 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical ammonia production on molybdenum nitride nanoclusters†
J. G.
Howalt
ab and
T.
Vegge
*a
aDepartment of Energy Conversion and Storage, Technical University of Denmark, DK-4000 Roskilde, Denmark. E-mail: teve@dtu.dk
bCenter for Atomic-scale Materials Design, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark
First published on 2nd October 2013
Abstract
Theoretical investigations of electrochemical production of ammonia at ambient temperature and pressure on nitrogen covered molybdenum nanoparticles are presented. Density functional theory calculations are used in combination with the computational hydrogen electrode approach to calculate the free energy profile for electrochemical protonation of N2 and N adatoms on cuboctahedral Mo13 nanoparticles. Pathways for electrochemical ammonia production via direct protonation of N adatoms and N2 admolecules with an onset potential as low as −0.5 V and generally lower than −0.8 V on both a nitrogen covered or a clean Mo nanoparticle. Calculations presented here show that nitrogen dissociation at either nitrogen vacancies on a nitrogen covered molybdenum particle or at a clean molybdenum particle is unlikely to occur under ambient conditions due to very high activation barriers of 1.8 eV. The calculations suggest that the nitrogen will be favored at the surface compared to hydrogen even at potentials of −0.8 V and the Faradaic losses due to HER should be low.
I. Introduction
Ammonia is a chemical compound of great interest and versatility, which is primarily used for making fertilizers; ultimately sustaining roughly one-third of the World's population.1,2 In terms of reducing the carbon footprint, ammonia is interesting for a number of reasons. Improving the sustainability of the already huge industrial catalytic production of ammonia, which is on the order of over 100 million metric tons annually and responsible for 1–2% of the global energy consumption would reduce cost of producing ammonia. Ammonia is also becoming increasingly interesting as a potential transportation fuel.3 As an energy carrier, ammonia has the benefit that it can be used in very energy efficient fuel cells, such as solid oxide fuel cells (SOFCs) or direct ammonia fuel cells (DAFCs).4 Furthermore, it has the interesting feature of not emitting CO2 while having a high energy density that is comparable with traditional fossil fuels, both volumetric and gravimetric.3,5 A highly energy-efficient method for the synthesis of ammonia (NH3) from molecular nitrogen (N2) is therefore desirable. Currently, ammonia synthesis is achieved by the Haber–Bosch process, in which N2 is initially dissociated and subsequently each nitrogen atom is protonated,6i.e. the dissociative mechanism. The Haber–Bosch process is energy-intensive and centralized due to the required high temperature and pressure and it is associated with a high capital cost to construct the production plants.Over the years, numerous experimental7–21 and theoretical22–34 studies have examined ammonia synthesis and they offer excellent insight into the challenges faced when developing new catalytic materials for ammonia synthesis. It has been shown in previous studies that ammonia synthesis is very structure sensitive on metal surfaces and primarily occurs on the surface steps of Fe and Ru.22,35,36 Whereas, the competing hydrogen evolution reaction (HER) is structure insensitive.37 Nanoclusters offer a way to increase the selectivity for NH3 production.
The natural enzymatic process for ammonia production in Nitrogenase takes place by initially weakening the N–N bond through successive electrochemical protonation, until the dissociation barrier is low enough to break the N–N bond; this process is referred to as the associative mechanism.38 In this paper, we present a pathway for electrochemical reduction of nitrogen into ammonia on molybdenum nanoclusters, which could ultimately become sustainable through utilization of renewable electricity sources like windmills or solar cells.
We have previously identified Mo nanoclusters as primary candidates for electrochemical production of NH3via the associative mechanism39 and shown that at potentials relevant for NH3 production, nitrogen will adsorb preferentially over hydrogen,35 thereby minimizing the traditional Faradaic losses due to the competing HER.37 These findings render nitrogen covered Mo nanoclusters as prime candidates for electrochemical ammonia production.
In this paper, we investigate nitride formation and reduction on molybdenum nanoclusters as well as the competing hydrogen adsorption process. The potentials required for ammonia production through direct protonation of adsorbed nitrogen adatoms and molecules through the associative mechanism are presented. The dissociation barrier for N2 molecules at various nitrogen coverages will also be presented.
II. Computational methods
A. DFT calculations
The calculations were carried out with density functional theory (DFT) calculations40,41 using the RPBE exchange correlation functional42 along with the projector augmented wave method43,44 as implemented in the GPAW code.45–47 A grid of (3,3) for the finite difference stencils has been used together with a grid spacing of 0.18 Å, at least 20 free bands above the Fermi level and a Monkhorst–Pack48k-point sampling of 2 × 2 × 2. A 7 Å vacuum layer around the nanocluster has been applied. When solving the electronic density self-consistently, the convergence criteria have been chosen such that the changes were ≤10−5 eV for the energy and 10−4 electrons per valence electron for the density. In all calculations, a Fermi smearing of 10−4 eV has been used. The atomic simulation environment ASE49 was used to set up the atomic structure of these systems. All structural (and atomic) relaxations of the adsorbates (N, H, NH, etc.) attached on the Mo13Nx nanocluster were carried out using the BFGS and FIRE50 optimizers within ASE. In the determination of transition states and activation barriers the nudged elastic band (NEB) method51–53 has been used with a spring constant of 1 eV Å−2. To precisely determine the transition state configuration and the corresponding minimum energy pathway between initial and final states, the climbing image method53 was used as the final step in the NEB calculations.B. The Mo13Nx cluster
The system of interest is the cuboctahedral molybdenum particle containing 13 atoms. The clean molybdenum structure is shown in Fig. 1a. It has a molybdenum atom at the center and a shell of 12 molybdenum atoms.54–56 The particle was allowed to relax to find its optimum lattice constant and is through the whole study allowed to fully relax in all directions. The Mo13 particle is of particular interest because it is highly undercoordinated and molybdenum binds nitrogen stronger than hydrogen; hence giving rise to a nitrogen coverage on the nanocluster.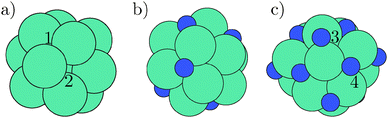 | ||
| Fig. 1 (a) The clean Mo13 nanocluster. (b) The Mo13N7 nanocluster after adsorption of seven nitrogen atoms (the dark atoms). (c) The Mo13N14 with a filled nitrogen skin. The clean cuboctahedral nanoparticle (a) has two special adsorption sites, where the three-fold hollow site is marked with 1 and the four-fold hollow site is marked with 2. In the filled nitrogen skin, the four-fold hollow adsorption sites have changed into a bridge site and are now marked 3 in (c), while the geometry is kept for the adsorbed nitrogen atoms in the original three-fold hollow sites (marked with 4). | ||
There are two relevant adsorption sites on the molybdenum particle surface. The first adsorption site is the three-fold hollow site with three nearest metal neighbors, marked with 1, and the second adsorption site is a four-fold hollow site with four closest metal neighbors, marked with 2. Two images for the filling of the nitrogen skin are shown in (1b) (half-filled skin) and (1c) (filled skin).
In the case of a full nitrogen skin, two types of bonding exist for the nitrogen adatoms. One is the three-fold hollow site, e.g., the atom marked 3 in Fig. 1c. The other adsorption site is a bridge site with two molybdenum atoms (marked 4 in Fig. 1c). In the process of filling the nitrogen skin, the adsorption sites change from a four-fold hollow site to a bridge site; the change will be discussed further in a later section.
C. Reaction pathways
In the process of describing the production of ammonia electrochemically, it is convenient to model the anode reaction| H2 ⇌ 2H+ + 2e− | (1) |
| N2 + 6H+ + 6e− → 2NH3 | (2) |
| N + 3H+ + 3e− → NH3. | (3) |
In this study, formation of ammonia through these pathways has been investigated at different nitrogen coverages. The reduction processes on the molybdenum cluster have been simulated using the Heyrovski-type57 reaction for both the protonation of nitrogen adatoms and nitrogen admolecules; the Heyrovski-type mechanism is a process where the proton is directly attached to an adatom or an admolecule from the electrolyte and the electron comes from the surface and merges with the proton to create a hydrogen atom bonded to the molecule. In this study, the adsorbed species of nitrogen adatoms or nitrogen admolecules are directly protonated and the following species are created on the surface; NHx or N2Hx (x = {0,1,2,3}). In principle, a Tafel-type reaction also exists, but it requires the reaction barriers for the hydrogenation steps34,58 and will therefore require a higher temperature to drive the process forward. This is due to the fact that the Tafel-type reaction59 requires that the proton and the electron to first merge on the surface to form a hydrogen adatom, and then the hydrogen adatom reacts with the adsorbed species of NHx or N2Hx (x = {0,1,2,3}).
The calculated reaction pathways for the electrochemical ammonia production are presented below. First, the reaction pathway of direct protonation of the nitrogen adatoms on the surface is considered. Here, a nitrogen adatom is directly protonated and after successive protonations, the formed ammonia is released from the surface. This process removes nitrogen atoms from the surface and would therefore require a regeneration of the nitrogen adatoms on the surface to retain the skin. Therefore, this presented pathway below does not describe the full catalytic cycle; only the nitride reduction.
| Mo13Nx-N + 3(H+ + e−) ⇌ Mo13Nx-NH + 2(H+ + e−) | (4a) |
| Mo13Nx-NH + 2(H+ + e−) ⇌ Mo13Nx-NH2 + (H+ + e−) | (4b) |
| Mo13Nx-NH2 + (H+ + e−) ⇌ Mo13Nx-NH3 | (4c) |
| Mo13Nx-NH3 ⇌ Mo13Nx + NH3(g) | (4d) |
Regeneration of the nitride will have to happen through N2 dissociation on the surface.
| Mo13Nx + N2(g) ⇌ Mo13Nx-N2 | (5) |
| Mo13Nx-N2 ⇌ Mo13Nx+2 | (6) |
This reaction is a heterogeneous reaction and will require low activation barriers to allow for preferential nitride regeneration over ammonia production.
Next, the associative Heyrovski mechanism is considered, where a nitrogen molecule is directly protonated until it splits into two molecules in the form of NHx species that later form gaseous ammonia. In the equations below, information from the calculations has been used, where the addition of the fourth H to the molecule N2H3* weakens the internal N–N bond sufficiently such that the molecule readily dissociates into NHx species on the surface.
| Mo13Nx-N2 + 6(H+ + e−) ⇌ Mo13Nx-N2H + 5(H+ + e−) | (7a) |
| Mo13Nx-N2H + 5(H+ + e−) ⇌ Mo13Nx-N2H2 + 4(H+ + e−) | (7b) |
| Mo13Nx-N2H2 + 4(H+ + e−) ⇌ Mo13Nx-N2H3 + 3(H+ + e−) | (7c) |
| Mo13Nx-N2H3 + 3(H+ + e−) ⇌ Mo13Nx-NH3-NH + 2(H+ + e−) | (7d) |
| Mo13Nx-NH3-NH + 2(H+ + e−) ⇌ Mo13Nx-NH3-NH2 + (H+ + e−) | (7e) |
| Mo13Nx-NH3-NH2 + (H+ + e−) ⇌ Mo13Nx-NH3-NH3 | (7f) |
| Mo13Nx-NH3-NH3 ⇌ Mo13Nx + 2NH3(g) | (7g) |
The reaction steps described above are either purely electrochemical or thermal steps and together they describe the full catalytic cycle of the Mo13Nx particle for ammonia synthesis.
D. Electrochemical modelling
With DFT it is possible to calculate the reaction energy, ΔE, for each of the reaction intermediates described above in the direct protonation of nitrogen adatoms and nitrogen admolecules following the Heyrovski type protonation process. This reaction energy is calculated with respect to the gas phase molecules of hydrogen and nitrogen and the corresponding Mo13Nx nanocluster: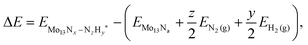 | (8) |
The reaction energies provide information about the catalytic properties for ammonia formation. However, for a thorough understanding, free energy corrections for each reaction intermediate need to be determined and included in the analysis. The expression of the free energy relative to the gas phase of molecular nitrogen and hydrogen is
| ΔG = ΔE + ΔEZPE − TΔS, | (9) |
The corrections for the zero point energy and entropy have been taken from a previous study,39 where the harmonic approximation was applied, the vibrational frequencies calculated for the reaction intermediates studied in this work and the corrections (ZPE and S)60,61 under ambient conditions.
In addition to the entropy and zero point energy corrections, an applied potential driving the electrochemical reaction will influence the free energy of the reactions. To include the effect of an applied potential, the computational hydrogen electrode62 has been employed, which has previously been very successful in describing a number of electrochemical reactions, including the trends in oxygen63–65 and nitrogen35,39 and CO2 reduction.66 The procedure of the computational standard hydrogen electrode is briefly outlined below.
The standard hydrogen electrode (SHE) is chosen as the reference potential. The chemical potential (the free energy per H) of H+ + e− is related to that of ½H2(g), see eqn (1). For an applied potential of U = 0 V relative to the SHE and a partial pressure of 1 bar of H2 in the gas phase at 298 K and pH = 0, the reaction free energy of eqn (1) is equal to the net reaction of eqn (4a)–(c) and (7a)–(f) at an electrode.
The next step is to incorporate the effects of an applied potential in all reactions involving an electron transfer and for the protons the pH. The free energy shift for a part reaction involving n electrons is −neU and hence the change in free energy reads
| ΔG = ΔE + ΔEZPE − TΔS − neU, | (10) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[10]. All calculations presented in the present study are for a pH value of 0.
ln[10]. All calculations presented in the present study are for a pH value of 0.
To drive the electrochemical reaction forward, the reaction should be exergonic, i.e. the change in free energy for each part reaction described in eqn (4a)–(c) and (7a)–(f) has to be exothermic. From eqn (10), it is evident that the applied potential can be tuned such that the reaction steps involving a proton transfer can be made exothermic and the specific applied potential ensuring this criterion is denoted the “onset” potential. As an example, the onset potential for part reaction 4b is determined in the following way:
| ΔGdis,2 = ΔGNH2* − ΔGNH* = ΔENH2 + EZPE,NH2 − TΔSNH2 − eU − (ΔENH + EZPE,NH − TΔSNH − 2eU). | (11) |
| U = (ΔENH + EZPE,NH − TΔSNH) − (ΔENH2 + EZPE,NH2 − TΔSNH2). | (12) |
Applying the potential U ensures that the reaction occurs spontaneously, as long as the protonation barrier is low.
III. Results and discussion
A. Stability of the nitrogen skin
The addition of nitrogen adatoms to the molybdenum nanoparticle has been carried out adding the nitrogen adatoms to three-fold hollow sites. For the first additions of nitrogen adatoms the particle changes shape to optimize the adsorption of nitrogen adatoms. For the fifth nitrogen addition, the adsorption in the three-fold hollow site again follows the cuboctahedral shape. The adsorption free energies of the first eight nitrogen atoms are shown in Fig. 2 (dark filled line) where 0 is the free energy of the clean Mo13 nanocluster and the respective number of 1/2N2 in gas phase. The graph shows that the adsorption free energies are strongest for the first additions and are then reduced when more nitrogen are added to the surface. The average binding energy for the nitrogen adatoms is presented with a dotted dark line in Fig. 2. The higher the nitrogen coverage, the smaller the structural changes.
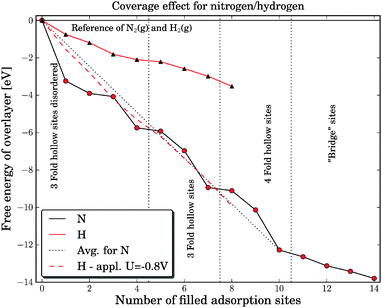 | ||
| Fig. 2 The free energy for covering the Mo13 cuboctahedral nanoparticle with either nitrogen or hydrogen. The filled dark line shows the filling of the nitrogen skin and the dark dotted line shows the average free energy of binding nitrogen to the surface. The light colored line shows the free energy of adsorbing hydrogen, while the dashed light colored line shows the free energy of adsorbed hydrogen with an external applied potential of −0.8 V. | ||
The adsorption of hydrogen on the cuboctahedral molybdenum nanoparticle is carried out by adding hydrogen to the three fold hollow sites. In Fig. 2, the adsorption free energy of hydrogen is plotted with a pale red color and forms almost a straight line. There is no observed restructuring of the nanocluster.
The results for molybdenum nanocluster covered with either nitrogen or hydrogen shows that it is indeed interesting to investigate the production of ammonia on this nitrogen skinned molybdenum particles because the particle should preferentially have nitrogen on the surface under reaction conditions. The stronger nitrogen bonds compared to hydrogen should subsequently result in reduced Faradaic losses due to lower hydrogen evolution on the molybdenum nanocluster under reaction conditions for ammonia production. The next step is to determine the electrochemical properties of the Mo13Nx nanoparticle with respect to the direct protonation of nitrogen adatoms or the associative pathway from an adsorbed N2 molecule at nitrogen vacancy sites.
B. High nitrogen coverage
Calculations performed on the filled skin are shown in Fig. 3 where the onset potential is the potential required to make each part reaction exothermic. The figure shows that the onset potential is close to −0.5 V for ammonia production with respect to SHE for both nitrogen adsorption sites. The geometry of the reaction intermediates for each of the reaction pathways is presented in Fig. 3. The geometries of NH are either a bridge site or a three-fold hollow site. For both NH2 species it is a bridge site, where the geometry of the NH2 in the three-fold hollow pathway has been moved from the three-fold hollow site to a bridge site during the relaxation of the system. The most stable adsorption site of NH3 for both studied reaction pathways is the on top sites. A comprehensive electronic charge analysis of the preferred protonation sites has not been performed due to the large associated variations in the geometrical relaxations of the small nanoclusters, but the observations of the adsorption geometries are in good agreement with observed geometries for NHx adsorption structures presented in the literature.32,35,39,67
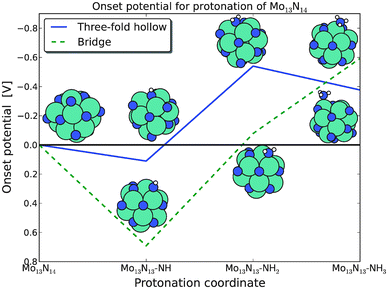 | ||
| Fig. 3 The onset potential for each protonation reaction of a nitrogen adatom originating from the Mo13N14 nanocluster structure. The onset potentials for the three-fold site, dark filled line, and the bridge site, light dashed line, are close to −0.5 V. | ||
For the further direct protonation of the Mo13Nx nanoclusters, similar onset potentials for reduction of the nitrogen skin are observed. For structures with high nitrogen coverage, the lowest onset potentials are on the order of −0.6 V to −0.8 V, see Fig. S1 in ESI.† These calculations indicate that the nitrogen skin can be protonated to create ammonia and a nitrogen vacancy site at the surface.
When a Mo13Nx nanocluster adsorbs a nitrogen molecule, there are two pathways for the further process. The nitrogen molecule can either go through the associative mechanism and create ammonia directly or dissociate into two nitrogen adatoms and hence regenerate the nitrogen skin. The dissociation will be discussed first.
The minimum energy path for the N2 dissociation on the Mo13N10 cluster can be seen in Fig. 4. Here, the initial and final state configuration together with the transition state is shown. The barrier for the splitting of N2 is found to be 1.72 eV, indicating that splitting of N2 is not possible on an almost filled nitrogen skin on the molybdenum particle.
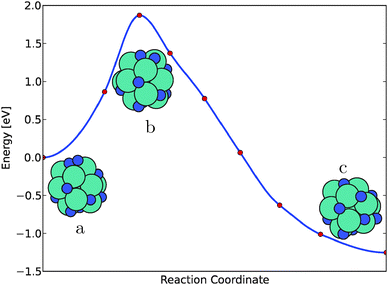 | ||
| Fig. 4 The dissociation of N2 on the Mo13N10 nanocluster calculated using the nudged elastic band method. The initial state of adsorbed N2, marked with (a), the transition state is marked with (b) and the final state is marked with (c). | ||
Mo13N12–N2 is the system with the highest nitrogen coverage that can adsorb a nitrogen molecule in a nitrogen vacancy site. The nitrogen molecule has two possible adsorption sites, one is a three-fold hollow site and the other is a bridge site. The vacancy site barely adsorbs the N2 molecule; the reaction free energy for adsorption is −0.07 eV with the three-fold hollow site as the most stable one. The reason for the weak adsorption of the nitrogen molecules is a steric hindrance caused by the nearby adsorbed nitrogen atoms. Fig. 5 shows the onset potentials for driving the protonation of N2. For both nitrogen pathways, the most endothermic reaction step is the first protonation. Here, the onset potential is around −0.6 V for the three-fold hollow site, marked by the dark filled line, and −0.75 V for the bridge site, marked by the light dashed line. For the pathway taking place on the bridge site, only the first three protonation steps are shown, since the onset potential for the pathway in the three-fold hollow site is lower. During the fourth protonation of N2H3 at the three-fold hollow site, the molecule prefers to dissociate into two NH2 molecules. The protonation and splitting of the N–N bond are strongly exothermic, where the reaction free energy of this step is 1 eV downhill. It was possible to find a semi-stable N2H4 configuration, but the creation of this reaction intermediate on the surface is 0.9 eV uphill.
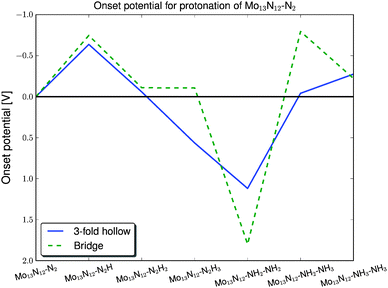 | ||
| Fig. 5 Onset potentials for all part reactions for protonation of an adsorbed nitrogen molecule on the Mo13N12 nanocluster. For the bridge site, the light dashed line, the onset potential is −0.75 V and for the three-fold site, the dark filled line, the onset potential is −0.6 V. | ||
The associative mechanism was also examined on a more reduced cluster (Mo13N10). The adsorption energy of the nitrogen molecule is stronger, −1.2 eV, and the onset potentials for the individual part reactions for ammonia formation are presented in Fig. S2 in the ESI† where an onset potential of −0.6 V is the best case.
With onset potentials of less than −0.8 V for all protonation processes, both the direct protonation of surface nitrogen and the associative pathways for N2 show that the protonation of nitrogen adatoms and admolecules into ammonia is possible.
C. The hydrogen competition
With an adsorption energy of −0.06 eV for N2, in the case of Mo13N12 and a corresponding adsorption energy of hydrogen adatoms of −0.65 eV, the hydrogen adatoms will preferentially bind to the nitrogen vacancy sites. Lower nitrogen coverage increases the adsorption energy of nitrogen molecules. It increases from −0.06 eV to −2.56 eV at the clean molybdenum particle, while the corresponding adsorption energies of hydrogen are almost constant, rising from −0.66 eV to −0.74 eV. The adsorption energies can be seen in Table 1.| System | ΔGN2 [eV] | ΔGH [eV] | ΔGH [eV] (U = −0.6 V) |
|---|---|---|---|
| Mo13N12 | −0.06 | −0.66 | −1.26 |
| Mo13N11 | −0.43 | −0.71 | −1.31 |
| Mo13N10 | −1.2 | −0.73 | −1.33 |
| Mo13N9 | −1.13 | −0.59 | −1.19 |
| Mo13 | −2.56 | −0.74 | −1.34 |
The pathways studied at the high nitrogen coverage show that the hydrogenation of the nitrogen adatoms and admolecules is possible at reasonable overpotentials, but the dissociation is an issue with a huge activation barrier. This will lead to formation of ammonia from adsorbed nitrogen, but the nitrogen skin will be reduced under reaction conditions because the dissociation will not lead to a regeneration of the nitrogen skin. Furthermore, the adsorption of nitrogen molecules with respect to hydrogen adatoms is not preferential at high nitrogen coverage.
At a coverage of 10 nitrogen atoms, N2 binds with −1.2 eV, compared to a hydrogen adsorption energy of ΔGH = −0.73 eV (see Table 1). Fig. S2 in the ESI† shows that the potential required for electrochemical ammonia production is −0.6 V. At this potential, the formation of H on the surface will have a reaction free energy of −1.33 eV, while the protonation of N2 will have a reaction free energy of only −0.4 eV. It is therefore expected that H will first cover the unoccupied nitrogen vacancy sites. When all sites are filled, the protonation of N2 will proceed because the formation of H2(g) will be 0.13 eV uphill and at the potential required for electrochemical ammonia production all reaction steps for ammonia production will be exergonic. Adsorbate–adsorbate interactions may however lower, e.g., the free energy barrier for producing gas phase hydrogen at high coverage, but quantification would require a more detailed analysis.
Overall, this will lead to reduction of the nitrogen skin and hydrogen adsorption at high nitrogen coverage. On the other hand, adsorption energies at low nitrogen coverage show that nitrogen will be preferred over hydrogen under these conditions.
D. Low nitrogen coverage
From the study of the N2 dissociation, the direct protonation of the nitrogen skin, the protonation of N2 and the competing adsorption of hydrogen, we find that the skin will most likely not be completely filled with nitrogen. Even at low nitrogen coverages the onset potential for the direct protonation is still less than −0.6 V, see Fig. S3 in the ESI.† Nitrogen adatoms on the surface for any given nitrogen coverage will be protonated into ammonia at potentials lower than −0.6 V. In the following, the dissociation and reduction of N2 molecules are carried out on a clean molybdenum surface with either N2 or two N adatoms adsorbed.As seen in Table 1, N2 adsorbs with 2.5 eV while two nitrogen atoms adsorb with 4 eV. In comparison the hydrogen binding energy is only −0.74 eV and with an applied potential of −0.8 V the hydrogen is bonded −1.54 eV and hence still weaker than N2.
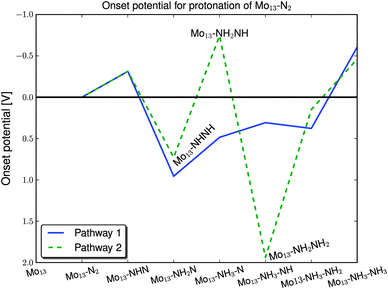 | ||
| Fig. 6 The onset potential of the protonation of the N2 molecule on the ‘clean’ Mo13 particle. | ||
The second route is going through NH–N, NH–NH and NH2–NH and then breaking of the N–N bond where NH and NH3 are formed on the surface. The issues for this route are both the formation of NH2–NH and the formation of NH3. These steps require an onset potential of −0.7 V and −0.45 V, respectively. The graph shows that the fourth protonation step, where the N–N breaking occurs, is very exothermic.
E. Desorption of ammonia
One issue that has not been illustrated with the use of the onset potential for the presented pathways of ammonia formation is desorption of ammonia from the surface. In this study the very reactive metal molybdenum is being studied and according to the literature and the Sabatier principle the best catalysts are the ones that have just the ‘right’ reactivity. Too reactive metals have an issue of getting products of the surface and too weak metals will have issues in the formation of the products.Two nitrogen coverages have been chosen to illustrate desorption of ammonia from the molybdenum nanocluster. One case is the high coverage of nitrogen (the Mo13N10 cluster) and the other case a low coverage of nitrogen (the Mo13 cluster). In both cases the molybdenum nanoclusters have two ammonia molecules adsorbed. The different ways of desorbing the two ammonia molecules were studied. The reference system for calculating the desorption energy of ammonia is the free ammonia particle and the respective ammonia molecule(s) in gas phase.
In the case of high nitrogen coverage the energy for desorption of ammonia from the surface is as low as 0.38 eV and in the worst-case scenario it is 0.51 eV. These desorption barriers will not be a major issue under ambient reaction conditions. Desorption of ammonia will be slightly hindered, but not the end that it will lower the activity of the catalyst by many orders of magnitude.
In the other case, the low nitrogen coverage, the values of the desorption energy of ammonia are in the best case 0.16 eV and in the worst case 0.40 eV. These are low thermodynamic barriers for desorption of ammonia from the surface. These calculations indicate that the electrochemical production of ammonia should not be thermally hindered.
IV. Conclusions
Our theoretical study of the nitrogen covered molybdenum nanoparticle at ambient temperature and pressure indicates that these particles have potential for creating ammonia with low onset potential both at low and high nitrogen coverage. Faradaic losses due to the competing HER are reduced compared to other model systems presented in the literature. On the molybdenum nanoparticles nitrogen should be favored on the surface with applied potentials as high as −0.8 V.At high nitrogen coverage, the pathways for creating ammonia are both the direct protonation of the nitrogen adatoms from the nitrogen coverage on the molybdenum nanoparticle and the protonation of a nitrogen molecule adsorbed in the created nitrogen vacancy. The required onset potentials for both reaction pathways are on the order of −0.7 V to −0.5 V. The competition between hydrogen adatoms and nitrogen admolecules will be an issue at high nitrogen coverage, where hydrogen is preferred.
At low nitrogen coverage, the associative mechanism should require onset potentials of −0.6 V. Nitrogen admolecules are preferred under these conditions compared to hydrogen adatoms.
The dissociation of nitrogen at both low and high nitrogen coverage has very high activation barriers of around 1.8 eV, effectively blocking the dissociative mechanism under ambient conditions.
Desorption energies of ammonia from the surface vary from 0.1 eV to 0.5 eV. These desorption barriers should not make the ammonia production on the molybdenum thermally hindered under ambient conditions and at room temperature.
The present study shows that molybdenum nanoclusters are promising electrocatalysts for ammonia production. Nitrogen molecules are found to bind preferentially over hydrogen on certain partially and fully reduced nitrogen skins on molybdenum nanoclusters at the potentials needed for electrochemical ammonia production through the associative mechanism.
Acknowledgements
The authors would like to acknowledge the Danish Center for Scientific Computing for supercomputer access. The Center for Atomic-scale Material Design (CAMD) and the Catalysis for Sustainable Energy (CASE) initiative is funded by the Danish Ministry of Science, Technology and Innovation.References
- V. Smil, Detonator of the population explosion, Nature, 1999, 415, 400 Search PubMed.
- V. Smil, Enriching the earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, Massachusetts Institute of Technology, 2001 Search PubMed.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, Ammonia for hydrogen storage: challenges and opportunities, J. Mater. Chem., 2008, 18(20), 2304–2310 RSC.
- T. Vegge, R. Z. Sørensen, A. Klerke, J. S. Hummelshøj, T. Johannessen, J. K. Nørskov and C. H. Christensen, Indirect hydrogen storage in metal ammines, British Welding Research Association, 2008, pp. 533–568 Search PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 90th edn, 2009 Search PubMed.
- F. Haber and G. van Oordt, Üher die Bildung von Ammoniak aus den Elementen, Z. Anorg. Chem., 1905, 47, 42–44 CrossRef CAS.
- G. Ertl, Z. Paal and S. B. Lee, Structure sensitivity in the iron single-crystal catalyzed synthesis of ammonia, Appl. Surf. Sci., 1981, 8, 231–249 CrossRef.
- G. Ertl, M. Weiss and S. B. Lee, Role of potassium in the catalytic synthesis of ammonia, Chem. Phys. Lett., 1979, 60, 391–394 CrossRef CAS.
- N. D. Spencer, R. C. Schoonmaker and G. A. Somorjai, Structure sensitivity in the iron single-crystal catalyzed synthesis of ammonia, Nature, 1981, 294, 643–644 CrossRef CAS.
- G. Ertl, S. B. Lee and M. Weiss, Kinetics of nitrogen adsorption on Fe (111), Surf. Sci., 1982, 114, 515–526 CrossRef CAS.
- G. Ertl, S. B. Lee and M. Weiss, Adsorption of nitrogen on potassium promoted Fe (111) and (100) surfaces, Surf. Sci., 1982, 114, 527–545 CrossRef CAS.
- N. D. Spencer, R. C. Schoonmaker and G. A. Somorjai, Iron single crystals as ammonia synthesis catalysts: effect of surface-structure on catalyst activity, J. Catal., 1982, 74, 129–135 CrossRef CAS.
- G. Ertl, Primary steps in catalytic synthesis of ammonia, J. Vac. Sci. Technol., A, 1983, 1, 1247–1253 CAS.
- L. Volpe and M. Boudart, Ammonia synthesis on molybdenum nitride, J. Phys. Chem., 1986, 90, 4874–4877 CrossRef CAS.
- D. R. Strongin, J. Carrazza, S. R. Bare and G. A. Somorjai, The importance of C7 sites and surface-roughness in the ammonia synthesis reaction over iron, J. Catal., 1987, 103, 213–215 CrossRef CAS.
- D. R. Strongin and G. A. Somorjai, The effects of potassium on ammonia synthesis over iron single-crystal surfaces, J. Catal., 1988, 109, 51–60 CrossRef CAS.
- J. A. Dumesic and A. A. Trevino, Kinetic simulation of ammonia synthesis catalysis, J. Catal., 1989, 116, 119–129 CrossRef CAS.
- L. M. Aparicio and J. A. Dumesic, Ammonia synthesis kinetics: surface chemistry, rate expressions and kinetic analysis, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- M. Boudart, Ammonia synthesis: the bellwether reaction in heterogeneous catalysis, Top. Catal., 1994, 1, 405–414 CrossRef CAS.
- A. Nielsen, Ammonia: catalysis and manufacture, Springer-Verlag, 1995 Search PubMed.
- G. Marnellos and M. Stoukides, Ammonia synthesis at atmospheric pressure, J. Mater. Chem., 1988, 282, 98–100 Search PubMed.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Role of steps in N2 activation on Ru(0001), Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- S. Dahl, E. Törnqvist and I. Chorkendorff, Dissociative adsorption of N2 on Ru(0001): a surface reaction totally dominated by steps, J. Catal., 2000, 192, 381–390 CrossRef CAS.
- S. Dahl, J. Sehested, C. J. H. Jacobsen, E. Törnqvist and I. Chorkendorff, Surface science based microkinetic analysis of ammonia synthesis over ruthenium catalysts, J. Catal., 2000, 192, 391–399 CrossRef CAS.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, Electrolytic synthesis of ammonia in molten salts under atmospheric pressure, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS PubMed.
- R. Kojima and K. Aika, Molybdenum nitride and carbide catalysts for ammonia synthesis, Appl. Catal., A, 2001, 219, 141–147 CrossRef CAS.
- T. H. Rod, A. Logadottir and J. K. Nørskov, Ammonia synthesis at low temperatures, J. Chem. Phys., 2000, 112, 5343–5347 CrossRef CAS.
- B. Hinnemann and J. K. Nørskov, Modeling a central ligand in the nitrogenase femo cofactor, J. Am. Chem. Soc., 2003, 125, 1466–1467 CrossRef CAS PubMed.
- A. Logadottir, T. H. Rod, J. K. Nørskov, B. Hammer, S. Dahl and C. J. H. Jacobsen, The Brønsted-Evans-Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts., J. Catal., 2001, 197(2), 229–231 CrossRef CAS.
- A. Logadottir and J. K. Nørskov, Ammonia synthesis over a Ru (0001) surface studied by density functional calculations, J. Catal., 2003, 220, 273–279 CrossRef CAS.
- A. Hellman, K. Honkala, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Ammonia synthesis and decomposition on a Ru-based catalyst modeled by first-principles, Surf. Sci., 2009, 603, 1731–1739 CrossRef CAS PubMed.
- A. Hellman, E. J. Baerends, M. Biczysko, T. Bligaard, C. H. Christensen, D. C. Clary, S. Dahl, R. van Harrevelt, K. Honkala, H. Jónsson, G. J. Kroes, M. Luppi, U. Manthe, J. K. Nørskov, R. A. Olsen, J. Rossmeisl, E. Skulason, C. S. Tautermann, A. J. C. Varandas and J. K. Vincent, Predicting catalysis: understanding ammonia synthesis from first- principles calculations, J. Phys. Chem. B, 2006, 110, 17719–17735 CrossRef CAS PubMed.
- T. Song and P. Hu, Insight into the adsorption competition and the relationship between dissociation and association reactions in ammonia synthesis, J. Chem. Phys., 2007, 127, 234706 CrossRef PubMed.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Ammonia synthesis from first-principles calculations, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- E. Skúlason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- C. J. H. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, H. Tøpsoe, D. V. Prip, P. B. Moenshaug and I. Chorkendorff, Structure sensitivity of supported ruthenium catalysts for ammonia synthesis, J. Mol. Catal. A: Chem., 2000, 163, 19–26 CrossRef CAS.
- R. Gomez, A. Fernandez-Vega, J. M. Feliu and A. Aldaz, Hydrogen evolution on platinum single crystal surfaces: effects of irreversibly adsorbed bismuth and antimony on hydrogen adsorption and evolution on platinum (100), J. Phys. Chem., 1993, 97(18), 4769–4776 CrossRef CAS.
- L. Stryer, Biochemistry, W. H. Freeman, New York, 4th edn, 1995 Search PubMed.
- J. G. Howalt, T. Bligaard, J. Rossmeisl and T. Vegge, Dft based study of transition metal nano-clusters for electrochemical NH3 production, Phys. Chem. Chem. Phys., 2013, 15, 7785–7795 RSC.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136(3B), B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev., 1965, 140(4A), A1133–A1138 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew–Burke–Ernzerhof functionals, Phys. Rev., 1999, 59(11), 7413–7421 Search PubMed.
- P. E. Blöchl, C. J. Först and J. Schimpl, Projector augmented wave method: ab-initio molecular dynamics with full wave functions, Bull. Mater. Sci., 2003, 26(33), 33–41 CrossRef.
- P. E. Blöchl, Projector Augmented Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Real-space grid implementation of the projector augmented wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(33), 035109 CrossRef.
- J. Enkovaara, et al. Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method, J. Phys.: Condens. Matter, 2010, 25, 253202 CrossRef PubMed.
- The GPAW code is available as a part of the CAMPOS software: http://www.camd.dtu.dk/Software.
- H. J. Monkhorst and J. D. Pack, Special points for brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192 CrossRef.
- The ASE code is available as a part of the CAMPOS software: http://www.camd.dtu.dk/Software.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Structural relaxation made simple, Phys. Rev. Lett., 2006, 97(12), 170201 CrossRef.
- H. Jónsson, G. Mills and K. W. Jacobsen, Nudged elastic band method for finding minimum energy paths of transitions, in Classical and Quantum Dynamics in Condensed Phase Simulations, ed. B. J. Berne, G. Ciccotti and D. F. Coker, World Scientific, Singapore, 1998, p. 385 Search PubMed.
- G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- A. A. Peterson, L. C. Grabow, T. P. Brennan, B. Shong, C. Ooi, D. M. Wu, C. W. Li, A. Kushwaha, A. J. Medford, F. Mbuga, L. Li and J. K. Nørskov, Finite-size effects in O and CO adsorption for the late transition metals, Top. Catal., 2012, 55(19–20), 1276–1282 CrossRef CAS.
- S. H. Huh, H. K. Kim and G. H. Lee, Critical cluster size of metallic Cr and Mo nanoclusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62(4), 2937–2943 CrossRef CAS.
- T. Vystavel, S. A. Koch, G. Palasantzas and J. Th. M. De Hosson, In situ transmission electron microscopy studies on structural dynamics of transition metal nanoclusters, J. Mater. Res., 2005, 20(7), 1785–2943 CrossRef CAS.
- J. Heyrovski, A theory of overpotential, Recl. Trav. Chim. Pays-Bas, 1927, 46(8), 582–585 CrossRef.
- S. Wang, V. Petzold, V. Tripkovic, J. Kleis, J. G. Howalt, E. Skulason, E. M. Fernandez, B. Hvolbaek, G. Jones, A. Toftelund, H. Falsig, M. Björketun, F. Studt, F. Abild-Pedersen, J. Rossmeisl, J. K. Nørskov and T. Bligaard, Universal transition state scaling relations for (de)hydrogenation over transition metals, Phys. Chem. Chem. Phys., 2011, 13(46), 20760–20765 RSC.
- J. Tafel, On the polarization during cathodic hydrogen evolution, Z. Phys. Chem., 1905,(50), 641 CAS.
- R. J. Silbey, R. A. Alberty and M. G. Bawendi, Physical Chemistry, Wiley, 4th edn, 2005 Search PubMed.
- C. J. Cramer, Physical Chemistry, John Wiley & Sons, Ltd., 2nd edn, 2004 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of water on (oxidized) metal surfaces, Chem. Phys., 2005, 319, 178–184 CrossRef CAS PubMed.
- J. Rossmeisl, Z.-W. Qu, G.-J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS PubMed.
- J. S. Hummelshøj, J. Blomquist, S. Datta, T. Vegge, J. Rossmeisl, K. S. Thygesen, A. C. Luntz, K. W. Jacobsen and J. K. Nørskov, Communications: elementary oxygen electrode reactions in the aprotic Li-air battery, J. Chem. Phys., 2010, 132(7), 071101 CrossRef PubMed.
- A. A. Peterson, F. Abild-Pedersen, J. Rossmeisl, F. Studt and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- F. Abild-Petersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Nørskov, Scaling properties of adsorption enemies for hydrogen containing molecules on transition metal surfaces, Phys. Rev. Lett., 2007, 99, 016105–016108 CrossRef.
- S. Wang, B. Temel, J. Shen, G. Jones, L. C. Grabow, F. Studt, T. Bligaard, F. Abild-Pedersen, C. H. Christensen and J. K. Nørskov, Universal Brønsted-Evans-Polanyi relations for C–C, C–O, C–N, N–O, N–N and O–O dissociation reactions, Catal. Lett., 2011, 141(3), 370–373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53160k |
| This journal is © the Owner Societies 2013 |