Elucidating the backbone conformation of photoswitchable foldamers using vibrational circular dichroism
Received 31st July 2013, Accepted 19th August 2013
First published on 19th August 2013
1 Introduction
Molecular systems with the ability to undergo conformational changes triggered by light have inspired the design of light-activated molecular machinery.1–6 An interesting class of such artificial molecular architectures is formed by photoswitchable amphiphilic oligo(azobenzene) foldamers, which can be manipulated to undergo a helix–coil conformational transition upon photoisomerization around the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, with extraordinary folding–unfolding efficiencies.2,7–9 The backbone of these foldamers consists of azobenzene monomeric units, which can be assembled to obtain molecular systems of various lengths (Fig. 1). The photo-induced trans-to-cis isomerization triggers a conformational change in the backbone of the Xn azo helix (where X denotes the number of phenylene units, and n indicates the number of azobenzene moieties), causing it to unfold.7–9 Refolding to the helical conformation can be achieved by irradiation with visible light or alternatively by thermal relaxation.7–9
N bonds, with extraordinary folding–unfolding efficiencies.2,7–9 The backbone of these foldamers consists of azobenzene monomeric units, which can be assembled to obtain molecular systems of various lengths (Fig. 1). The photo-induced trans-to-cis isomerization triggers a conformational change in the backbone of the Xn azo helix (where X denotes the number of phenylene units, and n indicates the number of azobenzene moieties), causing it to unfold.7–9 Refolding to the helical conformation can be achieved by irradiation with visible light or alternatively by thermal relaxation.7–9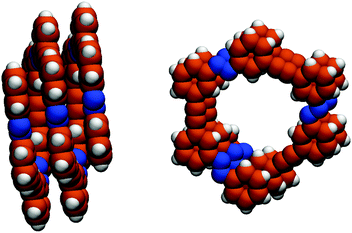 |
| | Fig. 1 Side view along the y axis (left) and top view along the z axis (right) of the backbone structure of an Xn azobenzene foldamer in its folded conformation. In this case, X = 14 and n = 7. The structure is optimized with the MM210 force field. | |
As yet, the folding and unfolding of synthetic foldamers are generally investigated using electronic circular dichroism (CD),11 which provides a rather indirect probe of the helical backbone conformation. Here, we investigate the conformation and photo-induced conformational changes in azo-foldamers using vibrational circular dichroism (VCD) spectroscopy12,13 on a vibrational mode involving the NN-stretch and in-plane vibrations of the aromatic rings of the azobenzene units in the foldamer backbone. In the last decade VCD has emerged as a powerful structure-resolving tool to study chiral molecules. In most cases, the interpretation of VCD spectra relies on complementary density-functional theory (DFT) calculations, and combined with such calculations VCD provides detailed information on the configuration and conformation of chiral molecules in the condensed phase.14–23 In particular, studies that combine VCD spectroscopy and ab initio calculations show convincing results regarding the preferred secondary structures of foldamers.24–26 As an alternative to spectral interpretation using DFT calculations, the simpler, semiquantitative coupled-oscillator method, in which the molecular vibrations are treated as localized, interacting transition dipoles, is attractive because of its transparency and computational ease,27–30 in particular for larger systems such as foldamers. Since we find that the NN-stretch VCD signal of azo-foldamers arises predominantly from through-space interaction between the NN-stretch transition-dipole moments, we can use this coupled-oscillator approach to interpret the observed foldamer VCD spectra. We find that the NN groups give rise to unique VCD signatures, which not only provide direct access to the twist sense of the helical structure, but also contain more structural information than the conventional (electronic) CD spectrum.
2 Materials and methods
The synthesis and purification of the foldamers have been reported elsewhere.7 Fourier-transform infrared (FTIR) and VCD spectra were obtained using a Bruker Vertex 70 spectrometer in combination with a PMA 50 module for polarization modulation measurements. Samples of the 147 foldamer were prepared in CD3CN and CD2Cl2 and kept in sealed infrared cells with CaF2 windows. Photo-switching of the 147 foldamer was performed using a 450 W Xe lamp (Müller Elektronik) and a notch filter with T = 12% at λ = 358 nm (FWHM = 45 nm). The cartesian coordinates of the nitrogen atoms in the backbone of the 147 foldamer were retrieved from an optimized structure obtained using the MM210 force field (Fig. 1).3 Mode assignment
The folded conformation of a 147 azo-foldamer is structurally organized as follows: an alternating sequence of seven azobenzene units is connected through ethynylene bridges. The side chains consist of oligo(ethylene glycol) moieties, which are attached to the main chain azobenzene phenyl rings and thus provide the necessary solvophobic driving force for folding in a polar medium.7 In addition, the presence of the side-chains causes a symmetry breaking around the NN bond. This lower symmetry as compared to that of non-substituted azobenzene gives rise to infrared activity of the vibrational mode involving mainly the stretching of the NN bond. For simplicity, we will from now on refer to this mode (which also involves the phenyl ring vibration) as the NN-stretch mode. The infrared activity of the NN-stretching mode in such non-symmetrically substituted trans-azobenzene systems has been convincingly demonstrated for a series of compounds in a recent combined experimental and theoretical study.31 We confirmed our assignment of the IR band at ≈ 1450 cm−1 to this mode by quantum-chemical calculations on an isolated substituted azobenzene unit (Fig. 2). The geometry optimization and calculation of harmonic vibrational frequencies were performed at the DFT/BLYP/6-31G(d)35–37 level of theory for a simplified model of the system, where we substitute the oligo(ethylene glycol) branches by ethyl-ester groups. Fig. 2 displays the calculated displacement vectors and the direction of the transition-dipole moment for the NN-stretching mode. We find the transition dipole to be approximately collinear (≈15°) with the NN bond.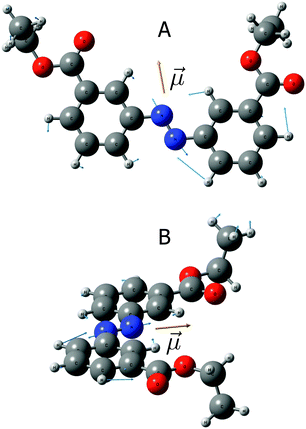 |
| | Fig. 2 Displacement vectors (in blue) and transition dipole moment vector (![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) i, in yellow) for the NN-stretching mode (ν = 1450 cm−1) of asymmetrically substituted azobenzene calculated at the DFT/BLYP/6-31G(d) level of theory. A – top view; B – side view. i, in yellow) for the NN-stretching mode (ν = 1450 cm−1) of asymmetrically substituted azobenzene calculated at the DFT/BLYP/6-31G(d) level of theory. A – top view; B – side view. | |
4 N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N-stretch VCD spectra
N-stretch VCD spectra
Initially we investigate to what extent the observed VCD signal arises from through-space interactions between the NN-stretch transition-dipole moments. Previous studies with electronic CD have shown that the helical content of 147 in CD3CN solution progressively vanishes upon addition of CDCl3 to the solution due to the reduction of the solvophobic effect. We therefore measured the IR and VCD spectra of a 10−4 M solution of 147 in CD3CN and CD2Cl2 (Fig. 3; CD2Cl2 rather than CDCl3 was used as a solvent because of strong infrared absorption of CDCl3 in the NN-stretch region). We find no NN-stretch VCD signal arising from the denatured, optically-inactive conformation of the foldamer in apolar media. This proves that the VCD signals observed for the folded conformation (the black solid line in Fig. 3) arise mainly from the helical arrangement and through-space coupling of the NN-stretch vibrational chromophores.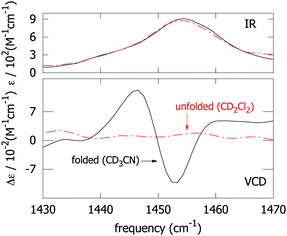 |
| | Fig. 3 Infrared absorption (upper panel) and VCD (lower panel) spectra of the 147 azo-foldamer in the folded (CD3CN, solid black lines) and unfolded conformation (CD2Cl2, dashed red lines). | |
4.1 Coupled-oscillator model for the NN-stretch modes
To calculate the vibrational frequencies and the vibrational circular dichroism of a helical azo-foldamer containing n NN groups using the coupled-oscillator model, we use the formalism derived by Diem et al.32 Due to the interaction between the NN-stretch modes, the NN-stretch normal modes will be delocalized over the helix, i.e., they will involve the vibrational motion of more than one chromophore. These delocalized normal modes are the eigenstates of the excitonic Hamiltonian| | 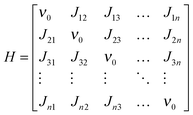 | (1) |
where ν0 is the frequency of an unperturbed NN-stretch mode, Jij the coupling between NN-stretch modes i and j. The Hamiltonian H is a symmetric (Hermitian and real) matrix so that Jij = Jji. The interaction between the NN groups is described by transition-dipole coupling:| | 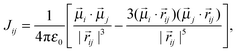 | (2) |
where ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) ij is the distance vector between dipoles i and j (Fig. 4). Note that the meta-linkages between the individual azobenzene repeated units prevent any through-bond coupling due to cross-conjugation.
ij is the distance vector between dipoles i and j (Fig. 4). Note that the meta-linkages between the individual azobenzene repeated units prevent any through-bond coupling due to cross-conjugation.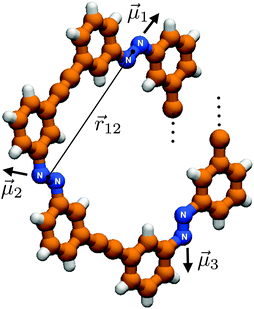 |
| | Fig. 4 Schematic representation of an Xn foldamer backbone with three monomeric NN units. Each transition dipole is considered to be collinear with the respective NN bond. | |
The dipole–dipole interaction lifts the degeneracy of the NN-stretching frequencies, and gives rise to as many delocalized normal modes as there are interacting NN oscillators. The vibrational frequencies of these delocalized normal modes (excitons) are the eigenvalues of the excitonic Hamiltonian matrix H. The dipole (D) and rotational (R) strengths of the kth exciton are given by32
| | 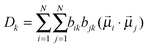 | (3) |
and
| | 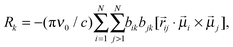 | (4) |
where
c is the velocity of light in vacuum, and
bij are the components of the eigenvectors of the Hamiltonian. The infrared absorption and vibrational circular dichroism spectra are calculated using
eqn (3) and (4), respectively, and convoluted with a Lorentzian band profile.
4.2 147 azo-foldamer
In Fig. 5a and b (solid lines) we show experimental FTIR and VCD spectra of the 147 azo-foldamer, respectively, in the NN-stretch spectral region. Using the experimental infrared absorption spectrum (Fig. 5a, solid line), we determine the magnitude of from the integrated absorption coefficient A = ∫ε(ν)dν. This value is used to calculate the coupling terms in eqn (2) and to construct the Hamiltonian with solely ν0 as a free parameter. Fig. 5c and d show simulated infrared absorption and VCD spectra (with || = 0.12 Debye), respectively, for the NN-stretch modes calculated using the coupled-oscillator formalism described above. The experimental VCD pattern, which exhibits a characteristic bisignate signal (positive couplet), is nicely reproduced in the simulated spectrum. The three most IR-intense calculated eigenmodes have vibrational frequencies of 1452, 1451 and 1450 cm−1. The remaining four predicted eigenmodes have extremely weak intensities (less than 5% of the total intensity) and are therefore neglected in the following analysis.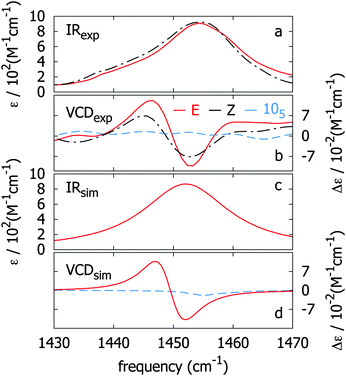 |
| | Fig. 5 Observed (a, b) and calculated (c, d) infrared absorption and VCD spectra of the NN-stretching mode for the 147 azo-foldamer in its folded conformation (E form, red curves). Infrared absorption and VCD spectra of the 147 azo-foldamer upon photo-excitation are depicted in dashed lines (Z form, black curves). Observed (b) and calculated (d) VCD spectra of a native 105 azo-foldamer (dashed blue curves). | |
Fig. 6 displays the three most intense normal modes as color coding on schematic representations of the 147 backbone structure. The color code is used to illustrate the relative amplitudes (eigenvector coefficients bij) of the NN oscillators in the eigenmodes that constitute the VCD spectrum (Fig. 5d). Eigenmode I (Fig. 6–I), which gives rise to the negative VCD peak (≈1452 cm−1), has its origin in a strong coupling (with negative amplitudes) of the three vertically aligned NN units (terminal and central units), while the interior units are not strongly involved. On the other hand, eigenmode II (Fig. 6–II), which gives rise to the positive VCD peak (≈1448 cm−1), originates from strong coupling (with positive amplitudes) of the terminal and central NN units (as opposite to eigenmode I) in addition to a strong coupling (with negative amplitudes) between the remaining interior NN groups. Finally, eigenmode III arises from strong contributions with opposite signs from the interior NN units (the NN units involved are depicted with the same color code in Fig. 6), while both terminal and central units have negligible contributions (Fig. 6–III).
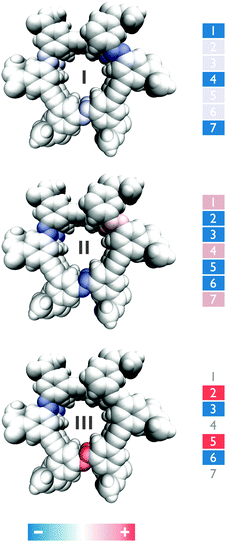 |
| | Fig. 6 Schematic representation of the relative amplitudes (eigenvector coefficients bij) of the NN oscillators in the eigenmodes I – (1452 cm−1), II – (1451 cm−1) and III – (1450 cm−1) respectively. | |
It is thus clear that the most intense VCD peaks arise from normal modes involving triply stacked terminal NN groups. This is confirmed by measurement on a 105 foldamer which lacks such triple-stacked arrangement. The VCD spectrum of a native-helical 105 foldamer is shown in Fig. 5b (dashed blue curve). The lack of signal intensity is consistent with a helical conformation lacking triple-stacked NN units when compared with that of the 147 foldamer (with 7 NN units). Moreover, in Fig. 5d (dashed blue curve) we show the simulated VCD spectrum of a 105 foldamer obtained with || = 0.12 Debye as an input parameter in the model. The signal magnitudes are extremely weak compared with those of the 147 foldamer, which confirms our conclusion that the VCD arises mainly from the triple-stacked NN bonds.
Clearly, the coupled-oscillator formalism applied to the backbone NN units successfully describes the observed features in the VCD spectrum of the 147 azo-foldamer. The good agreement of simulated and observed spectral features confirms the conclusions from the previously reported ECD spectra regarding the native-folded conformation of the azo-foldamer backbone.7–9 The good agreement between the observed and the modeled VCD spectra furthermore nicely corroborates the right-handed helix twist sense (P-helicity), which was previously assigned based on the shape of the exciton couplet.33,34
4.3 Photo-induced 147 unfolding
To determine the effect of azobenzene E → Z photoisomerization on the VCD response we irradiated a 10−4 M solution of 147 in CD3CN at λ = 358 nm using a 500 W Xe lamp equipped with a notch filter for a period of 24 h to assure reaching the photostationary state (PSS). Upon irradiation, the 147 azo-foldamer undergoes a helix–coil transition, leading to a PSS in which a fraction of the NN units is in the Z-configuration, leading to partial denaturation of the folded helical state. Previous studies have shown that in the PSS approximately 40% of the NN units that are in the Z-form belong to the termini.7 The IR and VCD spectra of the PSS are shown in Fig. 5a and b (dashed lines). While the IR spectrum shows only a slight red-shift, the VCD couplet is reduced in intensity by approximately 50%. This decrease in signal magnitude corroborates the previous conclusions based on electronic circular dichroism experiments,7 where the decrease in signal intensity was assigned to the loss of excess helicity of the azo-foldamer. The observed decrease in intensity of the VCD bisignate signal (Fig. 5b) is consistent with the predicted role of eigenmodes I and II as local probes in the coupling of the NN termini and core units, which is partially disrupted upon photoisomerization. Moreover, the partial denaturation of the foldamer, i.e., the reorientation of the termini and subsequent loss of helical content, effectively leads to the formation of a 105-type foldamer, in which the backbone still adopts a helical structure, but now with fewer transoid backbone NN units. This observation is in agreement with the unfolding mechanism proposed in which unfolding occurs predominantly starting from the termini of the foldamer.75 Conclusion
Our results demonstrate that VCD can be used to probe conformational changes during the folding and unfolding of azobenzene foldamers. The fact that the measurements can be interpreted using a simple coupled-oscillator approach ensures a straightforward relationship between observed VCD spectra and the foldamer conformation. Much more detailed information can be obtained from VCD measurements on foldamers in which specific NN bonds have been isotope-labeled. These labeled NN bonds will have a different local-mode stretching frequency, and will be spectrally isolated from the other NN bonds. They can therefore be described by a smaller (ideally 2 × 2, if two NN bonds are labeled) Hamiltonian. Such experiments should make it possible to probe conformational changes at specific sites in the foldamer backbone, and the interpretation should be just as straightforward as for the unlabeled foldamers studied here.Acknowledgements
The authors gratefully acknowledge Michiel F. Hilbers, Hans J. Sanders, Jan-Hein Hooijschuur and Freek Ariese for technical support. We would also like to thank Wybren Jan Buma for critically reading the manuscript. S.R.D. acknowledges financial support from the Portuguese Foundation for Science and Technology (FCT) under the fellowship SFRH/BD/48295/2008. S.A. would like to thank the Deutsche Akademie der Naturforscher Leopoldina – German National Academy of Sciences for a Leopoldina research fellowship (grant number LPDS 2011-18). S.H. gratefully acknowledges generous support from the German Research Foundation (DFG via SFB 658), the European Research Council (ERC) via ERC-2012-STG 308117 (Light4Function), and the European Science Foundation (ESF via P2M). S.W. acknowledges the European Research Council (ERC) for funding through Grant No. 210999.References
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- D. Bléger, Z. Yu and S. Hecht, Chem. Commun., 2011, 47, 12260–12266 RSC.
- A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505–10509 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- S. Braun, M. Bockmann and D. Marx, ChemPhysChem, 2012, 13, 1440–1443 CrossRef CAS PubMed.
- M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, A. M. Brouwer, W. J. Buma, D. A. Leigh and S. Woutersen, Science, 2010, 328, 1255–1258 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 1640–1643 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, Chem.–Eur. J., 2012, 18, 10519–10524 CrossRef CAS PubMed.
- Z. Yu, S. Weidner, T. Risse and S. Hecht, Chem. Sci., 2013 10.1039/C3SC51664D.
- N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS.
- R. B. Prince, L. Brunsveld, E. W. Meijer and J. S. Moore, Angew. Chem., Int. Ed., 2000, 39, 228–230 CrossRef CAS.
- L. A. Nafie, T. A. Keiderling and P. J. Stephens, J. Am. Chem. Soc., 1976, 98, 2715–2723 CrossRef CAS.
- P. J. Stephens, J. Phys. Chem., 1985, 89, 748–752 CrossRef CAS.
- T. B. Freedman, X. Cao, R. K. Dukor and L. A. Nafie, Chirality, 2003, 15, 743–758 CrossRef CAS PubMed.
- S. R. Domingos, M. R. Panman, B. H. Bakker, F. Hartl, W. J. Buma and S. Woutersen, Chem. Commun., 2012, 48, 353–355 RSC.
- S. R. Domingos, P. S. P. Silva, W. J. Buma, M. H. Garcia, N. C. Lopes, J. A. Paixão, M. R. Silva and S. Woutersen, J. Chem. Phys., 2012, 136, 134501 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin and J.-J. Pan, Chirality, 2008, 20, 643–663 CrossRef CAS PubMed.
- C. Merten and Y. Xu, ChemPhysChem, 2013, 14, 213–219 CrossRef CAS PubMed.
- C. Merten and Y. Xu, Angew. Chem., Int. Ed., 2013, 52, 2073–2076 CrossRef CAS PubMed.
- T. J. Measey and R. Schweitzer-Stenner, J. Am. Chem. Soc., 2011, 133, 1066–1076 CrossRef CAS PubMed.
- E. De Gussem, P. Bultinck, M. Feledziak, J. Marchand-Brynaert, C. V. Stevens and W. Herrebout, Phys. Chem. Chem. Phys., 2012, 14, 8562–8571 RSC.
- D. Kurouski, R. K. Dukor, X. F. Lu, L. A. Nafie and I. K. Lednev, Biophys. J., 2012, 103, 522–531 CrossRef CAS PubMed.
- V. P. Nicu, E. Debie, W. Herrebout, B. van der Veken, P. Bultinck and E. J. Baerends, Chirality, 2010, 21, E287–E297 CrossRef PubMed.
- T. A. Martinek, I. M. Mandity, L. Fulop, G. K. Toth, E. Vass, M. Hollosi, E. Forro and F. Fulop, J. Am. Chem. Soc., 2006, 128, 13539–13544 CrossRef CAS PubMed.
- L. Ducasse, F. Castet, A. Fritsch, I. Huc and T. Buffeteau, J. Phys. Chem. A, 2007, 111, 5092–5098 CrossRef CAS PubMed.
- G. Longhi, S. Abbate, F. Lebon, N. Castellucci, P. Sabatino and C. Tomasini, J. Org. Chem., 2012, 77, 6033–6042 CrossRef CAS PubMed.
- R. Schweitzer-Stenner, J. Phys. Chem. B, 2004, 108, 16965 CrossRef CAS.
- R. Schweitzer-Stenner, F. Eker, K. Griebenow, X. Cao and L. A. Nafie, J. Am. Chem. Soc., 2004, 126, 2768–2776 CrossRef CAS PubMed.
- T. Measey and R. Schweitzer-Stenner, Chem. Phys. Lett., 2005, 408, 123–127 CrossRef CAS PubMed.
- E. Schwartz, S. R. Domingos, A. Vdovin, M. Koepf, W. J. Buma, J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and S. Woutersen, Macromolecules, 2010, 43, 7931–7935 CrossRef CAS.
- N. Biswas and S. Umapathy, J. Phys. Chem. A, 2000, 104, 2734–2745 CrossRef CAS.
- T. Xiang, D. J. Goss and M. Diem, Biophys. J., 1993, 65, 1255–1261 CrossRef CAS.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, Wiley, New York, 2nd edn, 2000 Search PubMed.
- D. A. Lightner and J. E. Gurst, Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy, Wiley, New York, 2000, p 423, and references therein Search PubMed.
- M. J. Frischet al., Gaussian 09, Revision C.02, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a