 2100–13200 g mol−1 (ĐM = 1.22–1.92), respectively.146 Thermal analysis of the polymers revealed that poly(70) displayed a Tg = 128 °C145 whereas poly(71) was also found to be semicrystalline and displayed a Tg = 128 °C.146 Deprotection of ketal group of polymers containing monomer 71 was successfully achieved by using trifluoroacetic acid. Unfortunately however, deprotection resulted in some degradation due to the hydrolysis of the pendant ester bonds that link the carbohydrate to the carbonate. While deprotection of the homopolymer of 70 has not been reported to date, conversely, removal of a benzyl protecting group in copolymers containing monomer 70 can be achieved selectively by hydrogenation over palladium/carbon (90% removal) however again, acetonide deprotection resulted in some degradation. Deprotection of its copolymer with lactide showed a lowering in glass transition after both deprotection steps.
| Monomer | Number | Ref. |
|---|---|---|
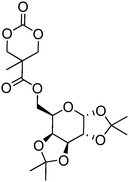
|
67 | 147 |

|
68 | 147 |
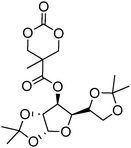
|
69 | 147 |
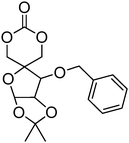
|
70 | 53 and 146 |
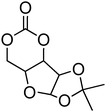
|
71 | 54, 145 and 146 |
Homopolymerisation of cyclic carbonates 67, 68 and 69 with pendant protected saccharides was carried out at room temperature using the TU/DBU organocatalytic system.147 Homopolymers were obtained with molecular weights between 17000 and 19000 g mol−1 and dispersities of 1.20–1.28. Recovery of the hydroxyl groups was carried out by deprotection of the acetonide groups using formic acid solution. Interestingly, full deprotection was achieved and no degradation of the polycarbonate backbone was observed. Block copolymers of 67–69 with TMC were reported for the preparation of micelles in aqueous solutions, and were prepared by sequential addition of TMC to poly(67), poly(68) and poly(69).
Macromonomers, bis- and tris-carbonates
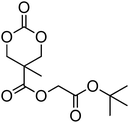
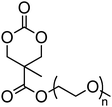
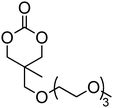
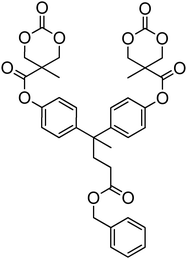
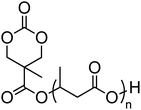
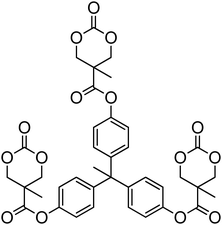
A number of cyclic carbonate-containing macromonomers and cross-linkers have been reported.33,37,44,70,77,87,142 In particular those containing ethylene glycol moieties (72–74) have received increased interest in recent years (Table 7). PEO-functional cyclic carbonate macromonomer (72), prepared from methoxy poly(ethylene oxide)s, have been reported with molecular weights ranging from 350 g mol−1 to 1900 g mol−1 (ĐM = 1.03–1.14). While homopolymerisation of 72 in methylene chloride solution at room temperature using DBU as a catalyst was possible (with Mn = 1900 g mol−1 and ĐM = 1.03), the reaction showed a maximum at 55% monomer conversion.33 The limited, low concentration of the cyclic carbonate (0.2 M), due to the large PEO pendant chain, was thought to cause the ring-chain equilibrium to dominate such that a high molecular weight polymers could not be obtained. To obtain higher molecular weight polymers, the cyclic carbonate concentration was increased by copolymerisation of 72 with TMC.33,44 Random copolymerisation of 72 with dimethyltrimethylene carbonate (DTC) from a poly(ε-caprolactone) macroinitiator at 130 °C in bulk using tin octanoate as catalyst or with dodecyl-functional monomer 8 in CH2Cl2 at room temperature using organic catalysts were further reported.70,77 The resulting block/graft copolymers could be self-assembled with the latter example displaying thermoresponsive behaviour owing to the combination of hydrophilic/hydrophobic groups in the second block. An organocatalytic approach to hydrogels was presented by Nederberg et al. by the synthesis of a bifunctional cyclic carbonate macromonomer from poly(ethylene oxide) diols.33 Poly(ethylene oxide)-α,ω-methylcarboxytrimethylene carbonate crosslinkers, 74, were prepared with Mn = 3400 g mol−1, 8000 g mol−1 and 18500 g mol−1 and copolymerised with TMC in CH2Cl2 at room temperature in the presence of DBU to form a poly(carbonate)/poly(ethylene oxide) cross-linked hydrogel. Hydrogels prepared with increased amounts of TMC showed hydrophobic PTMC domains which contribute to an increase of the toughness of the resulting hydrogels.33,142 This led to further investigation by synthesis of telechelic, carbonate functional poly(ε-caprolactone), 75, with a molecular weight of 8000 g mol−1 (ĐM = 1.40; DP = 70). Copolymerisation of this hydrophobic crosslinker with 74 (8000 g mol−1) and TMC to introduce hydrophobic segments resulted in particularly tough materials. Furthermore hydrogels prepared by copolymerisation from a poly(carbonate) macroinitiator bearing pendant urea functionalities (i.e. poly(66)) are presented as another method to strengthen the resulting network via hydrogen bonding with the poly(ethylene oxide) crosslinkers.33,87,142 Very recently, Akashi et al. developed macromonomers based on oligo poly(ethylene oxide) connected to the cyclic carbonate by an ether linker (73).148 Interestingly, the resulting homopolymers (Mn = 960–11000 g mol−1, ĐM = 1.06–1.42) displayed tunable thermoresponsive properties around body temperature.
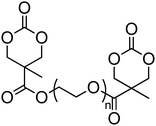

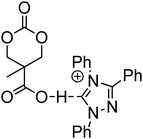
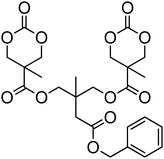

The synthesis of a triazolium-based cyclic carbonate, 76, that can act as an initiator for the anionic ROP of β-butyrolactone (BL) at elevated temperature was reported by Dubois and coworkers.149 To prove that the carbene catalyst was selective for the polymerisation of BL, its polymerisation from 76 was carried out at 90 °C in bulk. The resulting poly(β-butyrolactone) PBL cyclic carbonate macromonomer (77) was characterised by low dispersity (ĐM = 1.21) and preservation of the cyclic carbonate end-group as attested by 1H NMR spectroscopy. As an alternative, 76 was copolymerised with TMC at room temperature with a (−)-sparteine/4-chlorophenol cocatalyst system to give a co-poly(carbonate) with pendant triazolium groups (Mn = 4100 g mol−1; ĐM = 1.19) which, in a subsequent step could then be used as initiator for the ROP of BL in the preparation of graft copolymers.
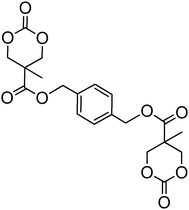
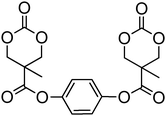
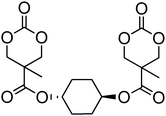
A range of other bis-cyclic carbonate cross-linkers and one tris-cyclic carbonate cross-linker have also been reported (78–84; Table 7) for the preparation of biodegradable cross-linked networks.44,150 Monomer 81 exists in both the trans- and cis-isomers which could be separated by column chromatography and isolated in a 1:1 ratio. Cross-linked networks were obtained by copolymerisation of the bifunctional monomers with TMC and ε-caprolactone at 140 °C in bulk in the presence of tin octanoate. As expected and in agreement with Flory's theory,151 the as-obtained cross-linked networks displayed increased glass transition temperatures with increased amount of cross-linker.
Other functional monomers
Several other six-membered cyclic carbonate monomers have been reported. Azidation of the 1,3-diol precursor to 46, followed by carbonate formation using ethyl chloroformate yielded diazido-functional 85. Poly(85) could be obtained at elevated temperatures under bulk conditions with tin-based catalysts (Mn = 10700–57900 g mol−1; ĐM = 1.20–1.41) or using DBU in methylene chloride at room temperature resulting in polymers with low dispersities (ĐM < 1.1). Poly(85) was also shown to be a semicrystalline material with a melting temperature ranging from 50 to 100 °C and a glass transition temperature that is slightly below 0 °C.136,137 Furthermore, 85 was reported in copolymerisations with DTC, lactide and in the preparation of block copolymers, by initiation from PEO macroinitiators. These copolymers were subsequently studied in hydrogel formation by “Click reaction”.57,58,136,137 In a comparable manner, Mindemark and Bowden described the azidation of 44 and 45 to yield the azide-functional analogues 86 and 87. While the ROP of these monomers was not reported, a range of triazole-functional monomers (88–97; Table 8) were prepared by Cu(I)-catalysed Huisgen dipolar cycloaddition with terminal alkynes. ROP of 89, 91, 93 and 95 were reported (target DP = 50) using the TU/DBU organocatalyst system to yield polymers with low dispersities (1.13–1.25) and Mn between 2300 and 5600 g mol−1.152
| Monomer | Number | Ref. | Monomer | Number | Ref. |
|---|---|---|---|---|---|
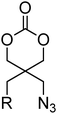
|
85 (R = N3) | 57, 58, 136, 137 and 152 |
|
102 | 45 |
| 86 (R = H) | |||||
| 87 (R = Me) | |||||
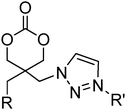
|
88 (R = H, R′ = Ph) | 152 |
|
103 | 44 |
| 89 (R = Me, R′ = Ph) | |||||
| 90 (R = H, R′ = C3H5) | |||||
| 91 (R = Me, R′ = C3H5) | |||||
| 92 (R = H, R′ = C3H7) | |||||
| 93 (R = Me, R′ = C3H7) | |||||
| 94 (R = H, R′ = C( = CH2)CH3) | |||||
| 95 (R = Me, R′ = C( = CH2)CH3) | |||||
| 96 (R = H, R′ = C(OH)Me2) | |||||
| 97 (R = Me, R′ = C(OH)Me2) | |||||
|
98 | 65 |
|
104 | 37 |
|
99 | 65 |
|
105 | 37 and 44 |
|
100 | 160 |
|
106 | 155 |
|
101 | 153 and 154 |
|
107 | 156 |
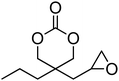
Other functional monomers reported include epoxide-functional cyclic carbonate, 98, which was converted to bifunctional cyclic carbonate 5-(2-oxo-1,3-dioxolan-4-yl)methyl-5-propyl-1,3-dioxan-2-one (99, Table 8).65 The selective polymerisation of the six-membered cyclic carbonate at 60 °C with DBU was achieved and the resulting homopolymer, with the pendant five-membered cyclic carbonate intact, was obtained in 50% yield (Mn = 14900 g mol−1; ĐM = 1.41). Hydroxyl-functional monomers 101 and 102 are incompatible for the controlled ROP process as the hydroxyl functionality acts as an initiating group. However, advantage was taken of this fact to prepare biodegradable, hydrophilic, hyperbranched hydroxyl-enriched aliphatic polycarbonates.45,153,154 Attaching PEG arms onto the hyperbranched structure led to self-assembly into supramolecular micelles with excellent stability and good cell biocompatibility.117
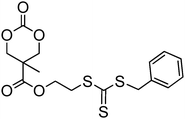
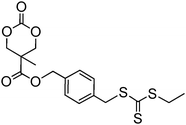
In a recent report by Lee et al., a cyclic carbonate with a pendant cholesterol group, cholesteryl-2-(5-methyl-2-oxo-1,3-dioxane-5-carboxyloyloxy) ethyl carbamate (100) was homopolymerised (DP 11) and copolymerised with TMC from a PEO macroinitiator using organic catalysts DBU and TU.160 Micelles prepared from the abovementioned block copolymers showed increased loading of the anticancer drug paclitaxel. It was hypothesised that this excellent compatibility is a result of the rigid aromatic structure of cholesterol and its available stereocenters that may interact with those in paclitaxel. Sulphide and disulphide functional cyclic carbonates 103, 104 and 105 (Table 8) have been reported, but their polymerisation has not yet been reported.37,44 Other sulfur-containing monomers have been recently reported, including those containing pendant groups capable of acting as chain-transfer agents (CTAs) in reverse addition-fragmentation chain transfer (RAFT) polymerisations.155,156 Mespouille et al. reported the synthesis of a bis-MPA-based cyclic carbonate bearing a 2-(benzylsulfanylthiocarbonylsulfanyl)ethyl ester, 106.155 ROP of 106 was investigated using both DBU and TU/(−)-sparteine catalysts with the latter system yielding polymers with lower dispersities (ĐM = 1.11–1.78). The polymerisation of N-isopropylacrylamide (NiPAAM) from the resulting polymers in a Z-group approach (in which the CTA remains bound to the backbone) in DMF at 70 °C for 8 h using AIBN as coinitiator resulted in successful chain grafting from the poly(carbonate) backbone. More recently, Williams, O'Reilly and Dove reported the synthesis of poly(carbonate) graft copolymers using ROP and an R-group RAFT approach in order that the CTA moiety be located at the graft chain termini.156 Here, the ROP of an analogous dual-functional monomer, 107, was undertaken simply using DBU as a catalyst to yield polymers with predictable molecular weights (in comparison to [M]/[I]) and low dispersities (ĐM < 1.16). Graft copolymers with NiPAAM, styrene and methyl acrylate were prepared in CHCl3 at 65 °C using AIBN as a coinitiator. Notably, the NiPAAM-grafted poly(carbonate)s displayed altered Tg's and lower critical solution temperatures that were in line with those expected based on their structure. Additionally, direct dissolution of these polymers in nanopure water at 3 °C resulted in the isolation of degradable nanoparticles.
Post-polymerisation modification of functional poly(carbonate)s
Deprotection of aryl- and alkyl-functional poly(carbonate)s
Alkyl- and aryl-functional groups are inert during most polymerisation conditions and are therefore often used to protect functional groups such as hydroxyl and carboxylic acid groups which may be incompatible in a ROP process. Aliphatic poly(carbonate)s containing pendant hydroxyl and carboxylic acid functions can be obtained by introducing benzyl ether or ester protecting groups respectively such that deprotection can be performed directly following the polymerisation process. Deprotection of the benzyl groups of poly(carbonate)s such as poly(MBC) and poly(17) has been reported by catalytic hydrogenolysis over palladium on carbon or palladium hydroxide on carbon to lead to carboxyl- and hydroxyl-functional poly(carbonate)s.42,78,90,92,93,95–102,104,157–159 In both cases the reduction in the molecular weight was confirmed by a shift to higher retention time observed by GPC analysis. Poly(17) and poly(MBC) are highly hydrophobic materials before deprotection, but the resulting hydroxyl and carboxylic acid functional polymers are hydrophilic materials that show enhanced biocompatibility and hydrosolubility compared to the benzyl-containing material.95,102 Homopolymers and copolymers of MBC also reveal a change in Tg after deprotection with poly(MBC) being isolated as a sticky amorphous polymer with Tg = ca. 3 °C. After deprotection, the polymer was isolated as a semicrystalline white powder with a higher Tg (47–60 °C) and a Tm = ca. 170 °C. Poly(17) homopolymers displayed an impressive 64-fold increase in water absorption after debenzylation. The bulk autodegradation process of poly(lactide) was increased in copolymers with 17 after benzyl deprotection due to the presence of the hydroxyl groups. The rate of degradation was found to be even faster under acidic conditions (pH = 4.4) than in neutral conditions (pH = 7.4).100 In a similar manner, hydroxyl-functional polymers were obtained by removal of the benzylidene group in polymers prepared from 19.50 In contrast to poly(17), poly(19) reveals two hydroxyl groups per repeat unit due to the double hydroxyl protection of the benzylidene group. Although to date no thermal properties are reported for poly(19), copolymers of lactide with 10% incorporation of deprotected 19 already show a decrease in Tg of 14 °C due to the increased flexibility of the polymer chains after removal of the rigid benzylidene groups. Interestingly, these lactide copolymers showed surface degradation in contrast to pure poly(lactide) which usually displays a bulk degradation profile. Facile deprotection of the benzylidene group could be achieved under acidic conditions in case of block copolymers of PEO-b-P(20) or PEO-b-P(21). In poly(20) and poly(21) the benzylidene groups are substituted with methoxy groups at the para-position and the ortho- and para-positions, respectively.105 Despite the high number of hydroxyl groups on the polymer, the deprotected poly(20/21) block is still insoluble in water and hence the block copolymers self-assemble into stable micelles at pH 7.4 and also show altered degradation profiles in slightly acidic conditions (pH 4–5). This polymer was successfully employed for the encapsulation of anti-cancer drugs and their subsequent pH-controlled release.105,161,162Fukushima et al.74 reported another example of a poly(carbonate) that deprotects under acidic conditions. In this contribution, deprotection of tetrahydropyranyloxy protecting groups in poly(10) was carried out at 50 °C using an ion-exchange resin to reveal hydroxyl functional polymers. The hydroxyl functionalities then acted as initiating sites in further ROP of stereopure poly(lactide) to form graft-copolymers with a poly(carbonate) backbone and stereopure poly(lactide) grafts. After mixing these PEO-poly(carbonate) block-copolymers grafted with PLA of opposite chirality (PLLA and PDLA), micelles could be formed at extremely low concentrations owing to the stereocomplexation driving force between PDLA and PLLA in the core of the micelles.
Jing et al.106,107 reported recently that the nitrobenzyl lateral groups in PEO-b-P(16) block copolymers could be deprotected under UV irradiation at 365 nm to convert the poly(carbonate) block from a hydrophobic block to a hydrophilic carboxylic acid-functional block. Micelles prepared from these PEO-b-P(16) block copolymers were shown to dissociate readily when irradiated due to the conversion of the amphiphilic block copolymers into hydrophilic copolymers.106,107
The newly available carboxylic acid and hydroxyl groups after deprotection are also open to further reactions. Further modification of carboxylic acid functional polymers were reported via N,N′-diisopropylcarbodiimide (DIC), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) or DCC couplings with free alcohol groups present in biologically active molecules or with N-hydroxysuccinimide (NHS) or hydroxybenzotriazole (HOBt) for further reaction with amines.95,96,98–100 Conjugation using these methods is limited since quantitative conversion of the acid groups into esters is rarely observed and can be as low as 20% in some reported cases. Examples of post-polymerisation modifications of acid-functional polymers obtained after deprotection included conjugation with aliphatic amines in the formation of DNA/polymer nanoparticles, the grafting of PEO-amines98 to form amphiphilic copolymers with a hydrophobic backbone and hydrophilic grafts that self-assemble in aqueous environments and conjugation with peptides95 or drugs such as heparin,93 docetaxel95 and paclitaxel.96 Further modifications of hydroxyl-functional monomers include reaction of the hydroxyl groups with acid chlorides (e.g. 4-isobutylmethylphenyl acetic acid).100
Post-polymerisation modifications of alkene-functional poly(carbonate)s
Alkenes participate in many reactions and are for that reason utilised extensively. In the post-polymerisation functionalisation of biodegradable polymers, the presence of carbon–carbon double bonds in the polymer chain113 or as lateral group have been utilised in cross-linking reactions and in the addition of new pendant functional moieties.44–48,60,65,68,71,76,115–134 Reported post-polymerisation functionalisations of alkenyl-functional poly(carbonate)s can divided into three general categories: cross-linking, thiol additions and epoxidations. Cross-linking of the hydrophobic core of micelles prepared from block copolymers with hydrophilic PEO blocks and hydrophobic poly(ester-carbonate) blocks with pendant allyl- or acryloyl-groups was realised by heating in the presence of azobisisobutyronitrile (AIBN) at 60 °C and by UV irradiation of the micelles in the presence of a biocompatible photoinitiator respectively.115,116,126 Micelles with cross-linked poly(ester-carbonate) cores, loaded with anti-cancer drug paclitaxel and with galactose groups at the micelle surface for specific targeting displayed enhanced antitumor efficacy in in vivo studies. The acroyl-functional monomer, 34, has also been copolymerised to form triblock PEG-b-Poly(34)-b-PLA copolymers.163 Here, the micellization in the presence of paclitaxel was followed by irradiation with UV light to crosslink the acroyl groups and stabilize the particle to prevent premature release of drug. In an extension to this work, synthesis of PEG-initiated statistical copolymers of 34 with 21 enabled the synthesis of crosslinked nanoparticles that were able to be deprotected upon lowering of the pH to enable a triggered release of the cargo.164 In another example, the radical cross-linking of a poly(carbonate) homopolymer with pendant styrene groups (poly(36)) was realised in the presence of the radical initiator benzoyl peroxide.48 The cross-linking was carried out in the presence of styrene to yield a poly(carbonate)/poly(styrene) network. Notably, the resulting cross-linked networks could be unlinked in the presence of t-BuOK, resulting in poly(styrene) with pendant six-membered cyclic carbonates. Photo-cross-linking of poly(ester-carbonates) (copolymers of L-lactide and 32) with pendant cinnamate functionalities could be cross-linked by UV irradiation at 365 nm providing biodegradable networks.124 This type of cross-linking occurs via dimerisation through the [2+2] cycloaddition reaction of two cinnamate moieties upon irradiation with UV light.The reaction of thiols and alkenes (‘thiol–ene’) has been utilised extensively as an efficient method in the preparation of functional polymers. Reactions proceed either via the free-radical addition of thiols to carbon–carbon double bonds or via the Michael-addition of thiols to electron-deficient carbon–carbon double bonds.111 Radical-type addition of thiols to the poly(carbonate) backbone has been reported for polymers containing pendant allyl functionalities.45,116,118,121 Homopolymers of MAC (29) were readily functionalized under radical ‘thiol–ene’ conditions (>2 eq. thiol, 90 °C, 24 h) in 1,4-dioxane with a range of thiol-containing species including some with functional groups that are not compatible with ring-opening polymerization such as alcohol and carboxylic acid groups.121 As expected the changes to the side-chain led to changes in the polymer Tg being observed, although the thermal degradation behaviour was largely unchanged. Copolymers prepared via the ROP of lactide and allyl-functional monomers (MAC, 29) could be functionalised with a number of thiols by UV irradiation at 254 nm in the presence of 1-hydroxycyclohexylphenyl ketone as radical initiator or without radical initiator.45,118 Although the reaction proceeds to high conversion at equimolar ene-thiol ratios, again an excess was necessary to achieve full conversion (5:1). Functionalisation of poly(ester-carbonates) with folic acid via radical thiol addition resulted in polymers which displayed increased cell affinity.116 Similarly, a range of alkene functional poly(carbonate)s could be modified with thiols via Michael-addition.47 PLA- or PCL-based polymers with low contents of acryloyl or methacryloyl functional cyclic carbonate monomers (34 and 35), were further functionalised in dimethylformamide at room temperature with a number of thiols.47 Quantitative conversions (using 10 equivalents of thiol) of the pendant (meth)acrylates was obtained for functionalisation with mercaptoethanol, cysteine and 2-mercaptoethylamine, but conjugation with a peptide only proceeded to 58% conversion whilst mercaptopropanoic acid only displayed a conversion of 30%. Conjugation of block copolymers composed of PEO and a small acryloyl-functional poly(carbonate) blocks (DP < 20) was performed with thiolated chitosan that led to in situ hydrogel formation.127 Such methodology was also extended to functionalise statistical copolymers of 34 with ε-caprolactone with cysteamine that was subsequently converted into a CTA for the RAFT polymerisation of hydroxyethylmethacrylate. In turn, these copolymers were assembled into graft copolymer micelles that showed pH-responsive release of doxorubicin.165 Vinyl sulfone-functional copoly(carbonate)s and poly(ester-carbonate)s, obtained from the copolymerisation of 38 with other cyclic monomers, could also be quantitatively functionalised via the Michael addition of thiols to the vinyl group without catalyst and under aqueous conditions.64 Another example of functionalisation of poly(carbonate)s and poly(ester-carbonate)s via Michael-addition of thiols was presented by Onbulak et al.129 Polymers with pendant furan-protected maleimides were deprotected via thermal retro-Diels Alder to give maleimide functional polymers that were further functionalised with the thiol-containing molecules, 6-(ferrocenyl)hexanethiol and 1-hexanethiol, in the presence of triethylamine. Notably, both deprotection and thiol addition proceeded without degradation of the polymer backbone.
Finally, alkene functional poly(carbonate)s could also be epoxidised using meta-chloroperoxybenzoic acid (m-CPBA). Epoxidation of functional aliphatic poly(carbonate)s and poly(ester-carbonate)s was reported for pendant allyl groups and pendant cyclohexenyl groups with efficiencies ranging from 35 to 99%. Hydrolysis of the epoxide groups was noted to occur in some cases.46,76,119–123 In case of epoxidation of PMAC homopolymers an increase in glass transition temperature from −12 °C to 14 °C was observed. The introduction of this new reactive functionality was applied in the grafting of poly(ethylene imine) to the poly(carbonate) backbone in the formation of biodegradable polycations and polycation/DNA polyplexes by Zhuo and coworkers.119
Post-polymerisation modifications of poly(carbonate)s via dipolar cycloadditions
Alkyne- and azide-functional poly(carbonate)s or poly(ester-carbonate)s were successfully functionalised via 1,3-dipolar cycloaddition reactions.57,58,69,73,150,126–129 Modifications of poly(ester-carbonates) obtained from copolymerisation of MPC (41) with lactide were successfully functionalised with azido-containing molecules using copper(II) sulfate pentahydrate and sodium ascorbate in water–DMSO mixtures.130,133,134 Modifications with glucose and galactose azides were shown to lead to increased biocompatibility and enhanced hydrophilicity and cellular affinity of the resulting polymers.134 Similarly, some polymers were also modified with CO-protected hemoglobin and proteins including Testis-specific protease 50 (TSP50), a breast cancer sensitive protease, and bovine serum albumin (BSA). The latter achieved only low grafting efficiencies of 0.7–1.6 mol%.130,133 Alternatively, azide-functional poly(carbonate)s could be obtained via polymerisation of azide functional cyclic carbonates or via modification of bromide functional poly(carbonate)s with sodium azide in DMF at various temperatures.57,58 The latter method, however, has limitations as azidation of bromide-functional polymers did not exceed 25% for homopolymers and degradation of the poly(carbonate) backbone was observed in both bromide functional homopolymers and copolymers.57,58 Successful cycloaddition of alkyne-functional molecules to azido-functional poly(carbonate)s via copper catalysed methods (CuAAC) and via strain-promoted methods (SPAAC) was reported by Song and coworkers and was used in the preparation of hydrogels by cross-linking of poly(carbonate)s with DABCO-terminated poly(ethylene oxide)s.136,137Quaternisation of halogen-functional poly(carbonate)s
A range of polymers containing quaternary amines have shown to exhibit antimicrobial activity. These polymers are, however, often not degradable which limits their use. The quaternisation of amines by reaction with halogen-containing compounds enables poly(carbonate) homopolymers and copolymers bearing pendant halogen functionalities to be reacted with tertiary amines such as trimethylamine or N,N,N′,N′-tetramethylethylenediamine. Recently, Yang and coworkers reported the first example of quaternised poly(carbonate) nanoparticles prepared from block copolymers of PTMC and a chloro-functional poly(carbonate) prepared from monomer 48.139 Investigation of quaternisation conditions revealed a dependence on the halogen substituent on the pendant chain. While chloro-functional poly(carbonate)s required heating at 90 °C, bromo- and iodo-functional poly(carbonate)s could be fully converted at room temperature. Another application for these cationic poly(carbonate)s was found in the synthesis of poly(carbonate)/DNA complexes.69,72,138,139Deprotection of carbamate-functional poly(carbonate)s
Poly(carbonate)s with pendant primary amines can be prepared via the ROP of monomers in which the primary amines are protected by conversion to benzyl- or tert-butyl-carbamate groups. Deprotection of the benzyl carbamate (Z) groups is carried out via hydrogenation catalysed by palladium while the tert-butyl-carbamate (Boc) groups can easily be removed using trifluoroacetic acid.52,55 Resulting primary amine-functional poly(carbonate)s and poly(ester-co-carbonate)s have been obtained whilst retaining low dispersities. As expected, a reduction in molecular weight was observed after deprotection and polymers were highly hydrophilic in contrast to the protected polymers. Primary amine functional poly(ester-carbonate)s could be further modified by reaction with peptides in the presence of CDI. Films of the resulting polymers displayed increased cell adhesion and multiplication.55 In another report, two Boc-protecting groups were removed to reveal pendant guanidinium groups. Hydrogels prepared from bis(carbonate) poly(ethylene oxide) cross-linker, Boc protected guanidinium functional cyclic carbonate and tert-butoxy protected carboxylic acid functional cyclic carbonate revealed additional stabilisation of the hydrogel network after removal of the protecting groups with trifluoroacetic acid as a result of the hydrogen bonding between the free guanidinium and carboxylic acid groups.87Other post-polymerisation modifications of functional poly(carbonate)s
Putnam and coworkers has reported a post-polymerisation conversion of dimethoxy-functional poly(carbonate), poly(4), and its copolymers into dihydroxy acetone-based poly(carbonate)s.63 The conversion to dihydroxyacetone functional poly(carbonate)s was accomplished by refluxing poly(4) with iodine in acetone. The resulting polymer presents an increase in glass transition temperature compared to the precursor from 45 to 68 °C. In addition, the resulting polymers showed very low solubility in common organic solvents. Moreover, copolymers composed of lactide and dihydroxyacetone-functional carbonate showed increasing degradation kinetics in copolymers enriched in functional carbonate.Conclusions
The combination of the many versatile methods reported for the synthesis of functional cyclic carbonate monomers from a range of biocompatible scaffolds and the excellent control in their ring-opening polymerisation, have led to preparation of a multitude of functional poly(carbonate)s with controlled molecular weights, low dispersities and high end-group fidelity. Despite the development of the large range of functional aliphatic poly(carbonate)s, their specific design to achieve better-controlled drug release and advanced tissue engineering scaffolds is very rare. Further development of controlled poly(carbonate) synthesis in combination with efficient post-polymerisation modifications with a specific focus on application could open up new ways to achieve their widespread biomedical application.Acknowledgements
The Research Councils UK (RCUK) are acknowledged for funding a fellowship to A.P.D. We gratefully acknowledge financial support from EPSRC (EP/F068808/1) for funding a studentship (S.T.). This work was supported by the European Commission and Région Wallonne FEDER program (Materia Nova) and OPTI²MAT program of excellence, by the Interuniversity Attraction Pole program of the Belgian Federal Science Policy Office (PAI 6/27) and by FNRS-FRFC. O. C. is a FNRS Research Associate.Notes and references
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- H. Keul, Polycarbonates, Wiley-VCH Verlag GmbH & Co. KGaA, New York, 2009, pp. 307–327 Search PubMed.
- Z. Zhang, D. W. Grijpma and J. Feijen, J. Controlled Release, 2006, 111, 263–270 CrossRef CAS.
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS.
- C. G. Pitt and Z. W. Gu, J. Controlled Release, 1987, 4, 283–292 CrossRef CAS.
- K. Fu, D. W. Pack, A. M. Klibanov and R. Langer, Pharm. Res., 2000, 17, 100–106 CrossRef CAS.
- A. P. Pêgo, M. J. A. Van Luyn, L. A. Brouwer, P. B. Van Wachem, A. A. Poot, D. W. Grijpma and J. Feijen, J. Biomed. Mater. Res., Part A, 2003, 67A, 1044–1054 CrossRef.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS.
- B. Ochiai and T. Endo, Prog. Polym. Sci., 2005, 30, 183–215 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS.
- J. Feng, R. X. Zhuo and X. Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237–266 CrossRef CAS.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- S. Matsumura, K. Tsukada and K. Toshima, Macromolecules, 1997, 30, 3122–3124 CrossRef CAS.
- R. F. Storey, B. D. Mullen, G. S. Desai, J. W. Sherman and C. N. Tang, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3434–3442 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153–160 CrossRef CAS.
- O. Coulembier, D. P. Sanders, A. Nelson, A. N. Hollenbeck, H. W. Horn, J. E. Rice, M. Fujiwara, P. Dubois and J. L. Hedrick, Angew. Chem., Int. Ed., 2009, 48, 5170–5173 CrossRef CAS.
- M. Pastusiak, P. Dobrzynski, B. Kaczmarczyk and J. Kasperczyk, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2504–2512 CrossRef CAS.
- Y. Yamamoto, S. Kaihara, K. Toshima and S. Matsumura, Macromol. Biosci., 2009, 9, 968–978 CrossRef CAS.
- S. Matsumura, K. Tsukada and K. Toshima, Macromolecules, 1997, 30, 3122–3124 CrossRef CAS.
- K. Odelius and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1249–1264 CrossRef CAS.
- B. L. Dargaville, C. Vaquette, H. Peng, F. Rasoul, Y. Q. Chau, J. J. Cooper-White, J. H. Campbell and A. K. Whittaker, Biomacromolecules, 2011, 12, 3856–3869 CrossRef CAS.
- S. Dai, L. Xue and Z. Li, ACS Catal., 2011, 1, 1421–1429 CrossRef CAS.
- M. Normand, E. Kirillov, J. F. Carpentier and S. M. Guillaume, Macromolecules, 2012, 45, 1122–1130 CrossRef CAS.
- J. Liu, C. Zhang and L. Liu, J. Appl. Polym. Sci., 2008, 107, 3275–3279 CrossRef CAS.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, J. Pharm. Sci., 2012, 101, 588–597 CrossRef CAS.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS.
- M. Helou, G. Moriceau, Z. W. Huang, S. Cammas-Marion and S. M. Guillaume, Polym. Chem., 2011, 2, 840–850 RSC.
- F. Nederberg, V. Trang, R. C. Pratt, A. F. Mason, C. W. Frank, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 3294–3297 CrossRef CAS.
- O. Thillaye du Boullay, N. Saffon, J.-P. Diehl, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2010, 11, 1921–1929 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS.
- J. Mindemark and T. Bowden, Polymer, 2011, 52, 5716–5722 CrossRef CAS.
- P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 5775–5777 RSC.
- M. Malkoch, E. Malmstrom and A. Hult, Macromolecules, 2002, 35, 8307–8314 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, Ph. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- T. F. Al-Azemi and K. S. Bisht, Macromolecules, 1999, 32, 6536–6540 CrossRef CAS.
- K. D. Weilandt, H. Keul and H. Hocker, Macromol. Chem. Phys., 1996, 197, 3851–3868 CrossRef CAS.
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- P. G. Parzuchowski, M. Jaroch, M. Tryznowski and G. Rokicki, Macromolecules, 2008, 41, 3859–3865 CrossRef CAS.
- F. He, Y.-P. Wang, G. Liu, H.-L. Jia, J. Feng and R.-X. Zhuo, Polymer, 2008, 49, 1185–1190 CrossRef CAS.
- W. Chen, H. Yang, R. Wang, R. Cheng, F. Meng, W. Wei and Z. Zhong, Macromolecules, 2010, 43, 201–207 CrossRef CAS.
- T. Miyagawa, M. Shimizu, F. Sanda and T. Endo, Macromolecules, 2005, 38, 7944–7949 CrossRef CAS.
- J. Yang, Q. Hao, X. Liu, C. Ba and A. Cao, Biomacromolecules, 2004, 5, 209–218 CrossRef CAS.
- Z. Xie, C. Lu, X. Chen, L. Chen, Y. Wang, X. Hu, Q. Shi and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1737–1745 CrossRef CAS.
- S.-H. Pyo, P. Persson, S. Lundmark and R. Hatti-Kaul, Green Chem., 2011, 13, 976–982 RSC.
- F. Sanda, J. Kamatani and T. Endo, Macromolecules, 2001, 34, 1564–1569 CrossRef CAS.
- R. Kumar, W. Gao and R. A. Gross, Macromolecules, 2002, 35, 6835–6844 CrossRef CAS.
- X. Chen and R. A. Gross, Macromolecules, 1999, 32, 308–314 CrossRef CAS.
- X. Hu, X. Chen, Z. Xie, H. Cheng and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7022–7032 CrossRef CAS.
- R. D. Youssefyeh, J. P. H. Verheyden and J. G. Moffatt, J. Org. Chem., 1979, 44, 1301–1309 CrossRef CAS.
- X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755–1759 CrossRef CAS.
- W. Zhu, Y. Wang, Q. Zhang and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4886–4893 CrossRef CAS.
- H.-F. Wang, W. Su, C. Zhang, X.-H. Luo and J. Feng, Biomacromolecules, 2010, 11, 2550–2557 CrossRef CAS.
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1997, 30, 3470–3476 CrossRef CAS.
- D. J. Darensbourg, A. I. Moncada and S. H. Wei, Macromolecules, 2011, 44, 2568–2576 CrossRef CAS.
- M. Helou, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2011, 2, 2789–2795 RSC.
- J. R. Weiser, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2011, 12, 977–986 CrossRef CAS.
- R. Wang, W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2011, 44, 6009–6016 CrossRef CAS.
- T. Endo, K. Kakimoto, B. Ochiai and D. Nagai, Macromolecules, 2005, 38, 8177–8182 CrossRef CAS.
- A. E. van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef CAS.
- A. E. van der Ende, J. Harrell, V. Sathiyakumar, M. Meschievitz, J. Katz, K. Adcock and E. Harth, Macromolecules, 2010, 43, 5665–5671 CrossRef CAS.
- R. M. Burk and M. B. Roof, Tetrahedron Lett., 1993, 34, 395–398 CrossRef CAS.
- Y. Qiao, C. Yang, D. J. Coady, Z. Y. Ong, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 1146–1153 CrossRef CAS.
- S. H. Kim, J. P. K. Tan, K. Fukushima, F. Nederberg, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomaterials, 2011, 32, 5505–5514 CrossRef CAS.
- S.-H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y. Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS.
- Z. Y. Ong, K. Fukushima, D. J. Coady, Y. Y. Yang, P. L. R. Ee and J. L. Hedrick, J. Controlled Release, 2011, 152, 120–126 CrossRef CAS.
- J. P. K. Tan, S. H. Kim, F. Nederberg, K. Fukushima, D. J. Coady, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromol. Rapid Commun., 2010, 31, 1187–1192 CrossRef CAS.
- K. Fukushima, R. C. Pratt, F. Nederberg, J. P. K. Tan, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2008, 9, 3051–3056 CrossRef CAS.
- F. He, Y. Wang, J. Feng, R. Zhuo and X. Wang, Polymer, 2003, 44, 3215–3219 CrossRef CAS.
- F. He, C. F. Wang, T. Jiang, B. Han and R. X. Zhuo, Biomacromolecules, 2010, 11, 3028–3035 CrossRef CAS.
- X. Zhang, F. Chen, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2010, 31, 2155–2159 CrossRef CAS.
- X. Zhang, H. Mei, C. Hu, Z. Zhong and R. Zhuo, Macromolecules, 2009, 42, 1010–1016 CrossRef CAS.
- N. Nemoto, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1305–1317 CrossRef CAS.
- O. Coulembier, D. P. Sanders, A. Nelson, A. N. Hollenbeck, H. W. Horn, J. E. Rice, M. Fujiwara, P. Dubois and J. L. Hedrick, Angew. Chem., Int. Ed., 2009, 48, 5170–5173 CrossRef CAS; O. Coulembier, D. P. Sanders, A. Nelson, A. N. Hollenbeck, H. W. Horn, J. E. Rice, M. Fujiwara, P. Dubois and J. L. Hedrick, Angew. Chem., 2009, 121, 5272–5275 CrossRef.
- T. Ariga, T. Takata and T. Endo, Macromolecules, 1997, 30, 737–744 CrossRef CAS.
- J. Ling, Z. Shen and Q. Huang, Macromolecules, 2001, 34, 7613–7616 CrossRef CAS.
- H. Peng, J. Ling, J. Liu, N. Zhu, X. Ni and Z. Shen, Polym. Degrad. Stab., 2010, 95, 643–650 CrossRef CAS.
- B. Liu and D. Cui, J. Appl. Polym. Sci., 2009, 112, 3110–3118 CrossRef CAS.
- J. Cai, K. J. Zhu and S. L. Yang, Polymer, 1998, 39, 4409–4415 CrossRef CAS.
- M. Helou, O. Miserque, J. M. Brusson, J. F. Carpentier and S. M. Guillaume, Chem.–Eur. J., 2010, 16, 13805–13813 CrossRef CAS.
- C. Bartolini, L. Mespouille, I. Verbruggen, R. Willem and Ph. Dubois, Soft Matter, 2011, 7, 9628–9637 RSC.
- A. N. Zelikin, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2006, 7, 3239–3244 CrossRef CAS.
- P. N. Zawaneh, A. M. Doody, A. N. Zelikin and D. Putnam, Biomacromolecules, 2006, 7, 3245–3251 CrossRef CAS.
- P. Brignou, J. F. Carpentier and S. M. Guillaume, Macromolecules, 2011, 44, 5127–5135 CrossRef CAS.
- J. Ling, Y. Dai, Y. Zhu, W. Sun and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3807–3815 CrossRef CAS.
- T. F. Al-Azemi, J. P. Harmon and K. S. Bisht, Biomacromolecules, 2000, 1, 493–500 CrossRef CAS.
- F. Gong, X. Cheng, S. Wang, Y. Zhao, Y. Gao and H. Cai, Acta Biomater., 2010, 6, 534–546 CrossRef CAS.
- Y. Yuan, X. Jing, H. Xiao, X. Chen and Y. Huang, J. Appl. Polym. Sci., 2011, 121, 2378–2385 CrossRef CAS.
- Z. Xie, X. Hu, X. Chen, T. Lu, S. Liu and X. Jing, J. Appl. Polym. Sci., 2008, 110, 2961–2970 CrossRef CAS.
- M. Danquah, T. Fujiwara and R. I. Mahato, Biomaterials, 2010, 31, 2358–2370 CrossRef CAS.
- W. Lu, F. Li and R. I. Mahato, J. Pharm. Sci., 2011, 100, 2418–2429 CrossRef CAS.
- J. Lu and M. S. Shoichet, Macromolecules, 2010, 43, 4943–4953 CrossRef CAS.
- W. Y. Seow and Y. Y. Yang, J. Controlled Release, 2009, 139, 40–47 CrossRef CAS.
- W. C. Ray III and M. W. Grinstaff, Macromolecules, 2003, 36, 3557–3562 CrossRef.
- X. L. Wang, R. X. Zhuo, L. J. Liu, F. He and G. Liu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 70–75 CrossRef CAS.
- J. Feng, X. L. Wang, F. He and R. X. Zhuo, Macromol. Rapid Commun., 2007, 28, 754–758 CrossRef CAS.
- L. L. Mei, G. P. Yan, X. H. Yu, S. X. Cheng and J. Y. Wu, J. Appl. Polym. Sci., 2008, 108, 93–98 CrossRef CAS.
- H. Chen, G. P. Yan, L. Li, C. W. Ai and X. H. Yu, J. Appl. Polym. Sci., 2009, 114, 3087–3096 CrossRef CAS.
- W. Chen, F. Meng, F. Li, S. J. Ji and Z. Zhong, Biomacromolecules, 2009, 10, 1727–1735 CrossRef CAS.
- Z. Xie, X. Hu, X. Chen, G. Mo, J. Sun and X. Jing, Adv. Eng. Mater., 2009, 11, B7–B11 CrossRef.
- Z. Xie, X. Hu, X. Chen, J. Sun, Q. Shi and X. Jing, Biomacromolecules, 2008, 9, 376–380 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS.
- Z. Ates, P. D. Thornton and A. Heise, Polym. Chem., 2011, 2, 309–312 RSC.
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jérôme, R. Jérôme and P. Lecomte, Chem. Commun., 2005, 5334–5336 RSC.
- X. Hu, X. Chen, Z. Xie, S. Liu and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5518–5528 CrossRef CAS.
- X. Hu, X. Chen, S. Liu, Q. Shi and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852–1861 CrossRef CAS.
- X. Hu, X. Chen, J. Wei, S. Liu and X. Jing, Macromol. Biosci., 2009, 9, 456–463 CrossRef CAS.
- J. Yue, X. Li, G. Mo, R. Wang, Y. Huang and X. Jing, Macromolecules, 2010, 43, 9645–9654 CrossRef CAS.
- C. F. Wang, Y. X. Lin, T. Jiang, F. He and R.-X. Zhuo, Biomaterials, 2009, 30, 4824–4832 CrossRef CAS.
- B. D. Mullen, C. N. Tang and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1978–1991 CrossRef CAS.
- S. Tempelaar, L. Mespouille, P. Dubois and A. P. Dove, Macromolecules, 2011, 44, 2084–2091 CrossRef CAS.
- W. Zhao, Y. Wang, X. Liu and D. Cui, Chem. Commun., 2012, 48, 4588–4590 RSC.
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1998, 31, 662–668 CrossRef CAS.
- X. Hu, X. Chen, H. Cheng and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 161–169 CrossRef CAS.
- P. Lundberg, A. Bruin, J. W. Klijnstra, A. M. Nyström, M. Johansson, M. Malkoch and A. Hult, ACS Appl. Mater. Interfaces, 2010, 2, 903–912 CAS.
- R. Yang, F. Meng, S. Ma, F. Huang, H. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 3047–3055 CrossRef CAS.
- Y. Yu, C. Deng, F. Meng, Q. Shi, J. Feijen and Z. Zhong, J. Biomed. Mater. Res., Part A, 2011, 99A, 316–326 CrossRef CAS.
- M. Murayama, F. Sanda and T. Endo, Macromolecules, 1998, 31, 919–923 CrossRef CAS.
- S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS.
- T.-H. Lia, X.-B. Jing and Y.-B. Huang, Polym. Adv. Technol., 2011, 22, 1266–1271 CrossRef.
- Y. Han, Q. Shi, J. Hu, Q. Du, X. Chen and X. Jing, Macromol. Biosci., 2008, 8, 638–644 CrossRef CAS.
- Q. Shia, X. Chena, T. Lua and X. Jing, Biomaterials, 2008, 29, 1118–1126 CrossRef.
- Q. Shi, Y. Huang, X. Chen, M. Wu, J. Sun and X. Jing, Biomaterials, 2009, 30, 5077–5085 CrossRef CAS.
- C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Polym. Chem., 2010, 1, 1205–1214 RSC.
- J. Xu, T. M. Filion, F. Prifti and J. Song, Chem.–Asian J., 2011, 6, 2730–2737 CrossRef CAS.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS.
- C. Yang, Z. Y. Ong, Y. Y. Yang, P. L. R. Ee and J. L. Hedrick, Macromol. Rapid Commun., 2011, 32, 1826–1833 CrossRef CAS.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y. Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS.
- C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401–16403 CrossRef CAS.
- X. Zhang, M. Cai, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2012, 33, 693–697 CrossRef CAS.
- F. Nederberg, V. Trang, R. C. Pratt, S. H. Kim, J. Colson, A. Nelson, C. W. Frank, J. L. Hedrick, P. Dubois and L. Mespouille, Soft Matter, 2010, 6, 2006–2012 RSC.
- M. Fujiwara, J. L. Hedrick, D. P. Sanders, Y. Terui and M. Yasumoto, (International Buisness Machines, USA; Central Glass Co. Ltd. Japan). Polymers bearing pendant pentafluorophenyl ester groups, and methods of synthesis and functionalization thereof. US Pat., 8,143,369 B2, March 27, 2012 Search PubMed.
- S. Slavin, J. Burns, D. M. Haddleton and C. R. Becer, Eur. Polym. J., 2011, 47, 435–446 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 3891–3897 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799–2802 CrossRef CAS.
- F. Suriano, R. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y. Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS.
- H. Ajiro, Y. Takahashi and M. Akashi, Macromolecules, 2012, 45, 2668–2674 CrossRef CAS.
- O. Coulembier, S. Moins and P. Dubois, Macromolecules, 2011, 44, 7493–7497 CrossRef CAS.
- T. F. Al-Azemi and K. S. Bisht, Polymer, 2002, 43, 2161–2167 CrossRef CAS.
- P. J. Flory and P. Rehner, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- J. Mindemark and T. Bowden, Polym. Chem., 2012, 3, 1399–1401 RSC.
- X.-H. Luo, F.-W. Huang, S.-Y. Qin, H.-F. Wang, J. Feng, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2011, 32, 9925–9939 CrossRef CAS.
- W. Su, X.-H. Luo, H.-F. Wang, L. Li, J. Feng, X. Z. Zhang and R.-X. Zhuo, Macromol. Rapid Commun., 2011, 32, 390–396 CrossRef CAS.
- L. Mespouille, F. Nederberg, J. L. Hedrick and P. Dubois, Macromolecules, 2009, 42, 6319–6321 CrossRef CAS.
- R. J. Williams, R. K. O'Reilly and A. P. Dove, Polym. Chem., 2012, 3, 2156–2164 RSC.
- F. Li, M. Danquah and R. I. Mahato, Biomacromolecules, 2010, 11, 2610–2620 CrossRef CAS.
- J. Lu, M. Shi and M. S. Shoichet, Bioconjugate Chem., 2009, 20, 87–94 CrossRef CAS.
- X. Hu, S. Liu, X. Chen, G. Mo, Z. Xie and X. Jing, Biomacromolecules, 2008, 9, 553–560 CrossRef CAS.
- A. L. Z. Lee, S. Venkataraman, S. B. M. Sirat, S. Gao, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2012, 33, 1921–1928 CrossRef CAS.
- E. J. Vandenberg and D. Tian, Macromolecules, 1999, 32, 3613–3619 CrossRef CAS.
- W. Chen, F. Meng, R. Cheng and Z. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS.
- J. Xiong, F. Meng, C. Wang, R. Cheng, Z. Liu and Z. Zhong, J. Mater. Chem., 2011, 21, 5786–5794 RSC.
- Y. Wu, W. Chen, F. Meng, Z. Wang, R. Cheng, C. Deng, H. Liu and Z. Zhong, J. Controlled Release, 2012 DOI:10.1016/j.jconrel.2012.07.011.
- J. Cheng, X. Wang, W. Chen, F. Meng, C. Deng, H. Liu and Z. Zhong, J. Mater. Chem., 2012, 22, 11730–11738 RSC.
| This journal is © The Royal Society of Chemistry 2013 |