 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal decomposition of carboxylate ionic liquids: trends and mechanisms†
Matthew T.
Clough
a,
Karolin
Geyer
*b,
Patricia A.
Hunt
*a,
Jürgen
Mertes
b and
Tom
Welton
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: p.hunt@imperial.ac.uk; t.welton@imperial.ac.uk
bBASF SE, Ludwigshafen, Germany. E-mail: karolin.geyer@basf.com
First published on 22nd October 2013
Abstract
The thermal stability of a series of dialkylimidazolium carboxylate ionic liquids has been investigated using a broad range of experimental and computational techniques. Ionic liquids incorporating fluoroalkyl carboxylate anions were found to have profoundly differing thermal stabilities and decomposition mechanisms compared with their non-fluorinated analogues. 1-Ethyl-3-methylimidazolium acetate was observed to largely decompose via an SN2 nucleophilic substitution reaction when under inert gas conditions, predominantly at the imidazolium methyl substituent. The Arrhenius equations for thermal decomposition of 1-ethyl-3-methylimidazolium acetate, and the C2-methylated analogue 1-ethyl-2,3-dimethylimidazolium acetate, were determined from isothermal Thermogravimetric Analysis experiments. The low thermal stability of 1-ethyl-3-methylimidazolium acetate has important implications for biomass experiments employing this ionic liquid. For these two ionic liquids, ion pair and transition state structures were optimised using Density Functional Theory. The activation barriers for the SN2 nucleophilic substitution mechanisms are in good agreement with the experimentally determined values.
Introduction
Ionic liquids are versatile materials, with applications as electrolytes,1 engineering fluids,2 pre-treatment agents for wood grinding/biomass extraction3 and, fundamentally, as solvents and catalysts for synthetic chemical reactions.4 They are liquid at, or near, room temperature.5 Fully composed of cationic and anionic species, the unsymmetrical and charge-diffuse nature of the ions contributes to their low melting points. More recently, it has been suggested that there may be an entropic factor that also contributes to the low melting points of ionic liquids.6 Dialkylimidazolium ionic liquids are generally preferred for their low melting points and ease of handling and preparation.7The ionic liquid 1-ethyl-3-methylimidazolium acetate, [C2C1im][OAc], has been popular in biomass pre-treatment studies.8 These processes are carried out at elevated temperatures (120–170 °C) for extended periods of time (up to 24 h).9 Degradation of the ionic liquid will reduce the efficacy of the process, and the build up of volatile or corrosive decomposition products poses a hazard, particularly in large-scale industry.
In addition, such processes are only economic if the ionic liquid can be recycled and used multiple times. Hence, a thorough understanding of the long-term thermal decomposition behaviour of [C2C1im][OAc], and related compounds, including knowledge of the upper operational temperature limit and the precise mechanisms that are occurring, is crucial.10
Thermogravimetric Analysis (TGA) has been used extensively to study the thermal stability of ionic liquids.11 TGA experiments can be broadly divided into two categories: (i) ‘temperature-ramped’ or ‘scanning’ TGA experiments measure weight % as a function of sample temperature, with a constant heating rate (typically 10 °C min−1 or 20 °C min−1); (ii) ‘isothermal’ TGA experiments record weight % as a function of time, whilst the sample is maintained at a constant temperature.
Temperature-ramped TGA experiments allow rapid comparison of thermal stability between compounds in a series. The Tonset value is determined from extrapolation of the steepest point of the thermograph, and has been used to quantify the thermal stabilities of ionic liquids.11d,12 However, the Tonset value is known to vary with experimental conditions, including heating rate, sample quantity and pan material.11c,13 Nevertheless, temperature-ramped TGA is a suitable technique for assessing the relative thermal stabilities of a series of compounds, when care is taken to keep experimental conditions uniform. It is well established that the Tonset value provides a large overestimate of ionic liquid thermal stability.14 The ‘Tx/z’ parameter was proposed, where x represents a given decomposition extent (e.g. 1%) in z length of time (e.g. 10 hours). Hence, the T0.01/10 parameter represents the temperature at which exactly 1% weight loss occurs in 10 hours. MacFarlane and co-workers determined the T0.01/10 values for several imidazolium, pyrrolidinium and phosphonium ionic liquids, and found them to be lower by typically 110 °C compared to the Tonset temperatures.15
To date, the thermal decomposition of [C2C1im][OAc] has been partially characterised.16 In this study, we investigate the thermal decomposition of [C2C1im][OAc] and related compounds (Fig. 1) using a variety of experimental and computational techniques. The emphasis is on determining the long-term thermal stability of the ionic liquids, and understanding the thermodynamics, kinetics, and chemical mechanisms of degradation.
![Ionic liquids investigated in this study, incorporating the 1-ethyl-3-methylimidazolium [C2C1im]+ cation (1–12) and 1-ethyl-2,3-dimethylimidazolium [C2C1C1im]+ cation (13 and 14).](/image/article/2013/CP/c3cp53648c/c3cp53648c-f1.gif) | ||
| Fig. 1 Ionic liquids investigated in this study, incorporating the 1-ethyl-3-methylimidazolium [C2C1im]+ cation (1–12) and 1-ethyl-2,3-dimethylimidazolium [C2C1C1im]+ cation (13 and 14). | ||
Experimental
The ionic liquids investigated in this study are shown in Fig. 1. With the exception of 3c, which was purchased, all compounds were prepared in our laboratory from commercial starting materials, taking care to ensure purity at each step. The synthesis and characterisation of ionic liquids 1–14 is described in the ESI.† Compounds incorporating the bromide, 1, and bis(trifluoromethanesulfonyl)imide, 2, anions have been previously studied, and are included in the study for comparative purposes.14b,17 Each of the carboxylate ionic liquids 4–12 can be regarded as an analogue of [C2C1im][OAc], 3, involving systematic functionalisation of the acetate anion by fluorination (4 and 5), alkyl chain extension (6–9), including branching (10), a strained cyclopropane ring (11), and by incorporating a sulfur atom into the carboxylate group to give the thioacetate anion (12). Three separate samples of 1-ethyl-3-methylimidazolium acetate, [C2C1im][OAc], denoted 3a–c, represent the same compound prepared by three different synthetic procedures. A metathesis reaction involving [C2C1im]Br, 1, and silver(I) acetate in aqueous solution yielded 3a. Compound 3b was prepared by the neutralisation of an aqueous solution of [C2C1im][OH] with acetic acid. The aqueous [C2C1im][OH] was generated from a metathesis of [C2C1im][HSO4] with barium hydroxide in aqueous solution, which precipitates insoluble barium sulfate.18 Ionic liquid 3c was a purchased commercial sample of [C2C1im][OAc].19Thermogravimetric Analysis (TGA) was carried out on a PerkinElmer ‘Pyris 1’ TGA apparatus. Temperature-ramped TGA experiments were performed on 4–8 mg of the ionic liquid, using a heating rate of 10 °C min−1, and platinum pans. Unless otherwise stated, nitrogen was used as the inert carrier gas. Isothermal TGA was performed on 7–8 mg of the ionic liquid. Thermogravimetric Analysis–Mass Spectrometry (TGA-MS) experiments were performed on 20–60 mg of the ionic liquid, using ceramic pans. A heating rate of 10 °C min−1 and a CP Grade helium flow of 20 ml min−1 were employed. The ‘Pyris 1’ TGA was connected to a ‘Hiden Analytical HPR 20’ Mass Spectrometer with a ceramic heated capillary. Electron Ionisation (EI) mass spectrometry was employed, with an ionisation energy of 70 eV.
Differential Scanning Calorimetry (DSC) experiments were performed on 1–9 mg of the ionic liquid, using aluminium pans. In order to allow volatile degradants to escape, and to avoid the risk of the pans bursting under pressure, a small incision was made in the top of both the sample and the reference pan. The pan was heated initially to 100 °C for 30 minutes, in order to remove water. The sample pan was then re-weighed and the DSC program was initiated. DSC traces were collected in the range 30–410 °C, with a heating rate of 2.5 °C min−1. After the first heating step, the sample was cooled to 30 °C at a rate of 20 °C min−1. The sample was then maintained at 30 °C for 15 minutes, followed by a second heating step using an identical program to the first heating step. The second heating step would indicate the phase behaviour of any residual decomposition products present in the sample pan following the first heating stage.
Full TGA, TGA-MS and DSC experimental conditions are described in the ESI.†
Results and discussion
The thermal stabilities of ionic liquids 1–14 were compared using temperature-ramped TGA (Fig. 2). The temperature-ramped TGA experiments were performed in triplicate for each ionic liquid, in order to assess the reproducibility of the Tonset measurement. For all ionic liquids in this investigation, the Tonset values were reproducible to within ±2 °C. The middle value for each ionic liquid is tabulated in Table 1. The three samples of [C2C1im][OAc], 3a–c, give identical Tonset values within the experimental error (Table 1). This implies that the commercial sample, 3c, generated on large scale, is as stable at high temperatures as the samples prepared in the laboratory either via silver(I) or barium metathesis reactions, 3a and 3b respectively.![Temperature-ramped TGA thermographs for selected [C2C1im]+ and [C2C1C1im]+ ionic liquids, 1, 2, 3a, 4 and 14, with heating rate 10 °C min−1.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f2.gif) | ||
| Fig. 2 Temperature-ramped TGA thermographs for selected [C2C1im]+ and [C2C1C1im]+ ionic liquids, 1, 2, 3a, 4 and 14, with heating rate 10 °C min−1. | ||
| Ionic liquid | T onset ± 2 (°C) | |
|---|---|---|
| 1 | [C2C1im]Br | 301 |
| 2 | [C2C1im][NTf2] | 436 |
| 3a | [C2C1im][OAc] Ag route | 214 |
| 3b | [C2C1im][OAc] Ba route | 216 |
| 3c | [C2C1im][OAc] Commercial | 216 |
| 4 | [C2C1im][CF3CO2] | 172 |
| 5 | [C2C1im][CHF2CO2] | 263 |
| 6 | [C2C1im][CH3(CH2)2CO2] | 213 |
| 7 | [C2C1im][CH3(CH2)4CO2] | 212 |
| 8 | [C2C1im][CH3(CH2)6CO2] | 213 |
| 9 | [C2C1im][CH3(CH2)14CO2] | 216 |
| 10 | [C2C1im][C(CH3)3CO2] | 212 |
| 11 | [C2C1im][c-(C3H5)CO2] | 219 |
| 12 | [C2C1im][SAc] | 188 |
| 13 | [C2C1C1im]Br | 299 |
| 14 | [C2C1C1im][OAc] | 221 |
Trifluorination of the acetate anion in compound 4 leads to lower thermal stability than the non-fluorinated compound 3a–c (Fig. 2). Pibiri and co-workers examined the thermal stabilities of a series of protic perfluoroalkyl-carboxylate ionic liquids using TGA, and found these to be low.20
By contrast, ionic liquid 5, which incorporates the difluoroacetate anion, is found to be significantly more stable than both the non-fluorinated and trifluorinated compounds, 3 and 4, with a Tonset of 263 °C. Therefore, [C2C1im][CHF2CO2], 5, was found to be an analogue of the acetate ionic liquid, 3, with improved thermal stability.
Extension of the carboxylate alkyl chain, in compounds 6–9, has a minimal effect on the Tonset thermal decomposition temperature of the ionic liquid (Fig. 2). Surprisingly, even the 16-carbon palmitate compound, [C2C1im][CH3(CH2)14CO2], 9, gave a thermograph very similar to the acetate ionic liquid, 3a. Branching of the carboxylate alkyl chain, illustrated by compounds 10 and 11, also has a very small effect on the Tonset value. There is a small increase in the basicity of the carboxylate anion with the addition of electron-donating alkyl substituents, indicated by the pKa values of the conjugate carboxylic acids (acetic acid 4.76; butyric acid 4.83; cyclopropanecarboxylic acid 4.83; hexanoic acid 4.85; octanoic acid 4.89; trimethylacetic/pivalic acid 5.03).21 Nevertheless, this brings about no substantial change in the Tonset value of the ionic liquid. Therefore, temperature-ramped TGA experiments indicate that there is no substantial difference in the thermal stability of [C2C1im]+ carboxylate ionic liquids with differing length/branched alkyl chains, and that the decomposition mechanism involves the reactive carboxylate functional group.
The sulfur analogue, [C2C1im][SAc], 12, is substantially less thermally stable than the acetate equivalent, 3. Thermal decomposition of 12 occurs through a nucleophilic substitution mechanism (Fig. 7, below), because of the high nucleophilicity and low basicity of the thioacetate anion (pKa thioacetic acid = 3.33).21 The negative charge on the thioacetate anion is known to be centred almost entirely on the sulfur atom, due to the large difference in the energies of the thiol and thione forms.22 It was observed that over a period of six months, the yellow thioacetate ionic liquid, 12, gradually darkened when stored at 4 °C under nitrogen (Fig. 3).
![(a) Observed decomposition of [C2C1im][SAc], 12, after six months, when stored under nitrogen at 4 °C. (b) Aliphatic region of the 1H NMR spectrum of [C2C1im][SAc], 12, after six months, demonstrating the disappearance of the thioacetate singlet at 2.12 ppm, and the appearance of a peak at 1.75 ppm.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f3.gif) | ||
| Fig. 3 (a) Observed decomposition of [C2C1im][SAc], 12, after six months, when stored under nitrogen at 4 °C. (b) Aliphatic region of the 1H NMR spectrum of [C2C1im][SAc], 12, after six months, demonstrating the disappearance of the thioacetate singlet at 2.12 ppm, and the appearance of a peak at 1.75 ppm. | ||
The colour change suggests poor long-term stability. Moreover, the 1H NMR spectrum of the darkened liquid showed a decrease in the integration of the thioacetate methyl peak, accompanied by the appearance of a new small singlet at 1.75 ppm. 1H NMR and LSIMS mass spectrometry experiments revealed that partial hydrolysis of thioacetate to acetate had occurred. The convoluted synthesis and offensive odour of 12 indicate that it will not be a suitable candidate for many ionic liquid applications.
The identity of the flow gas could potentially affect the thermal stabilities of ionic liquids 1–14. Therefore, subsequent temperature-ramped TGA experiments were performed for the archetypal ionic liquid [C2C1im][OAc], 3c, under compressed air and CO2 flow gases, in addition to the usual inert nitrogen. A TGA thermograph for 3c under a CO2 carrier gas is displayed in the ESI† (Fig. E2). Prior to each scan, the ionic liquid was heated to 80 °C for 10 hours in the TGA apparatus under a gentle flow of each gas, to ensure equilibration of the dissolved gases in the ionic liquid. The presence of oxygen gas in compressed air introduces the possibility of oxidation or combustion occurring. Moreover, employing CO2 as the carrier gas may indicate the presence of an N-heterocyclic carbene (NHC) species forming in the ionic liquid, since CO2 could act as a carbene trap.23 Interestingly, no change in the thermal stability of 3c was observed under the compressed air environment and a Tonset value of 216 °C was recorded, suggesting that neither oxidation or combustion are thermal decomposition mechanisms. However, for the experiment employing a CO2 carrier gas, an initial gain of mass was observed for the first two hours of the 10 hour 80 °C equilibration period, followed by gradual loss of mass for the remainder of the equilibration step. The sample then gave a Tonset value of 225 °C in the subsequent temperature-ramped step, slightly higher than for 3c in the nitrogen experiment. This result suggests the formation of the [C2C1im+-CO2−] complex previously observed by Rogers and co-workers.23 Therefore, it is crucial to consider the nature of the gaseous environment when employing ionic liquid 3c.
The Tonset temperature, as determined by a step-tangent method, is well known to overestimate the thermal stability of ionic liquid compounds.14,15 Therefore, the T0.01/10 value was determined for [C2C1im][OAc], 3a, and [C2C1C1im][OAc], 14, from isothermal TGA experiments using the method of MacFarlane and co-workers.14b The procedure is described in the ESI.† Isothermal TGA graphs for 3a are shown below (Fig. 4); graphs for 14 are displayed in the ESI† (Fig. E4).
![(a) Isothermal TGA analysis of [C2C1im][OAc], 3a. (b) The t0.99 (time taken for 1% decomposition to occur) values were plotted against temperature (T) in Kelvin, giving an exponential fit. The fitted curve was extrapolated backwards to find T0.01/10 corresponding to t0.99 = 600 min/10 hours. T0.01/10 was determined as 102 °C.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f4.gif) | ||
| Fig. 4 (a) Isothermal TGA analysis of [C2C1im][OAc], 3a. (b) The t0.99 (time taken for 1% decomposition to occur) values were plotted against temperature (T) in Kelvin, giving an exponential fit. The fitted curve was extrapolated backwards to find T0.01/10 corresponding to t0.99 = 600 min/10 hours. T0.01/10 was determined as 102 °C. | ||
For 3a and 14, the T0.01/10 parameter was measured as 102 °C and 99 °C, respectively. For 3a, this value is significantly lower than the operating temperature of many biomass processes employing this ionic liquid (typically upwards of 120 °C).9 In addition, biomass procedures are often carried out for periods substantially longer than 10 hours. Therefore, these procedures are likely to lead to substantial degradation of this acetate ionic liquid.
The T0.01/10 value is a reasonable approximation of long-term thermal stability, although the ability to accurately predict the rate of decomposition at any given temperature is necessary. The Arrhenius equation, (1), representing the ionic liquid decomposition, can be determined from the isothermal TGA data.24 Hence, the activation energy, Ea, and the pre-exponential factor, A, can be calculated. This allows rapid and accurate prediction of the decomposition rate at any temperature. The Arrhenius equations representing decomposition of [C2C1im][OAc], 3a, and [C2C1C1im][OAc], 14, have been determined using this isothermal method.
Where ionic liquids thermally decompose via the reaction of one cation with one anion, thereby generating volatile degradants, the concentration of cations and anions in the bulk ionic liquid does not change. Therefore in these circumstances, decomposition is pseudo zeroth order. The rate of a decomposition of this type can be expressed in terms of the molar proportion of the ionic liquid that has decomposed as a function of time, dα/dt (1). Plotting ln(dα/dt) against 1/T gives a linear fit with a gradient equal to −Ea/R and a y-axis intercept equal to ln(A).
dα/dt = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT) exp(−Ea/RT) | (1) |
![Thermal decomposition kinetics for [C2C1im][OAc], 3a, determined from isothermal TGA experiments. Ea = 115.8 kJ mol−1, A = 2.98 × 109.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f5.gif) | ||
| Fig. 5 Thermal decomposition kinetics for [C2C1im][OAc], 3a, determined from isothermal TGA experiments. Ea = 115.8 kJ mol−1, A = 2.98 × 109. | ||
The activation energy, Ea, was found to be 115.8 kJ mol−1 for 3a and 99.6 kJ mol−1 for 14. Although the T0.01/10 values for ionic liquids 3a and 14 are very similar (102 and 99 °C, respectively), the activation energies differ by approximately 16 kJ mol−1. However, the pre-exponential factors differ by two orders of magnitude (2.98 × 109 for 3a, 2.19 × 107 for 14). The pre-exponential factor, A, often referred to as the ‘frequency factor’, is intrinsically linked to the frequency of molecular collisions occurring, whether or not the collision leads to a reaction. The differing values of A suggest significantly fewer collisions are occurring for 14 than for 3a, although the lower Ea value for 14 indicates that a greater proportion of collisions will result in a decomposition reaction. The differing values of A may be a result of the reduced mobility of the 1-ethyl-2,3-dimethylimidazolium cation, caused by the additional steric bulk and van der Waals interactions of the C2-methyl substituent.
Ionic liquid 14 has a measured melting point of 90–92 °C, whereas 3a is a liquid at room temperature. Therefore, at temperatures high enough for 14 to be in the liquid state, decomposition will already be occurring at a rate of approximately 1% in 24 hours. This will restrict the suitability of ionic liquid 14 for many applications.
When describing thermal decomposition in an open system, such as in the TGA apparatus, the contribution to mass loss from evaporation of the ionic liquid must also be considered. Despite once being heralded as completely non-volatile, ionic liquids are now known to exert a vapour pressure.25 Successful distillation of high thermal stability [NTf2]-containing ionic liquids has been demonstrated, by employing ultra-high vacuum (UHV) conditions.25b,d Heym and colleagues observed that for [C4C1im][NTf2], evaporation dominated thermal decomposition at low heating rates in thermogravimetric experiments.25e The enthalpy of vaporisation at 298 K, ΔvapH298, for this ionic liquid has been measured as 133–140 kJ mol−1, depending on the chosen experimental method.25a
For the dialkylimidazolium and trialkylimidazolium carboxylate ionic liquids investigated here, it is unclear whether thermal decomposition is occurring exclusively, or whether vaporisation of the intact ionic liquid is also involved. However, Shimizu and co-workers have calculated the ΔvapH value for [C4C1im][OAc] (using the relationship ΔvapH = Ucohesive + RT), and found this value to be 100 kJ mol−1 higher than for [C4C1im][NTf2].25j Furthermore, Chandran et al. obtained a ΔvapH value of 187.6 kJ mol−1 for [C4C1im][OAc] from MD simulations.25g The Tonset temperatures for all alkyl and fluoroalkyl carboxylate ionic liquids here are at least 170 °C lower than for [C2C1im][NTf2], 2 (Table 1).
Therefore, there is likely to be a contribution from ionic liquid vaporisation for ionic liquid 2.25e However, on the basis of low thermal decomposition temperatures and high vaporisation enthalpies, the contribution to mass loss from vaporisation is likely to be negligible for all alkyl and fluoroalkyl carboxylate ionic liquids, 3–14, in this study. Instead, all mass loss is attributed to the evaporation of volatile thermal decomposition products.
An understanding of the safe operating temperatures of an ionic liquid is crucial. In addition, knowledge of the thermal decomposition products is important; the production of volatile or corrosive degradants represents a safety hazard, and may lead to a reduction in the efficacy of a process. Certain ionic liquids are known to thermally decompose by nucleophilic or basic attack of the anion on the cation,11a,15,17b,26 or by initial degradation of the anion into reactive species, which subsequently react with the cation.17a,c,27 The formation of NHC species from dialkylimidazolium ionic liquids is recognised as another potential decomposition route.28 Ionic liquids incorporating the tetrafluoroborate, [BF4]−, and hexafluorophosphate, [PF6]−, anions are susceptible to hydrolysis, liberating highly corrosive hydrogen fluoride, HF.29 Carboxylate anions are both moderately nucleophilic and basic, and several chemically-plausible decomposition mechanisms can therefore be proposed for the 1-ethyl-3-methylimidazolium carboxylate ionic liquids (A–E, Fig. 6).
![Postulated decomposition mechanisms for [C2C1im]+ ionic liquids 3a, 4, 8 and 12: (A) N-heterocyclic carbene (NHC) formation; (B) + (C) SN2 nucleophilic substitution; (D) E2 Hofmann elimination; (E) decarboxylation of the anion.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f6.gif) | ||
| Fig. 6 Postulated decomposition mechanisms for [C2C1im]+ ionic liquids 3a, 4, 8 and 12: (A) N-heterocyclic carbene (NHC) formation; (B) + (C) SN2 nucleophilic substitution; (D) E2 Hofmann elimination; (E) decarboxylation of the anion. | ||
Thermogravimetric Analysis (TGA) coupled with Mass Spectrometry (MS) allows the stream of inert gas to be analysed, and chemical products from the ionic liquid thermal decomposition can be detected. The detection of peaks at m/z values corresponding to parent ions or fragment ions of certain decomposition products is indicative of a particular mechanism or mechanisms occurring.
Dialkylimidazolium and trialkylimidazolium ionic liquids incorporating bis(trifluoromethanesulfonyl)imide (2), acetate (3a, 14), trifluoroacetate (4), octanoate (8) and thioacetate (12) anions have been investigated in this TGA-MS study. In order for the mass spectral peaks to be of sufficient intensity, a larger quantity (20–60 mg) of ionic liquid was used than in the regular temperature-ramped TGA experiments. High purity helium was used as the carrier gas. Electron Ionisation (EI) mass spectra were recorded at intervals throughout the experiment, in the range m/z 10–300. A 70 eV ionisation energy was employed. The main peaks of interest were of m/z 40 and above.
Fragmentation of small-molecule decomposition products is possible under the harsh conditions of electron ionisation mass spectrometry (EIMS). Therefore, the parent ion corresponding to a given thermal decomposition product may not always be observed, even if the decomposition product is present in the inert flow gas. Data from the ‘National Institute of Standards and Technology’ (NIST) Chemistry WebBook was used to identify expected fragmentation of small-molecule decomposition products.30
For each ionic liquid, a single mass spectrum was selected from the TGA-MS data, recorded at a time point during the main thermal decomposition period of the ionic liquid. Major peaks were then assigned as parent peaks, or fragments, of decomposition products. Example MS histograms for [C2C1im][OAc], 3a, [C2C1im][CF3CO2], 4, and [C2C1im][SAc], 12, are shown in Fig. 7. Subsequently, the intensity of particular m/z peaks could be plotted as a function of temperature, and overlaid with the TGA thermograph. An example TGA-MS graph of this type for 3a is shown in Fig. 8. Additional TGA-MS spectra are shown in the ESI† (Fig. E5 and E6).
![TGA-MS data for ionic liquids [C2C1im][OAc], 3a, (a), [C2C1im][CF3CO2], 4, (b), and [C2C1im][SAc], 12, (c). The displayed MS histograms were recorded during the main thermal decomposition period of the ionic liquid. Peaks were assigned to parents or fragments of expected decomposition products. Spectra are shown in the region 40–150 m/z, for clarity.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f7.gif) | ||
| Fig. 7 TGA-MS data for ionic liquids [C2C1im][OAc], 3a, (a), [C2C1im][CF3CO2], 4, (b), and [C2C1im][SAc], 12, (c). The displayed MS histograms were recorded during the main thermal decomposition period of the ionic liquid. Peaks were assigned to parents or fragments of expected decomposition products. Spectra are shown in the region 40–150 m/z, for clarity. | ||
![TGA-MS investigation of [C2C1im][OAc], 3a. The solid black line represents the TGA thermograph; coloured dashed lines represent the MS intensities for selected m/z values; the assigned decomposition products are shown.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f8.gif) | ||
| Fig. 8 TGA-MS investigation of [C2C1im][OAc], 3a. The solid black line represents the TGA thermograph; coloured dashed lines represent the MS intensities for selected m/z values; the assigned decomposition products are shown. | ||
The thermal decomposition of [C2C1im][NTf2], 2, has been previously studied in detail, and a decomposition mechanism was suggested.17c Chen and colleagues observed a peak at m/z 64, which was assigned as [SO2]+˙. In addition, their experiments showed smaller peaks corresponding to a number of other degradants, including 1-methylimidazole, 1-ethylimidazole, ethene, CH3NH2 and CHF3. They therefore proposed a mechanism for the decomposition of 2, involving an anion-centred decomposition to generate reactive radical intermediates (CF3˙, NH2˙, F˙), which then react with the [C2C1im]+ cation. The results of our TGA-MS studies on compound 2 (shown in the ESI†) are consistent with their findings; peaks at m/z 64 and m/z 69 were seen during the main decomposition period, which were assigned as [SO2]+˙ and [CF3]+, respectively. Therefore, the presence of these peaks suggests degradation of the [NTf2]− anion during the decomposition of ionic liquid 2.
In the TGA-MS experiments for the archetypal carboxylate ionic liquid [C2C1im][OAc], 3a, the onset of thermal decomposition is accompanied by the detection of a large peak at m/z 74 in the mass spectrometer, which is assigned to the parent ion of methyl acetate, [CH3CO2CH3]+˙ (Fig. 7a and 8). This strongly suggests an SN2 mechanism occurring at the methyl substituent of the [C2C1im]+ cation, because it is unlikely that any other mechanism could produce substantial amounts of this degradant (Fig. 6). The peak at m/z 59 was assigned to [CH3CO2]+, a known fragment of methyl acetate, but not of acetic acid or ethyl acetate in EI mass spectrometry.30 This is further evidence for the SN2 mechanism occurring at the methyl substituent.
A small peak corresponding to ethyl acetate, [CH3CO2CH2CH3]+˙ (m/z 88), was also observed, indicative of nucleophilic attack at the more sterically crowded ethyl substituent (Fig. 8). These mechanisms are consistent with the nucleophilic character of the acetate anion. An acetic acid parent peak (m/z 60) is detected as a minor decomposition product (Fig. 8). This suggests that either an E2 Hofmann elimination mechanism or NHC formation is occurring to a small extent. No fragment at m/z 60 is expected in the EI mass spectrum of either methyl acetate or ethyl acetate,30 therefore our observed peak at m/z 60 cannot be regarded as a fragment of either ester decomposition product.
Interestingly, no significant peaks were observed for the molecular ion of 1-methylimidazole (m/z 82), 1-ethylimidazole (m/z 96), or for the N-heterocyclic carbene 1-ethyl-3-methylimidazol-2-ylidene (m/z 110) that would form in the event of NHC decomposition (mechanism A, Fig. 6). The neutral heterocycle 1-ethylimidazole should be present as a product of the SN2 thermal decomposition mechanism occurring at the methyl substituent of the 1-ethyl-3-methylimidazolium cation of 3a. Attempts to confirm 1-ethylimidazole as a decomposition product of 3a, by high-temperature 1H NMR experiments, are described below.
The low Tonset value for [C2C1im][CF3CO2], 4, can be understood in terms of an anion-centred decarboxylation mechanism, enabled by the stability of the CF3− anion or CF3˙ radical that would be formed. The TGA-MS data for this compound supports this hypothesis, as indicated by the presence of peaks at m/z 44, [CO2]+˙, m/z 69, [CF3]+, and m/z 51, [CHF2]+. The peak at m/z 69 could be attributed to multiple plausible decomposition products of [C2C1im][CF3CO2], 4, including CHF3, trifluoroacetic acid, methyl trifluoroacetate and ethyl trifluoroacetate.30 However, the fragment at m/z 51, [CHF2]+, strongly suggests the presence of CHF3 as a major decomposition product of ionic liquid 4, since it is not a known fragment of the potential acid or ester products generated by SN2 or E2 mechanisms.30 Although the peak at m/z 44 is observed throughout the duration of the TGA-MS experiment (CO2 is a minor contaminant of the helium carrier gas), there is a visible spike in intensity corresponding to the onset of mass loss. The TGA-MS data strongly suggests an anion-centred decarboxylation mechanism for the thermal decomposition of ionic liquid 4.
The TGA-MS results for the octanoate ionic liquid, 8, were compared with those for the acetate ionic liquid, 3a, in order to understand the effect of alkyl chain extension on the prevailing mechanisms of thermal decomposition. For 8, a peak was observed at m/z 158, which has been assigned as the parent peak of methyl octanoate, [CH3(CH2)6CO2CH3]+˙. This suggests an SN2 nucleophilic attack of the octanoate anion at the methyl substituent of the [C2C1im]+ cation (mechanism B, Fig. 6). Lower intensity peaks were observed that can be assigned to fragments of octanoic acid and ethyl octanoate. Unlike the TGA-MS experiment for 3a, the parent peaks of 1-methylimidazole (m/z 82) and 1-ethylimidazole (m/z 96) were observed in the spectrum of [C2C1im][CH3(CH2)6CO2], 8. The presence of these two neutral imidazole products is further evidence for SN2-type decomposition mechanisms.
For the ionic liquid [C2C1im][SAc], 12, a large peak at m/z 90 was observed, corresponding to the parent ion of methyl thioacetate, [CH3COSCH3]+˙. This is strong evidence of an SN2 mechanism occurring at the methyl substituent of the [C2C1im]+ cation, analogous to the decomposition of 3a. This is unsurprising, given the known high nucleophilicity of the thioacetate anion.
For the C2-methyl substituted ionic liquid, [C2C1C1im][OAc], 14, the NHC decomposition route (mechanism A, Fig. 6) is not possible. Instead, a proton may be abstracted from the C2 methyl group, generating acetic acid and a neutral species, 1-ethyl-3-methyl-2-methylene-2,3-dihydroimidazole (m/z 124). However, no detectable peak was observed at m/z 124 during the decomposition of ionic liquid 14, suggesting that this mechanism does not occur to any measurable extent. Instead, the major observed decomposition product in the TGA-MS experiment for ionic liquid 14 was methyl acetate, [CH3CO2CH3]+˙, at m/z 74. Therefore, it is likely that the decomposition of both acetate ionic liquids, 3a and 14, occurs predominantly via an SN2 nucleophilic attack at the imidazolium methyl substituent.
The TGA-MS results therefore indicate the occurrence of SN2 nucleophilic substitution decomposition mechanisms (3a, 8, 12 and 14), or anion-centred decomposition, generating reactive intermediates which subsequently attack the [C2C1im]+ cation (2 and 4). For each of the ionic liquids investigated in this TGA-MS study, there was no substantive evidence of the anion behaving as a base.
The decomposition of [C2C1im][OAc], 3a, was explained in terms of an SN2 nucleophilic substitution mechanism at the methyl substituent of the [C2C1im]+ cation, on the basis of the large observed peak for methyl acetate in the TGA-MS experiment. However, the other major expected decomposition product, 1-ethylimidazole, was not observed.
In an attempt to monitor the formation of 1-ethylimidazole and other decomposition products, a neat sample of 3a was heated to 120 °C for 12 hours in a sealed, thick-walled NMR tube. A 1H NMR spectrum was recorded every 30 minutes. The high-temperature 1H NMR experiment was performed unlocked, in the absence of any NMR solvent. Therefore, to determine the chemical shifts of the ionic liquid and decomposition product peaks, two separate control NMR spectra were obtained, whereby pure [C2C1im][OAc] had been deliberately spiked with a small quantity of an expected decomposition product, 1-methylimidazole and 1-ethylimidazole respectively. A DMSO-d6 capillary was used as a reference in each reference sample.
During the 12-hour experiment, no additional peaks were observed. It is possible that the broadened ionic liquid peaks engulfed the decomposition product peaks, or that the decomposition products were partially in the gas phase. However, upon cooling 3a to room temperature, new peaks not present in the ionic liquid prior to the high-temperature experiment were observed in the 1H NMR spectrum (Fig. 9). Several peaks in the region 7.4–6.1 ppm were observed at approximately 1–2% of the integration of the ionic liquid aromatic peaks. The chemical shifts of these aromatic peaks matched the neutral heterocyclic molecules 1-methylimidazole and 1-ethylimidazole (Fig. 9a). In addition, a triplet peak was observed at 0.69 ppm, which has been assigned to the terminal –CH3 unit of the ethyl chain of 1-ethylimidazole (Fig. 9b). A considerably smaller peak was observed for the aliphatic methyl group of 1-methylimidazole. This lends evidence towards an SN2 nucleophilic substitution reaction occurring at both alkyl substituents of the 1-ethyl-3-methylimidazolium cation, but predominantly at the less hindered methyl substituent.
![Expanded regions of the 1H NMR spectrum of [C2C1im][OAc] at room temperature, after the 12 h experiment at 120 °C. (a) New aromatic peaks were observed in the region 7.4–6.1 ppm, which were assigned to 1-alkylimidazole decomposition products. (b) A triplet peak was observed at 0.69 ppm; the chemical shift and splitting pattern indicate 1-ethylimidazole.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f9.gif) | ||
| Fig. 9 Expanded regions of the 1H NMR spectrum of [C2C1im][OAc] at room temperature, after the 12 h experiment at 120 °C. (a) New aromatic peaks were observed in the region 7.4–6.1 ppm, which were assigned to 1-alkylimidazole decomposition products. (b) A triplet peak was observed at 0.69 ppm; the chemical shift and splitting pattern indicate 1-ethylimidazole. | ||
Subsequently, thermal decomposition of the fluoroalkyl carboxylate ionic liquids [C2C1im][CF3CO2], 4, and [C2C1im][CHF2CO2], 5, was investigated using 1H and 19F NMR. Small samples (ca. 20 mg) of 4 and 5 were placed in 25 ml sealed glass pressure vessels, and maintained at 100 °C for 18 hours, before being allowed to cool to room temperature. The residues were dissolved in DMSO-d6, and 1H and 19F NMR spectra were recorded.
The 19F NMR experiments did not reveal any fluorine-containing thermal decomposition products for either ionic liquid 4 or 5. Two expected decomposition products from the anion decarboxylation of 4 are CHF3 and CO2, which are both gaseous products (Fig. 6). Therefore, if formed, these thermal decomposition products would have escaped when the pressure tubes were opened. The high Tonset value for [C2C1im][CHF2CO2], 5, may explain why no fluorine-containing products were observed for this ionic liquid after the 18 hour 100 °C heating period.
By contrast, small peaks were observed in the 1H NMR spectrum for 4 that were not present in the pure ionic liquid before commencing the experiment. These peaks, at 7.18, 6.87 and 3.64 ppm, were subsequently assigned to the C4, C5 and –CH3 protons on the neutral heterocycle 1-methylimidazole, by comparing the spectrum of 4 after the heating period against a reference 1H NMR spectrum of 1-methylimidazole in DMSO-d6. These 1H and 19F NMR results are compatible with the previous observation that [C2C1im][CF3CO2], 4, decomposes via an anion-centred decarboxylation (Fig. 7b). Therefore, it is likely that the CF3− anion, generated by the decarboxylation mechanism, then removes a β-proton from the ethyl substituent on the [C2C1im]+ cation in an E2-type mechanism, generating the observed 1-methylimidazole.
Smaller peaks were observed at the same chemical shifts in the 1H NMR of 5, following the 18 hour heating period at 100 °C, indicating that a very small quantity of 1-methylimidazole had also been generated from the thermal decomposition of this ionic liquid. 1-Methylimidazole was not employed at any stage of the synthesis of compounds 4 and 5, therefore it cannot be regarded as an impurity.
The thermal decomposition of ionic liquids 3b, 4, 11 and 14 was subsequently investigated using Differential Scanning Calorimetry (DSC), and ΔHdecomposition values were measured. In order to correlate the DSC peaks to specific decomposition processes observed in the TGA, a temperature-ramped TGA experiment was performed for each of the four ionic liquids at the same slow heating rate of 2.5 °C min−1. These TGA thermographs were subsequently superimposed on the DSC traces. DSC procedures are described in the experimental section of this contribution and in the ESI.† An example graph for [C2C1im][OAc], 3b, is displayed in Fig. 10. Graphs for ionic liquids 4, 11 and 14 are displayed in the ESI† (Fig. E7). ΔHdecomposition data for the four ionic liquids is shown in Table 2.
![DSC trace for the decomposition of [C2C1im][OAc], 3b, shown in the range 100–300 °C (blue). A temperature-ramped TGA thermograph is superimposed (black).](/image/article/2013/CP/c3cp53648c/c3cp53648c-f10.gif) | ||
| Fig. 10 DSC trace for the decomposition of [C2C1im][OAc], 3b, shown in the range 100–300 °C (blue). A temperature-ramped TGA thermograph is superimposed (black). | ||
| Ionic liquid | ΔHdecomposition (kJ mol−1) | |
|---|---|---|
| 3b | [C2C1im][OAc] | 51.9 |
| 4 | [C2C1im][CF3CO2] | 15.7, 10.5 |
| 11 | [C2C1im][c-(C3H5)CO2] | 64.7 |
| 14 | [C2C1C1im][OAc] | 18.0 |
The DSC profile for the first heating step of 3b displayed one broad endothermic peak, between approximately 200–250 °C. This endothermic peak occurs in the same temperature range as the TGA weight loss, and has been assigned as decomposition of the ionic liquid, with ΔHdecomposition = +51.9 kJ mol−1 (Fig. 10).
The DSC profile representing the decomposition of [C2C1im][CF3CO2], 4, demonstrated a two-step process, corresponding to the two visible decomposition stages in the temperature-ramped TGA thermograph. Both steps were endothermic in nature and of similar magnitude, +15.7 and +10.5 kJ mol−1, respectively.
Ionic liquid 11, containing the strained cyclopropane ring, exhibits endothermic decomposition in a similar fashion to the acetate ionic liquid 3b. The measured ΔHdecomposition value of +64.7 kJ mol−1 for 11 is comparable to the value for 3b. No additional peaks were observed in the DSC decomposition profile for 11, suggesting that opening of the cyclopropane ring does not occur. A similar shallow, broad endothermic decomposition DSC trace was obtained for the C2-methyl ionic liquid, [C2C1C1im][OAc], 14, and ΔHdecomposition was measured as +18.0 kJ mol−1.
The four ionic liquids investigated by DSC (3a, 4, 11 and 14) appeared to thermally decompose entirely to volatile products; the aluminium DSC pans were opened after the experiments, and there was no observable decomposition residue. Accordingly, the second heating scan of these DSC experiments displayed no noticeable peaks. However, one very small endothermic peak was observed in the cooling stage for [C2C1im][CF3CO2], 4, at 247 °C. This suggests that a small quantity of decomposition residue was present after the first heating stage for this ionic liquid.
In summary, the DSC experiments indicated that for the four investigated imidazolium ionic liquids incorporating alkyl (3a, 11, 14) and fluoroalkyl (4) carboxylate anions, thermal decomposition is an endothermic event with positive values of ΔHdecomposition. In addition, there was an absence of any decomposition residue for the three non-fluorinated ionic liquids investigated, although a small amount of residue may have been formed from the decomposition of [C2C1im][CF3CO2], 4.
Computational results
The liquid structure of 1-ethyl-3-methylimidazolium acetate, [C2C1im][OAc], and the interaction of this ionic liquid with water and CO2, have been previously explored using experimental and computational techniques.31In this investigation, DFT was used in order to understand the ion pair structuring and, subsequently, the decomposition mechanisms of the two acetate ionic liquids, [C2C1im][OAc] and [C2C1C1im][OAc]. All calculations were performed at the B3LYP-D/6-311++G(d,p) level of theory. Full details of the computational methods are described in the ESI.†
Fig. 11 and 12 represent the low energy ion pair conformers for [C2C1im][OAc] and [C2C1C1im][OAc] obtained in this study, and the naming convention of the ion pairs used in the following discussion. Tables 3 and 4 summarise the ΔG and ΔE (BSSE and ZPE corrected) energies of these ion pair conformers. The ΔH, TΔS ΔZPE and BSSE energies are listed in the ESI† (Tables E2 and E3). In addition, the NBO charges for the lowest energy ion pair of [C2C1im][OAc] and [C2C1C1im][OAc] are listed in the ESI† (Fig. E8 and E9, respectively).
![(a) Diagram representing the location of primary cation–anion interaction for [C2C1im][OAc]. (b) Structure of the lowest energy ‘Front Methyl + Ethyl’ ion pair, showing primary H-bond interactions. (c) Diagram representing a dimer conformer of [C2C1im][OAc].](/image/article/2013/CP/c3cp53648c/c3cp53648c-f11.gif) | ||
| Fig. 11 (a) Diagram representing the location of primary cation–anion interaction for [C2C1im][OAc]. (b) Structure of the lowest energy ‘Front Methyl + Ethyl’ ion pair, showing primary H-bond interactions. (c) Diagram representing a dimer conformer of [C2C1im][OAc]. | ||
![(a) Diagram representing the location of primary cation–anion interaction for [C2C1C1im][OAc]. (b) Structure of the lowest energy ‘Top Methyl 1’ ion pair, showing primary H-bond interactions.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f12.gif) | ||
| Fig. 12 (a) Diagram representing the location of primary cation–anion interaction for [C2C1C1im][OAc]. (b) Structure of the lowest energy ‘Top Methyl 1’ ion pair, showing primary H-bond interactions. | ||
| Ion pair [C2C1im][OAc] | ΔE (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|
| From methyl + ethyl | 0.0 | 0.0 |
| Front ethyl | 4.9 | 3.2 |
| Front methyl | 4.9 | 3.5 |
| Top | 9.4 | 11.0 |
| Side ethyl (down) | 41.4 | 42.0 |
| Side methyl | 46.0 | 43.8 |
| Side ethyl (up) | 44.9 | 46.9 |
| Back methyl | 59.6 | 57.2 |
| Back ethyl | 55.7 | 57.3 |
| Ion pair [C2C1C1im][OAc] | ΔE (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|
| Top methyl 1 | 0.0 | 0.0 |
| Top methyl 2 | −1.8 | 0.3 |
| Bottom | −0.6 | 1.0 |
| Top ethyl | −1.0 | 5.8 |
| Side ethyl | 29.9 | 24.9 |
| Side methyl | 33.2 | 34.1 |
| Back ethyl | 46.4 | 41.9 |
| Back methyl | 48.7 | 43.1 |
The results suggest that the acetate anion is preferentially located in the plane of the imidazolium ring, in agreement with previous studies on this ionic liquid.31a,d Anion sites for our ion pair conformers of [C2C1im][OAc] resemble those previously determined for the prototypical ionic liquid 1-butyl-3-methylimidazolium chloride, [C4C1im]Cl.32 Whereas the chloride anion is monoatomic, acetate can bind strongly through the two oxygen atoms of the carboxylate group. As a result, the low energy ‘Front’ conformers are more centralised, with the acetate binding both to the C2 proton and α-/β-protons on the methyl and ethyl side chains. In the front and back ion pair conformers, both oxygen atoms of the acetate anion remain essentially in the plane of the imidazolium ring, whereas in the top and side conformers the acetate lies out of the plane of the ring. The two distinct ‘Side Ethyl’ conformers are denoted ‘up’ (acetate and ethyl chain both lie above the plane of the imidazolium ring) and ‘down’ (acetate and ethyl chain on opposite sides of the plane of the ring).
The lowest energy conformer (Fig. 11) located in this investigation is the ‘Front Methyl + Ethyl’ structure, similar to the structure reported by Hollóczki and co-workers.28d In this conformer, the ethyl chain rotates forward in order to bind to the acetate anion through a β-proton. Two other ‘Front’ conformers, ‘Front Methyl’ and ‘Front Ethyl’ (Table 3), are approximately 3.5 kJ mol−1 higher in energy, and lack this interaction with the ethyl β-proton. A ‘Top’ ion pair structure was obtained, whereby the acetate anion lies above the plane of the imidazolium ring, with an energy approximately 10 kJ mol−1 greater than the ‘Front Methyl + Ethyl’ structure. Attempts to optimise the top conformer at the B3LYP/6-311++G(d,p) level of theory were unsuccessful, indicating that the inclusion of dispersion is necessary for locating this ion pair.
Bowron and co-workers observed a high probability of the acetate anion lying in the plane of the imidazolium ring for [C2C1im][OAc], which resulted in a decrease in anion density above and below the ring.31d As a result, cation–cation π-stacking was possible. Interestingly, in our investigation no front ion pair conformers were located where the acetate lies perpendicular to the plane of the imidazolium ring.
Conformational analysis of the ethyl side chain was performed for the low energy ‘Front Methyl + Ethyl’ and ‘Top’ ion pair conformers of [C2C1im][OAc]. In each case, the C2–N–Cα–Cβ dihedral angle was rotated in increments of 30°, and all other coordinates were optimised. The change in energy and structure of the ion pair was recorded as a function of the dihedral angle. Graphs representing the conformational analysis around the ‘Front Methyl + Ethyl’ and ‘Top’ conformers are displayed in the ESI† (Fig. E10). The structure of the front ion pair was largely unchanged upon rotation of the ethyl chain, with the acetate anion remaining bound to the C2 proton and α-proton of the methyl side chain. Small rotational energy barriers of <10 kJ mol−1 were observed for ethyl rotation of the front conformer. For the top ion pair conformer, rotation of the ethyl chain carried the acetate anion towards the front of the imidazolium ring, giving structures analogous to the ‘Front Methyl’ and ‘Front Methyl + Ethyl’ minima.
In a recent molecular dynamics study, Brehm and co-workers investigated the preferred location of the acetate anion, relative to the [C2C1im]+ cation, by examining the angle between the ring normal vector and the connecting line from the centre of the ring to the position of the acetate carboxylate carbon atom.31i Their results indicated that in the neat ionic liquid, the acetate is largely associated with the plane of the ring (angle approximately 90°). However, their results also showed some probability of the acetate being located out of the ring plane by up to approximately 30°. To this end, we have investigated a sample of dimer structures for [C2C1im][OAc] (stable geometries involving two cations and two anions). Structures were optimised as for the ion pairs, at the B3LYP-D/6-311++G(d,p) level of theory. Although our calculations have not yet yielded an exhaustive list of dimer structures, the results do indicate the possibility of the acetate anions being located out of the plane of the imidazolium rings. An example optimised dimer structure is displayed in Fig. 11c, and in the ESI† (Fig. E11).
For the ionic liquid 1-ethyl-2,3-dimethylimidazolium acetate, [C2C1C1im][OAc], the C2 proton has been replaced by a methyl substituent, and thus there are no equivalent stable ‘Front’ ion pair conformers. Instead, the low energy conformers are ‘Top’ and ‘Bottom’ structures, whereby the acetate anion sits above or below the plane of the imidazolium ring. Four distinct ‘Top’ and ‘Bottom’ structures were located, with relative energies within a 6 kJ mol−1 range. The lowest energy ion pair structure obtained, shown in Fig. 12b, involves the H-bond interaction of the acetate anion with protons on the C2 methyl substituent and α-protons on both the methyl and ethyl side chains. The ethyl side chain is rotated almost into the plane of the ring to facilitate this interaction.
Plausible thermal decomposition mechanisms of [C2C1im][OAc] and [C2C1C1im][OAc] have been explored using DFT. The decomposition routes under investigation are displayed in Fig. 13. Mechanisms A–D are analogous to those shown for the TGA-MS results (Fig. 6). For each of the two ionic liquids, possible decomposition mechanisms include the SN2 attack of the acetate anion at either the N-methyl (mechanisms B1 and B2) or N-ethyl (mechanisms C1 and C2) substituents on the imidazolium cation, or the E2 Hofmann elimination mechanism (mechanisms D1 and D2) occurring at the ethyl substituent. The decomposition mechanisms at the C2 position (mechanisms A1 and A2) are different for the two acetate ionic liquids; for [C2C1im][OAc], the formation of an NHC is possible, whereby the C2 proton is removed by the basic acetate anion (mechanism A1). For [C2C1C1im][OAc], the carbene-forming mechanism is not possible. Instead the basic acetate anion may remove a proton from the C2 methyl substituent, generating acetic acid and the neutral species, 1-ethyl-3-methyl-2-methylene-2,3-dihydroimidazole (mechanism A2).
![Decomposition routes investigated for the two acetate ionic liquids in this computational study: [C2C1im][OAc], A1–D1, and [C2C1C1im][OAc], A2–D2.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f13.gif) | ||
| Fig. 13 Decomposition routes investigated for the two acetate ionic liquids in this computational study: [C2C1im][OAc], A1–D1, and [C2C1C1im][OAc], A2–D2. | ||
Decomposition reaction profiles have been studied starting from the lowest energy ion pair of [C2C1im][OAc] and [C2C1C1im][OAc], (‘Top Methyl + Ethyl’ and ‘Top Methyl 1’, respectively). The respective SN2 and E2 decomposition mechanisms (B1–D1, B2–D2) are analogous for the two acetate ionic liquids, and are therefore discussed first. The transition state structures for mechanisms B1, C1 and D1 are shown in Fig. 14, and the relevant ΔE, ΔG, ΔH and TΔS energies are displayed in Table 5. The decomposition mechanisms at the C2 position (A1 and A2) are less trivial, and are discussed separately shortly.
![Transition states for [C2C1im][OAc], representing the SN2 nucleophilic substitution (B1 and C1) and E2 Hofmann elimination (D1) mechanisms, atomic distances are in angstroms.](/image/article/2013/CP/c3cp53648c/c3cp53648c-f14.gif) | ||
| Fig. 14 Transition states for [C2C1im][OAc], representing the SN2 nucleophilic substitution (B1 and C1) and E2 Hofmann elimination (D1) mechanisms, atomic distances are in angstroms. | ||
| Mechanism | Transition state (kJ mol−1) | Product (kJ mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE | ΔG | ΔH | TΔS | ΔE | ΔG | ΔH | TΔS | |
| Bl | 139.1 | 134.6 | 139.5 | 4.9 | 4.0 | −35.3 | 0.6 | 35.9 |
| C1 | 135.6 | 135.4 | 136.2 | 0.8 | 0.2 | −39.6 | −3.0 | 36.7 |
| D1 | 156.4 | 156.1 | 156.8 | 0.7 | 61.3 | −25.8 | 63.3 | 89.1 |
| B2 | 119.8 | 118.5 | 119.1 | 0.5 | 2.3 | −41.1 | −1.0 | 40.1 |
| C2 | 116.7 | 119.3 | 114.8 | −4.6 | −0.1 | −41.7 | −5.2 | 36.5 |
| D2 | 138.6 | 137.7 | 138.6 | 0.9 | 61.0 | −27.8 | 61.1 | 88.9 |
For both acetate ionic liquids, the ΔHactivation energies for the two SN2 nucleophilic substitution mechanisms are approximately 20 kJ mol−1 lower than for the E2 Hofmann elimination mechanism (Table 5). In addition, the calculated ΔHactivation energies for the SN2 mechanisms are 15–24 kJ mol−1 higher than the experimentally determined Ea values from the isothermal TGA experiments (Ea = 115.8 kJ mol−1 for [C2C1im][OAc], 99.6 kJ mol−1 for [C2C1C1im][OAc]). The TGA-MS data (Fig. 7 and 8) suggests that SN2 attack occurs predominantly at the methyl substituent of the cation, however the calculations find little difference in the activation barriers for attack at the N-methyl or N-ethyl substituents. These results indicate that there is no inherent large energetic difference between these reactions and that steric effects are most likely responsible for the more facile reactivity at the N-methyl substituent.
The DSC investigation of [C2C1im][OAc] and [C2C1C1im][OAc] indicated that thermal decomposition was an endothermic event (ΔHdecomposition = +51.7 and +18.0 kJ mol−1, respectively, Fig. 10). By contrast, our gas phase calculations suggested that the ΔHproduct-reactant values for the SN2 mechanisms of these two ionic liquids are close to zero. However, the DSC experiments involve both the thermal decomposition of the ionic liquid and an associated phase change of the decomposition products from the ionic liquid to gaseous molecules. Therefore the difference in ΔHproduct-reactant values from the gas phase calculations and DSC experiments give an indication as to the magnitude of ΔvapH associated with the vaporisation of neutral ester and imidazole products from the SN2 thermal decomposition mechanisms. It is unclear whether thermal decomposition occurs from within the liquid phase, followed by subsequent vaporisation of the neutral species, or whether decomposition is occurring primarily at the surface of the ionic liquid.
The potential for N-heterocyclic carbene (NHC) formation in dialkylimidazolium ionic liquids is now widely recognised.28 This phenomenon has been exploited in catalytic applications.33 In the case of [C2C1im][OAc], removal of the C2 proton by the basic acetate anion would generate acetic acid and the NHC species, 1-ethyl-3-methylimidazol-2-ylidene (mechanism A1, Fig. 13). A stable minimum exists involving the interaction of acetic acid and the NHC, which is referred to here as the ‘Carbene-Acid Pair’ (CAP). Hollóczki and colleagues performed DFT calculations on the ion pair and CAP structure of [C2C1im][OAc] using a range of functionals and basis sets, and found the energy difference to be less than 20 kJ mol−1 in each case.28d Their calculations indicated that the C2 proton is very labile in the gas phase. Moreover, mass spectrometry experiments indicated the presence of acetic acid (m/z 60) and the isolated neutral carbene species (m/z 110) as decomposition products of [C2C1im][OAc] in the gas phase.
In our investigation, structures relating to the NHC decomposition route were optimised, and are shown in Fig. 15. The C2–O(acetate) distances, displayed in blue, are not exactly equal to the sum of the C2–H and O–H distances because of the slightly non-linear bond angle. Bond distances are in angstroms.
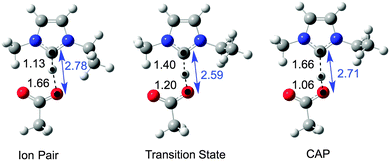 | ||
| Fig. 15 Gas phase ‘Front Methyl + Ethyl’ ion pair, transition state and carbene-acid pair (CAP) structures for mechanism A1. | ||
The SCF energies showed the TS to be lower in energy than the CAP product; 0.0, 12.6 and 13.7 kJ mol−1 for the ion pair, TS and CAP respectively. When ZPE corrections were taken into account this effect was enhanced; 0.0, 1.7 and 11.5 kJ mol−1 for the ion pair, TS and CAP respectively. These energies suggest that the transition state is lower in energy than the CAP, which is implausible.
However, the optimised TS structure has a single strong negative frequency of −728 cm−1. Moreover, the intrinsic reaction coordinate (IRC) protocol was employed, and demonstrated that this TS structure does link the ion pair and CAP.
In order to understand why the TS falls at a lower energy than the CAP product, the structures in Fig. 15 were investigated further. The gas phase calculations (Fig. 15) indicate a contraction of the C2–O(acetate) distance in the TS (2.59 Å) relative to the ion pair (2.78 Å) and CAP (2.71 Å). Thus, the C2-carbon–oxygen distance was fixed at a range of values between 2.4 and 2.8 angstroms (Å), and the hydrogen atom moved in increments of 0.05 Å, computing the potential energy surface (PES) shown in Fig. 16.
![3D energy surface for the proton transfer between the imidazolium C2 carbon atom and the acetate oxygen atom of [C2C1im][OAc] (mechanism A1). The white line represents the lowest energy pathway from the ion pair through the transition state to the CAP (critical points shown as green circles).](/image/article/2013/CP/c3cp53648c/c3cp53648c-f16.gif) | ||
| Fig. 16 3D energy surface for the proton transfer between the imidazolium C2 carbon atom and the acetate oxygen atom of [C2C1im][OAc] (mechanism A1). The white line represents the lowest energy pathway from the ion pair through the transition state to the CAP (critical points shown as green circles). | ||
This PES (Fig. 16) reveals that at short C2–O distances, (y) in the range 2.4–2.6 Å, a very broad flat single energy well exists spanning all of the ion-pair, TS and CAP structures (rear blue line in Fig. 16). As the C2–O distance is increased towards 2.8 Å, the ion pair is stabilised and a barrier develops between the ion-pair and CAP. The barrier increases as C2–O increases in the range 2.6–2.8 Å.
A scan performed with a fixed C2–O distance, ‘y’, of 2.8 Å shows the presence of a true TS (26.5 kJ mol−1), front blue line in Fig. 16. The CAP is approximately 9 kJ mol−1 higher in energy than the ion pair. Thus, a more reasonable reaction profile has been obtained.
The white line on Fig. 16 shows the likely reaction path, starting from the ion pair minimum through the lowest energy TS (which occurs in the region of the PES where the barrier is just developing) and finishing in the CAP product. Thus, the TS essentially ‘vanishes’ before a CAP minimum has fully developed.
The energy, Eν, of the transition state vibrational mode (−728 cm−1 or 8.7 kJ mol−1) is comparable to the energy differences between the ion pair, transition state and CAP (<14 kJ mol−1). To compare the energies of these structures on a more equal footing the vibrational energy associated with the reaction coordinate has been removed from the zero point corrected energies for the reactant and product. (Because the TS vibration is imaginary it has already been excluded from the zero point corrected energy of the TS.) This corresponds to the C–H vibration in the ion pair (2507 cm−1 or 30.0 kJ mol−1) and the O–H vibration in the CAP (2139 cm−1 or 25.6 kJ mol−1). The resulting effective energies (ΔEeff) are 0.0 kJ mol−1 for the ion-pair, 31.7 kJ mol−1 for the TS and 15.9 kJ mol−1 for the CAP (ESI,† Table E5). The relative magnitudes of these effective energies provide a more reasonable reaction profile for mechanism A1, with a transition state of higher energy than the ion pair and CAP.
Altogether, these results show that a TS exists at the C2–O distances of the ion-pair and CAP (∼2.76 Å), but that the two ions must contract significantly (reduce C2–O distance, ‘y’) to follow the lowest energy path for the reaction.
Despite the known labile nature of the C2 proton of [C2C1im][OAc] in the gas phase, no strong evidence for an NHC decomposition pathway under inert conditions was observed in our experimental investigation. Acetic acid and compounds relating to the NHC species were observed as only very minor peaks in the TGA-MS experiments; instead, the major observed decomposition products were indicative of SN2 nucleophilic substitution mechanisms.
To further explore NHC formation in the liquid phase, mechanism A1 was investigated using DFT and a Conductor-type Polarisable Continuum Model (CPCM).34 The CPCM calculations approximate the liquid phase environment and provide a rudimentary approximation of the bulk polar liquid and hence stabilisation due to a polar environment. Solvents benzene (C6H6, εr = 2.28), dichloromethane (CH2Cl2, εr = 8.93) and methanol (CH3OH, εr = 33.0) were used to demonstrate the effects of a medium increasingly capable of stabilising an ionic solute.21,34 The values of the dielectric constant for the molecular solvents CH3OH and CH2Cl2 cover the range anticipated to be typical for ionic liquids.35
No stable structures were obtained using the CPCM environment for the TS or CAP structures. This is consistent with recent ab initio MD simulations of the ionic liquid [C2C1im][OAc].28h Subsequently, scans were performed with a fixed C2–O distance, ‘y’, of 2.76 Å in the gas phase and using CPCM conditions, Fig. 17. The fixed coordinate prevents contraction of the ‘y’ distance, and therefore these scans do not perfectly represent the proton transfer mechanism. However, frequency analysis of the maximum structure in the gas phase scan (Fig. 17) revealed the expected (single imaginary) vibrational mode corresponding to mechanism A1.
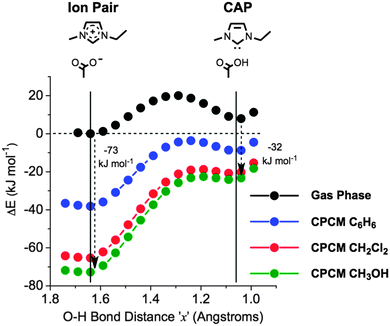 | ||
| Fig. 17 Change in ΔE during the proton shift between the C2 carbon atom and the acetate oxygen atom (mechanism A1), in the gas phase, and with CPCM solvent environments of C6H6 (εr = 2.28), CH2Cl2 (εr = 8.93) and CH3OH (εr = 33.0). The C2–O distance, ‘y’, is fixed at 2.76 Å. | ||
The CPCM scans of Fig. 17 suggest that the energy of the ion pair is stabilised to a far greater extent than the neutral CAP and TS in a polarised medium. These results are supported by the ‘charge-screening’ effects observed in ab initio MD simulations of the ionic liquid [C2C1im][OAc].28h In the gas phase scan, the CAP is ∼8 kJ mol−1 higher in energy than the ion pair. Introducing a weakly polar solvent environment (εr = 2.28) lowers the ion pair energy by 38 kJ mol−1 relative to the gas phase calculation, compared to approximately 17 kJ mol−1 for the CAP. For the more polar solvent environment (εr = 33.0), the energy of the ion pair is lowered by ∼73 kJ mol−1 relative to the gas phase calculation, and in contrast the energy of the CAP is lowered by only 32 kJ mol−1.
The potential energy surface within the polar medium is extremely shallow in the region of the CAP, with a very small energy barrier for the reverse reaction to re-form the ion pair (for example, ΔEactivation = <2 kJ mol−1 for the CH3OH solvent, Fig. 17). Thus, the PES has a ‘shelf-like’ feature that develops into a small barrier in less polar environments; such a low barrier will be difficult to observe experimentally, and will result in a highly reversible mechanism. This feature may explain why the CAP can be trapped with CO2,23 but is not observed as a primary decomposition route at high temperatures under inert (nitrogen, helium or compressed air) conditions.
In addition, these results indicate that it may be possible to encourage NHC formation by diluting the ionic liquid with a neutral non-polar solvent, reducing the ‘charge-screening effect’. Alternatively, forcing the C2–O distance to contract, perhaps under high pressure, may facilitate carbene formation.
For [C2C1C1im][OAc], the abstraction of a proton from the C2-methyl group by the acetate anion generates acetic acid and 1-ethyl-3-methyl-2-methylene-2,3-dihydroimidazole (mechanism A2, Fig. 13). This mechanism was also initially characterised by gas phase DFT calculations, and subsequently using CPCM. Reaction profiles and a transition state structure for decomposition mechanism A2 are displayed in the ESI† (Fig. E13).
The calculations suggest that even in the gas phase, the ion pair is of lower energy than the neutral products. Again, the CPCM calculations using CH2Cl2 and CH3OH solvents bring about a large stabilisation of the ion pair, and a significantly smaller stabilisation of the transition state and product energies. Similar to [C2C1im][OAc], the CPCM calculations for [C2C1C1im][OAc] indicate that the ion pair is thermodynamically more stable than the neutral products when in a polarised medium. Moreover, the very small energy barrier for the reverse reaction (ΔGactivation ≤ 3 kJ mol−1, CPCM calculation using a CH3OH solvent) indicates that the reaction is likely to be highly reversible.
Therefore, for the two decomposition mechanisms occurring at the front of the imidazolium ring (A1 and A2, Fig. 13), the CPCM calculations indicate stabilisation of the ion pair relative to the neutral products in an ionic environment. In addition, for each mechanism there is a minimal energy barrier (<3 kJ mol−1) for the reverse reaction. These results suggest that mechanisms A1 and A2, if occurring in the bulk liquid, are likely to be highly reversible. Our TGA-MS experimental data corroborates with this observation, because the expected neutral decomposition products generated by these two mechanisms were not observed.
Moreover, the measured activation energies, Ea, for thermal decomposition of both acetate ionic liquids were in reasonable agreement with the calculated ΔHactivation values for the SN2 thermal decomposition mechanisms. Ester products corresponding to the SN2 mechanisms were detected as major decomposition products in the TGA-MS experiments, and neutral imidazole products were clearly observed in the high-temperature 1H NMR experiment.
Decomposition mechanism A1 of [C2C1im][OAc] is likely to occur in the gas phase, as observed by the mass spectrometry experiment of Hollóczki and colleagues.28d In addition, studies indicate that the NHC is highly accessible in the liquid phase.23 However, our combined experimental and computational investigation suggests that thermal decomposition of [C2C1im][OAc] from the liquid phase does not primarily occur via the NHC route, when under inert conditions (nitrogen, helium or compressed air gases).
Conclusions
A broad series of [C2C1im]+ and [C2C1C1im]+ carboxylate ionic liquids has been synthesised, and the thermal stabilities were compared using temperature-ramped TGA. The difluoroacetate ionic liquid, [C2C1im][CHF2CO2], was found to be the most stable imidazolium carboxylate ionic liquid of the series, with a Tonset value approximately 50 °C higher than the non-fluorinated ionic liquid, [C2C1im][OAc]. The high thermal stability of [C2C1im][CHF2CO2] suggests that it may be a suitable replacement for the trifluoroacetate analogue in applications operating at high temperatures.36 Extension and branching of the anion alkyl chain, from two to 16 carbon atoms, had a minimal effect on the thermal stability of the ionic liquid, as determined from temperature-ramped TGA.The Arrhenius parameters for decomposition of [C2C1im][OAc] and [C2C1C1im][OAc] have been experimentally determined, and allow rapid prediction of the rate of decomposition of the ionic liquid at any temperature, in an open system. The safe operating temperature limit for [C2C1im][OAc] was found to be substantially lower than previously thought, and significantly lower than the operating temperature of many literature biomass procedures. In light of this result, certain biomass experiments that employ [C2C1im][OAc] should be modified. If the basic acetate anion is essential for the efficacy of the process, then the procedure must be carried out at a reduced temperature. Alternatively, low-melting inorganic mixtures of lithium, potassium and caesium acetates have been recently reported, which have improved thermal stability.37 Where the acetate anion is not essential, a more stable ionic liquid incorporating a different anion may be employed.
Continuing to operate biomass processing procedures using [C2C1im][OAc] at temperatures of 120 °C and above for extended periods of time will result in the build-up of volatile neutral products, posing a hazard and preventing effective reuse/recycle of the ionic liquid. Our results demonstrating the lower than previously believed maximum operating temperatures for this ionic liquid have important implications for these experiments and procedures will need to be modified in light of these.
It has been demonstrated that ionic liquids incorporating the acetate, octanoate and thioacetate anions decompose primarily via SN2-type nucleophilic substitution mechanisms, largely at the imidazolium N-methyl substituent. A high-temperature 1H NMR experiment showed the formation of the expected neutral 1-alkylimidazole products. However, the ionic liquids incorporating the fluorinated anions were observed to decompose by initial decarboxylation of the anion.
Ion pair structures and plausible decomposition mechanisms of [C2C1im][OAc] and [C2C1C1im][OAc] were explored using Density Functional Theory methods. The activation barriers for the SN2 nucleophilic decomposition mechanisms were found to be 20 kJ mol−1 lower than the E2 Hofmann elimination reactions, and the calculated ΔHactivation energies were in reasonable agreement with the experimentally determined Ea values from the isothermal TGA experiments. The CPCM calculations for decomposition mechanisms at the front of the imidazolium rings suggest that each ionic liquid is thermodynamically more stable than the neutral products that form, and that these mechanisms are likely to be highly reversible. Whilst the NHC species may be highly accessible in [C2C1im][OAc] and other dialkylimidazolium carboxylate ionic liquids, our experimental and computational results suggest that NHC formation is not a significant decomposition mechanism at elevated temperatures and under inert conditions.
Acknowledgements
The authors gratefully acknowledge the help of Dr Richard L. Thompson and W. Douglas Carswell from the University of Durham with the TGA-MS experiments, and Prof. John M. Seddon and Dr Arwen I. I. Tyler for assistance with the DSC experiments. We are thankful to Claire Ashworth, Dr Jason P. Hallett, Dr Kevin R. J. Lovelock and Dr Klemens Massonne for valuable conversations. The authors are thankful to BASF SE for the sponsorship of Matthew T. Clough.References
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- H. Zhao, Chem. Eng. Commun., 2006, 193, 1660 CrossRef CAS.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489 RSC.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS PubMed.
- (a) I. Krossing, J. M. Slattery, C. Daguenet, P. J. Dyson, A. Oleinikova and H. Weingärtner, J. Am. Chem. Soc., 2006, 128, 13427 CrossRef CAS PubMed; (b) P. A. Hunt, J. Phys. Chem. B, 2007, 111, 4844 CrossRef CAS PubMed.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965 RSC.
- (a) A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672 RSC; (b) A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- (a) W. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038 RSC; (b) D. B. Fu and G. Mazza, Bioresour. Technol., 2011, 102, 8003 CrossRef CAS PubMed.
- (a) C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963 RSC; (b) S. Sowmiah, V. Srinivasadesikan, M.-C. Tseng and Y.-H. Chu, Molecules, 2009, 14, 3780 CrossRef CAS PubMed.
- (a) B. K. M. Chan, N.-H. Chang and M. R. Grimmett, Aust. J. Chem., 1977, 30, 2005 CrossRef CAS; (b) M. R. R. Prasad, K. Krishnan, K. N. Ninan and V. N. Krishnamurthy, Thermochim. Acta, 1997, 297, 207 CrossRef CAS; (c) H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357–358, 97 CrossRef CAS; (d) C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954 CrossRef CAS; (e) D. M. Fox, J. W. Gilman, H. C. De Long and P. C. Trulove, J. Chem. Thermodyn., 2005, 37, 900 CrossRef CAS PubMed; (f) A. Seeberger, A.-K. Andresen and A. Jess, Phys. Chem. Chem. Phys., 2009, 11, 9375 RSC; (g) L. A. Daily and K. M. Miller, J. Org. Chem., 2013, 78, 4196 CrossRef CAS PubMed.
- (a) J. M. Crosthwaite, M. J. Muldoon, J. K. Dixon, J. L. Anderson and J. F. Brennecke, J. Chem. Thermodyn., 2005, 37, 559 CrossRef CAS PubMed; (b) I. H. J. Arellano, J. G. Guarino, F. U. Paredes and S. D. Arco, J. Therm. Anal. Calorim., 2011, 103, 725 CrossRef CAS.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47 CrossRef CAS PubMed.
- (a) D. M. Fox, W. H. Awad, J. W. Gilman, P. H. Maupin, H. C. De Long and P. C. Trulove, Green Chem., 2003, 5, 724 RSC; (b) K. J. Baranyai, G. B. Deacon, D. R. MacFarlane, J. M. Pringle and J. L. Scott, Aust. J. Chem., 2004, 57, 145 CrossRef CAS.
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691 RSC.
- (a) F. Wendler, L.-N. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76 CrossRef CAS PubMed; (b) F. Wendler, L.-N. Todi, F. Meister, Thermal investigations of polysaccharide blend solutions in imidazolium ionic liquids, 241st ACS National Meeting, March 27–31, Anaheim, USA, 2011; (c) A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, RSC Adv., 2012, 2, 8020 RSC.
- (a) M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Thermochim. Acta, 2007, 465, 40 CrossRef CAS PubMed; (b) S. D. Chambreau, J. A. Boatz, G. L. Vaghjiani, C. Koh, O. Kostko, A. Golan and S. R. Leone, J. Phys. Chem. A, 2012, 116, 5867 CrossRef CAS PubMed; (c) Y. Chen, Y. Cao, Y. Shi, Z. Xue and T. Mu, Ind. Eng. Chem. Res., 2012, 51, 7418 CrossRef CAS.
- J. L. Ferguson, J. D. Holbrey, S. Ng, N. V. Plechkova, K. R. Seddon, A. A. Tomaszowska and D. F. Wassell, Pure Appl. Chem., 2012, 84, 723 CrossRef CAS.
- 1-Ethyl-3-methylimidazolium acetate, BASF Basionics™ ‘BC 01’, CAS-No. 143314-17-4.
- I. Pibiri, A. Pace, S. Buscemi, V. Causin, F. Rastrelli and G. Saielli, Phys. Chem. Chem. Phys., 2012, 14, 14306 RSC.
- CRC Handbook of Chemistry and Physics, Internet Version, ed. D. R. Lide, Taylor and Francis, Boca Raton, FL, 87th edn, 2007, http:/www.hbcpnetbase.com Search PubMed.
- R. Fausto, L. A. E. Batista De Carvalho and J. J. C. Teixeira-Dias, THEOCHEM, 1990, 207, 67 CrossRef.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024 CrossRef CAS PubMed.
- J. H. Flynn and L. A. Wall, Polym. Lett., 1966, 4, 323 CrossRef CAS.
- (a) J. M. S. S. Esperança, J. N. Canongia Lopes, M. Tariq, L. M. N. B. F. Santos, J. W. Magee and L. P. N. Rebelo, J. Chem. Eng. Data, 2010, 55, 3 CrossRef; (b) M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831 CrossRef CAS PubMed; (c) A. Deyko, K. R. J. Lovelock, J.-A. Corfield, A. W. Taylor, P. N. Gooden, I. J. Villar-Garcia, P. Licence, R. G. Jones, V. G. Krasovskiy, E. A. Chernikova and L. M. Kustov, Phys. Chem. Chem. Phys., 2009, 11, 8544 RSC; (d) A. W. Taylor, K. R. J. Lovelock, A. Deyko, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2010, 12, 1772 RSC; (e) F. Heym, B. J. M. Etzold, C. Kern and A. Jess, Phys. Chem. Chem. Phys., 2010, 12, 12089 RSC; (f) K. R. J. Lovelock, A. Deyko, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2010, 12, 8893 RSC; (g) A. Chandran, K. Prakash and S. Senapati, Chem. Phys., 2010, 374, 46 CrossRef CAS PubMed; (h) A. Deyko, K. R. J. Lovelock, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2011, 13, 16841 RSC; (i) S. P. Verevkin, D. H. Zaitsau, V. N. EmeÌyanenko and A. Heintz, J. Phys. Chem. B, 2011, 115, 12889 CrossRef CAS PubMed; (j) K. Shimizu, M. Tariq, M. F. Costa Gomes, L. P. N. Rebelo and J. N. Canongia Lopes, J. Phys. Chem. B, 2010, 114, 5831 CrossRef CAS PubMed; (k) S. P. Verevkin, D. H. Zaitsau, V. N. Emel’yanenko, A. V. Yermalayeu, C. Schick, H. Liu, E. J. Maginn, S. Bulut, I. Krossing and R. Kalb, J. Phys. Chem. B, 2013, 117, 6473 CrossRef CAS PubMed.
- (a) A. G. Glenn and P. B. Jones, Tetrahedron Lett., 2004, 45, 6967 CrossRef CAS PubMed; (b) M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Thermochim. Acta, 2007, 465, 40 CrossRef CAS PubMed; (c) H. Ohtani, S. Ishimura and M. Kumai, Anal. Sci., 2008, 24, 1335 CrossRef CAS; (d) N. Meine, F. Benedito and R. Rinaldi, Green Chem., 2010, 12, 1711 RSC.
- J. Huang and A. F. Hollenkamp, J. Phys. Chem. C, 2010, 114, 21840 CAS.
- (a) J. Dupont and J. Spencer, Angew. Chem., Int. Ed., 2004, 43, 5296 CrossRef CAS PubMed; (b) T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366 CrossRef CAS PubMed; (c) S. T. Handy and M. Okello, J. Org. Chem., 2005, 70, 1915 CrossRef CAS PubMed; (d) O. Hollóczki, D. Gerhard, K. Massonne, L. Szarvas, B. Németh, T. Veszprémi and L. Nyulászi, New J. Chem., 2010, 34, 3004 RSC; (e) O. Hollóczki and L. Nyulászi, Org. Biomol. Chem., 2011, 9, 2634 RSC; (f) Z. Kelemen, O. Hollóczki, J. Nagy and L. Nyulászi, Org. Biomol. Chem., 2011, 9, 5362 RSC; (g) H. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222 RSC; (h) O. Hollóczki, D. S. Firaha, J. Friedrich, M. Brehm, R. Cybik, M. Wild, A. Stark and B. Kirchner, J. Phys. Chem. B, 2013, 117, 5898 CrossRef PubMed.
- (a) R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361 RSC; (b) M. G. Freire, C. M. S. S. Neves, I. M. Marrucho, J. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2010, 114, 3744 CrossRef CAS PubMed; (c) S. Steudte, J. Neumann, U. Bottin-Weber, M. Diedenhofen, J. Arning, P. Stepnowski and S. Stolte, Green Chem., 2012, 14, 2474 RSC.
- National Institute of Standards and Technology (NIST) Chemistry WebBook, http://webbook.nist.gov/chemistry/.
- (a) N. R. Dhumal, H. J. Kim and J. Kiefer, J. Phys. Chem. A, 2009, 113, 10397 CrossRef CAS PubMed; (b) J. Kiefer, K. Obert, J. Fries, A. Bosmann, P. Wasserscheid and A. Leipertz, Appl. Spectrosc., 2009, 63, 1041 CrossRef CAS PubMed; (c) M. B. Shiflett and A. Yokozeki, J. Chem. Eng. Data, 2009, 54, 108 CrossRef CAS; (d) D. T. Bowron, C. D’Agostino, L. F. Gladden, C. Hardacre, J. D. Holbrey, M. C. Lagunas, J. McGregor, M. D. Mantle, C. L. Mullan and T. G. A. Youngs, J. Phys. Chem. B, 2010, 114, 7760 CrossRef CAS PubMed; (e) W. Shi, K. Damodaran, H. B. Nulwala and D. R. Luebke, Phys. Chem. Chem. Phys., 2012, 14, 15897 RSC; (f) S. Stevanovic, A. Podgoršek, A. A. H. Pádua and M. F. Costa Gomes, J. Phys. Chem. B, 2012, 116, 14416 CrossRef CAS PubMed; (g) M. Brehm, H. Weber, A. S. Pensado, A. Stark and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 5030 RSC; (h) S. Zhang, Y. Zhang, X. Ma, L. Lu, Y. He and Y. Deng, J. Phys. Chem. B, 2013, 117, 2764 CrossRef CAS PubMed; (i) M. Brehm, H. Weber, A. S. Pensado, A. Stark and B. Kirchner, Z. Phys. Chem., 2013, 227, 177 CrossRef CAS.
- (a) P. A. Hunt, B. Kirchner and T. Welton, Chem.–Eur. J., 2006, 12, 6762 CrossRef CAS PubMed; (b) P. A. Hunt and I. R. Gould, J. Phys. Chem. A, 2006, 110, 2269 CrossRef CAS PubMed; (c) P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9 CrossRef CAS.
- M. E. M. Berger, D. Assenbaum, N. Taccardi, E. Spiecker and P. Wasserscheid, Green Chem., 2011, 13, 1411 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) C. Wakai, A. Oleinikova, M. Ott and H. Weingärtner, J. Phys. Chem. B, 2005, 109, 17028 CrossRef CAS PubMed; (b) H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654 CrossRef PubMed.
- C. Shi, Q. Ge, F. Zhou, M. Cai, X. Wang, X. Fang and X. Pan, Sol. Energy, 2009, 83, 108 CrossRef PubMed.
- S. Bajus, A. Deyko, A. Bösmann, F. Maier, H.-P. Steinrück and P. Wasserscheid, Dalton Trans., 2012, 41, 14433 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53648c |
| This journal is © the Owner Societies 2013 |