
Optimizing nanoporous materials for gas storage†
Abstract
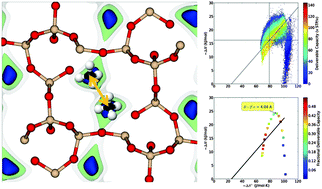
In this work, we address the question of which thermodynamic factors determine the deliverable capacity of methane in nanoporous materials. The deliverable capacity is one of the key factors that determines the performance of a material for methane storage in automotive fuel tanks. To obtain insights into how the molecular characteristics of a material are related to the deliverable capacity, we developed several statistical thermodynamic models. The predictions of these models are compared with the classical thermodynamics approach of Bhatia and Myers [Bhatia and Myers, Langmuir, 2005, 22, 1688] and with the results of molecular simulations in which we screen the International Zeolite Association (IZA) structure database and a hypothetical zeolite database of over 100 000 structures. Both the simulations and our models do not support the rule of thumb that, for methane storage, one should aim for an optimal heat of adsorption of 18.8 kJ mol−1. Instead, our models show that one can identify an optimal heat of adsorption, but that this optimal heat of adsorption depends on the structure of the material and can range from 8 to 23 kJ mol−1. The different models we have developed are aimed to determine how this optimal heat of adsorption is related to the molecular structure of the material.
 Please wait while we load your content...
Please wait while we load your content...

