 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of electrolyte constituents on the motion of ionic species and recombination kinetics in dye-sensitized solar cells†
Shota
Kuwahara
*a,
Soichiro
Taya
a,
Naoya
Osada
ab,
Qing
Shen
bc,
Taro
Toyoda
b and
Kenji
Katayama
*a
aDepartment of Applied Chemistry, Chuo University, 1-13-27, Kasuga, Bunkyo, Tokyo 112-8551, Japan. E-mail: kkata@kc.chuo-u.ac.jp; Fax: +81-3-3817-1913; Tel: +81-3-3817-1913
bDepartment of Engineering Science, The University of Electro-Communications, 1-5-1 Chofugaoka, Chofu, Tokyo 182-8585, Japan
cPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
First published on 19th December 2013
Abstract
The dynamic motion of ions in electrolyte solutions and its effect on recombination was investigated by the heterodyne transient grating method in addition to transient absorption and transient photocurrent methods in dye sensitized solar cells. Realignment of ionic species at the electrode/electrolyte interface was observed after the electron injection in TiO2 on the order of μs. The process was affected by the total quantity of ionic species as well as cation species in the electrolyte. The recombination processes of the electrons were also affected by the constituents; the probability of the electron–electrolyte recombination decreased with decrease in I2 concentration; the dominant recombination process changed from the electron–electrolyte to the electron–dye recombination by decreasing I− concentration. It is concluded that sufficient I− is necessary for the suppression of the electron–dye recombination and that sufficient I2 is necessary for an efficient redox cycle, while low concentration of I3− ions at the electrolyte/TiO2 interface is preferable to suppress the electron–electrolyte recombination. The effect of the cation size in an electrolyte solution on the charge dynamics was also investigated, and it was revealed that the steric hindrance of cations changed the penetration of ionic species into the nanoporous dye/TiO2 electrode, causing a change in the electrostatic properties at the interface. The cation dependence indicated that the presence of large-sized cations suppressed the electron–electrolyte recombination by disturbing the approach of I3− paired with the cations.
Introduction
Dye sensitized solar cells (DSSCs) typically consist of a nanoporous layer of semiconductor materials such as a titanium oxide (TiO2) layer, dyes, an electrolyte solution and a counter electrode. This simple configuration for light harvesting is strongly attractive, and much effort has been made on a search for new materials to achieve higher light harvesting efficiency by increasing light absorption in broader wavelength regions and reducing the loss in the photon-to-current conversion processes.1–3 Understanding of the working mechanism of the devices is also necessary, and has been studied especially for the last 20 years.4–8 DSSCs work in the following steps: absorption of light by dyes, electron injection from the dyes to a semiconductor layer, dye regeneration by conversion from the dye cations reduced by redox species, electron diffusion in a semiconductor layer or recombination of the remaining electrons with electrolytes and dyes. Most of these charge transport processes occur at the semiconductor/electrolyte solution interface, and it was reported that the constituents in the electrolyte solution such as cation and redox species affect various processes and the performance of DSSCs as a result.9–14It is generally known that the constituents in an electrolyte solution have an influence on the yield of electron injection, open-circuit voltage, electron diffusion coefficient and the rate of dye cation regeneration.4,6 As for the effect of additives, by putting 4-tert-butyl pyridine (TBP) in an electrolyte solution, the open circuit voltage increases, while the electron injection yield decreases. This is because the conduction band edge of the semiconductor rises close to the LUMO of the dye molecules.10,15–17 It was reported on the cation effects that the flatband potential changes depending on the cation species, and that electron injection efficiency increased with increase in the charge/radius ratio of the cations9,10,14,17 due improvement of the overlap between the conduction band state of the semiconductor and the LUMO orbital of the dye molecules based on the Marcus–Gerischer theory.18 The dye regeneration was accelerated using small cations,19 and the addition of appropriate cations in an electrolyte solution improved the performance of DSSCs.
As for the effect of I3−/I− redox species, the recombination of electrons is caused predominantly by I3− ions rather than dye cations with sufficient redox concentration. The electron lifetime decreased with increase in I3−, proportional to the I2 concentration, because it works as an electron acceptor via the electron–electrolyte recombination process.10 The effect of I− concentration was also investigated,10,20 and significant increase in the electron lifetime was observed by increasing the lithium iodide (LiI) concentration from 10 to 20 mM. The effect was explained by the model that the dominant recombination channel of electrons was changed from the electron–dye recombination to the electron–electrolyte recombination within this concentration range.10
To clarify the role of the constituents in electrolyte solutions, the electron lifetimes or electron diffusion times were measured by intensity-modulated or time-resolved photocurrent or photovoltage techniques,4,10 and the recombination processes were discussed.21–26 The transient absorption (TA) method has also been utilized to obtain various decay kinetics via absorption changes in the dye, dye cation.17,21,25–32 However, the discussions have been made based on the electron lifetime and diffusion coefficient only in a semiconductor, and the effect of the species on the liquid side have not been clarified fully. We need to get more information on the liquid side to understand the effect, because there are various possible recombination pathways depending on the species on the liquid side.
Recently, we demonstrated a new approach for investigating carrier dynamics in DSSCs via a refractive index change by using the heterodyne transient grating (HD-TG) method, combined with TA and transient photocurrent (TP) methods. We observed the dynamics after initial electron injection from dyes to a semiconductor, and the time range observed was on the order of nanoseconds to seconds.33,34 HD-TG provides information on the photo-induced physical or chemical changes via a change in the refractive index,35–37 and many studies on the carrier dynamics at solid/liquid interfaces were reported, for example, proton transfer, hole transfer and motion of liquid crystal molecules, etc.38–40 In a previous paper, the charge dynamics at the TiO2/solution interface in DSSCs were observed and there were some processes, observed only by the HD-TG method, not by the TA method; especially on the dynamics in which charged species on the liquid side were involved. The corresponding processes were as follows; (1) rearrangement of charged ion species on the liquid side to stabilize a charged TiO2 surface due to electron trapping, (∼10−5 s), (2) the electron–electrolyte recombination and the following escape of redox species from the interface (10−3 to 10−1 s). We found that these charge dynamics were sensitive to the existence of TBP, which implied the interaction between TBP and redox species.33
Here we report on the dependence of the constituents in an electrolyte solution, such as cations and redox species (I2 and I−), on the charge dynamics at the interface observed by the HD-TG, TA and TP methods. These constituents are frequently used as components in the electrolyte solutions, and are known to affect the performance of DSSCs. We discussed the ionic dynamics and effect on the recombination processes, utilizing the HD-TG responses with a combination of the information obtained from TA and TP. We could successfully show how to utilize these combination techniques to distinguish electron–dye and electron–electrolyte recombination, and how to study the effect of the ionic species on such recombination processes.
Experimental
Nanostructured TiO2 films were prepared on fluorine-doped tin oxide (FTO) substrates by the doctor-blade technique.35,41 A colloidal TiO2 suspension was prepared as described in a previous paper,41 and was spread on the FTO substrate by using a glass rod with adhesive tapes with a thickness of ∼60 μm as spacers. Briefly, commercial TiO2 powder (1 g, P25, Degussa) was ground in a mortar with a small amount of water (0.33 ml) including acetylacetone (33 μl) to prevent aggregation of the particles. After viscous TiO2 paste was prepared, it was diluted by adding water (1 ml) slowly under continuous grinding. The film was sintered at 450 °C for 1 h and the final TiO2 thickness was about 10 μm. It was then immersed in the N3 dye (cis-bis(iso-thiocyanato)bis(2,2′-bipyridyl-4,4′-di-carboxylato) ruthenium(II)) bath for 40 h. The prepared sample was used as a working electrode, and an electrochemical cell was prepared by putting another glass plate together with a silicon rubber spacer, and the spacing between the working electrode and the glass plate was less than 1 mm. A platinum wire was put in the cell as a counter electrode. Electrochemical measurements were made by the two-electrode system, and the platinum wire was used as a reference/counter electrode. The electrochemical cell was kept under the short or open circuit conditions, and no external bias was applied to the cell (all results under the open circuit conditions are in the ESI†). The composition of the electrolyte was I2 (0.3–30 mM) and LiI (3–300 mM) in acetonitrile (ACN). When an electrolyte solution was replaced, the electrochemical cell was kept immersed for at least 30 min in a new electrolyte solution. To study the effect of the concentration of I−, LiClO4 was added to keep the Li+ concentration constant so that the cation effect was negligible. To study the cation effect, 1,2-dimethyl-3-propylimidazolium iodide (DMPII) and tetra-n-butylammonium iodide (TBAI) were dissolved in electrolyte instead of LiI while the concentrations of I− and I2 were kept constant (30 mM for I2, 300 mM for I−).The principle and the setup of the HD-TG method were reported in detail in previous papers.33,36,42 In this study, the pump light source was the second harmonic of an Nd:YAG laser (Surelite, Continuum, Electro-Optics Inc.). The pump pulse had a wavelength of 532 nm, a pulse width of 5 ns, an intensity of ∼0.5 mJ per pulse, and the probe light was a CW semiconductor laser with a wavelength of 635 nm. The pump and probe lights were incident from the FTO substrate side to avoid absorption of the pump light by iodine redox species. TP was measured using a potentiostat with a time resolution of 10 μs (Nikko Keisoku), and was recorded on an oscilloscope at the same time as the HD-TG measurement. TA was measured at the same probe wavelength just by removing a plate of the transmission grating used for the HD-TG measurement. In addition, TA was measured at 785 nm, too (ESI†). Each response was recorded with an interval of 20 s to avoid the effect of the previous pump pulse. In the HD-TG and TA measurements, the probe light at 635 nm was kept irradiated, which corresponded to an intensity of about 0.3 sun, and the cell showed almost the same Voc under 1 sun condition. In the TP response, there was a constant background current due to irradiation of the CW probe light, corresponding to a short-circuit current for a wavelength of 635 nm. To compare the results with a previous work, all data in this paper were taken under the short-circuit condition (the data taken under the open circuit condition is in the ESI†). The TP response was obtained under a constant background current. Thus, the TP response corresponds to a short circuit current response generated by additional irradiation of the pump pulse with a wavelength of 532 nm.
Results and discussion
I2 concentration dependence
Fig. 1(a) shows HD-TG and TA responses in LiI (300 mM)/I2 (30 mM)/ACN. Four components appeared in the HD-TG response in the time region of 0.4 μs to 1 s as was previously reported.33 The first component (I) with a decay time less than 1 μs was assigned to the disproportionation reaction of I2−. The second component (II) is a rising component observed in the time region of 10−6 to 10−5 s. This component is due to rearrangement of charged species on the liquid side in an electric double layer, which was induced by the electron trap in TiO2 after the injection of electrons from photoexcited dyes to TiO2. The third component (III) is a decaying component observed in the time region of 10−5 to 10−3, corresponding to the thermal diffusion response typically observed by the HD-TG method. The fourth component (IV) is a negative signal, which went back to the original baseline observed in the time region of 10−3 to 10−1 s, caused by the electron–electrolyte recombination and the following escape of I− from the electric double layer. The second (II) and fourth (IV) components were referred to as Arearrange and Ae/electrolyte, respectively, in the following.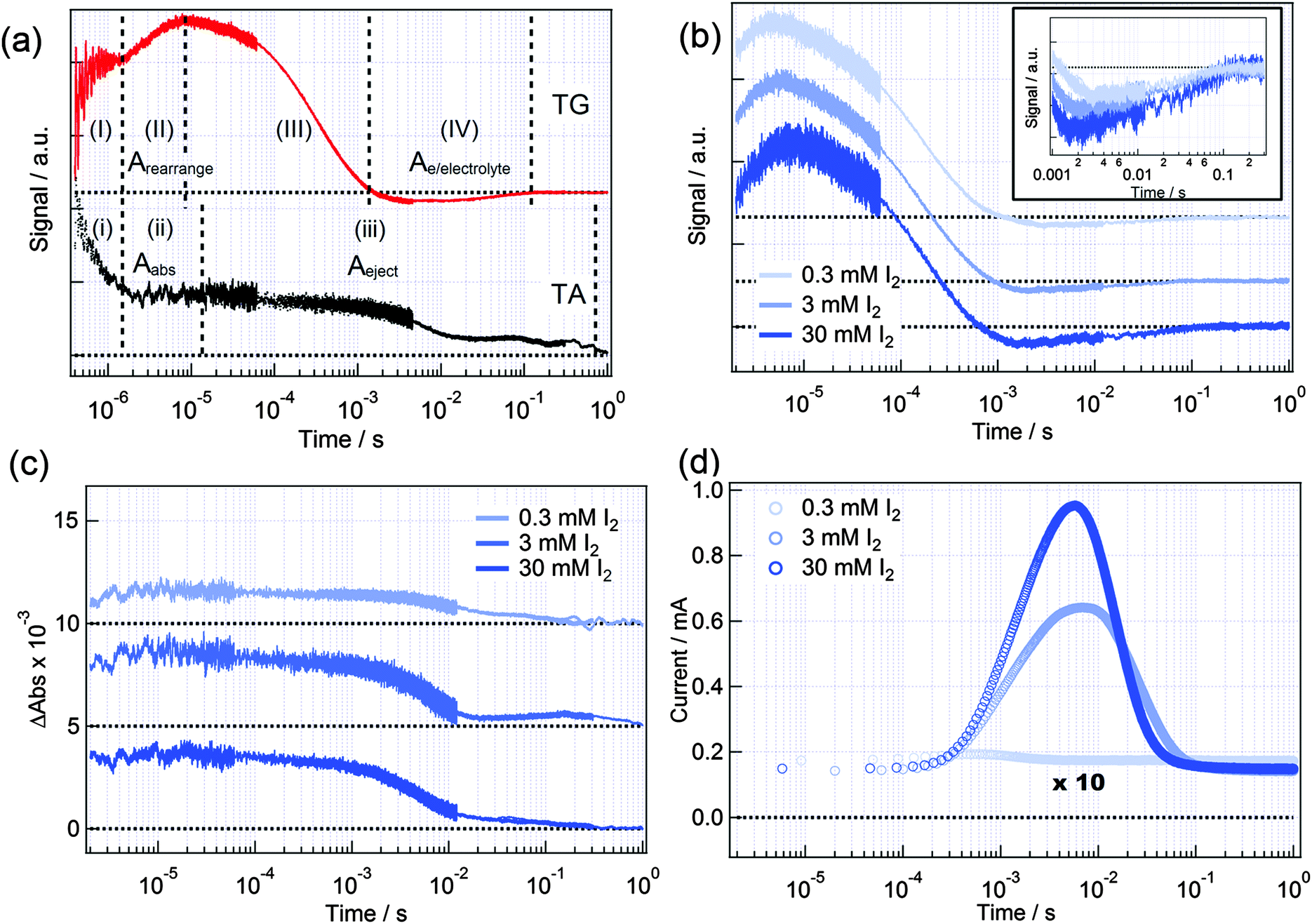 | ||
| Fig. 1 (a) Typical HD-TG and TA responses for a DSSC in LiI (300 mM)/I2 (30 mM)/ACN. (b) HD-TG, (c) TA and (d) TP responses for a DSSC with 300 mM LiI/ACN in the presence of 0.3, 3.0, 30 mM I2. The horizontal axis was plotted on a logarithmic scale. Each horizontal black dot line shows the background of signals, which were vertically displaced for clarity. The black dot line in (d) shows zero current. The HD-TG responses were normalized at the maximum signal intensity. The inset in (b) enlarges the HD-TG responses in the time range of 10−3 to 10−1 s, where the background of signals set to the same line. | ||
In the TA response shown in Fig. 1(a), three components were observed in the same time region above, as previously reported.33 The first component (i) with a decay time less than 1 μs was assigned to the disproportionation reaction of I2−, the same as the first HD-TG component. The second rising component (ii) in 10−6–10−4 s corresponds to the electro-absorption effect of the dyes due to the Stark effect28,30 induced by ionic rearrangement,28,30 and the third decaying component (iii) in 10−4 to 1 s corresponds to electron loss by the diffusion from the TiO2 layer to the electrode or by recombination processes. The second (ii) and the third (iii) components were denoted as Aabs and Aeject, respectively, in the following. Hereinafter, (I) in HD-TG and (i) in TA components were not discussed in this paper because of ambiguity of the assignment and overlap with the instrumental noise.
Generally, addition of I2 to a LiI solution generates I3− in the following equilibrium.
| I3− ⇄ I2 + I− | (1) |
Fig. 1(b) shows the HD-TG responses, and three components appeared in all responses. The rise of the Arearrange component was delayed (the time constant increased from 4 to 7 μs) as the I2 concentration increased. It indicates that the rearrangement of charged species became slower for higher I2 concentration. On the other hand, the decay of the Ae/electrolyte component was accelerated (the time constant decreased from 4 to 2 ms) with the I2 concentration, and it is understood that the electron–electrolyte recombination was accelerated by increasing the electron acceptors, namely I3− (or I244,45).
The TA responses (Fig. 1(c)) had 2 components for all electrolyte solutions. The rising component, Aabs around ∼10−5 s did not show any dependence. Since it was reported that the electro-absorption effect is caused by Li+, it is reasonable that the response did not change by variation of the I3− concentration under constant Li+ concentration. The Aeject intensity was reduced much for 0.3 mM, and it indicated that the quantity of originally injected electrons decreased.
The TP decreased as decrease in I2 concentration, and much reduced for 0.3 mM (Fig. 1(d)). This would be caused by the lower efficiency of redox cycling at the counter electrode.
There are two possible reasons to explain the I2 concentration dependence on the rearrangement response observed for the Arearrange component; one reason is that the change in the quantity of the originally injected electrons on the TiO2 side caused variation of the electrostatic force applied to the charged species and the other is that the concentration of the charged species affected the diffusion property of the charged species. The results of the TA and TP indicated that the injected electrons in TiO2 increased with increase in the redox concentration, and if the former were the reason, the rearrangement process would be accelerated by a stronger electrostatic interaction between the charged TiO2 surface and ions. Thus the latter reason is true, and the ionic motion became slower by interaction between the ions, which was supported by the fact of the smaller diffusion coefficient of ions for higher concentrations.46,47 However, the total amount of ions was almost the same and only the ratio between I3− and I− context was varied in this case. Thus, this result can be explained by the diffusion coefficient dependence of the mixed ratio in the electrolyte solution.
The electron–electrolyte recombination was accelerated by increasing the acceptor of electrons. This means that the low probability of the electron encounter with I3− (or I2) is effective for reducing the electron recombination. However, the TP decreased with the decrease in I2 concentration, and, that is to say, a sufficient quantity of I3− is needed to keep the short-circuit current. Therefore, much I3− is necessary in the bulk electrolyte solution but it should be reduced at the electrolyte/TiO2 interface.
I− concentration dependence
The influence of I− concentration was investigated while maintaining the same ionic strength of the electrolyte solution by introducing LiClO4 as a supporting electrolyte. As shown in Fig. 2(a), the response of the Arearrange component did not depend on the I− concentration. From the intensity dependence of the Ae/electrolyte, the electron–electrolyte recombination was not almost observed for concentration lower than 30 mM. | ||
| Fig. 2 (a) HD-TG, (b) TA and (c) TP responses for a DSSC with 30 mM I2/ACN in the presence of 3, 30, 300 mM LiI. LiClO4 acetonitrile solution was introduced as supporting electrolyte to maintain nearly the same ionic strength and cation concentration. The horizontal axis was plotted on a logarithmic scale. Each horizontal black dot line shows the background of signals, which were vertically displaced for clarity. The black dot line in (c) shows zero current. | ||
The TA responses shown in Fig. 2(b) had a decay component in the time region from 10−6 to 10−3, and the Aabs component was not observed except for 300 mM I− concentration. The probe wavelength (λ = 635 nm) is in the region where the dye and the dye cation have a similar absorption strength, and TA monitors both the bleach recovery of the dye absorption (MLCT) and the decay of the dye cation absorption (LMCT).30,31,48,49 It was reported that regeneration of dye cations typically occurs on the order of nanoseconds but it becomes slower, extending to the order of milliseconds under lower I− or I2 concentrations.12,13,21 We considered that slower regeneration of the dye cations (electron–dye recombination) was observed for the lower I−,20,50 and it is supported that the TA responses at 785 nm showed a similar tendency (ESI†). Hereinafter, the decaying component was referred to as Ae/dye in the following.
The Aeject component in 10−3 to 10−1 s showed a decrease in the signal intensity with decreasing the I− concentration, and was hardly observed for less than 30 mM. This indicates a decrease in the number of electrons in the TiO2 layer as a decrease in I− concentration.
The TP decreased as decreasing the I− concentration. This is because the number of electron donors, I−, was decreased and the dye regeneration did not proceed swiftly. This is consistent with the result of the Ae/dye component in the TA response.
As shown in Fig. 2(a), the Arearrange component did not depend on the I− concentration in the same ionic strength of the electrolyte solution. This process was dominated by the diffusion coefficient of ions, which was controlled by the ionic strength, namely the total ion number. It is likely that this response was affected more by I3−, considering the dependence on the ratio of I3−/I− (Fig. 1(b)) and no dependence on I− concentration (Fig. 2(a)).
The electron–electrolyte recombination was reduced by decreasing the I− concentration. It is supposed that this decrease was caused by a decrease in the electron acceptor, I3− (or I2), or reduction of the originally injected electrons in TiO2. Although I3− concentration depended on the I− concentration due to the equilibrium shown in (1), it remained 30 mM for 30 and 300 mM of I− because of the small equilibrium constant. However, the Ae/electrolyte component was almost lost for a concentration less than 30 mM. Thus, the reason for decrease in the electron–electrolyte recombination could not be explained with the I3− concentration dependence. On the other hand, the I− concentration dependence on the TA responses indicated a slow decay of dye cations for concentrations less than 30 mM, which caused electron–dye recombination. Considering these HD-TG and TA results, it is concluded that the decrease in electron–electrolyte recombination under low I− concentration was due to reduction of the accumulated electrons in TiO2, caused by the electron–dye recombination process. Thus, the TP decrease for lower I− concentrations was not caused by the electron–electrolyte recombination but by increase in the electron consumption by the electron–dye recombination prior to electron–electrolyte recombination.
Cation dependence
Cation species, which influence the electrolyte/TiO2 interface, are also an important factor to control the charge dynamics and the performance of DSSCs. To clarify the effect, different cations, Li+, DMPI+ and TBA+ were compared. The order of the ionic radius for the cations is Li+ < DMPI+ < TBA+.9,10,14Fig. 3 shows the cation dependence on the HD-TG and TP responses. The response of the Arearrange component for TBA+ was slower than those for Li+ and DMPI+. It indicates that TBA+ delayed the rearrangement process of the charged species as compared to the cases for Li+ and DMPI+. The cations used in this study had a different size and different adsorption ability on TiO2.9,10,14 TBA+ is bulky and less adsorptive than Li+ and DMPI+. It is supposed that the steric hindrance of TBA+ interrupted the ionic species exchange, causing slower rearrangement for TBA+ than those for Li+ or DMPI+.
 | ||
| Fig. 3 (a) HD-TG and (b) TP responses for a DSSC with 30 mM I2/ACN in the presence of 300 mM LiI, DMPII and TBAI. The horizontal axis was plotted on a logarithmic scale. Each horizontal black dot line shows the background of signals. The black dot line in (b) shows the zero current. | ||
The intensity of the Ae/electrolyte component also depended on the cation species. The signal intensity decreased in the order of Li+, DMPI+, TBA+, and was totally lost for TBA+. Since the order is same as the size of the cations, it is supposed that the steric hindrance for larger cations restricted their penetration into the dye/TiO2 electrode, which lowered the screening effect of the negative charge accumulated at the TiO2/solution interface. It is assumed that this hindrance of cation penetration reduces the local concentration of the counter anions, I3− (or I2), causing less probability of electron–electrolyte recombination.
The background current, corresponding to the short-circuit current generated by the constant probe light intensity, was increased in the order of Li+ > DMPI+ > TBA+, as previously reported.9,10 With regard to the TP, the total transient current, equal to the area of the TP peaks, was in the order of Li+ ≥ DMPI+ > TBA+, although the peak current for DMPI+ was larger than that for Li+. The result of the Ae/electrolyte component indicated that the probability of electron–electrolyte recombination was in the order of Li+, DMPI+, TBA+, which means the electron–electrolyte recombination was not a dominant cause for the decrease in the TP or the background current. Therefore, it is supposed that the current reduction for the larger cations was caused by decrease in the injection yield, due to the flatband potential change. As described above, the cations used in this study have a different adsorption ability on TiO2. Besides, our results indicated that the extent of the penetration into the dye/TiO2 electrode was different depending on the cation. Thus, it is supposed that ionic organization at the electrolyte/TiO2 interface induced a change in the flatband potential of TiO2, causing a change in the injection yield. There is another possibility that electron–dye recombination was increased with the cation size. This is because anions are paired with cations, similarly as the I3− (or I2), and dye regeneration may be delayed by a decrease in I− concentration at the interface.
Discussion and summary
From the results on the Arearrange component, the total amount of ionic species affected the rearrangement of the charged species at the electrolyte/TiO2 interface because this component did not change under the same ionic strength, and it is likely that I3− is mainly involved in this process. The rearrangement was delayed as an increase in the amount of ionic species, which was caused by diffusivity decrease of the involved ions with increase in the ion concentration.The size of the ionic species also affects the rearrangement process because of difference in the ion penetration into the dye/TiO2 electrode due to steric hindrance of the ions. It is well known that Li+ screen the negative charge of the injected electrons at the TiO2 surface,10 and the effect of the screening was lowered for larger cations due to their steric hindrance, where the electrostatic force applied to ions would be increased. In such electrostatic interactions between the electrons in TiO2, cations and anions affected the rearrangement of ionic species by changing the cation size.
The redox (I−, I3−) concentrations and the cation species also had an influence on the recombination processes, by changing the ratio of electron–electrolyte recombination and electron–dye recombination, or by increasing or decreasing the probability of each process. The electron–electrolyte and electron–dye recombination were observed by the Ae/electrolyte component in the HD-TG response, and the Ae/dye component in the TA response, respectively. From the I2 concentration dependence (Fig. 1(b)), the probability and the rate of electron–electrolyte recombination increased as the I2 concentration increased, which indicates that I3− (or I2) is the main acceptor for the electron capture. This electron–electrolyte recombination process was totally lost by decreasing I− concentration as shown in Fig. 2(a), because the dominant recombination process changed from the electron–electrolyte to electron–dye recombination, which is a faster recombination process than the former process, as was confirmed from Fig. 2(b). It is noted that I2 or I− concentration dependences were studied under the almost constant Li+ concentration and ionic strength, and it is considered that the redox potential of the electrolytes and the flatband potential were not changed.44 Thus, the driving force for the dye regeneration caused by I− and electrons in TiO2 was not affected, and the probability of electron–dye recombination was increased due to the long lifetime of the dye cation under low I− concentration.
The cation properties such as size, affected the electron–electrolyte recombination process, too, as shown in Fig. 3, because the cation size influenced the penetration of cations into nanoporous dye/TiO2 electrode. For the case of a larger cation, such as TBA+, steric hindrance prevented the cation approach to the interface and lowered the screening effect of the negative charge at the dye/TiO2 electrode surface, and, as a result, the approach of I3− (or I2) to the interface was prohibited. This is why the electron–electrolyte recombination process was not observed for TBA+. For DMPI+, the probability for electron–electrolyte recombination was smaller than that for Li+ but not totally lost.
As a result, it is supposed that the presence of large-sized cations suppresses electron–electrolyte recombination by disturbing the approach of I3− (or I2) to the interface due to steric hindrance of the cations. However, since it is difficult for large cations to approach the interface, the conduction band energy of TiO2 is increased, causing increase in the open-circuit voltage and decreases in the short-circuit current as was previously reported.9,10,14 To avoid decrease in the short-circuit current and to suppress electron–electrolyte recombination, it is possible that a mixture of large and small cations is used at the same time, where large cations work to prevent I3− (or I2) penetration and small cations work for the increase in the short-circuit current. Since the steric hindrance of the large cations depends on the structure of the nanoporous dye/TiO2 electrode, the ratio of the large and small cation species should be optimized. Moreover, it is suggested that I− concentration should be sufficiently high to prevent the electron–dye recombination process, and also lower I2 concentration is preferable to decrease in the probability of electron–electrolyte recombination as long as the quantity of I3− in the bulk is sufficiently high for the redox cycling.
Conclusions
We investigated the dependence of the constituents and their concentrations in the electrolyte solution on the dynamics of electrons and ion species in DSSCs, which are known to have a large influence on the performance of DSSCs. By combination of the HD-TG, TA and TP methods, we could obtain a microscopic point of view on the interface between the working electrode and an electrolyte solution. The concentration dependences of I− and I3− (or I2) on the carrier dynamics indicated that the decrease in the I3− (or I2) concentration at the dye/TiO2 electrode was effective for reducing the probability of electron–electrolyte recombination, while sufficient I− is necessary to inhibit the electron–dye recombination and sufficient I3− ions are necessary to cycle the redox reaction efficiently.The cation dependence indicated that the cation size influenced penetration of the cations into a nanoporous dyes/TiO2 electrode. It is understood that the steric hindrance disturbed the screening effect for negative charge at the dye/TiO2 electrode interface, and the hindrance also prevented I3− (or I2) approach to the interface, paired with the cations. These results indicate the possibility to control the probability of electron–electrolyte recombination by the cation size and its concentration in an electrolyte solution.
HD-TG can detect the dynamics of charged species on the liquid side in an electric double layer, including the penetration of ionic species into nanoporous dyes/TiO2 electrode. Since prevention of the penetration into the interface for the electron acceptor in the electrolyte, such as I3− (or I2) is one of the key issues for improvement of the performance of DSSCs, this approach is useful for estimation of the effect of the acceptor penetration. The electron–dye recombination can be estimated by TA, and the output current was obtained by TP, and injection efficiency can be estimated. Since we can investigate the effect of the constituents in an electrolyte solution by combination of these techniques, we expect that our approach was efficient to optimize the electrolyte, and it would accelerate the development of new DSSCs.
References
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- Z. J. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170–1181 CAS.
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- P. R. F. Barnes, A. Y. Anderson, J. R. Durrant and B. C. O'Regan, Phys. Chem. Chem. Phys., 2011, 13, 5798–5816 RSC.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- J. Bisquert, F. Fabregat-Santiago, I. Mora-Sero, G. Garcia-Belmonte and S. Gimenez, J. Phys. Chem. C, 2009, 113, 17278–17290 CAS.
- R. Katoh, A. Furube, A. V. Barzykin, H. Arakawa and M. Tachiya, Coord. Chem. Rev., 2004, 248, 1195–1213 CrossRef CAS PubMed.
- H. X. Wang and L. M. Peter, J. Phys. Chem. C, 2012, 116, 10468–10475 CAS.
- S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3480–3487 CrossRef CAS PubMed.
- C. L. Olson, J. Phys. Chem. B, 2006, 110, 9619–9626 CrossRef CAS PubMed.
- A. N. M. Green, R. E. Chandler, S. A. Haque, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2005, 109, 142–150 CrossRef CAS PubMed.
- S. Martiniani, A. Y. Anderson, C. Law, B. C. O'Regan and C. Barolo, Chem. Commun., 2012, 48, 2406–2408 RSC.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047–7054 CrossRef CAS.
- G. Boschloo, L. Haggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS PubMed.
- M. Durr, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2006, 89, 061110 CrossRef PubMed.
- R. Katoh, M. Kasuya, S. Kodate, A. Furube, N. Fuke and N. Koide, J. Phys. Chem. C, 2009, 113, 20738–20744 CAS.
- D. F. Watson and G. J. Meyer, Coord. Chem. Rev., 2004, 248, 1391–1406 CrossRef CAS PubMed.
- S. Pelet, J. E. Moser and M. Gratzel, J. Phys. Chem. B, 2000, 104, 1791–1795 CrossRef CAS.
- I. Montanari, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2002, 106, 12203–12210 CrossRef CAS.
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2010, 114, 1953–1958 CAS.
- P. R. F. Barnes, A. Y. Anderson, S. E. Koops, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2009, 113, 1126–1136 CAS.
- J. Bisquert and V. S. Vikhrenko, J. Phys. Chem. B, 2004, 108, 2313–2322 CrossRef CAS.
- A. Zaban, M. Greenshtein and J. Bisquert, ChemPhysChem, 2003, 4, 859–864 CrossRef CAS PubMed.
- B. C. O'Regan and F. Lenzmann, J. Phys. Chem. B, 2004, 108, 4342–4350 CrossRef CAS.
- M. Wang, P. Chen, R. Humphry-Baker, S. M. Zakeeruddin and M. Gratzel, ChemPhysChem, 2009, 10, 290–299 CrossRef CAS PubMed.
- S. A. Haque, Y. Tachibana, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 1998, 102, 1745–1749 CrossRef CAS.
- J. G. Rowley, S. Ardo, Y. L. Sun, F. N. Castellano and G. J. Meyer, J. Phys. Chem. C, 2011, 115, 20316–20325 CAS.
- J. G. Rowley and G. J. Meyer, J. Phys. Chem. C, 2011, 115, 6156–6161 CAS.
- S. Ardo, Y. Sun, A. Staniszewski, F. N. Castellano and G. J. Meyer, J. Am. Chem. Soc., 2010, 132, 6696–6709 CrossRef CAS PubMed.
- J. Kallioinen, G. Benko, V. Sundstrom, J. E. I. Korppi-Tommola and A. P. Yartsev, J. Phys. Chem. B, 2002, 106, 4396–4404 CrossRef CAS.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Gratzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS.
- S. Kuwahara, H. Hata, S. Taya, N. Maeda, Q. Shen, T. Toyoda and K. Katayama, Phys. Chem. Chem. Phys., 2013, 15, 5975–5981 RSC.
- N. Maeda, H. Hata, N. Osada, Q. Shen, T. Toyoda, S. Kuwahara and K. Katayama, Phys. Chem. Chem. Phys., 2013, 15, 11006–11013 RSC.
- H. Hata, Q. Shen, T. Toyoda and K. Katayama, Jpn. J. Appl. Phys., 2012, 51, 042601 Search PubMed.
- Q. Shen, Y. Ayuzawa, K. Katayama, T. Sawada and T. Toyoda, Appl. Phys. Lett., 2010, 97, 263113 CrossRef PubMed.
- Q. Shen, K. Katayama, T. Sawada, M. Yamaguchi, Y. Kumagai and T. Toyoda, Chem. Phys. Lett., 2006, 419, 464–468 CrossRef CAS PubMed.
- A. Harata, T. Edo and T. Sawada, Chem. Phys. Lett., 1996, 249, 112–116 CrossRef CAS.
- T. Morishita, A. Hibara, T. Sawada and I. Tsuyumoto, J. Phys. Chem. B, 1999, 103, 5984–5987 CrossRef CAS.
- M. Terazima, Y. Kojima and N. Hirota, Chem. Phys. Lett., 1996, 259, 451–458 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphrybaker, E. Muller, P. Liska, N. Vlachopoulos and M. Gratzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. Okuda and K. Katayama, Chem. Phys. Lett., 2007, 443, 158–162 CrossRef CAS PubMed.
- I. V. Nelson and R. T. Iwamoto, J. Electroanal. Chem., 1964, 7, 218–221 CAS.
- J. G. Rowley, B. H. Farnum, S. Ardo and G. J. Meyer, J. Phys. Chem. Lett., 2010, 1, 3132–3140 CrossRef CAS.
- C. E. Richards, A. Y. Anderson, S. Martiniani, C. H. Law and B. C. O'Regan, J. Phys. Chem. Lett., 2012, 3, 1980–1984 CrossRef CAS.
- J. F. Dufreche, O. Bernard, P. Turq, A. Mukherjee and B. Bagchi, Phys. Rev. Lett., 2002, 88, 095902 CrossRef.
- Y. Aihara, S. Arai and K. Hayamizu, Electrochim. Acta, 2000, 45, 1321–1326 CrossRef CAS.
- S. Das and P. V. Kamat, J. Phys. Chem. B, 1998, 102, 8954–8957 CrossRef CAS.
- A. Reynal, A. Forneli, E. Martinez-Ferrero, A. Sanchez-Diaz, A. Vidal-Ferran, B. C. O'Regan and E. Palomares, J. Am. Chem. Soc., 2008, 130, 13558–13567 CrossRef CAS PubMed.
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2011, 115, 2439–2447 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53964d |
| This journal is © the Owner Societies 2014 |