Cation exchange on the nanoscale: an emerging technique for new material synthesis, device fabrication, and chemical sensing
Jessy B.
Rivest
a and
Prashant K.
Jain
*b
aThe Molecular Foundry, Materials Science Division, Lawrence Berkeley National Lab, Berkeley, CA 94720
bDepartment of Chemistry, of Physics, and the Beckman Institute of Advanced Science and Technology, University of Illinois, Urbana-Champaign, IL 61801. E-mail: jain@illinois.edu
First published on 11th September 2012
Abstract
Cation exchange is an age-old technique for the chemical conversion of liquids or extended solids by place-exchanging the cations in an ionic material with a different set of cations. The technique is undergoing a major revival with the advent of high-quality nanocrystals: researchers are now able to overcome the limitations in bulk systems and fully exploit cation exchange for materials synthesis and discovery via rapid, low-temperature transformations in the solid state. In this tutorial review, we discuss cation exchange as a promising materials synthesis and discovery tool. Exchange on the nanoscale exhibits some unique attributes: rapid kinetics at room temperature (orders of magnitude faster than in the bulk) and the tuning of reactivity via control of nanocrystal size, shape, and surface faceting. These features make cation exchange a convenient tool for accessing nanocrystal compositions and morphologies for which conventional synthesis may not be established. A simple exchange reaction allows extension of nanochemistry to a larger part of the periodic table, beyond the typical gamut of II–VI, IV–VI, and III–V materials. Cation exchange transformations in nanocrystals can be topotactic and size- and shape-conserving, allowing nanocrystals synthesized by conventional methods to be used as templates for production of compositionally novel, multicomponent, or doped nanocrystals. Since phases and compositions resulting from an exchange reaction can be kinetically controlled, rather than governed by the phase diagram, nanocrystals of metastable and hitherto inaccessible compositions are attainable. Outside of materials synthesis, applications for cation exchange exist in water purification, chemical staining, and sensing. Since nanoscale cation exchange occurs rapidly at room temperature, it can be integrated with sensitive environments such as those in biological systems. Cation exchange is already allowing access to a variety of new materials and processes. With better mechanistic understanding and control, researchers may be able to advance the field to a stage where a custom nanostructure of arbitrary complexity would be achievable by simple cation exchange chemistry and a basic understanding of the periodic table.
 Jessy B. Rivest | Jessy Baker Rivest earned her undergraduate degree at the Massachusetts Institute of Technology. After working in the photovoltaics industry, she obtained her PhD at UC Berkeley in 2011 under A. Paul Alivisatos, investigating self-assembly and cation exchange of nanocrystals. She is currently a postdoctoral scholar with Delia Milliron at the Molecular Foundry, LBNL. Jessy's research interests surround the materials science and interfacial chemistry of clean energy technologies. |
 Prashant K. Jain | Prashant K. Jain is an Assistant Professor of Chemistry and IACAT faculty fellow at UIUC with affiliations in Physics and the Beckman Institute. His laboratory research is focussed on nano-optics, plasmonics, and super-resolution chemical imaging, areas in which his work has been cited over 4000 times. Prashant obtained his PhD from Georgia Tech in 2008 followed by postdoctoral work at Harvard and a Miller fellowship at UC Berkeley from 2009 to 2011. His lab webpage can be found at http://nanogold.org |
1. Introduction
Major advances in our understanding of optical and electronic behavior on the nanoscale, and fundamental size scaling laws, have been catapulted by the ability to synthesize inorganic nanocrystals with exquisite size and shape control.1–3 This is exemplified by the case of binary nanocrystals (II–VI, IV–VI, and III–V type), for which colloidal synthetic techniques are often most advanced,4–8 though other high-temperature techniques such as vapor–liquid–solid and lithography are also popular for nanostructure fabrication.9,10The traditional method for colloidal synthesis of nanocrystals is hot-injection: organometallic or metal–organic precursors are allowed to react at high temperatures (ca. 300 °C) to form nuclei of the binary compound, which further grow into nanocrystals.4 Kinetics of crystal growth – tunable by presence of coordinating ligands – govern nanocrystal size and shape,7 whereas thermodynamics often dictates the phase and/or chemical composition of the nanocrystals. Nanocrystals of tunable shape and composition can be routinely synthesized via hot injection, making it the de facto method for colloidal semiconductor nanocrystal synthesis.
Recent literature has brought to the fore a new strategy for the fabrication of ionic nanocrystals: cation exchange, a phenomenon in which the cations comprising an ionic solid can be place-exchanged with different cations. While solid-state cation exchange has been known in extended solids for decades,11 the more recent cation exchange in nano-sized crystals shows attributes unique to the nanoscale, notably remarkable reaction speeds.12 Whereas cation exchange in a bulk, or extended solid, crystal can take weeks at elevated temperatures, the reaction in nanocrystals is typically complete within seconds, likely due to enhanced surface access and lower activation barriers to diffusing ions.
Because of facile reaction conditions, cation exchange has been quickly identified as a strategy for post-synthetic chemical modification of nanocrystals. With one simple and rapid room temperature chemical step, one can achieve nanocrystal compositions outside of the typical gamut of easily-synthesized II–VI, III–V, and IV–VI. Cation exchange enables the exploration of new materials by allowing access to nanoparticles of compositions or morphologies that are difficult to achieve by traditional hot-injection techniques. By definition, cation exchange requires nanocrystals synthesized using another technique; one can consider the nanocrystal as a solid-state template for fabricating new nanocrystals – in fact, the size and shape of the nanocrystal template is conserved in a properly-designed exchange reaction.12,13 Cation exchange also enables the building of complexity in nanostructures;14–17 depending on the extent of the exchange – from a few % to complete conversion – it is possible to achieve, in progression, a doped nanocrystal, a heterostructure, or a compositionally and/or structurally new nanocrystal.
Cation exchange is not simply an alternative nanofabrication tool, but a mechanistically unique one. In hot injection synthesis, there is typically a dynamic equilibrium between the growing nanocrystal and the monomer in solution, which often constrains the phase and composition of the product nanocrystals to be the most stable one.5 Cation exchange, on the other hand, yields phases and compositions that are often kinetically, rather than thermodynamically, controlled. It is possible, in such a low-temperature and fast place-exchange of ions, to arrive at phases and compositions that are metastable, not otherwise accessible, or even undiscovered. This is a paradigm shift from conventional synthesis.
In the following sections, we provide a perspective on the promise of cation exchange using recent examples from our work and related work in the literature. We emphasize the abundant opportunities to utilize cation exchange as a tool for custom design and discovery of novel nanostructures; for accessing non-equilibrium phases and compositions; for fabrication of heterostructures or nano-scale devices; or for chemical sensing. An ultimate dream for the scientific community is to be able to arrive at a nanocrystal of arbitrarily desired structure and composition simply by using cation exchange chemistry and knowledge of the periodic table.
2. Cation exchange in bulk crystals: mechanistic insights
Cation exchange, simply put, involves replacing the cations in an ionic crystal while the anionic framework remains intact. This happens by exposing the parent ionic crystal to new cations, either in solid or solvated liquid form. The new cations enter the parent crystal as the original cations diffuse out of the crystal and into the reaction solvent or matrix (see Fig. 1). | ||
| Fig. 1 Guidelines for achieving exchange, and schematic of reaction. | ||
The analogous anion exchange has also been reported,18,19 but due to the generally larger radius of an anion (extra valence electrons), anions often have low mobilities that necessitate high reaction temperatures or long reaction times, resulting in poor-quality product materials. In fact, it is the very slow anionic diffusivity that ensures the structural rigidity of the solid lattice during cation exchange, enabling high-quality materials. For this reason, we will restrict our discussion here to cation exchange, although a few examples of high-quality anion exchange syntheses are also known.20
The mechanism for crystal cation exchange has been investigated in detail in order to understand geological mineral replacement reactions such as calcite (CaCO3) or aluminosilicate (Al2SiO5) feldspar structures, which can encode fine features of fossilized remains or can preserve the crystallographic information of the parent crystal, including grain sizes and orientations.21
The overall cation exchange reaction is
| Mn+(liquid) + C–A(crystal) → Cn+(liquid) + M–A(crystal). | (1) |
C–A → C + A (dissociation, i.e. 160 kJ/mol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22) 22) | (2) |
| Mn+ → M (desolvation, i.e. 400 kJ/mol23) | (3) |
| M + A → M–A (association, i.e. −130 kJ/mol22) | (4) |
| C → Cn+ (solvation, i.e. −1575 kJ/mol23) | (5) |
Eqn (2) and (4) describe the crystal energy, composed of Born–Lande lattice energy and surface energy. Strain and dislocation energies (i.e. 0.2 eV per unit cell to >1.5 eV per unit cell14) between the parent and product phases of ionic crystal also contribute to the thermodynamics and kinetics of the cation exchange reaction. Therefore, topotactic exchange reactions, which map anions from the parent to the product lattice, tend to be more favorable than non-topotactic exchange reactions.
Eqn (3) and (5) describe the desolvation and solvation energies of cations. The ease with which the parent (ingoing) metal is dissociated (associated) from the lattice and subsequently solvated (desolvated) dictate the energy balance of cation exchange. The reaction is more likely to proceed if the parent cation is more strongly solvated in the reaction solvent than the ingoing cation.
The thermodynamic driving force can therefore be determined by summing contributions from the crystal lattice energy, interfacial strain energy, dislocation energy, dissociation and solvation energy, all of which are crucial for cation exchange reactions (Fig. 1). Such thermodynamic information aids prediction of likelihood of an exchange reaction. It can also give detailed information about the nature of the exchange. For instance, the occurrence of a solid solution (evenly mixed new and parent cations)24,25versus a two-component crystal (phase separated parent and product crystals)15,16 would be determined by the mutual solubility limits of the two phases as well as interfacial energies.
Each cation exchange reaction has a unique fingerprint of thermodynamic parameters that dictate the conditions under which it must be performed. It is clear from the free energies listed for the example above (eqn (2)–(5)) that the Cu+ exchange of CdS is strongly driven by the preferential solvation of the cadmium ions relative to the copper ions. Reactions in other systems, e.g. isovalent exchanges, may be dictated less by the solvation environment, but more by lattice energy or ionic size considerations. In cases where differences in lattice or solvation energies are small, the law of mass action can be used to drive the reaction.
Exchange reactions at ambient temperatures are seldom under thermodynamic equilibrium; therefore kinetic factors such as nucleation energy barriers play a key role in determining the outcome of the reaction. The kinetics of the cation exchange reaction is driven by the activation energy of each sub-reaction. Because they are inherently linked – the products of one sub-reaction are the reactants of the next sub-reaction – the reactions must proceed concertedly. Ion exchange is sometimes described as having a reaction zone, in which dissolution and re-precipitation occur simultaneously over a small length scale, allowing communication between the parent and product crystals, and thereby preserving crystallographic information.21 The nature of the reaction front is of great importance: diffusion rates of interstitials, dislocations, and vacancies control the communication between the parent and product crystals, which can manifest in the final crystallinity and porosity of the product.
The kinetics of cation exchange can be accelerated by elevated temperature, but often at the expense of material stability or manufacturability. In fact, ion exchange in extended solids often involves a tradeoff between kinetics and thermal stability. Early exchange reactions in extended solids were performed at temperatures of 100–800 °C for long reaction times, sometimes multiple days. Long processing times are obvious limitations to this synthetic technique for bulk materials, and any further elevation in temperature is constrained by material stability, as anionic diffusivity is increased. Equally importantly, at high temperatures, exchange will proceed close to thermodynamic equilibrium, preventing kinetically controlled access to metastable or non-equilibrium structures, which is a salient feature of cation exchange as a synthetic tool.
3. Cation exchange in nanocrystals
The fundamental limitations of solid-state exchange discussed above are mitigated in nanometer-scale ionic crystals, whose high specific surface area and low atomic count enable dramatically accelerated kinetics: room-temperature and sub-second reaction times are commonly reported for nanocrystal cation exchange reactions – up to 100 times faster than bulk in the case of Ag+ exchange of CdSe26 – likening them more to molecular exchange reactions than to solid state reactions. The reaction barrier for nucleation may be reduced on the nanoscale, where high curvature surfaces and low co-ordination facets could serve as high-energy sites ideal for nucleation. Interfacial strain may also be significantly relaxed in a smaller nanocrystal. Undoubtedly, the kinetics of exchange can vary widely depending on nanocrystallite sizes, shapes, and compositions. In addition, the energetics of the exchange reaction is also modulated by nanocrystal size, since surface energy begins to have an increasingly dominant contribution to lattice energy as the nanocrystal size becomes smaller.From a mechanistic standpoint, the high surface area of tiny crystals may reduce sub-reaction activation barriers27 and enable access to unique or metastable phases. Reduced activation barriers allow the reaction direction to be changed with little effort, enabling almost completely reversible cation exchange reactions.12,15,28 With nanocrystals, either a large excess of the incoming cation and/or preferential solvation of the parent cation appears sufficient to drive the reaction at room temperatures. Differential solvation is especially easy to achieve for exchange from a divalent cation such as Cd2+ to a monovalent cation such as Cu+ or Ag+.15,26 Divalent cations, due to their higher charge/volume classify as hard acids and can be solvated by the presence of a hard base such as methanol. Conversely, the reverse cation exchange from Cu2S to CdS can be driven at room temperature simply by a huge excess of Cd2+ and by providing ligands with soft base character, such as t-butylphosphonic acid, that preferentially solvate the soft acid Cu+. In the case of isovalent systems, solvation energies of the two ions are comparable, and therefore the direction of exchange would be dictated by the formation of the more stable lattice, or in cases where the difference in lattice stabilities is small, by providing an excess of ions.
The high surface area and small number of atoms may also allow easy crystal reorganization, influencing the surface faceting or the porosity/morphology of the product crystal.27,29 The very small scale of nanocrystals provides strain relief and may prevent dislocations.30,31 Exchange-dominant crystal facets may be selected by tuning the original nanocrystal shape, thereby imparting controlled anisotropy to cation diffusion in systems with crystallographic anisotropies.
The fast kinetics makes nanoscale cation exchange a simple single-step room-temperature process for converting, for instance, CdX (X = S, Se, Te) nanocrystals into a wide variety of nanocrystals, such as those of Ag, Cu, Pb, Zn, and Hg chalcogenides.12,19,32,33 The template (i.e. CdX) nanocrystals can be synthesized by hot injection with exquisite size and shape control;4,7 cation exchange allows one to exploit established Cd chalcogenide chemistry to make other nanocrystals, which may not be as easy to synthesize directly with similar size and shape control. The ability of nanocrystals to undergo cation exchange reactions rapidly at room temperature opens up a much wider array of applications at the nano scale than we have seen in bulk systems. There are several examples of nanocrystal cation exchange requiring high temperatures and/or long reaction times.34,35 We have, however, limited our discussion here to rapid room-temperature colloidal transformations, which hold more general potential for materials synthesis by virtue of their gentle reaction conditions, an attribute that is unique when compared to conventional solvothermal techniques.
4. Building complexity into nanocrystals: heterostructures
In addition to post-synthetic transformation of nanocrystal compositions, cation exchange allows added complexity in synthesized nanocrystals by their partial transformation.14–16 The resulting multi-cation profile is likely dictated by the mechanism of the exchange, loosely classified by us as either “homogeneous” or “heterogeneous”“Homogeneous” ion exchange would yield a solid solution, i.e., a nanocrystal doped with the foreign cation, possibly the case in a recent synthesis of heavily doped nanocrystals of InAs25 or CuInZnS nanocrystals.24 While doping is not often associated with cation exchange, an exchange reaction with the appropriate driving forces can be employed to dope a nanocrystal in a controlled fashion post synthesis at room temperature.36 Currently, chalcogenide nanocrystals are doped by introduction of the dopant precursor in the hot-injection step, however this method offers poor stoichiometric control and limits the doped materials to chemistries and crystallographies stable at elevated temperature. In contrast, doping via low amounts of “homogeneous” cation exchange, with strong thermodynamic driving conditions, can ensure controlled stoichiometry and temperatures mild enough to maintain any desired metastability.
At the other end of the spectrum lie several cases where the new phase nucleates at a surface facet, followed by growth of the phase topotaxially towards the interior of the nanocrystal. Such a “heterogeneous” mechanism allows the use of partial exchange to introduce sharp interfaces or heterojunctions within a nanocrystal. Multicomponent nanocrystals with heterojunctions have been of particular interest since they allow electron and hole transport and confinement to be controlled independently.37–40 Such band engineering in heterostructures forms the basis of several optoelectronic applications such as high-efficiency light emitting diodes and photovoltaic cells.
As one example, PbX (X = S, Se, Te) nanocrystals have been exchanged with Cd2+, where the reaction is limited to the thin outermost shell of the nanocrystal.41,42 In the resulting PbSe/CdSe core/shell heterostructure (type I), electron and hole carriers are confined within the lower band-gap PbSe core, resulting in high quantum yield excitonic emission from the core and improved oxidative stability. Traditionally, epitaxial growth of a semiconductor onto seed nanocrystals is achieved via multistep hot injection, however cation exchange is simpler and more flexible. The topotaxial nature of the exchange reaction may also facilitate interfaces with relatively few defects.
Certain complex heterostructure morphologies are possible due to the unique nanoscale attributes of exchange reactions such as facet dependence, asymmetry, and anisotropy. For instance, Cu+ exchange of a CdS nanorod proceeds selectively from the end facets, such that Cu2S grows inwards from the ends.14 Additionally, since the two end facets of wurtzite CdS nanorods are crystallographically nonequivalent, Cu+ exchange proceeds preferentially from one end. This yields binary CdS–Cu2S nanorods, which represent nanoscale heterojunctions (Fig. 4), with sharp epitaxial interfaces resulting from the 30× faster Cu diffusion along the a-axis than along the c-axis of the CdS wurtzite crystal.
A stark contrast is seen in the case of partial exchange with a different cation such as Ag+. In this case, interfacial strain between Ag2S and CdS causes a significant repulsive elastic interaction, leading to formation of equi-spaced domains of Ag2S segmented along the CdS rod, akin to droplets of oil in water. In the case of partial exchange of CdSe with Pb2+, a range of complex morphologies, such as core–shell nanorods and CdSe nanorods with embedded PbSe dots become possible via formation of stable {111}/{111} PbSe/CdSe interfaces.17,41,42 Since Cu2S or Ag2S can be easily exchanged with divalent cations such as Pb2+ and Zn2+ in the presence of a soft base, the Cu2S segment can be used as a sacrificial segment to synthesize other binary rods (e.g. PbS–CdS) through a multi-step exchange.14,15
Both the preservation of nanocrystal size/shape in an exchange reaction and the ability to perform partial transformations are possible due to the solidarity of the anionic framework, a hallmark of cation exchange in chalcogenides.13,17 The important structural role of the rigid anionic framework was recently demonstrated: exchange of CdSe/CdS dot/rod heterostructures with Cu+ at room temperature resulted in Cu2Se/Cu2S dot/rod heterostructures, with no inter-diffusion of the S and Se atoms across the dot/rod heterojunction (see Fig. 2). Thus, in addition to nanocrystal size and shape, compositional interfaces within a heterostructure are preserved with high fidelity during cation exchange.13,16 This allows multicomponent/heterostructured nanocrystals to also be used as templates, just like single-phase nanocrystals. It is highly facile to transform CdSe/CdS dot–rods (heterostructures for visible band-gap engineering) to ZnSe/ZnS (ultraviolet band-gap)43 or PbSe/PbS (near-infrared band-gap) dot-in-rods,13 the latter being relatively difficult to synthesize via hot-injection due to the symmetric rocksalt crystallography of lead chalcogenides.
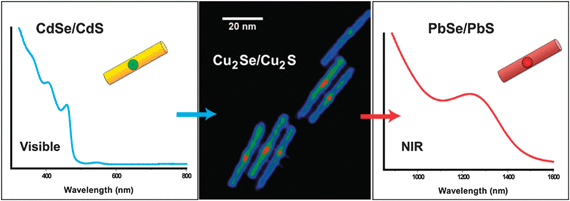 | ||
| Fig. 2 Cation exchange in nanoheterostructures enabled by preservation of the anionic framework. CdSe/CdS dot/rods can serve as templates to synthesize Cu2Se/Cu2S or PbSe/PbS dot/rods otherwise difficult to synthesize via conventional hot injection. Reprinted with permission from ref. 13. Copyright 2010 American Chemical Society. | ||
Researchers will benefit from improved understanding of the nucleation and growth in exchange reactions, in order to determine reaction conditions suited for production of monodisperse samples of multicomponent nanocrystals.
The quality of quantum dots synthesized using hot injection is typically very high: they are ultrapure and defect-free. On the other hand, nanocrystals made via cation exchange have been found to have kinetically frozen defects such as stacking faults, grain boundaries, and remnant impurities of the outgoing cation, consequently resulting in poor opto-electronic performance.28 Cationic impurities, in particular, can serve as recombination centers, leading to severely deteriorated quantum yields, even for type-I heterostructures (e.g. Cu+ in CdSe/CdS).28 However, mild thermal annealing can aid removal of the impurities, restoring quantum yields to levels exhibited by quantum dots and heterostructures made via hot injection. Such high quality heterostructures fabricated using cation exchange are enabling the fundamental study of quantum transport of charge carriers. Studies may range from exciton splitting between two semiconductors (PbS/PbSe)13 or the coupling between excitons of a semiconductor with free carriers of a semi-metal (e.g. Cu2−xSe/CdSe).
Cation exchange has already enabled the production of a diverse set of heterostructures, yet there is no limit to the number of additional complex structures that could be accessed. For instance, it would be possible to engineer a series of band-aligned materials into a nanowire or nanorod to achieve highly customized carrier confinement profiles. Goals could range from efficient charge separation across two or more materials (artificial photosynthesis) to enhanced electron-hole overlap and radiative recombination (light emitting diode). As another example, a finely graded index of refraction could be achieved by controlling the degree of phase separation vs. solid solution.
5. Accessing metastable phases and compositions
As indicated by some of the examples above, nanostructure morphologies resulting from exchange are often dictated by the parent template and the kinetics of exchange rather than being simply constrained by the thermodynamically stable crystallography of the final product. In contrast, in hot-injection synthesis, the product is determined almost exclusively by the thermodynamically stable crystal phase, although, some level of kinetic control of morphology/shape is also possible via strongly coordinating ligands. The cation exchange reaction is by its inherent nature a non-equilibrium method with the outcome being primarily kinetically controlled. Particularly unique to cation exchange is the ability to access metastable compositions and phases.A classic example is the case of Cu2S, the most stable form of which in nature is the highly copper-deficient djurleite phase with a stoichiometry of Cu1.93–1.96S.44,45 Accordingly, Cu2S nanocrystals made via solvothermal methods are mostly, with a few exceptions,46,47 in the copper-deficient djurleite form.44,45 In contrast, Cu2S nanocrystals obtained from Cu+ exchange of CdS are fully stoichiometric, known by the low-chalcocite phase.45 Copper deficient Cu2−xS nanocrystals possess a high concentration of hole carriers, as a result of which they exhibit a near-infrared plasmon resonance, a feature markedly absent in cation exchange-obtained Cu2S nanocrystals. However, the Cu2S nanocrystals obtained from exchange, upon exposure to oxygen or iodine, controllably develop copper deficiencies, switching on a plasmon resonance.45 Such a demonstration of active switching of plasmon resonances would not have been possible without access to the metastable fully stoichiometric form of copper sulfide. Chalcocite Cu2S nanocrystals made via cation exchange have also enabled systematic studies of the nanosize-dependence of the solid–solid transformation from low- to high-chalcocite.48,49
The starting nanocrystal acts not only as a shape/morphology template but also a crystallographic template. For instance, CdSe nanorods obtained by cation exchange of PbSe nanorods emerge in the zincblende form.17 Zincblende CdSe, although less stable than the hexagonal wurtzite CdSe, is closer in crystallographic symmetry to the lattice structure of the starting template (rocksalt PbSe). Manna and coworkers gave an exemplary demonstration of this phenomenon (Fig. 3).50 Cubic zincblende CdSe nanocrystals exchanged with Cu yielded Cu2Se in the closely allied fcc or tetragonal phase and with Zn yielded ZnSe in the cubic zincblende phase. On the other hand, hexagonal wurtzite CdSe nanocrystals upon exchange yield hexagonal phase Cu2Se and ZnSe nanocrystals.
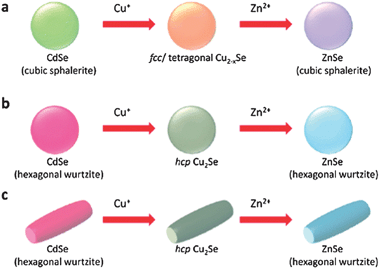 | ||
| Fig. 3 Cation exchange of CdSe nanocrystals showing that the starting nanocrystal acts not only as a shape/morphology template but also a crystallographic template. The product ZnSe nanocrystals are in a crystallographic phase closely related to the starting lattice; not necessarily the most thermodynamically stable one. Reprinted with permission from ref. 50. Copyright 2011 American Chemical Society. | ||
Nanocrystals in a metastable phase may relax to a more stable phase with or without external perturbations, enabling study of the fundamental dynamics of nanoscale solid-state transformations. However, most exciting is the possibility of arriving at previously undiscovered compositions and phases, hitherto not accessible via the thermodynamic phase diagram.
6. Cation exchange for device applications
The ability to synthetically access multi-component heterostructures and metastable morphologies and compositions makes cation exchange powerful as a manufacturing technique for electronic and other devices.Indeed, cation exchange was used 50 years ago for the manufacture of the first thin-film solar cell: the Cu2S–CdS heterojunction photovoltaic device.51 Cation exchange allowed the formation of an epitaxial heterojunction. Recently, it was proposed that such a scheme may be enhanced using nanocrystals, where strain relief would allow a defect-free heterojunction to be formed despite the lattice mismatch between the two phases.30,31 The low-temperature rapid fabrication step enables in-situ junction formation under gentle conditions, while allowing asymmetric structures, such as the one shown in Fig. 4. Cation exchange techniques may also allow for enhanced control of doping and graded compositions in thin film photovoltaics.
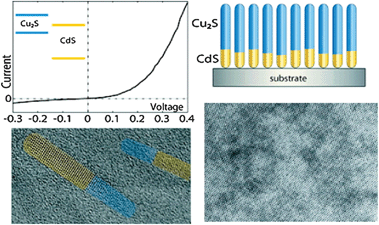 | ||
| Fig. 4 Cation exchange as a device fabrication technique. An array of nanoscale heterojunctions fabricated by facile Cu+ exchange of a self-assembled monolayer of CdS nanorods. Reprinted with permission from ref. 31. Copyright 2011 American Chemical Society. | ||
Cation exchange has been used in extended solids to access crystallographic phases that allowed for facile intercalation of lithium ions for battery applications52,53 and improved metal–organic frameworks for gas adsorption.54 Nanoparticles, in particular, are appealing in battery electrodes, as they allow higher surface areas resulting in rapid intercalation enabling high rate capabilities. Cation exchange studies can shed fundamental light on cation mobility, which is crucial to enhancing battery operation. Additionally, cation exchange may enable discovery of novel and metastable crystallographic phases55 that are predicted to serve as high-capacity electrodes or other functional materials.
In particular, computationally-predicted materials with desirable properties that would otherwise be discarded due to metastability may now be synthetically accessible using cation exchange. In-situ cation exchange for devices may also allow selective swelling or porosity control desirable in the formation of optimal nanostructured electrodes. Surface exchange can aid passivation against detrimental interfacial effects. The attraction of cation exchange for device fabrication lies in its ability for gentle, low-temperature processing, allowing materials production at a low monetary and environmental cost, and access to new and higher-performing materials.
7. Applications of cation exchange outside materials synthesis
The speed and completion of the cation exchange reaction in nanosized crystals has motivated some interesting applications. For instance, purification of heavy metal ions from water is often carried out via ion exchange resins. Brock and coworkers56 recently demonstrated that ZnS nanoparticle gels could be employed for removal of Pb2+ and Hg2+ from aqueous solutions. The released Zn2+ has relatively lower toxicity than the heavy metal ions. Exploitation of the nanoscale morphology enabled a record water remediation capacity of 14.2 mmol Pb2+ per gram of ZnS aerogel.Antibody-conjugated ZnSe and CdSe-based quantum dots are being employed in the area of cellular labeling and sensing. At ultralow concentrations of cellular analytes, unstable intermittent emission from the low number of bound quantum dot probes hinders sensitive or quantitative detection. However, cation exchange of the CdSe quantum dots can serve as a tool for fluorescence signal amplification.58 By fast cation exchange with Ag+, each bound quantum dot probe can release thousands of Zn2+ ions. These ions can be quantitatively detected by a fluorogenic Zn2+ sensing dye such as Fluo-4 or Rhod-5N, generating a highly amplified and quantitative emission signal (Fig. 5). Using this scheme, microRNAs have been detected down to a detection limit of 35 femtomolar and specificities of one or two-nucleotide sequences.59
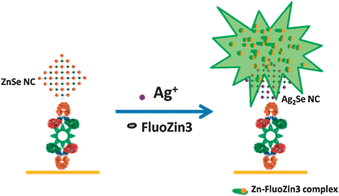 | ||
| Fig. 5 Signal amplification via cation exchange. Reprinted with permission from ref. 57. Copyright 2011 American Chemical Society. | ||
Another potential application of cation exchange is in electron microscopy as a tool for chemical-selective staining. By controlled exchange of selective components of a composite material with a heavier cation (e.g. Pb2+), the target regions can be effectively “stained” and rendered to exhibit high contrast in microscopy. This would allow nanometer resolution of compositional profiles in a heterostructure, otherwise difficult due to low differential contrast between the original components of the heterostructure.
8. Outlook
The advent of well-controlled ionic nanostructures brings renewed excitement to cation exchange, which can now be explored through a new set of lenses. Using cation exchange, it will be possible in the future to make a wide variety of templated nanomaterials. Carefully doped semiconductors, complex heterostructures, metastable phases, and hitherto undiscovered compositions now appear achievable with simple low temperature transformation.Several open questions must be addressed in order to expand the scope of the cation exchange technique to a larger part of the periodic table (Fig. 6). Is there a limit on the size/diffusivity of the cations being exchanged? Or is the nature of bonding (degree of ionicity or covalency) more critical? Are chalcogenides more amenable to cation exchange than oxides due to inherent bonding differences? How general are these mechanistic aspects across different materials? What is the effect of the crystal surface on the cation exchange reaction overpotentials and nucleation rates? What role do the surface passivating ligands play in ion exchange?
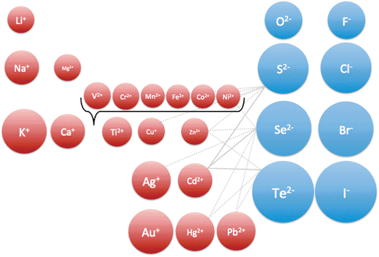 | ||
| Fig. 6 Periodic arrangement of ions, with proportional ionic radii. Connecting lines show representative cation exchange pairs. Some pairs may be uninvestigated as yet. | ||
Answers to these and more questions will enable a more complete mechanistic understanding of the cation exchange reaction, allowing its use in synthesis of new materials and devices. The rapidity and selectivity of the nanoscale cation exchange will allow for detailed studies of the kinetics, lending insights into the parameter space that controls cation exchange reactions. The ability to subject nanostructures to high-resolution microscopy, especially in situ, promises to revolutionize our mechanistic understanding of solid-state transformations on the nanoscale. Further study in this field should continue to build the library of materials, incorporating novel pairs of anions and cations to modulate composition and crystallography. With these advances, cation exchange can become a new paradigm for solid-state materials discovery and analysis.
Acknowledgements
P. K. Jain thanks UIUC Beckman Institute for support. We acknowledge the research contributions of A. P. Alivisatos, L.-K. Fong, B. Sadtler, B. Beberwyck, L. Amirav, and J. Luther. G. Girolami and E. Chan provided helpful reading of the manuscript.References
- M. Bawendi, W. Wilson, L. Rothberg, P. Carroll, T. Jedju, M. Steigerwald and L. Brus, Phys. Rev. Lett., 1990, 65, 1623–1626 CrossRef CAS PubMed
.
- A. Alivisatos, Science, 1996, 933–937 CAS
- J. Brennan, T. Siegrist and P. Carroll, J. Am. Chem. Soc., 1989, 111, 4141–4144 CrossRef CAS
- C. Murray, D. Norris and M. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS
- X. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS
- D. V. Talapin, A. L. Rogach, E. V. Shevchenko, A. Kornowski, M. Haase and H. Weller, J. Am. Chem. Soc., 2002, 124, 5782–5790 CrossRef CAS PubMed
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed
- J. E. Murphy, M. C. Beard, A. G. Norman, S. P. Ahrenkiel, J. C. Johnson, P. Yu, O. I. Mićić, R. J. Ellingson and A. J. Nozik, J. Am. Chem. Soc., 2006, 128, 3241–3247 CrossRef CAS PubMed
- K. Kolasinski, Curr. Opin. Solid State Mater. Sci., 2006, 10, 182–191 CrossRef CAS
- R. Ito and S. Okazaki, Nature, 2000, 406, 1027–1031 CrossRef PubMed
- H. Jenny, J. Phys. Chem., 1932, 36, 2217–2258 CrossRef CAS
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed
- P. K. Jain, L. Amirav, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 9997–9999 CrossRef CAS PubMed
- B. Sadtler, D. O. Demchenko, H. Zheng, S. M. Hughes, M. G. Merkle, U. Dahmen, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 5285–5293 CrossRef CAS PubMed
- J. M. Luther, H. Zheng, B. Sadtler and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 16851–16857 CrossRef CAS PubMed
- K. Miszta, D. Dorfs, A. Genovese, M. R. Kim and L. Manna, ACS Nano, 2011, 5, 7176–7183 CrossRef CAS PubMed
- M. Casavola, M. A. van Huis, S. Bals, K. Lambert, Z. Hens and D. Vanmaekelbergh, Chem. Mater., 2012, 24, 294–302 CrossRef CAS
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. M. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed
- L. Dloczik and R. Koenenkamp, J. Solid State Electrochem., 2004, 8, 142–146 CrossRef CAS
- M. Saruyama, Y.-G. So, K. Kimoto, S. Taguchi, Y. Kanemitsu and T. Teranishi, J. Am. Chem. Soc., 2011, 133, 17598–17601 CrossRef CAS PubMed
- A. Putnis, Mineral. Mag., 2002, 66, 689–708 CrossRef CAS
- J. N. Plendl and P. J. Gielisse, Z. Kristallogr., 1963, 118, 404–421 CrossRef CAS
- Y. Marcus, Faraday Trans., 1991, 87, 2995 RSC
- L. De Trizio, M. Prato, A. Genovese, A. Casu, M. Povia, R. Simonutti, M. J. P. Alcocer, C. D'Andrea, F. Tassone and L. Manna, Chem. Mater., 2012, 24, 2400–2406 CrossRef CAS
- D. Mocatta, G. Cohen, J. Schattner, O. Millo, E. Rabani and U. Banin, Science, 2011, 332, 77–81 CrossRef CAS PubMed
- E. M. Chan, M. A. Marcus, S. Fakra, M. ElNaggar, R. A. Mathies and A. P. Alivisatos, J. Phys. Chem. A, 2007, 111, 12210–12215 CrossRef CAS PubMed
- S. H. Tolbert and A. P. Alivisatos, Science, 1994, 265, 373–376 CAS
- P. K. Jain, B. J. Beberwyck, L.-K. Fong, M. J. Polking and A. P. Alivisatos, Angew. Chem., Int. Ed., 2012, 51, 2387–2390 CrossRef CAS PubMed
- Y. Yu, J. Zhang, X. Wu, W. Zhao and B. Zhang, Angew. Chem., Int. Ed., 2012, 51, 897–900 CrossRef CAS PubMed
- R. D. Robinson, B. Sadtler, D. O. Demchenko, C. K. Erdonmez, L. W. Wang and A. P. Alivisatos, Science, 2007, 317, 355–358 CrossRef CAS PubMed
- J. B. Rivest, S. L. Swisher, L.-K. Fong, H. Zheng and A. P. Alivisatos, ACS Nano, 2011, 5, 3811–3816 CrossRef CAS PubMed
- A. Mews, A. Eychmüller and M. Giersig, J. Phys. Chem., 1994, 98, 934–941 CrossRef CAS
- P. H. C. Camargo, Y. H. Lee, U. Jeong, Z. Zou and Y. Xia, Langmuir, 2007, 23, 2985–2992 CrossRef CAS PubMed
- S. Deka, K. Miszta, D. Dorfs, A. Genovese, G. Bertoni and L. Manna, Nano Lett., 2010, 10, 3770–3776 CrossRef CAS PubMed
- M. V. Kovalenko, D. V. Talapin, M. A. Loi, F. Cordella, G. Hesser, M. I. Bodnarchuk and W. Heiss, Angew. Chem., Int. Ed., 2008, 47, 3029–3033 CrossRef CAS PubMed
- A. W. Wills, M. S. Kang, K. M. Wentz, S. E. Hayes, A. Sahu, W. L. Gladfelter and D. J. Norris, J. Mater. Chem., 2012, 22, 6335–6342 RSC
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS
- S. Kim, B. Fisher, H. Eisler and M. Bawendi, J. Am. Chem. Soc., 2003, 125, 11466–11467 CrossRef CAS PubMed
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS PubMed
- A. Sitt, F. Della Sala, G. Menagen and U. Banin, Nano Lett., 2009, 9, 3470–3476 CrossRef CAS PubMed
- K. Lambert, B. D. Geyter, I. Moreels and Z. Hens, Chem. Mater., 2009, 21, 778–780 CrossRef CAS
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS PubMed
- H. Li, R. Brescia, R. Krahne, G. Bertoni, M. J. P. Alcocer, C. D'Andrea, F. Scotognella, F. Tassone, M. Zanella, M. De Giorgi and L. Manna, ACS Nano, 2012, 6, 1637–1647 CrossRef CAS PubMed
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef CAS PubMed
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed
- M. Lotfipour, T. Machani, D. P. Rossi and K. E. Plass, Chem. Mater., 2011, 23, 3032–3038 CrossRef CAS
- M. B. Sigman, A. Ghezelbash, T. Hanrath, A. E. Saunders, F. Lee and B. A. Korgel, J. Am. Chem. Soc., 2003, 125, 16050–16057 CrossRef CAS PubMed
- H. Zheng, J. B. Rivest, T. A. Miller, B. Sadtler, A. Lindenberg, M. F. Toney, L. Wang, C. Kisielowski and A. P. Alivisatos, Science, 2011, 333, 206–209 CrossRef CAS PubMed
- J. B. Rivest, L. K. Fong, P. K. Jain, M. F. Toney and A. P. Alivisatos, J. Phys. Chem. Lett., 2011, 2, 2402–2406 CrossRef CAS
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Nano Lett., 2011, 11, 4964–4970 CrossRef CAS PubMed
- D. Reynolds, G. Leies, L. Antes and R. Marburger, Phys. Rev., 1954, 96, 533–534 CrossRef CAS
- J. Tarascon and G. Hull, Mater. Res. Bull., 1984, 19, 915–924 CrossRef CAS
- J. L. C. Rowsell, N. J. Taylor and L. F. Nazar, J. Am. Chem. Soc., 2002, 124, 6522–6523 CrossRef CAS PubMed
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef CAS PubMed
- E. Dilena, D. Dorfs, C. George, K. Miszta, M. Povia, A. Genovese, A. Casu, M. Prato and L. Manna, J. Mater. Chem., 2012, 22, 13023–13031 RSC
- I. R. Pala and S. L. Brock, ACS Appl. Mater. Interfaces, 2012, 4, 2160–2167 CAS
- J. Yao, S. Schachermeyer, Y. Yin and W. Zhong, Anal. Chem., 2011, 83, 402–408 CrossRef CAS PubMed
- J. Li, T. Zhang, J. Ge, Y. Yin and W. Zhong, Angew. Chem., Int. Ed., 2009, 48, 1588–1591 CrossRef CAS PubMed
- J. Li, S. Schachermeyer, Y. Wang, Y. Yin and W. Zhong, Anal. Chem., 2009, 81, 9723–9729 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2013 |