 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Large amplitude motion in cold monohydrated dihydrogen phosphate anions H2PO4−(H2O): infrared photodissociation spectroscopy combined with ab initio molecular dynamics simulations†
Ling
Jiang‡
a,
Shou-Tian
Sun
b,
Nadja
Heine
a,
Jian-Wen
Liu
c,
Tara I.
Yacovitch§
d,
Torsten
Wende¶
a,
Zhi-Feng
Liu
*b,
Daniel M.
Neumark
*de and
Knut R.
Asmis
*af
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, D-14195 Berlin, Germany. E-mail: asmis@fhi-berlin.mpg.de
bDepartment of Chemistry and Centre for Scientific Modeling and Computation, Chinese University of Hong Kong, Shatin, Hong Kong, China. E-mail: zfliu@cuhk.edu.hk
cNational Supercomputing Center in Shenzhen, Shenzhen, China
dDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: dneumark@berkeley.edu
eChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
fWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstrasse 2, D-04103 Leipzig, Germany
First published on 15th November 2013
Abstract
The vibrational spectroscopy of monohydrated dihydrogen phosphate anions, H2PO4−(H2O), is studied in the O–H stretching (2700–3900 cm−1) and the fingerprint regions (600–1800 cm−1). Assignment of the experimental infrared multiple photon photodissociation spectra based on the predicted harmonic spectra of energetically low-lying 0 K structures is not conclusive. Ab initio molecular dynamics simulations reveal that the water molecule undergoes large amplitude motion, even at low internal temperatures, and that the dipole time correlation function qualitatively captures the anharmonic effects of the low-barrier isomerization reaction on the infrared intensities.
Phosphate anions play a key role in biological and agricultural systems.1–3 They are found in various esters, e.g. in adenosine phosphates, and in aqueous solution in the form of conjugate base anions H3−xPO4x− with x = 1–3 (inorganic phosphates). At physiological pH, H2PO4− and HPO42− are abundant and important in acid–base equilibria involved in metabolic pathways. Loss of water from dihydrogen phosphate (H2PO4−) leads to metaphosphate (PO3−), which is proposed to be a key intermediate in the aqueous hydrolysis of phosphate monoesters.1 However, its identification in solution remains elusive. How phosphate ions are hydrated (and dehydrated) at the molecular level is thus crucial for a mechanistic understanding of hydrolysis reactions, but difficult to extract from condensed phase measurements. Here, we use gas phase vibrational spectroscopy of isolated monohydrated dihydrogen phosphate in combination with molecular dynamics simulations to extract structural and dynamical information on the H2PO4−(H2O) complex.
Early studies on the PO3− + H2O reaction in the gas phase4 found that metaphosphate is unreactive, even though H2PO4− is thermodynamically more stable, owing to a high activation barrier. High pressure mass spectroscopic investigations by Keesee and Castleman,5 supported by electronic structure calculations,6,7 suggested that formation of dihydrogen phosphate does occur in the third hydration step and that PO3−(H2O)3 is in equilibrium with H2PO4−(H2O)2 with a four-center transition-state structure.7 Later, Kebarle and coworkers determined hydration energies of H2PO4− with up to two water molecules using electrospray-ionization mass spectrometry,8 but found no evidence for the formation of PO3−(H2O)3 from dehydrated dihydrogen phosphate. Fourier-transform infrared (FTIR) spectra of the H2PO4− anions in aqueous solution have been recorded at 300 K9 and interpreted by comparison to first-principles molecular dynamics simulations.10 Electronic structure calculations predict that the global ground state of H2PO4−(H2O) contains water in a double donor (DD) configuration bound to the two unprotonated phosphoryl O-atoms.7,11–13 An alternative arrangement with the water in an acceptor–donor (AD) motif is found to be higher in energy.11 However, no experimental gas-phase data regarding the cluster structures are available.
Infrared multiple photon dissociation (IRMPD) spectroscopy of size-selected ion–solvent complexes has emerged as a powerful tool for the structural characterization of microhydrated ions in the gas phase.14–24 It is becoming increasingly evident that the interpretation of IRMPD spectra, in particular when hydrogen bonding is involved, requires theoretical tools beyond the harmonic approximation.25–31 Here, IRMPD spectra of bare and monohydrated H2PO4− clusters are presented. We first try to understand the spectra based on a comparison to simulated harmonic IR spectra. However, the spectra of H2PO4−(H2O) exhibit pronounced anharmonic effects that can only be understood on the basis of the results derived from ab initio molecular dynamics (AIMD) simulations.
In order to check that dihydrogen phosphate anions are actually formed by electrospray, we recorded the IRMPD spectrum of H2PO4− in the fingerprint region by monitoring the H2O loss channel (Fig. 1). Comparison of the experimental band positions to those in the simulated MP2/aug-cc-pVDZ harmonic vibrational spectra of H2PO4− and PO3−(H2O) yields satisfactory agreement only with the spectrum of dihydrogen phosphate, allowing assignment of the four IR-active features to the antisymmetric (H, 1299 cm−1) and symmetric (I, 1094 cm−1) P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, POH bending (J, 1049 cm−1) and antisymmetric P–OH stretching (K, 770 cm−1) modes. Poorer agreement between 600–1400 cm−1 as well as the lack of any signal in the water bending region (∼1700 cm−1) rules out any contribution from the monohydrated metaphosphate anion, which is predicted to lie +37.4 kJ mol−1 higher in energy. Discrepancies regarding the IRMPD vs. the linear harmonic intensities are attributed to the high predicted dissociation energy of H2PO4− of 49.7 kJ mol−1 for water loss and the even higher calculated barrier of 122.1 kJ mol−1 for this channel (MP2/aug-cc-pVDZ level including zero point energy and BSSE corrections). Hence, absorption of many IR photons is required for IRMPD.32
O stretching, POH bending (J, 1049 cm−1) and antisymmetric P–OH stretching (K, 770 cm−1) modes. Poorer agreement between 600–1400 cm−1 as well as the lack of any signal in the water bending region (∼1700 cm−1) rules out any contribution from the monohydrated metaphosphate anion, which is predicted to lie +37.4 kJ mol−1 higher in energy. Discrepancies regarding the IRMPD vs. the linear harmonic intensities are attributed to the high predicted dissociation energy of H2PO4− of 49.7 kJ mol−1 for water loss and the even higher calculated barrier of 122.1 kJ mol−1 for this channel (MP2/aug-cc-pVDZ level including zero point energy and BSSE corrections). Hence, absorption of many IR photons is required for IRMPD.32
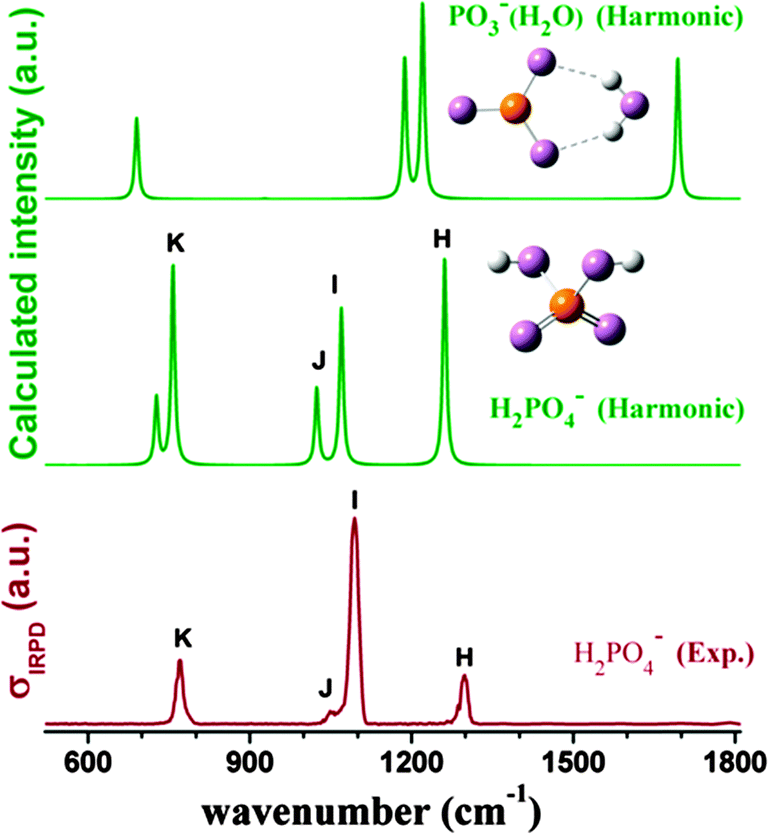 | ||
| Fig. 1 Comparison of the experimental (red) IRMPD spectrum of [H2PO4−] (bottom) and simulated (green) MP2/aug-cc-pVDZ harmonic vibrational spectra of the global minimum-energy structures of H2PO4− (center) and PO3−(H2O) (top) (see Section 1B in the ESI† for scale factors and the method for convoluting the stick spectra of harmonic frequencies). | ||
The experimental IRMPD spectrum of H2PO4−(H2O), recorded from 550–1800 and 2600–3950 cm−1 and followed via H2O loss, is shown in Fig. 2. Six features, labeled A–F, are observed in the O–H stretching region (>2700 cm−1). Only band A lies above 3600 cm−1, which is in the region of the free O–H stretching modes.21 Consequently, bands B–F (<3600 cm−1) are attributed to progressively more strongly hydrogen-bonded O–H stretching modes, which is also reflected in the increasing width of these features with decreasing energy. In contrast, all bands below 1800 cm−1 (G–L) appear relatively narrow. The band around 1700 cm−1 (G) is assigned to the water bending mode.33 The bands below 1500 cm−1, similar to those observed for bare H2PO4− (Fig. 1), are due to P–O and PO stretches, as well as bending and other lower-frequency modes9 (see Table S1 for band positions and assignments in the ESI†).
 | ||
| Fig. 2 Comparison of the experimental IRMPD spectrum of H2PO4−(H2O) (bottom) and simulated MP2/aug-cc-pVDZ harmonic and DTCF spectra from 200 ps trajectories of isomers 1-1 and 1-2, and transition state 1-3 (see text). | ||
The two most stable predicted structures for H2PO4−(H2O) are the complexes containing either a DD- or an AD-water molecule, labeled 1-1 and 1-2, respectively, as shown in Fig. 3. In 1-1, the H2O molecule donates two HBs to the PO2− moiety, yielding a symmetric structure (C2v), in which the negative charge in H2PO4− is stabilized on the PO2− moiety. The HB distances of 2.07 Å are only slightly longer than the distance of 2.00 Å in the water dimer,34 implying that the interactions are of comparable strength. The structure of 1-2 is asymmetric. The H2O molecule donates a strong HB (1.64 Å) to one PO group, and accepts a weaker one (2.13 Å) from one of the hydroxyl groups. The other PO and P–OH groups do not interact with the water molecule, but form a weak internal hydrogen bond (2.48 Å).
 | ||
| Fig. 3 Minimum energy (1-1 and 1-2) and first-order transition state (1-3) structures for the H2PO4−(H2O) complex. B3LYP/aug-cc-pVDZ and MP2/aug-cc-pVDZ relative energies (in kJ mol−1) are listed with (inside round brackets) and without (inside square brackets) ZPE corrections. The MP2 distances of hydrogen bonds are given in angstrom. | ||
The energetic ordering of these two isomers depends on the model used. B3LYP predicts 1-2 as the global minimum energy structure and 1-1 +0.8 kJ mol−1 higher in energy, including zero point energies (ZPE). In contrast, MP2 places 1-2 +0.1 kJ mol−1 above 1-1. These two minimum energy structures are separated by a small barrier (B3LYP: +3.5 kJ mol−1; MP2: +6.5 kJ mol−1) at the first-order transition state (TS) 1-3 (Fig. 3), indicating a fairly flat potential energy surface. In structure 1-3, the H2O molecule forms only a single HB with a phosphoryl group. The MP2/aug-cc-pVDZ binding energies between H2PO4− and water in 1-1 and 1-2 are 52.8 and 50.0 kJ mol−1 including zero point energy and BSSE corrections, respectively, which are close to the experimental value of 58.6 kJ mol−1 determined mass spectrometrically.8
Simulated MP2 harmonic spectra of 1-1 and 1-2 are shown above the experimental IRMPD spectrum in Fig. 2 (see Fig. S1 and S2, ESI† for detailed assignments). Upon first glance, a satisfactory agreement between experiment and harmonic theory is observed for isomer 1-1, especially below 2000 cm−1. The harmonic spectra of 1-1 and 1-2 account for all the experimentally observed peaks (G–L), while the spectrum of 1-2 predicts additional intense bands at 1170 cm−1 (symmetric OPO stretch) and 966 cm−1 (water wag) that are not observed in the experiment. Above 2000 cm−1, the spectrum of 1-1 also accounts for peak A (free PO–H stretch) and the doublet C (hydrogen-bonded water symmetric and antisymmetric stretches) at ∼3450 cm−1. However, the harmonic spectrum of 1-1 leaves bands B and D–F unassigned. On the other hand, the harmonic spectrum of 1-2 yields reasonable assignments for bands A (free HO–H stretch), B (hydrogen bonded PO–H stretch) and F (hydrogen bonded H–OH stretch) in the O–H stretching region, leaving C–E unassigned. In particular, the strongest hydrogen bond in 1-2 (1.64 Å) nicely accounts for the characteristically red-shifted band F (2700–3100 cm−1), even though its width cannot be rationalized at the harmonic level. In summary, neither harmonic IR spectra of the two isomers nor a linear combination of the two can satisfactorily explain the experimental IRMPD spectrum shown in Fig. 2.
To disentangle the discrepancies between the harmonic and experimental IRMPD spectra, we performed ab initio molecular dynamics (AIMD) simulations (see Section 1B in the ESI† for details). Briefly, vibrational profiles at finite temperature are obtained by the Fourier transform of the dipole time correlation function (DTCF), which accounts for anharmonic as well as dynamic effects. Two sets of long AIMD simulations were performed at 140 K and 180 K for a more extensive sampling of the phase space. At each temperature, two trajectories were simulated, one starting with 1-1 and the other with 1-2, each lasting 200 ps. Each trajectory was then cut into 10 ps intervals that were Fourier-transformed, and all 40 frequency profiles were then added up to produce the DTCF spectrum at a specific temperature. The DTCF spectra from both trajectories differ only slightly from each other as shown in Fig. S3 (ESI†), indicating that 1-1 and 1-2 interconvert readily at these temperatures. DTCF spectra were also determined from AIMD simulations at 20 K (10 ps trajectory) starting from the two isomers. All simulations are shown in Fig. 2.
The AIMD simulations at 20 K are helpful to test the quality of the potential energy surface, but do not correspond to a physically achievable temperature since zero-point energies are not considered. The general appearance of the 20 K DTCF spectra is indeed similar to the previously discussed harmonic spectra, with the hydrogen-bonded O–H stretching modes showing the largest shifts due to the use of different methods (PBE vs. MP2). Interestingly, the relative band intensities of experimental features A–C and G–L, with the exception of band H, are reproduced better by the 1-1 simulation already at 20 K compared to the harmonic spectrum (Fig. 2). The spectrum of 1-2 at 20 K, on the other hand, still mainly reflects the harmonic intensities, but does capture the pronounced red-shift of band F. To determine ZPE and finite temperature effects, the simulation temperature is raised to 140 K and 180 K.
There is considerably better agreement between the experimental spectra and the DTCF spectra at higher simulation temperatures (140 K or 180 K) throughout the spectral range for band positions and relative intensities. At these simulation temperatures, isomers 1-1 and 1-2 are interconverting. The complexity of the features in the O–H stretching region as well as the number and relative intensities of the bands in the fingerprint region are qualitatively reproduced. The increased broadening of the hydrogen-bonded O–H stretching bands B–F with the strength of the hydrogen bonds, i.e., with the red shift of the corresponding IR band, is also captured.
In more detail, peak A remains sharp at 140 K and 180 K, indicative of a O–H stretching mode of a free PO–H group. Comparison of DTCF spectra from both trajectories in Fig. S3 (ESI†) shows that both 1-1 and 1-2 contribute to peak A. Peak B is due to the O5–H7 stretch in 1-2 (Fig. 3). The O8–H9 and O8–H10 stretches in 1-1 are responsible for the double peaks C. These three peaks are similar in width (around 50 cm−1) and the lengths of the respective hydrogen bonds involved are all predicted close to ∼2.1 Å, indicating comparable hydrogen bond strengths. Band F is the broadest predicted and observed feature and involves the strongest hydrogen bond (O3⋯H9 in 1-2). Consequently, the integrated intensity of the sharp peak F in the harmonic spectrum (and also in the 20 K spectrum) of 1-2 is distributed over a much larger energy range. Similar broadening has been observed for other cluster ions.35–41 The reduced relative intensity and broadening of the water bending mode (band G) relative to the harmonic spectra, is also reproduced by the DTCF spectra.
Between 1000 and 1300 cm−1, eight IR active PO stretching and the P–O–H bending modes, three for 1-1 and five for 1-2, are predicted by the harmonic analysis, while in the experimental spectrum only three bands, two intense bands (H and I in Fig. 2) and one weaker band (J), are observed. The DTCF spectra better reproduce this region too. The reason for this becomes apparent in the partial density of state (PDOS) plots shown in Fig. 4. The PDOS plots are based on the Fourier transform (see Section 1B in the ESI†) of the velocity correlation function for a specific atom. They also indicate that there is loss of the relative peak intensity due to broadening in the 900–1800 cm−1 region. However, the broadening is limited to the POH bending bands (H6 and H7 contributions in Fig. 4(a)), while the widths of the bands involving only the heavier atoms are not much affected by the elevated temperature in the simulations. Consequently, the two intense bands correspond predominantly to the antisymmetric (H) and symmetric (I) PO stretching modes, while the weaker feature J is attributed to the water wagging band.
 | ||
| Fig. 4 DTCF spectra (bottom) and PDOS plots (top) obtained from a 200 ps AIMD trajectory at 180 K for the specified atoms (Fig. 3) in H2PO4−(H2O). Representative structures depicted in the left column are captured at 0.3, 1.2, and 4.5 ps during the AIMD simulation. H10 and H9 readily interconvert. | ||
In the region below 900 cm−1, there are several bands related to wagging and rocking modes (harmonic analysis). The mode involving H atoms is broadened and smeared out at a simulation temperature above 140 K. Only the antisymmetric (K) and symmetric (L) stretching modes of P–OH bonds, are left, which is in good agreement with the experimental observation of peaks K and L.
The above analysis leaves bands D and E unassigned, as these cannot be attributed to normal modes of either structure 1-1 or 1-2. Is there a third species responsible for these absorptions? The DTCF spectrum at 180 K indeed reproduces a broad feature centered at 3161 cm−1, near the experimental peak E. This feature is also observed in the 140 K spectrum but with much less intensity. Interconversion between 1-1 and 1-2 involves considerable displacement of the water molecule (see the structures shown in the left column in Fig. 4) across a nearly flat potential energy surface with a barrier of less than 7 kJ mol−1. At 140 K, the cluster is mainly confined in the potential well of 1-1 or 1-2 and does not visit the transition region (1-3) much. Increasing the simulation temperature to 180 K leads to a different situation. The cluster spends considerably more time around 1-3. The transition region is loosely bound and favored by entropy. The clusters thus undergo large amplitude motion, and as a consequence vibrational frequencies associated with structure 1-3 contribute to the spectrum. Comparison of the DTCF spectrum and PDOS plots in Fig. 4(b) shows that this peak is indeed due to the O–H stretching of water in 1-3. This assignment is further supported by MP2 harmonic analysis on 1-3, which predicts this mode at 3220 cm−1 (Fig. 2). To sum up, these results indicate that peak E is due to the hydrogen-bonded O–H stretching mode of water in the transient structure 1-3. Similar signature of broken hydrogen-bond networks from finite ion temperature have been seen in Ar-tagged Br−(H2O)2,3 complexes.21 Note that the roaming water molecule observed in the present study is qualitatively different from the water migration reported for cold anion monohydrates in the excited OH stretching manifold.42 Here, water migration occurs on the vibrational ground state potential and over larger distances, involving hydrogen bond cleavage and formation.
Conclusions
In summary, the IRMPD spectra of H2PO4−(H2O) show evidence for isomerization even at cryogenic temperatures, wherein the clusters undergo large amplitude motion over a small barrier. As a result, key aspects of the spectra cannot be interpreted within the framework of the harmonic approximation. AIMD simulations provide insight into these effects and qualitatively reproduce the experimental IRMPD spectra. The remaining differences can be attributed to approximations in the simulations, including the limited sampling time, the use of pseudopotentials and the neglect of nuclear quantum effects. Experimentally, the measured IRMPD intensities deviate from the linear absorption cross sections. The isomerization at low temperatures observed here may be indicative of a highly functional water network around dihydrogen phosphate and therefore it will prove important to also study the larger hydrated clusters, work that is currently in progress. Such studies can then also resolve the questions regarding the interconversion of H2PO4−(H2O)n to PO3−(H2O)n+1 that is predicted for n > 1.Acknowledgements
We would like to thank the Stichting voor Fundamenteel Onderzoek der Materie (FOM) for beam time at FELIX and the FELIX-staff for excellent support and assistance. L. Jiang thanks the Alexander-von-Humboldt Foundation for a post-doctoral scholarship. We are grateful for the generous allocation of computer time at the National Supercomputing Center in Shenzhen and on the clusters of PCs at the Center for Scientific Modeling and Computation of the Chinese University of Hong Kong. The project is supported by a Direct Grant (2060427) from The Chinese University of Hong Kong. J.-W. Liu thanks Shenzhen Strategic Emerging Industry Special Fund Program of China (Grant No. GGJS 20120619101655715) for financial support. T. I. Yacovitch and D. M. Neumark were supported by the Air Force Office of Scientific Research under Grant No. FA9550-12-1-0160.Notes and references
- F. H. Westheimer, Chem. Rev., 1981, 81, 313–326 CrossRef CAS.
- Y. Nishizuka, Science, 1986, 233, 305–312 CAS.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- M. Henchman, A. A. Viggiano, J. F. Paulson, A. Freedman and J. Wormhoudt, J. Am. Chem. Soc., 1985, 107, 1453–1455 CrossRef CAS.
- R. G. Keesee and A. W. Castleman, J. Am. Chem. Soc., 1989, 111, 9015–9018 CrossRef CAS.
- B. Y. Ma, Y. M. Xie, M. Z. Shen and H. F. Schaefer, J. Am. Chem. Soc., 1993, 115, 1943–1951 CrossRef CAS.
- Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 1993, 115, 11997–12002 CrossRef CAS.
- A. T. Blades, Y. H. Ho and P. Kebarle, J. Am. Chem. Soc., 1996, 118, 196–201 CrossRef CAS.
- M. Klahn, G. Mathias, C. Kotting, M. Nonella, J. Schlitter, K. Gerwert and P. Tavan, J. Phys. Chem. A, 2004, 108, 6186–6194 CrossRef.
- E. Pluharova, M. Oncak, R. Seidel, C. Schroeder, W. Schroeder, B. Winter, S. E. Bradforth, P. Jungwirth and P. Slavicek, J. Phys. Chem. B, 2012, 116, 13254–13264 CrossRef CAS PubMed.
- L. D. Zhang, D. Q. Xie, D. G. Xu and H. Guo, J. Phys. Chem. A, 2005, 109, 11295–11303 CrossRef CAS PubMed.
- E. A. Ruben, M. S. Chapman and J. D. Evanseck, J. Phys. Chem. A, 2007, 111, 10804–10814 CrossRef CAS PubMed.
- S. A. Brandan, S. B. Diaz, R. C. Picot, E. A. Disalvo and A. B. Altabef, Spectrochim. Acta, Part A, 2007, 66, 1152–1164 CrossRef CAS PubMed.
- C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Science, 1997, 276, 1678–1681 CrossRef CAS.
- T. Ebata, A. Fujii and N. Mikami, Int. Rev. Phys. Chem., 1998, 17, 331–361 CrossRef CAS.
- M. Beyer, E. R. Williams and V. E. Bondybey, J. Am. Chem. Soc., 1999, 121, 1565–1573 CrossRef CAS PubMed.
- E. J. Bieske and O. Dopfer, Chem. Rev., 2000, 100, 3963–3998 CrossRef CAS PubMed.
- M. A. Duncan, Int. J. Mass Spectrom., 2000, 200, 545–569 CrossRef CAS.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1–18 RSC.
- M. A. Duncan, Int. Rev. Phys. Chem., 2003, 22, 407–435 CrossRef CAS.
- W. H. Robertson and M. A. Johnson, Annu. Rev. Phys. Chem., 2003, 54, 173–213 CrossRef CAS PubMed.
- A. Kamariotis, O. V. Boyarkin, S. R. Mercier, R. D. Beck, M. F. Bush, E. R. Williams and T. R. Rizzo, J. Am. Chem. Soc., 2006, 128, 905–916 CrossRef CAS PubMed.
- J. M. Lisy, J. Chem. Phys., 2006, 125, 132302 CrossRef PubMed.
- K. R. Asmis and D. M. Neumark, Acc. Chem. Res., 2012, 45, 43–52 CrossRef CAS PubMed.
- K. R. Asmis, N. L. Pivonka, G. Santambrogio, M. Brummer, C. Kaposta, D. M. Neumark and L. Woste, Science, 2003, 299, 1375–1377 CrossRef CAS PubMed.
- S. S. Iyengar, M. K. Petersen, T. J. F. Day, C. J. Burnham, V. E. Teige and G. A. Voth, J. Chem. Phys., 2005, 123, 084309 CrossRef PubMed.
- G. Gregoire, M. P. Gaigeot, D. C. Marinica, J. Lemaire, J. P. Schermann and C. Desfrancois, Phys. Chem. Chem. Phys., 2007, 9, 3082–3097 RSC.
- O. Vendrell, F. Gatti and H.-D. Meyer, Angew. Chem., Int. Ed., 2007, 46, 6918–6921 CrossRef CAS PubMed.
- M. Kaledin, A. L. Kaledin, J. M. Bowman, J. Ding and K. D. Jordan, J. Phys. Chem. A, 2009, 113, 7671–7677 CrossRef CAS PubMed.
- A. Cimas, P. Maitre, G. Ohanessian and M. P. Gaigeot, J. Chem. Theory Comput., 2009, 5, 2388–2400 CrossRef CAS.
- M. Baer, D. Marx and G. Mathias, Angew. Chem., Int. Ed., 2010, 49, 7346–7349 CrossRef CAS PubMed.
- T. I. Yacovitch, T. Wende, L. Jiang, N. Heine, G. Meijer, D. M. Neumark and K. R. Asmis, J. Phys. Chem. Lett., 2011, 2, 2135–2140 CrossRef CAS.
- N. I. Hammer, J. W. Shin, J. M. Headrick, E. G. Diken, J. R. Roscioli, G. H. Weddle and M. A. Johnson, Science, 2004, 306, 675–679 CrossRef CAS PubMed.
- X. Xu and W. A. Goddard, J. Phys. Chem. A, 2004, 108, 2305–2313 CrossRef CAS.
- R. S. Walters, E. D. Pillai and M. A. Duncan, J. Am. Chem. Soc., 2005, 127, 16599–16610 CrossRef CAS PubMed.
- M. F. Bush, R. J. Saykally and E. R. Williams, J. Am. Chem. Soc., 2008, 130, 15482–15489 CrossRef CAS PubMed.
- J. D. Rodriguez and J. M. Lisy, J. Phys. Chem. A, 2009, 113, 6462–6467 CrossRef CAS PubMed.
- J. D. Rodriguez, T. D. Vaden and J. M. Lisy, J. Am. Chem. Soc., 2009, 131, 17277–17285 CrossRef CAS PubMed.
- J. P. Beck and J. M. Lisy, J. Chem. Phys., 2011, 135, 044302 CrossRef PubMed.
- J. D. Rodriguez and J. M. Lisy, J. Am. Chem. Soc., 2011, 133, 11136–11146 CrossRef CAS PubMed.
- J. T. O'Brien and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 10228–10236 CrossRef CAS PubMed.
- E. M. Myshakin, K. D. Jordan, E. L. Sibert and M. A. Johnson, J. Chem. Phys., 2003, 119, 10138–10145 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp54250e |
| ‡ Current address: State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. |
| § Current address: Aerodyne Corporation, Billlerica, MA 01821, USA. |
| ¶ Current address: Physical and Theoretical Chemistry Laboratory, South Parks Road, Oxford OX1 3QZ, UK. |
| This journal is © the Owner Societies 2014 |