 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilized gold nanorod–dye conjugates with controlled resonance coupling create bright surface-enhanced resonance Raman nanotags†
Alison McLintocka, Hye Jin Leeb and Alastair W. Wark*a
aDepartment of Pure and Applied Chemistry, Centre for Molecular Nanometrology, WestCHEM, University of Strathclyde, Glasgow, UK G1 1XL. E-mail: alastair.wark@strath.ac.uk
bDepartment of Chemistry and Green-Nano Materials Research Centre, Kyungpook National University, 1370 Sankyuk-dong, Buk-gu, Daegu-city, 702-701, Republic of Korea
First published on 2nd October 2013
Abstract
The preparation and characterization of stable and non-aggregated colloidal suspensions of gold nanorod–molecular dye complexes which exhibit very bright surface-enhanced resonance Raman scattering (SERRS) signals is described. A systematic study was performed where both the localized surface plasmon resonance (LSPR) of the nanorod and the molecular resonance of dyes adsorbed onto the rod surface were selectively tuned with respect to the laser excitation wavelengths. Resonance coupling was found to be a significant factor in the overall SERRS enhancement. The polymer stabilized nanorod–dye conjugates were prepared without the added complexity of nanoparticle aggregation as well as having good control over the surface coverage and orientation of the dye molecules. Furthermore, we demonstrate that this new class of Raman nanotags greatly outperforms an approach based on quasi-spherical gold nanoparticles.
1. Introduction
Understanding and controlling the interaction between the localized surface plasmon resonance (LSPR) of gold and silver nanoparticles and light-absorbing molecular dyes is a rapidly emerging research area.1 Hybridization between plasmonic and molecular resonances can give rise to a variety of distinct optical properties which depend on the surface density and assembly behaviour of the dye adsorbed on the nanoparticle surface, the relative spectral overlap of both constituents as well as the distance between the dye layer and nanoparticle surface. A number of experimental2–11 and theoretical12–14 investigations have been recently reported characterising hybrid dye–metal nanoparticle structures. Of particular interest is the development of nanoparticle-enhanced spectroscopies including surface-enhanced Raman spectroscopy (SERS),15 resonance energy transfer,16 plasmon-enhanced fluorescence17 and fluorescence quenching.18Surface-enhanced resonance Raman scattering, SE(R)RS or SERRS, is a phenomenon that can occur when the laser excitation wavelength, the nanoparticle localized SPR and the electronic resonance of the molecular chromophore in contact with the nanoparticle surface all spectrally overlap to some extent.19,20 The relative importance of the multiplicative contributions to the overall SERRS signal from both the local electromagnetic field associated with LSPR and the molecular resonance Raman enhancement is an issue that continues to be debated in the literature. In addition, even further enhancement of the Raman signal from colloidal suspensions of gold and silver nanoparticles (NP's) is typically achieved via nanoparticle aggregation. The formation of hot-spots within small clusters of Ag quasi-spherical nanoparticles has enabled single molecule SERRS measurements to be demonstrated.21 However, aggregation also makes profiling the LSPR contributing to the enhanced SERRS response much more complex, with parameters such as cluster size, distribution in size, relative NP orientations and spacing within each cluster, location of probe molecules and changes in particle concentration all important. Consequently, the design of stable colloidal suspensions of bright SERRS-active probes which can be potentially used across a wide range of sensing and imaging applications has continued to be an area of intense research.22
In this report, we prepare a series of monodispersed and stable colloidal suspensions of gold nanorod–dye hybrid nanostructures and demonstrate their potential as a new class of bright SERRS-active substrates. A significant advantage of gold nanorods (NR's) is the ability to selectively tune the LSPR excitation across most of the visible and near-infrared (NIR) regions of the electromagnetic spectrum.23 In contrast, monodispersed gold quasi-spherical particles are limited to resonance wavelengths of ∼510–600 nm and aggregation is necessary to achieve significant SERS enhancements at common laser excitation wavelengths such as 633 and 785 nm. Furthermore, the nanorod–dye conjugate system also enables resonance coupling between the rod LSPR and dye to be explored over a range of wavelengths and a small number of studies have recently been reported by Wang et al.9–11 and others8,24,25 using extinction and fluorescence spectroscopy measurements.
However, even fewer investigations of the SERRS properties of gold nanorod–dye systems have been reported to date. Recently, von Maltzahn et al.26 compared a series of NIR dyes via the formation of mixed polyethylene glycol–dye monolayers on the nanorod surface to establish the brightest probe candidate for SERRS imaging in cells at 785 nm. Pal et al.27 performed SERRS measurements at 633 nm on rod aggregates prepared by drop-casting on a glass surface while Cai et al. have modelled the Raman spectrum of 3,3′-diethylthiadicarbocyanine iodide (DTTCI) enhanced by nanorod films.28 Gabudean et al. have recently demonstrated the dual use of metal-enhanced fluorescence and SERS on nanorods.29 Recently, Murphy et al., reported a SERS study utilizing a dye reporter molecule spaced away from the rod surface via adsorption on a polyelectrolyte-coated layer, followed by wrapping with another polymer layer to encapsulate the dye.30 They showed that the maximum SERS enhancement is obtained where the plasmon resonance is blue-shifted with respect to the single laser excitation wavelength used in their study with optimal signals reported when the LSPR was approximately 50 nm blue shifted from the laser excitation. This work was also transmission based, rather than a back-scattering configuration, therefore extinction losses along the sample path length are an issue. Also, an initial study by our group31 demonstrated that the side-by-side assembly of gold nanorods, which had a NIR dye sequestered into the surfactant bilayer surrounding each nanorod, could be controlled and also resulted in an enhanced SERRS response.
Here, we systematically prepare and characterize different combinations of gold nanorods and dyes whose target resonances strongly overlap at either 633 or 785 nm. As well as controlling factors such as dye surface coverage on the rod surface, the conjugates were wrapped with a polymer layer to achieve long-term stability and signal reproducibility. This enabled comparison between different nanoparticle conjugates and to demonstrate the effective use of the CTAB layer for both sequestering the dye molecules and maintaining their orientation and thus enhanced Raman spectral profile. In particular, the observation of large SERRS intensities while circumventing the need to induce particle aggregation and the generation of interparticle hot-spots represents a model system for promoting investigations of plasmon–dye resonance coupling and in the rational design of SERRS probes.
2. Experimental
2.1. Chemicals
Cetyltrimethylammonium bromide (CTAB), hydrogen tetrachloroaurate (HAuCl4), ascorbic acid, silver nitrate (AgNO3), sodium borohydride (NaBH4), sodium citrate tribasic dihydrate, 3,3′-diethylthiadicarbocyanine iodide (DTDCI), 3,3′-diethylthiatricarbocyanine iodide (DTTCI), poly(sodium 4-styrenesulfonate) (PSS) ∼70 kDa, sodium chloride (NaCl), poly(diallyldimethylammonium chloride) (PDDAC), cyclohexane and magnesium sulphate were all purchased from Sigma-Aldrich and used as received. All solutions were prepared using Millipore water.2.2. Nanorod synthesis
Two nanorod solutions were prepared using a modified version of the procedures developed by Sau and Murphy32 and Nikoobakht and El-Sayed.33 A key difference was that the reaction volume was scaled up to 1 L to give a large volume of stock NRs to avoid problems associated with batch to batch variations. Furthermore, the samples were centrifuged and resuspended four times in 1 mM CTAB. Each step dilutes the original 0.2 M CTAB stock ∼40-fold on replacing the supernatant following centrifugation such that we assume the CTAB concentration is sufficiently close to 1 mM. Establishing a consistent CTAB concentration was critical for the reproducible formation of mixed CTAB–dye layers around the nanorod surface. Samples were characterised using UV-vis measurements to determine the LSPR position, and to calculate the sample concentrations.2.3. Preparation of NR–dye conjugates
1 mM DTTCI and DTDCI stock solutions were prepared in MeOH, and further diluted in water immediately prior to addition of the desired volume of dye to 6 ml of stock NR solution to give a bulk dye concentration of 0.5, 1, 5, 10, 20 or 50 μM and the samples were left to equilibrate overnight (∼16 hours) at 27 °C. Next, PSS coating of the conjugates was carried out by adding 1.2 ml of 10 mg ml−1 PSS in 5 mM NaCl dropwise to the NR–dye conjugate solution while stirring rapidly. After a few minutes the samples were then centrifuged at ∼4774g for 20 min and resuspended in 6 ml of water; this was repeated a further three times to remove excess bulk dye and CTAB molecules. Additional characterization of the particle concentrations and aggregation state of the NR–dye conjugate concentrations was performed using a NanoSight LM20 and accompanying Nanoparticle Tracking Analysis software as well as UV-vis spectroscopy.2.4. Preparation of spherical gold nanoparticles
Gold nanoparticles were prepared by a variation of the Turkevich method.34 Here, 100 mg of HAuCl4 was added to 490 ml water, heated to boiling and then reduced using 105.7 mg of sodium citrate tribasic dihydrate in 10 ml water to afford colloidal gold with an average diameter of approximately 38 nm. An extinction coefficient of 7.8 × 109 M cm−1 was used to calculate the nanoparticle concentration.352.5. SERRS measurements
All SERRS measurements were performed using a Renishaw InVia Raman inverted microscope system, equipped with a 20× long working distance objective. Two excitation sources were used at 632.8 nm and 785 nm along with an 1800 or 1200 gr per mm grating respectively. The typical laser power at 633 nm was 1.0 mW while for 785 nm laser powers of 12.0 mW and 1.2 mW were used depending on the nanotag signal intensity obtained. Specifically, 12 mW was utilised at conjugates prepared using dye concentrations of 10 μM and below while 1.2 mW was used at higher concentrations. Also, sample spectra acquired at both laser powers and normalized with respect to a cyclohexane reference under identical conditions could be directly compared. Samples were analysed through transparent bottom micro-titre plates with 300 μl of the nanorod conjugate solution placed in each well. A signal collection time of 10 s per spectra was used. Cyclohexane was used to optimise the signal collection as well as to provide an intensity reference for data normalization. For each spectrum reported, a minimum of three sample aliquots in separate wells were acquired and compared. For experiments involving the controlled aggregation of quasi-spherical nanoparticles 1 μl of 1 M MgSO4 was added to 500 μl of colloid followed by mixing. All spectra were processed and background corrected using Grams/AI software (version 7.0).2.6. Scanning electron microscopy (SEM)
SEM images were obtained using an FEI Sirion 20 ultra-high resolution Schottky field emission scanning electron microscope with FEI software. Further details and representative images are provided in the ESI.†3. Results and discussion
3.1. Design and preparation of nanorod–dye conjugates
Gold nanorod (NR) sizes and dyes were selected which had resonances closely matching one of two common Raman excitation wavelengths (633 and 785 nm) and which were then selectively combined to create a range of NR–dye conjugates. An outline of the strategy used for the preparation of stable and SERRS-active NR–dye conjugates is shown in Fig. 1(a). The bilayer of CTAB surrounding the nanorod is responsible for both directing the rod shape during synthesis and maintaining colloidal stability. However, the presence of this bilayer also complicates subsequent surface functionalization, and this has previously hindered the use of nanorods for SERS-based applications. Furthermore, CTAB has a fairly weak affinity for the gold 〈110〉 longitudinal face and a net desorption from the rod surface starts to occur when the bulk concentration is lowered below ∼0.5 mM, which results in colloidal instability when cleaning. Consequently, the CTAB concentration in the prepared nanorod stock solutions was fixed at 1 mM by performing a minimum of four repeat centrifuge and resuspension (in 1 mM CTAB) steps after the NR synthesis. | ||
| Fig. 1 (a) Schematic showing the simple two-step preparation of the NR–dye conjugates. A mixed CTAB–dye layer is first formed before stabilizing the conjugate with a PSS polyelectrolyte wrapping. (b) Schematic overview of the various NR–dye combinations prepared. Extinction profiles of (c) nanorod stock solutions used and (d) 5 μM DTDCI and DTTCI in MeOH. The chemical structures of the (e) DTDCI and (f) DTTCI dyes are also shown. | ||
Following the preparation of the nanorod stock solutions, the first step in Fig. 1(a) shows the addition of a reporter dye, either DTDCI (Fig. 1(e)) or DTTCI (Fig. 1(f)), to form a mixed layer of CTAB and dye. Instead of replacing the CTAB, both our group31 and another36 have shown that hydrophobic molecules can be sequestered into the CTAB bilayer with high efficiencies. A further advantage of maintaining the CTAB bilayer is that it controls the orientation of the dye with respect to the NR surface, even at very low dye fractional surface coverages. The second preparative step involves wrapping the positively charged NR–dye assembly with a polyelectrolyte layer of PSS. The polymer wrapping has a number of roles: it encapsulates the conjugate and enables the subsequent removal of any excess dye and CTAB from the bulk solution, and also stabilises the conjugate, resulting in reproducible SERRS signals when comparing repeat preparations as well as after several months' storage.
Different combinations of nanorods and dyes were compared as summarised in Fig. 1(b) to observe and quantify the effect of altering the nanorod plasmon resonance and the dye molecular resonance. Fig. 1(c) and (d) show the extinction spectra for the NR and dye stock solutions used respectively. The first NR stock solution featured transverse and longitudinal plasmon peaks at 516 nm and 618 nm respectively, along with an average rod length of 16 nm and an aspect ratio of 2.0. The second NR sample had transverse and longitudinal peaks at 511 nm and 770 nm respectively, for which an average length of 47 nm and an aspect ratio of 4.3 were measured. For both stock solutions, the particle concentration was fixed at ∼1 nM based on extinction coefficients of 2.5 × 109 and 4.6 × 109 M−1 cm−1 respectively at the λmax of the longitudinal LSPR.37 The choice of DTDCI and DTTCI as Raman reporter molecules was based on the fact that they are both strongly absorbing (λmax = 646 nm and 754 nm respectively) close to the 633 and 785 nm laser excitation wavelengths. In addition, both dyes are structurally very similar differing only in the length of the polymethine chain and have poor water solubility.
The formation of a mixed CTAB–dye layer is supported by several experimental observations. These include comparing the improved stability of the dye absorbance spectrum in 1 mM CTAB solution compared to water only where the dye molecules form aggregates (see Fig. S2, ESI†). In addition, fluorescence spectroscopy (Fig. S3a and b, ESI†) shows quenching quickly occurring upon the addition of dye to the NR solutions compared to when nanorods are absent. The large Raman signals described later support the adsorption of dye molecules onto the nanorod surface, and with the bulk CTAB concentration fixed at 1 mM no destabilization of the colloid solution was observed at dye concentrations as high as 50 μM. When preparing the conjugates, each sample was left overnight to reach a steady-state fractional surface coverage of dye prior to PSS wrapping.
Considerable effort was made during the preparation of the NR–dye conjugates to ensure that no significant level of particle aggregation took place following the addition of the dyes and in the final conjugate solution as even a small fraction of aggregates can potentially have a significant impact on the bulk SERRS intensities obtained, as well as cause a shift in the nanorod LSPR.31 Also, in order to quantitatively compare different conjugate solutions, the particle concentrations were kept constant throughout the preparation and analysis processes. A number of complementary techniques were utilised to determine whether or not aggregates were present and to monitor the particle concentrations of the samples, including the analysis of extinction spectra, SEM (see Fig. S1, ESI†) and also the application of NanoSight video tracking analysis.38
3.2. Observation of resonance coupling
A series of NR–dye conjugates were prepared where both the NR and the dye have overlapping resonances, or the NR and dye have different resonances. A fixed aliquot volume taken from the same nanorod stock solutions was used for each preparation and to which a range of bulk dye concentrations (0.5–50 μM) were added and left overnight. The conjugates were then polymer wrapped followed by centrifugation and resuspension (3×) in water to ensure that excess bulk dye is removed from the solution and then characterized using UV-vis spectroscopy, with the results shown in Fig. 2. This approach ensures that there is no contribution from any remaining free dye whose concentration will be at least two orders of magnitude lower than the UV-vis detection limit of ∼0.5 μM, and that the spectral shift is due to dye adsorbed onto the nanorod surface. All four combinations of nanorods and dyes resulted in both dampening and a spectral shift of the longitudinal plasmon peak, with both effects increasing at higher dye concentrations. It can be seen in Fig. 2 that the relative positions of the dye and rod resonances are important. Both the size and direction of shift, either to the blue or red, depends on both the relative absorbance wavelengths of the dye and the nanorod.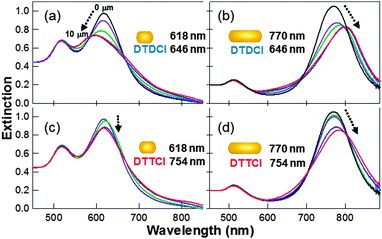 | ||
| Fig. 2 Extinction spectra of PSS-wrapped nanorod–dye conjugates prepared at a number of dye concentrations (0, 0.5, 1, 5, 10 μM) and a fixed nanorod concentration (arrow indicates direction of longitudinal plasmon resonance shift and also increasing dye concentration). (a) 618 nm NR with DTDCI; (b) 770 nm NR with DTDCI; (c) 618 nm NR with DTTCI; and (d) 770 nm NR with DTTCI. | ||
The size and direction of the longitudinal LSPR shift varies depending on the NR–dye combination in question. When DTTCI is adsorbed onto the 618 nm NR sample, the longitudinal plasmon undergoes a small red shift of ∼3 nm. When the same dye is adsorbed onto a 770 nm NR sample, the shift direction remains the same, but the size of shift increases up to approximately 20 nm for a 50 μM bulk dye concentration. DTDCI displays very different behaviour when it is adsorbed onto 618 nm NRs, the plasmon undergoes a blue shift of up to 24 nm. When adsorbed onto a 770 nm NR, the direction of shift changes to the red, and is still significant, at up to around 32 nm. In all cases, the amount of shift has been shown to be dependent upon the bulk dye concentration, with increased dye surface coverage resulting in increased plasmon shift and increased dampening of the signal. Similar trends have been reported by Ni et al.10 who reported a net blue shift in the longitudinal plasmon peak position when the λmax of the adsorbing dye was greater than the longitudinal plasmon peak position, and a net red shift when the initial longitudinal plasmon maximum was greater than the absorbance of the adsorbing dye.
A significant difference in our results compared to several recent studies on metal nanoparticle–dye systems is that no Rabi splitting was observed where the LSPR band develops into two bands. This effect appears to be associated with instances where dye molecule aggregates (which support excitons) are interacting with the nanoparticle surface, and is most dramatic where the LSPR and molecular resonance have the biggest overlap and also at larger dye fractional surface coverages.39–41 The absence of plasmon band splitting observed here despite a larger bulk excess of dye molecules is attributed to differences in control over the dye assembly around the nanorod involving a relatively uniform dispersion of non-aggregated single dye molecules. Maintaining a stable CTAB bilayer around each rod limits the extent to which both the DTTCI and DTDCI dyes used here assemble into J-aggregates. In pure water, both dyes quickly self-assemble, while in 1 mM CTAB solution the dye absorbance spectra are much more stable (see Fig. S2, ESI†). The observation of “weaker” dye–nanoparticle resonance coupling (i.e. smaller LSPR shifts with no plasmon band splitting) has also been reported3,42 in contrast to studies where the molecular stacking and orientation of the dye molecules on the metal nanoparticle surface is relatively uncontrolled10,39,40 resulting in “strong” exciton–plasmon coupling (i.e. larger LSPR shifts and band splitting). In this initial study we have focused on performing a quantitative SERRS study where the CTAB bilayer provides good control over the dye behaviour on the nanorod surface.
3.3. SERRS analysis
The SERRS signals of the different combinations of dyes and nanorods summarized previously in Fig. 1(b) were then systematically acquired at both 633 and 785 nm laser excitation frequencies. In each case, a fixed volume of the nanorod stock solution was exposed to dye concentrations ranging from 0.5–50 μM before polymer wrapping and subsequent removal of excess bulk dye, CTAB and PSS via repeat centrifugation and resuspension. A combination of extinction spectral monitoring, SEM and dynamic nanoparticle imaging and tracking measurements were performed for each of the samples to ensure that the nanorods were monodispersed with negligible levels of particle aggregation. Furthermore, the intensities of the SERRS spectra reported were normalized with respect to a cyclohexane reference acquired under identical conditions (i.e. laser power and focus, sample volume, integration time) as the sample measurement. This was to promote a direct comparison of spectral intensities acquired between samples at different laser excitation wavelengths and laser intensities.Fig. 3 clearly shows that the SERRS response increases when higher bulk concentrations of DTTCI dye are utilized to prepare the PSS-wrapped nanorod–dye conjugates due to an increase in the number of dye molecules encapsulated in the CTAB bilayer. At bulk dye concentrations above ∼20 μM, further changes in the SERRS intensity are much less which suggests that a maximum dye loading has been achieved at the steady-state conditions employed. In addition, Fig. 3 shows that the relative peak intensities in the spectral profile did not change as a function of dye concentration. This suggests the dye orientation on the nanorod surface is maintained by the CTAB bilayer unlike the data shown later (in Fig. 6) where the spectral profile changes as a function of dye surface coverage in the absence of CTAB. It is also important to point out that no difference in the spectral profile was observed when comparing suspensions before or after PSS wrapping.
 | ||
| Fig. 3 SERRS spectra obtained for a series of PSS-wrapped rod–dye conjugates prepared using a fixed concentration of NR's (λmax = 770 nm) and various concentrations of DTTCI dye. A bulk CTAB concentration of 1 mM is also maintained throughout. Laser excitation wavelength = 785 nm. The spectra have been vertically offset to allow comparison. | ||
The change in SERRS signal as a function of bulk dye concentration was found to fit a Langmuir adsorption isotherm (see Fig. S5a, ESI†) which gave an equilibrium affinity constant of Kads = 1.07 (±0.1) × 105 M−1. This is about an order of magnitude higher than the value obtained by Alkilany et al. for the partitioning of 1-napthol into the CTAB bilayer.36 Ideally, it would be preferable to also measure the ratio of dye molecules free in the bulk solution to that adsorbed on the nanorod surface using UV-vis spectroscopy, however we found that the tendency of the free dye molecules to aggregate over longer periods of time, especially for DTTCI, prevented sufficiently accurate measurements (see Fig. S2 in the ESI†). Repeat measurements where the DTDCI dye is instead loaded onto nanorods from the same stock solution (λmax = 770 nm) are also shown in the ESI† (Fig. S4 and S5b) indicating similar concentration dependent behavior was also observed.
A comparison of the normalized SERRS intensities acquired for different nanorod–dye combinations is summarized in Fig. 4 and 5, which allowed a number of observations to be made. Each sample was analyzed at both 633 and 785 nm excitation and the bulk dye concentration was fixed at 5 μM during the rod–dye conjugate preparation. In order to quantitatively compare the relative SERRS intensities for the different combinations, Fig. 5 summarizes different selected peak intensity values for each of the spectra shown in Fig. 4. The values were obtained from peak maxima and minima intensity positions (described in the Fig. 5 caption) extracted prior to baseline correction and used as a numerical indicator with which relative SERRS intensities could be compared.
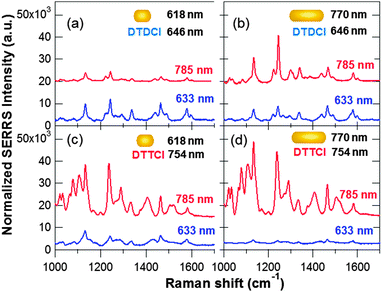 | ||
| Fig. 4 Normalized SERRS spectra obtained at an excitation wavelength of 785 nm and 633 nm. All the data shown is for conjugates prepared using a 5 μM bulk dye concentration: (a) 618 nm NR, DTDCI; (b) 770 nm NR, DTDCI; (c) 618 nm NR, DTTCI; and (d) 770 nm NR, DTTCI. Each spectrum is normalized with respect to a cyclohexane standard and baseline corrected. | ||
 | ||
| Fig. 5 Comparison of normalized SERRS intensities for the corresponding spectra and excitation wavelengths showed previously in Fig. 4(a)–(d). The intensity of a representative peak was obtained by finding the maximum peak height at 1245 (±2) cm−1 and subtracting the minimum at 1195 (±5) cm−1 for DTDCI. The process was repeated for DTTCI using the maximum peak height at 1241 (±2) cm−1 and subtracting the minimum peak height at 1195 (±5) cm−1. | ||
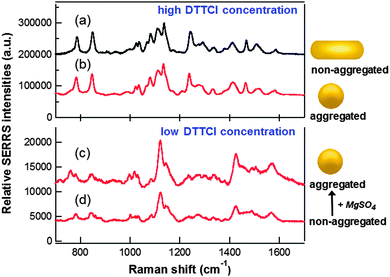 | ||
| Fig. 6 Normalized SERRS spectra at 785 nm excitation for (a) monodispersed solution of polymer-wrapped DTTCI–rod conjugates prepared with a 50 μM bulk dye concentration; (b) aggregated quasi-spherical Au nanoparticles in the presence of 2 μM DTTCI and MgSO4, representing the maximum SERRS obtained from this system. Measurements at a lower DTTCI concentration of 0.05 μM are shown for quasi-spherical Au particles (c) following aggregation induced by MgSO4 and (d) non-aggregated. | ||
Control measurements acquired in the absence of dye molecules for both the stock solution nanorods in 1 mM CTAB and for PSS-wrapped rods are provided in the ESI,† Fig. S7. Using identical laser powers, integration times and particle concentrations it can be clearly seen that both CTAB and PSS have a negligible contribution to the SERRS spectral profiles shown in Fig. 3–5. Also shown in the ESI† in Fig. S8 are resonance Raman spectra for both the DTDCI and DTTCI dyes deposited onto a planar gold film to quench the fluorescent background signal. Significantly greater laser powers and accumulation times were required than for the solution-based measurements. For DTDCI, no resonance Raman spectrum was obtained at 785 nm, only at 633 nm, while for DTTCI resonance Raman spectra at both excitation wavelengths were observed. The poorer signal-to-noise for the resonance Raman measurements indicate that the overall SERRS enhancement is due to a combination of molecular and plasmon resonance contributions.
The changes in SERRS intensities observed on switching between 633 and 785 nm excitation were observed consistently for conjugates prepared at all bulk dye concentrations (1–50 μM). Thus, with the exception of the rod–dye combination in sample (a), the relative SERRS intensities obtained for 785 nm excitation were consistently larger than that obtained at 633 nm excitation for samples (b–d).
A similar comparison can also be made between Fig. 5(b) and (d) involving the larger aspect ratio nanorods (λmax = 770 nm). For both dyes, the spectral intensities were greater at 785 nm laser excitation than 633 nm, even for DTDCI. This initially suggests that the rod LSPR is playing a slightly more dominant role in the overall SERRS enhancement than the dye. On the other hand, when comparing (c) and (d) in Fig. 4 and 5, the DTTCI signal from the short rod (off-resonance) is nearly the same as the longer rod (on-resonance) which strongly suggests the resonance contribution of the dye is the stronger effect. However, it is important to point out that comparing (b) and (d) with different rod sizes (stock solution concentrations are similar) involves differences in available surface areas and a comparison between (c) and (d) is more accurate since they were prepared from the same rod solution.
Ultimately, understanding of the relative importance of the dye and rod contributions to the overall SERRS signal also depends on knowing the distribution of dye molecules between the rod ends and sides and if this changes as a function of dye fractional surface coverage. The LSPR electromagnetic field enhancements are greatest at the rod ends, rather than along the particle sides44 and thus dye molecules located at the ends will have a relatively larger contribution to the SERRS intensities, however, the CTAB layer at the rod ends has a lower molecular density and this will affect the dye adsorption in this region since the strong surface affinity of the dye for the rod surface is based on the CTAB bilayer. Further work is underway involving a study at the single particle level including control of the incident light polarization parallel and perpendicular to both rod axes and will be reported in a future publication.
The calculated differential cross-section values are comparable with the brightest signals obtained in the recent study by Amendola and Meneghetti46 who quantified the SERS response for a range of spherical nanoparticle, dye-label combinations and which also contained a significant subpopulation of aggregated nanoparticles. To gain further insight, a direct comparison of the relative brightness of the nanorod–dye conjugates versus labelled spherical nanoparticles is described in the next section.
Fig. 6 compares normalized SERRS spectra obtained at 785 nm excitation for polymer-wrapped DTTCI–rod conjugates and quasi-spherical nanoparticles. When DTTCI was introduced to the citrate-stabilized Au colloid stock solution, nanoparticle aggregation started to occur at bulk dye concentrations above ∼0.05 μM (see Fig. S10 in the ESI†). Therefore, in order to first compare the signal from non-aggregated spherical particles, Fig. 6(d) shows the SERRS spectrum obtained at a bulk dye concentration of 0.05 μM. When an optimal amount of MgSO4 aggregating agent was added to this solution to maximise the SERRS enhancement, a relatively modest ∼2-fold increase in intensity was observed (see Fig. 6(c)). Comparison of the spectra in (c) and (d) show similar peak profiles, both of which are markedly different from the spectra obtained for the nanorod–dye complexes. However, when higher DTTCI concentrations are introduced to the quasi-spherical nanoparticle colloid, the resulting SERRS profile was very similar to that of the rod–dye conjugates (compare Fig. 6(a) and (b)). This emphasizes the role of the CTAB bilayer in controlling the orientation of the dye on the nanorod surface while the average orientation of the dye on the citrate-stabilized quasi-spherical particles must vary with dye fractional surface coverage. The relationship between the molecule orientation on a nanoparticle surface and the SERS spectral profile has been described elsewhere.47,48
The SERRS spectrum of the polymer-wrapped dye–rod conjugates in Fig. 6(a) represents the maximum signal obtained at a fixed nanorod concentration of ∼1.1 nM with no nanoparticle aggregation. The most intense peak in (a) is ∼24 fold greater than the most intense peak obtained for the non-aggregated quasi-spherical particles in Fig. 6(d). The role of nanoparticle aggregation to increase SERRS signals is well-established, and the spectrum in Fig. 6(b) represents the maximum response obtained following a series of repeat measurements involving the aggregation of the quasi-spherical nanoparticles at higher DTTCI concentrations (see Fig. S11 in the ESI† for additional spectra). Comparing (a) and (b), the signal for the non-aggregated rods is approximately 10% higher than for the aggregated spheres. The concentrations of the nanorod (∼1.1 nM) and quasi-spherical (∼0.26 nM) stock solutions are not identical while the surface area of the average spherical particle is ∼1.8× greater than that of the nanorods. It is also reasonable to assume a significantly higher dye fractional coverage on the citrate-stabilized spheres than the CTAB-coated rods. Thus the difference in the total number of dye molecules adsorbed onto a nanoparticle surface in both cases will be considerably less than a factor of two. Given that inducing aggregation results in orders of magnitude increase in the SERRS response of spherical particles38 and the fact that the normalized SERRS signal associated with the monodispersed nanorod–dye conjugates is comparable with the aggregated spherical system provides evidence that the nanorod–dye conjugates are significantly brighter. Recently, we have shown that the controlled side-by-side assembly of the nanorod–dye conjugates can result in up to a ∼4-fold increase in signal.31 Furthermore, when rod aggregation is performed in a random fashion in the presence of DTTCI, then an even larger increase in intensity is easily achieved, such as the example shown in Fig. S12 in the ESI.†
An absolute comparison between spheres and rods is difficult to achieve due to the complexity associated with factors such as dye surface coverage, orientation, available surface area and particle size distribution. However, these results clearly demonstrate nanorod–dye assemblies to be extremely promising SERRS substrates compared to more conventional spherical colloidal particles of similar dimensions. In the future, it will be interesting to compare the nanorod–dye system quantitatively with other single encoded SERS particles which typically use thiol-PEG and/or silica coatings for encapsulation of reporter molecules.49
4. Conclusion
A series of stable, monodisperse suspensions of polymer-wrapped gold nanorod–dye conjugates were prepared and their optical extinction and SERRS properties characterized by selectively tuning both the plasmon resonance of the nanorod and the molecular resonance of the dye with respect to the laser excitation wavelengths (633 nm and 785 nm). A particular advantage of our approach is its simplicity with bright Raman signals obtained without nanorod aggregation and good control over the surface coverage and orientation of reporter molecules on the nanorod surface. In comparison, most efforts at producing SERS probes have involved an aggregation step during their preparation which is relatively uncontrolled38,50,51 with a recent trend in the literature towards achieving greater aggregation control as part of the SERS optimization process.52,53 Further static and dynamic studies at the single nanoparticle level are currently underway to investigate the polarization dependence of the SERRS response and assess if there is a preferential adsorbance of dye, at least initially, on the rod ends or sides. This will also let us assess the percentage of particles that are SERRS active and compare with other substrates such as nanostars,54 capsules55,56 and aggregated systems. We have clearly shown the nanorod–dye platform to be a highly promising class of SERRS labels that offers a number of advantages. The polymer-wrapped conjugates were found to be very stable, allowing a significant Raman signal to be obtained after months of storage. The use of polyelectrolyte layers both as a platform for subsequent biofunctionalization57 and to also significantly reduce the cell toxicity of CTAB-coated nanorods58 has also been demonstrated. In particular, we hope the simplicity of the system will further promote the rational design of SERRS substrates for use in a wide range of applications.Acknowledgements
This work was supported by an Engineering Physical Sciences Research Council (EPSRC) Science and Innovation Award in Nanometrology and an EPSRC First Grant award (#EP/H030468/1). We would also like to thank Prof. WE Smith for valuable discussions over the course of the work.References
- H. Chen, T. Ming, L. Zhao, F. Wang, L. D. Sun, J. Wang and C. H. Yan, Nano Today, 2010, 5, 494–505 CrossRef CAS PubMed.
- N. Kometani, M. Tsubonishi, T. Fujita, K. Asami and Y. Yonezawa, Langmuir, 2001, 17, 578–580 CrossRef CAS.
- A. J. Haes, S. Zou, J. Zhao, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2006, 128, 10905–10914 CrossRef CAS PubMed.
- J. Zhao, L. Jensen, J. Sung, S. Zou, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2007, 129, 7647–7656 CrossRef CAS PubMed.
- G. P. Wiederrecht, G. A. Wurtz and J. Hranisavljevic, Nano Lett., 2004, 4, 2121–2125 CrossRef CAS.
- I. I. S. Lim, F. Goroleski, D. Mott, N. Kariuki, W. Ip, J. Luo and C.-J. Zhong, J. Phys. Chem. B, 2006, 110, 6673–6682 CrossRef CAS PubMed.
- N. T. Fofang, T.-H. Park, O. Neumann, N. A. Mirin, P. Nordlander and N. J. Halas, Nano Lett., 2008, 8, 3481–3487 CrossRef CAS PubMed.
- Y.-W. Hao, H.-Y. Wang, Y. Jiang, Q.-D. Chen, K. Ueno, W.-Q. Wang, H. Misawa and H.-B. Sun, Angew. Chem., Int. Ed., 2011, 50, 7824–7828 CrossRef CAS PubMed.
- W. H. Ni, H. J. Chen, J. Su, Z. H. Sun, J. F. Wang and H. K. Wu, J. Am. Chem. Soc., 2010, 132, 4806–4814 CrossRef CAS PubMed.
- W. H. Ni, Z. Yang, H. J. Chen, L. Li and J. F. Wang, J. Am. Chem. Soc., 2008, 130, 6692–6693 CrossRef CAS PubMed.
- W. Ni, T. Ambjornsson, S. P. Apell, H. Chen and J. Wang, Nano Lett., 2010, 10, 77–84 CrossRef CAS PubMed.
- A. M. Kelley, Nano Lett., 2007, 7, 3235–3240 CrossRef CAS PubMed.
- D. Gulen, J. Phys. Chem. C, 2010, 114, 13825–13831 CAS.
- S. M. Morton and L. Jensen, J. Chem. Phys., 2011, 135, 134103 CrossRef PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- G. L. Liu, Y.-T. Long, Y. Choi, T. Kang and L. P. Lee, Nat. Methods, 2007, 4, 1015–1017 CrossRef CAS PubMed.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308–1346 RSC.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS PubMed.
- D. Cunningham, R. E. Littleford, W. E. Smith, P. J. Lundahl, I. Khan, D. W. McComb, D. Graham and N. Laforest, Faraday Discuss., 2006, 132, 135–145 RSC.
- G. McNay, D. Eustace, W. E. Smith, K. Faulds and D. Graham, Appl. Spectrosc., 2011, 65, 825–837 CrossRef CAS PubMed.
- K. A. Willets, S. M. Stranahan and M. L. Weber, J. Phys. Chem. Lett., 2012, 3, 1286–1294 CrossRef CAS.
- I. A. Larmour, E. A. Argueta, K. Faulds and D. Graham, J. Phys. Chem. C, 2012, 116, 2677–2682 CAS.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS.
- B. K. Juluri, M. Q. Lu, Y. B. Zheng, T. J. Huang and L. Jensen, J. Phys. Chem. C, 2009, 113, 18499–18503 CAS.
- A. Yoshida, N. Uchida and N. Kometani, Langmuir, 2009, 25, 11802–11807 CrossRef CAS PubMed.
- G. von Maltzahn, A. Centrone, J.-H. Park, R. Ramanathan, M. J. Sailor, T. A. Hatton and S. N. Bhatia, Adv. Mater., 2009, 21, 3175–3180 CrossRef CAS PubMed.
- S. Pal, L. E. Depero and I. Alessandri, Nanotechnology, 2010, 21, 425701 CrossRef PubMed.
- H. Cai, J. Zhu, G. Chen, L. Liu, G. S. He and X. Zhang, J. Raman Spectrosc., 2011, 42, 1722–1727 CrossRef CAS.
- A. M. Gabudean, M. Focsan and S. Astilean, J. Phys. Chem. C, 2012, 116, 12240–12249 CAS.
- S. T. Sivapalan, B. M. DeVetter, T. K. Yang, T. van Dijk, M. V. Schulmerich, P. S. Carney, R. Bhargava and C. J. Murphy, ACS Nano, 2013, 7, 2099–2105 CrossRef CAS PubMed.
- A. McLintock, N. Hunt and A. W. Wark, Chem. Commun., 2011, 47, 3757–3759 RSC.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414–6420 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hiller, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- A. M. Alkilany, R. L. Frey, J. L. Ferry and C. J. Murphy, Langmuir, 2008, 24, 10235–10239 CrossRef CAS PubMed.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CrossRef CAS PubMed.
- A. W. Wark, R. J. Stokes, S. B. Darby, W. E. Smith and D. Graham, J. Phys. Chem. C, 2010, 114, 18115–18120 CAS.
- Y. B. Zheng, B. K. Juluri, L. L. Jensen, D. Ahmed, M. Lu, L. Jensen and T. J. Huang, Adv. Mater., 2010, 22, 3603–3607 CrossRef CAS PubMed.
- N. T. Fofang, N. K. Grady, Z. Fan, A. O. Govorov and N. J. Halas, Nano Lett., 2011, 11, 1556–1560 CrossRef CAS PubMed.
- H. Chen, L. Shao, K. C. Woo, J. Wang and H.-Q. Lin, J. Phys. Chem. C, 2012, 116, 14088–14095 CAS.
- Y. B. Zheng, B. Kiraly, S. Cheunkar, T. J. Huang and P. S. Weiss, Nano Lett., 2011, 11, 2061–2065 CrossRef CAS PubMed.
- J. Zhao, J. A. Dieringer, X. Zhang, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2008, 112, 19302–19310 CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CAS.
- V. Amendola and M. Meneghetti, Adv. Funct. Mater., 2012, 22, 353–360 CrossRef CAS.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
- J. C. S. Costa, R. A. Ando, P. H. C. Camargo and P. Corio, J. Phys. Chem. C, 2011, 115, 4184–4190 CAS.
- C. Fernandez-Lopez, C. Mateo-Mateo, R. A. Alvarez-Puebla, J. Perez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Langmuir, 2009, 25, 13894–13899 CrossRef CAS PubMed.
- X. Su, J. Zhang, L. Sun, T.-W. Koo, S. Chan, N. Sundararajan, M. Yamakawa and A. A. Berlin, Nano Lett., 2005, 5, 49–54 CrossRef CAS PubMed.
- G. Goddard, L. O. Brown, R. Habbersett, C. I. Brady, J. C. Martin, S. W. Graves, J. P. Freyer and S. K. Doorn, J. Am. Chem. Soc., 2010, 132, 6081–6090 CrossRef CAS PubMed.
- T. A. Laurence, G. Braun, C. Talley, A. Schwartzberg, M. Moskovits, N. Reich and T. Huser, J. Am. Chem. Soc., 2009, 131, 162–169 CrossRef CAS PubMed.
- N. Pazos-Perez, C. S. Wagner, J. M. Romo-Herrera, L. M. Liz-Marzan, F. J. Garcia de Abajo, A. Wittemann, A. Fery and R. A. Alvarez-Puebla, Angew. Chem., Int. Ed., 2012, 51, 12688–12693 CrossRef CAS PubMed.
- H. Yuan, Y. Liu, A. M. Fales, Y. L. Li, J. Liu and V.-D. Tuan, Anal. Chem., 2013, 85, 208–212 CrossRef CAS PubMed.
- J. Song, J. Zhou and H. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS PubMed.
- M. Sanles-Sobrido, W. Exner, L. Rodriguez-Lorenzo, B. Rodriguez-Gonzalez, M. A. Correa-Duarte, R. A. Alvarez-Puebla and L. M. Liz-Marzan, J. Am. Chem. Soc., 2009, 131, 2699–2705 CrossRef CAS PubMed.
- L. G. Xu, Y. Liu, Z. Y. Chen, W. Li, L. M. Wang, X. C. Wu, Y. L. Ji, Y. L. Zhao, L. Y. Ma, Y. M. Shao and C. Y. Chen, Nano Lett., 2012, 12, 2003–2012 CrossRef CAS PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization including UV-vis, Raman, SEM, and fluorescence measurements of the dyes and nanorod–dye conjugates as well as more details on the (dσR/dΩ) calculation are provided. See DOI: 10.1039/c3cp52946k |
| This journal is © the Owner Societies 2013 |