 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-mediated folding of dicarboxylate dianions: aliphatic chain length dependence and origin of the IR intensity quenching†
Marius
Wanko
a,
Torsten
Wende‡
b,
Marta Montes
Saralegui
a,
Ling
Jiang§
b,
Angel
Rubio
*ab and
Knut R.
Asmis
*bc
aNano-Bio Spectroscopy Group and ETSF Scientific Development Centre, Departamento de Física de Materiales, Universidad del País Vasco, Centro de Física de Materiales CSIC-UPV/EHU-MPC and DIPC, Av. Tolosa 72, 20018 San Sebastián, Spain. E-mail: angel.rubio@ehu.es
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: asmis@fhi-berlin.mpg.de
cWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstrasse 2, 04103 Leipzig, Germany
First published on 15th October 2013
Abstract
We combine infrared photodissociation spectroscopy with quantum chemical calculations to characterize the hydration behavior of microsolvated dicarboxylate dianions, (CH2)m(COO−)2·(H2O)n, as a function of the aliphatic chain length m. We find evidence for solvent-mediated folding transitions, signaled by the intensity quenching of the symmetric carboxylate stretching modes, for all three species studied (m = 2, 4, 8). The number of water molecules required to induce folding increases monotonically with the chain length and is n = 9–12, n = 13, and n = 18–19 for succinate (m = 2), adipate (m = 4), and sebacate (m = 8), respectively. In the special case of succinate, the structural transition is complicated by the possibility of bridging water molecules that bind to both carboxylates with merely minimal chain deformation. On the basis of vibrational calculations on a set of model systems, we identify the factors responsible for intensity quenching. In particular, we find that the effect of hydrogen bonds on the carboxylate stretching mode intensities is strongly orientation dependent.
1 Introduction
In the last few years, an increasing amount of microhydration studies focussed on the formation of extended hydrogen-bonded networks, their effect on the structure of organic and biomolecular ions (see for example ref. 1–5 and references therein), and the effect of messenger atoms on the isomer distribution of larger clusters (see ref. 6 and references in ref. 7). This interest was triggered in particular by advances in gas phase infrared spectroscopy and, more recently, ion mobility spectrometry. In this context, the stepwise hydration of dicarboxylate dianions serves as a model system for studying solvent-mediated folding processes.4,8–13These dianions consist of two charged carboxylate groups, separated by a hydrophobic, aliphatic (CH2)m chain, which also makes them interesting for studying solvent-mediated effects.11 Ion-trap experiments on microsolvated adipate (m = 4) and suberate (m = 6) dianions showed a characteristic change in the photoelectron spectrum (PES), which was assigned to a solvation-mediated folding of the dianion, supported by theoretical calculations.11,12 In the PES, a drop in the adiabatic detachment energy was observed when increasing the number of water molecules n to 13 and 16 for adipate and suberate, respectively. This drop was attributed to a transition from a linear configuration, with two water clusters separated by the hydrophobic chain, to a folded one, where the Coulomb repulsion between the two ion centers is overcome by merging of the two water clusters. The required number of water molecules was later shown to be temperature-dependent.9 For the smaller succinate and the larger tetradecandioic dianion, no such transition was detected for up to 18 water molecules.12
Similar folding transitions have been found very recently for other molecules. Demireva et al. detected the folding of the 1,7-diammoniumheptane dication (CH2)7(NH3+)2 microsolvated in water by using infrared photodissociation (IRPD) spectroscopy.3 Analysis of the intensities of water OH-stretching bands associated with free and hydrogen-bond donating OH groups showed a pronounced increase of the latter, which was absent in the equivalent monocation. This was interpreted as an indication of additional hydrogen bonds that are formed when the two water clusters merge due to the folding of the aliphatic chain. Murdachaew et al.14 studied complexes of alkali cations and dicarboxylate dianions with PES and suggested that the cations may bind to both carboxylates, inducing a folded chain configuration.
The solvent configuration can be influenced by quite subtle interactions. IRPD spectroscopic studies on protonated water clusters have demonstrated the coexistence of structural isomers and have shown that their distribution can be varied by tagging the cluster with different inert gas atoms (see ref. 7 for a review and references). Even without electrostatic interactions, linear molecules can undergo a folding transition. The Raman experiment of Lüttschwager et al.15 proved that neutral alkanes at low temperatures undergo a transition from a linear to a folded hair-pin structure, which is stabilized by dispersion forces between the two parallel chains.
These examples show that the interdependence of solute and solvent structure has various aspects and the different kinds of interactions range from dispersion forces, polarization, and steric strain to strong Coulomb interactions and hydrogen bonds. In most cases, it is difficult to find a rationale for the observed trends.
Recently, we have shown that the linear and folded configurations of the microsolvated suberate dianion can be distinguished for arbitrary number n of water molecules using IRPD spectroscopy. The folding transition is marked by a strong quenching of the symmetric CO stretching band intensity.4 Therefore, IRPD spectroscopy represents an ideal tool to study the folding transition of carboxylate dianions. The previous studies, however, raise a series of questions: (1) What is the origin of the intensity quenching? (2) What is the driving force for the folding transition? (3) How does the critical number of water molecules depend on the length of the aliphatic chain? (4) Does the smallest (succinate) dianion fold? To answer these questions, we apply here the same experimental technique to the shorter adipate (m = 4) and succinate (m = 2) dianions as well as to the longer sebacate (m = 8) dianions. We use molecular dynamics simulations and quantum chemical calculations to find the lowest energy minimum structures for linear and folded chain configurations, to explain the changes in the IR spectrum, and to analyze the energetics of the folding transition. Furthermore, we analyze in detail the various factors that affect the intensities of the carboxylate stretching modes, in particular the role of hydrogen bonds and their orientation.
2 Experimental details
The IRPD experiments are carried out using a ring-electrode ion-trap tandem mass-spectrometer,16,17 temporarily installed at the “Free Electron Laser for Infrared eXperiments” (FELIX) facility18 at the FOM institute Rijnhuizen. Microsolvated dicarboxylate dianions (CH2)m(COO−)2 with different chain lengths (m = 2, 4, 8) are produced in a commercial Z-spray source from a solution containing 1 mM of the respective acid and 2 mM NaOH in a water–acetonitrile mixture (20/80). Typical cluster distributions of succinate (m = 2) and adipate m = 4 dianions containing up to 40 water molecules is shown in Fig. S4 in the ESI.† Parent ions are mass-selected in a quadrupole mass-filter and focused into a cryogenically-cooled ring-electrode ion-trap. Here, the ions are accumulated and thermalized for 98 ms by multiple collisions with a helium buffer gas at an ion-trap temperature of 15 K. After filling the trap, the ion packet is extracted and focused both temporally and spatially into the center of the extraction region of an orthogonally mounted linear time-of-flight (TOF) mass spectrometer. Here, the ions interact with the IR laser pulse and high voltage extraction-pulses are applied for recording TOF mass spectra.Pulsed IR radiation is used from FELIX operated in the spectral region of the carboxylate stretching vibrations from 1250–1800 cm−1. IR pulses are provided with average pulse energies of 30 mJ and a bandwidth of approximately 0.3% root mean square (RMS) of the central wavelength. TOF spectra are recorded for each wavelength step and IRPD spectra are obtained by plotting the photodissociation cross-section, σ, according to σ = −ln[IP(ν)/(IP(ν) + IF(ν))]/P(ν), with the parent and photofragment ion intensities IP(ν) and IF(ν), respectively, and the frequency-dependent laser pulse energy P(ν). Correcting with P(ν) and not the laser fluence takes the multiple photon absorption character into account and allows for a better comparison with the calculated linear absorption cross-section.
3 Computational details
To obtain the minimum-energy structure for the folded, bridged, and linear configurations of dicarboxylate dianions of variable chain length, a multi-stage protocol was used. We did not consider all n, as the spectra for vicinal n are very similar. First, the configuration space was roughly sampled by performing 5 ns of molecular dynamics (MD) simulation, after 1 ns of equilibration, using CHARMM with the TIP3P water model and force field parameters adopted from the CHARMM27 parameters for the glutamic acid side chain. At a later stage, these were repeated with harmonic constraints on the distance between the carboxylate carbon atoms (D), to effectively sample relevant regions of the configuration space. Details are given in the ESI.†From each simulation, 500 random snapshots were optimized using the quantum mechanical (QM) SCC-DFTB method.19 Using ultra-tight optimization and appropriate energy thresholds, identical structures and local minima that are more than 5 kcal mol−1 higher in energy than the lowest one were discarded.
The resulting minima (typically 20–200) were optimized with DFT using the PBE functional,20 the aug-cc-pVDZ basis set,21 and the RI approximation within turbomole.22 To gain confidence in the PBE structures and energies, geometries were further optimized with B3LYP/aug-cc-pVDZ and MP2/TZVP and their energies evaluated with CC2/aug-cc-pVDZ within turbomole. The aug-cc-pVDZ basis set was found necessary to reproduce all stable water configurations with PBE. The PBE/aug-cc-pVDZ geometries were found sufficiently accurate to evaluate the total energy at higher levels of theory. Structures were inspected and an energy threshold of 3–5 kcal mol−1 was chosen to select the PBE/aug-cc-pVDZ structures for further evaluation. Vibrational spectra and zero-point vibration energies (ZPVE) were calculated using PBE/aug-cc-pVDZ and total energies were re-evaluated using the CEPA/1 method in ORCA,23 corrected by the PBE ZPVE's. For the succinate dianions and up to n = 10 water molecules, as well as for selected adipate clusters, total energies were obtained at the CEPA/1 level with aug-cc-pVTZ basis and ZPVE's from B3LYP/aug-cc-pVDZ (see ESI†).
4 Results and discussion
4.1 Experimental IRPD spectra
The experimental and computational results of the vibrational spectroscopy of the (CH2)m(COO−)2·(H2O)n complexes (m = 2, 4, 8) are summarized in Fig. 1. IRPD spectra were measured from 1300–1650 cm−1, covering the region of the symmetric (νS) and antisymmetric (νA) carboxylate stretching modes (the complete dataset is shown in Fig. S1 of the ESI†). From the experimental IRPD spectra band intensities (I) and band centroids (ν) were determined by integration (see ESI†). The normalized νS band intensities were then determined according to IS/(IS + IA). These results are plotted in the top and bottom parts of Fig. 1 as a function of the number of water molecules n. Extensive calculations were performed for m = 2 and m = 4. A summary of these results is included in Fig. 1, and they will be discussed in subsequent sections.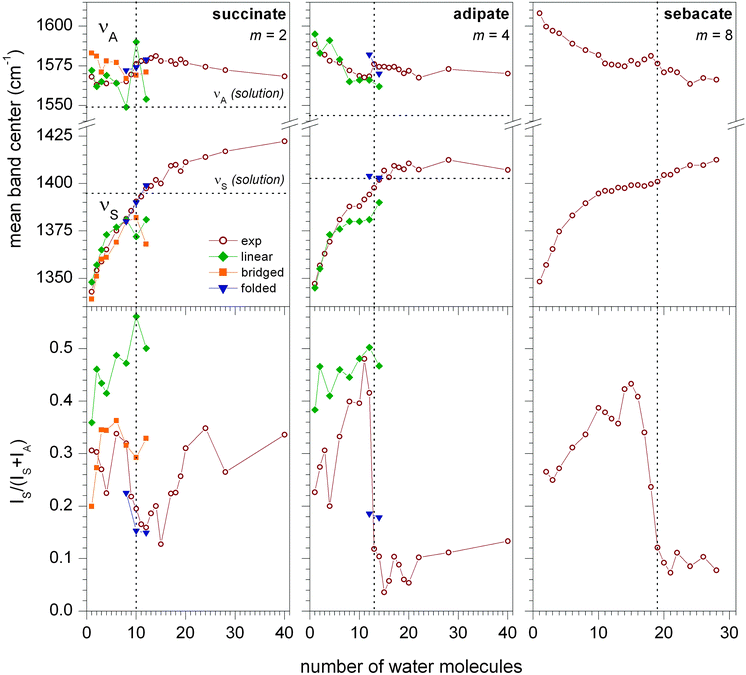 | ||
| Fig. 1 Center of the symmetric (νS) and antisymmetric (νA) CO stretching bands (top) and normalized intensity IS/(IS + IA) (bottom) for microsolvated (CH2)m(COO−)2·(H2O)n clusters (m = 2, 4, 8) as a function of n. Experimental data (open wine circles) as well as simulated data for linear (solid green diamonds), bridged (solid orange squares) and folded (solid blue triangles) geometries are shown (one selected representative for each structural motif). Lines connecting the symbols are drawn to guide the eye. Dotted vertical lines mark the critical solvent number where the normalized intensity drops below 0.2. Dotted horizontal lines indicate absorption frequencies in solution.24 | ||
The evolution of the band centroids of νS as a function of n for succinate (m = 2), adipate (m = 4), and sebacate (m = 8), see Fig. 1, is rather similar in nature, following the same trends as previously found for the suberate dianion (m = 6).4 A nearly monotonic increase of νS from below 1350 cm−1 for n = 1 to over 1400 cm−1 for n > 20 is observed for all systems, with a reduction in slope in the n = 4–14 region. The behavior is quite different for νA. The extent of the shift from small to large n is smaller and increases with m. In fact, for succinate and adipate, all values lie within 25 cm−1 of each other and no clear trend in a particular direction is observed. This suggests that νA is more sensitive to long-range effects, whereas νS probes more local changes in the immediate hydration environment. This short vs. long-range assumption is corroborated by the comparison with the solution-phase values for νS and νA. The curves for νS converge at this value for n > 20 for adipate (and suberate), whereas they do not reach the bulk limit yet for νA, even at the largest cluster size probed here. The behavior of succinate is exceptional in this regard. Here, νS overshoots the bulk-solution limit, suggesting that the hydration behavior of succinate is principally different from the longer-chain dicarboxylate dianions.
For all chain lengths, the evolution of the band centroids with n shows no clear evidence for the occurrence of a solvent-mediated conformation change. This is different for the relative band intensities IS and IA. Similar to the case of the suberate dianion,4 all three systems studied here show a characteristic drop in the normalized νS intensity as n increases (see Fig. 1 bottom). This drop occurs at n = 9–12, n = 13 and n = 18–19 for m = 2, m = 4 and m = 8, respectively, and, as we will show later, can be unambiguously assigned to the solvent-mediated folding transition. When n is increased further, the normalized intensity stays below 0.2 for m > 2. For succinate, however, a characteristic increase is observed at n > 16, suggesting that linear configurations are competitive with folded ones. This shows that the hydration behavior of succinate is fundamentally different from that of the longer carboxylate dianions and is consistent with the suggested bulk solvation of succinate vs. surface solvation of dianions with longer chains.12
4.2 Quantum chemical calculations
In the following sections, we present the results of our calculations and discuss what can be learned from the comparison of theoretical and experimental data. In separate sections, we discuss the most important aspects, including (1) the energetic selection of relevant structures, (2) the possibility of a linear chain configuration with bridging water molecules for the succinate dianion, (3) the relationship between structural motifs and the normalized νS band intensity, and (4) the occurrence of asymmetric solvation. We then proceed with a complete assignment of minimum-energy structures to the experimental IRPD spectra that we discussed above. To this end, Fig. 2 and 3 compare the IRPD spectra with calculated linear IR absorption spectra of selected structures. The central column shows the IR spectra of the most likely structures, which were selected in terms of energy, band position, band shape, and normalized intensity. The IR spectra of alternative structures are shown on the right hand side. A complete comparison including the spectra of all structures within 1 kcal mol−1 and their respective energies is provided in the ESI.† In the final part of the article, we explore the origin of the IS quenching, i.e., how structural parameters determine the carboxylate stretching band intensities.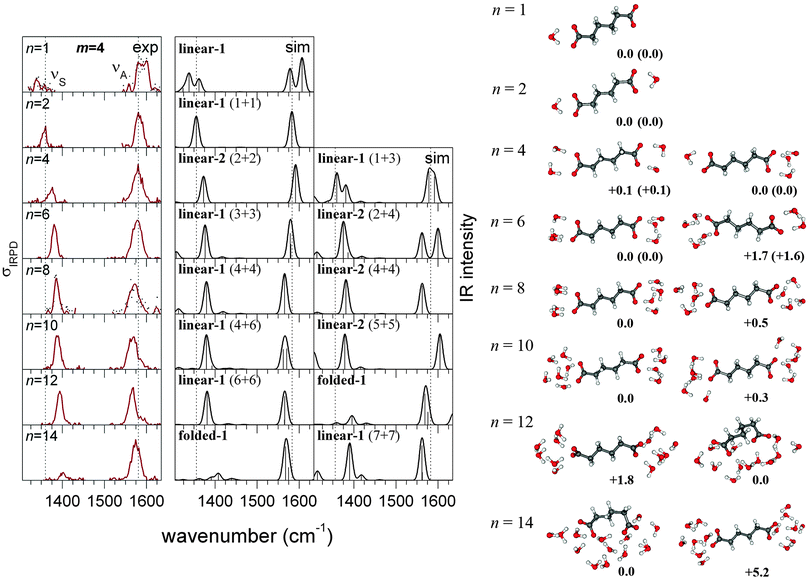 | ||
| Fig. 2 Experimental IRPD spectra (left) and simulated linear IR absorption spectra of the most likely (center) and alternative (right, see text) structures of microsolvated adipate dianions (CH2)4(COO−)2·(H2O)n. The corresponding geometries and relative energies (kcal mol−1) are shown next to the spectra. CEPA/1/aug-cc-pVDZ energies are corrected by ZPVE from PBE (energies from CEPA/1/aug-cc-pVTZ corrected by ZPVE from B3LYP are shown in parentheses). | ||
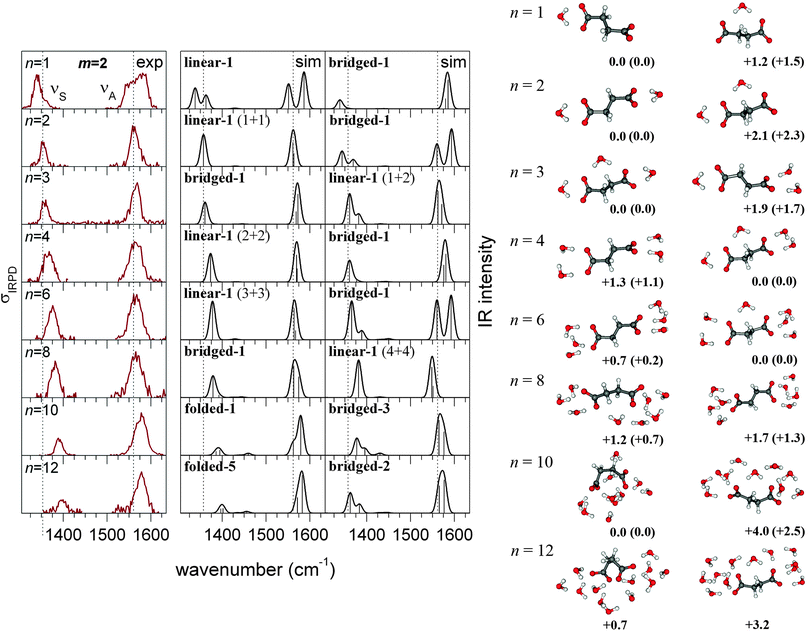 | ||
| Fig. 3 Experimental IRPD spectra (left) and simulated linear IR absorption spectra of the most likely (center) and alternative (right, see text) structures of microsolvated succinate dianions (CH2)2(COO−)2·(H2O)n. The corresponding geometries and relative energies are shown next to the spectra. CEPA/1/aug-cc-pVDZ energies are corrected by ZPVE from PBE (energies from CEPA/1/aug-cc-pVTZ corrected by ZPVE from B3LYP are shown in parentheses). | ||
4.3 Energetic selection
The goal of our computational approach is to find an ensemble of low-energy structures that contains the experimentally observed species with high probability. For the comparison of different water cluster configurations solvating the same isomer of the dianion, we assume an error of 1 kcal mol−1 for relative energies that were obtained with the CEPA/1 method and the aug-cc-pVDZ basis and were corrected for ZPVE from DFT using the PBE functional. This error estimation is based on benchmark calculations on the microsolvated formate anion (see Tables S5 and S6 in the ESI†). In the following, we mainly focus on the structures obtained within this 1 kcal mol−1 threshold. Larger errors are expected when comparing the energies of linear vs. folded chain isomers, as the basis set superposition error (BSSE) becomes more important here. Therefore, we will not use the energy criterion for the assignment of the linear or folded structural motif to the experimental IRPD spectra. For small numbers n of water molecules, a unique assignment is possible, as other local minima of the potential energy surface are higher in energy by significantly more than 1 kcal mol−1. This applies to n = 0, 1, 2, and 6 for the adipate dianion and n = 0, 1, 2, 3 for the succinate dianion, respectively. In other cases, the number of relevant structures can be reduced further by comparison of the experimental and simulated IR spectra in terms of band maxima (νS, νA), band intensities (IS, IA), and band shapes.4.4 Structural motifs, normalized νs intensity
The succinic acid dianions differ from the larger dianions in one essential aspect. The separation of the two carboxylate groups is sufficiently small to actually allow individual water molecules to form H-bonds with both of them, without significant chain deformation. In structures with such “bridging” water molecules, the central C–C bond torsion from co-planarity can be as small as 21°, with the C–C–C–C dihedral ranging from 129 to 159°. Our calculations support this structural motif in several cases. For n = 3, the structure with one bridging water is found to be 1.7 kcal mol−1 lower in energy than the asymmetrically (2 + 1) solvated linear complex and is therefore expected to be the one observed in the experiment. More surprisingly, we find bridged structures also plausible for n = 4 and 8, with energies competitive to those of linear structures and IR spectra that are consistent with experiments, as will be shown further below. Even for n = 10, we cannot exclude that bridged structures are relevant, although the folded structural motif is more likely to dominate. For n ≥ 8, we also obtain fully folded structures, which feature a C–C–C–C dihedral in the range 60–104° and a significantly reduced separation between the carboxylate groups.Our calculations show that each structural motif has a characteristic range of values for the normalized νS intensity (see Fig. 4). For adipate and longer chains (m ≥ 4), these regimes are well separated and can be used to distinguish unambiguously between linear and folded structures. Hence, the experimental IRPD spectra can pin-point the solvent-mediated folding transition at n = 13 for adipate. The situation is somewhat more complicated for succinate, due to the possibility of the bridged structural motif. The calculated intensities for the bridged structures lie in between the linear and the folded regimes. Taking into account further criteria (energy, band position and shape), the comparison of experimental and theoretical data can narrow down the folding transition to n = 9–12 for succinate.
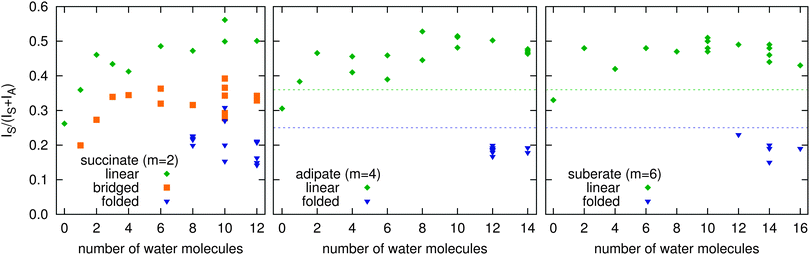 | ||
| Fig. 4 Normalized intensities of the νS band of the succinate (left), adipate (center), and suberate (right) dianion. The data of all calculated structures with energies predicted to be ≤1 kcal mol−1 above the global minimum (for each of the three structural motifs) are shown. | ||
4.5 Asymmetric solvation
Due to the strong solvent-induced shifts in the carboxylate stretching bands, in particular for small n (see Fig. 1), a splitting or broadening of the νS and νA bands can be expected for asymmetric solvation. The experimental spectra show this clearly for n = 1 (both succinate and adipate dianions, see Fig. 2 and 3), but not for any larger cluster. Our calculations not only reproduce this feature for n = 1, but also show that asymmetric solvation is energetically possible in some cases with even number of water molecules. This applies to the adipate dianion solvated by 10 water molecules, where only the asymmetrically (4 + 6) solvated complexes produce spectra that are consistent with experiment. Also for n = 4, asymmetric solvation is energetically plausible, but features a large splitting in the νS band, which is not supported by the experiment. For larger n, we did not find asymmetrically solvated adipate dianions with linear configurations that are competitive in energy. For the succinate (m = 2) dianions, asymmetric (symmetric) solvation is possible also for an even (odd) number of water molecules, respectively, due to the possibility of bridging water molecules. As the experimental spectra show band splittings only for n = 1, we can exclude asymmetric solvation in those cases where our calculations indicate a very strong splitting of the νA band. This applies to n = 2 and n = 6 (right column in Fig. 3). For n = 8, in contrast, an asymmetrically solvated succinate with bridging water is lowest in energy and produces the best matching spectrum. In conclusion, our calculations show that asymmetric solvation is energetically favorable in some unexpected cases and does not necessarily result in band splittings that are detectable by IRPD spectroscopy.4.6 Structural assignment (adipate dianion)
For n ≤ 2, the spectrum can be assigned to one specific structure, as other minima are predicted to be more than 1 kcal mol−1 higher in energy. The simulated spectra of these structures (see Fig. 2) reproduce the experimental band shifts well, but the calculated normalized νS intensities are larger (Fig. 1). The differences in the experimental and calculated intensities likely reflect the nonlinear (multiple photon) nature of the IRPD absorption process, which is not taken into account by the calculations. In the case of n = 4, the asymmetrically (1 + 3) solvated complex (linear-1) is predicted to be 0.1 kcal mol−1 more stable than the symmetrically solvated one (linear-2). The position of both stretching bands fits well to experiment for both structures, but the simulated spectrum of the 1 + 3 cluster shows a split νS band, which is not observed in the IRPD spectrum. Therefore, the linear-1 structure can be considered less probable than the linear-2 structure, but it cannot be excluded safely, based on the experimental data. The same situation was found for the corresponding cluster of the suberate dianion.4 For n = 6 water molecules, the assignment to the symmetrically solvated linear-1 structure is unique, as other minima are predicted to be more than 1 kcal mol−1 higher in energy. The most stable asymmetrically (2 + 4) solvated structure is predicted to be 1.6 kcal mol−1 higher in energy and its spectrum shows a clear splitting that is not supported by experiment. For clusters with n = 8 or more water molecules, the structural assignment is ambiguous, as several water configurations are energetically competitive and produce very similar spectra, which are in good agreement with experiment. The isomerization state of the dianion, however, can always be assigned with certainty, as the experimental band intensities are reproduced only by structures that are either linear (n ≤ 12) or folded (n ≥ 14), respectively. Also the energies calculated with a double-ζ basis set are consistent with a transition to folded structures for n ≥ 14. For n = 12, the folded structures are predicted to be up to 1.8 kcal mol−1 lower in energy than the linear-1 complex, but this is within the expected BSSE, as discussed in ref. 4. For n = 14, however, the folded structures are at least 5.2 kcal mol−1 lower in energy, which cannot be attributed to the BSSE.4.7 Structural assignment (succinate dianion)
For n = 1, the measured spectrum can clearly be assigned to the linear-1 structure. With the water in a symmetric bridging position (bridged-1), the energy is 1.5 kcal mol−1 higher and the experimentally observed broadening of the νA band is not reproduced (see Fig. 3). Also for n = 2, the assignment (linear-1) is unique, as a large splitting or broadening of both bands is predicted for the bridged-1 structure, which is in conflict with experiment. The case of two bridging water molecules can be excluded energetically (11 kcal mol−1 higher in energy). Also for n = 3 and n = 4, two alternative structures are found. The predicted band splittings for the asymmetric structures are much smaller and both bridged and unbridged structures produce IR spectra in agreement with experiment. For n = 3, the bridged-1 structure is predicted to be 1.7 kcal mol−1 lower in energy and therefore most likely the relevant one. For n = 4, the unbridged linear-1 structure is 1.1 kcal mol−1 higher in energy but reproduces better the νS − νA band separation, hence the assignment is not clear. For n = 6, only the spectrum of the linear-1 structure compares well with the experimental one. The asymmetrically solvated bridged-1 structure is predicted to be 0.2 kcal mol−1 lower in energy but shows a large splitting of the νA band. Another bridged-2 structure (see ESI†) is found to be 0.6 kcal mol−1 higher in energy, but its spectrum does not reproduce the experimental band separation. For n = 8, we find all three structural motifs, of which none can be excluded on energetic terms. The bridged-1 structure is predicted to be 0.6 kcal mol−1 lower in energy than the linear-1 structure and its spectrum reproduces the position of the experimental νA band more accurately. Moreover, the νS band intensity of the linear-1 structure appears too high, considering that for the longer dianions (m ≥ 4) the IRPD and simulated spectra show similar intensities for n = 8 (Fig. 1 and 2). The spectra of all folded structures show a quenched symmetric band and therefore clearly disagree with experiment. For n = 10, the quenching of IS is observed also in the experimental spectrum. We find four folded structures within the energy threshold of 1 kcal mol−1 with similar spectra (see, e.g., folded-1 in Fig. 3), which are all consistent with experiment. In addition, some of the unfolded structures with a bridging water (bridged-3 and bridged-4) feature a strong distortion of the C–C–C–C dihedral (ca. 130°) and, as a result, produce a weak symmetric band, as well. Their spectra are still reconcilable with experiment, but fit less well than those of the fully folded structures (folded-1, folded-2, folded-3, folded-4). Furthermore, the energies of the linear and bridged structures are obtained more than 2 kcal mol−1 higher than that of the folded-1 structure. Due to the larger BSSE of the folded structures, this energy difference depends strongly on the employed basis set (see Table S4 in the ESI†) and, considered alone, does not allow a safe exclusion of the unfolded structural motif. For n = 12, the spectra of all folded structures agree with experiment regarding the νS band position and intensity, in contrast to those of the unfolded ones (with or without bridging water).4.8 Origin of the frequency quenching
In our earlier work,4 we proposed that several mechanisms may contribute to the observed quenching of IS upon folding, i.e., the transition between linear and isomerized chain configuration, when a certain number of water molecules is exceeded. Here, we analyze in more detail how the intensities IS and IA are affected by (1) the chain configuration, (2) the ion–ion interaction, (3) charge screening, (4) hydrogen bonds, and (5) the chain length, and how these effects interfere/interact.In a first step to elucidate the mechanisms for the folding-induced quenching of IS, we consider in Fig. 5 the different geometrical situations that occur in the ensemble of minimum-energy structures of the microsolvated succinate dianion. These structures cover the situations found in the longer dianions, and furthermore provide the case of a linear chain with bridging water molecules. The normalized intensity IS/(IS + IA) in vacuo is 0.26 (top left in Fig. 5) and hardly changes when the geometry is constrained to reduce the separation of the carboxylates, like in the folded clusters. This result is independent of whether the C–C–C–C dihedral is constrained (center and right in Fig. 5), or the distance between the outermost carbon atoms is fixed, instead (not shown). Even when the dihedral is reduced to 60°, the normalized intensity is still 0.24 (not shown). It is drastically enhanced to 0.46, though, when adding one water molecule on both sides (2nd row in Fig. 5). Then again, the effect of further addition of water is small. Interestingly, the normalized νS intensity depends strongly on the number of water molecules binding to both carboxylates (bridging water molecules), whereas it is largely independent of the total number of water molecules and the particular structure of the water cluster. To rationalize this, we investigate simplified model systems to separate the different effects at work.
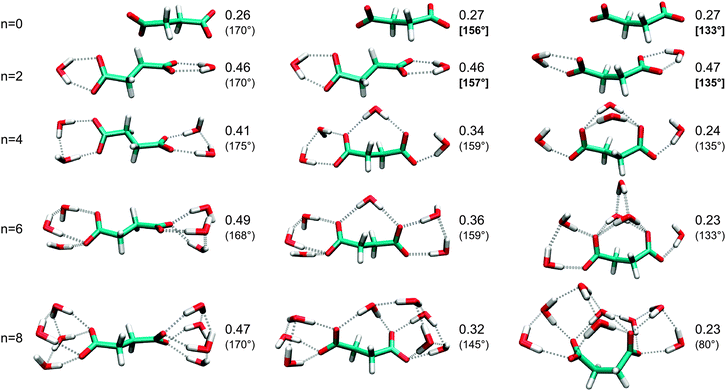 | ||
| Fig. 5 Normalized intensities IS/(IS + IA) for different configurations of the succinic acid dianion containing 0 (left), 1 (center), or 2 (right) water molecules bound to both carboxylates (bridging water molecules). The C–C–C–C dihedral is given in parentheses. In cases where bridging water molecules are not available (n = 0 and n = 2), the structures have been optimized under a constrained C–C–C–C dihedral (boldface value in brackets) that is typical for a cluster with 1 (center) or 2 (right) bridging water molecules. | ||
First, we substitute the second carboxylate by a methyl group. Table 1 demonstrates that folding, i.e., isomerization of the outermost C–C–C–C dihedral significantly reduces the normalized intensity, both in vacuo and after adding one water molecule (n = 1). The result is independent of the length of the saturated chain. For the longest chain, associated with the suberic acid, we also tested the effect of isomerizing the second outermost C–C–C–C dihedral, which corresponds to the situation found in the folded dianion clusters (n ≥ 12). The resulting normalized intensity increases to a value in between the linear and the singly isomerized chain (0.27 in vacuo, 0.41 for n = 1). The pronounced and systematic quenching of IS due to chain isomerization seems to contradict the invariance observed above for the succinic acid dianions. In fact, the presence of the second ion leads to a certain quenching in the linear case (but not in the folded one), which is significant for the succinic acid (see Table 1, lower part) but becomes irrelevant for longer chains. A similar quenching of the normalized intensity is obtained when adding a negative charge (Cl− at fixed 5 bohr distance to the Me carbon) in place of the second carboxylate (see Table 1). Hence, the quenching in the linear succinic acid is associated with polarization along the long molecular axis. Note that in the folded case, the normalized intensities are essentially the same for mono- and dianions. Therefore, the concept of charge screening, associated with the effective dielectric medium in solution, appears inappropriate for explaining the intensity quenching.
| m = 2 | m = 4 | m = 6 | ||
|---|---|---|---|---|
| a Local minimum with C–C–C–COO− dihedral between 68 and 70°. b Constrained O–O distance (see text). c The Cl− distance from the Me carbon is constrained to 5 bohr. | ||||
| Monoanion CH3(CH2)mCOO− | ||||
| n = 0 | Linear | 0.34 | 0.33 | 0.34 |
| Foldeda | 0.25 | 0.24 | 0.22 | |
| n = 1 | Linear | 0.46 | 0.47 | 0.47 |
| Foldeda | 0.38 | 0.36 | 0.37 | |
| Dianion (CH2)m(COO−)2 | ||||
| n = 0 | Linear | 0.26 | 0.31 | 0.33 |
| Foldedb | 0.26 | 0.24 | 0.24 | |
| Monoanion–Cl− complexc | ||||
| n = 0 | Linear | 0.29 | 0.32 | 0.33 |
In the next step, we analyze the role of hydrogen bonds in intensity quenching. Here, it is more useful to consider IS and IA individually instead of the normalized intensity IS/(IS + IA). While the chain configuration in the models above has no significant effect on IA, both intensities respond to hydrogen bonds and in a quite different way. Already in Fig. 3 and 5, we have shown that bridging water molecules affect intensities quite differently to those binding to only one carboxylate. Again for the monoanion, we vary the direction of the hydrogen bonds by constraining the position of a single or two water molecules (see Fig. 6). The enhancement of IS due to the water gradually turns into a reduction when moving the water out of the COO-plane. IA, in contrast, is quenched by the water, but the quenching reduces as the water is moved out of the plane. In consequence, the normalized νS intensity in the perpendicular orientation (0.36) is reduced to almost the vacuum value (0.34).
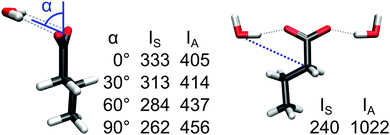 | ||
| Fig. 6 Intensities IS and IA (km mol−1) of the microsolvated butanoic acid anion (in vacuo IS and IA are 291 and 554 km mol−1, respectively). To achieve different hydrogen-bond orientations, the water oxygen positions were constrained during optimization by fixing the angle α (left) or the C–O distance (right). | ||
The anisotropy of the quenching is even stronger when the direction of the hydrogen bond is varied within the COO-plane. A lateral orientation of two water molecules (Fig. 6, right) results in a strongly quenched IS, whereas IA is almost twice as large as in the vacuum case, which leads to a very small normalized νS intensity of 0.19.
The two factors, chain folding and hydrogen-bond orientation, are exemplified in Fig. 7 for three different configurations of the same cluster of two butanoic acid anions and two water molecules. The chain isomerization affects primarily IS (compare Fig. 7a and b), whereas both IS and IA are strongly dependent on the direction of the hydrogen bonds (compare Fig. 7b and c). In this way, the normalized intensity can be varied in the range 0.16–0.44.
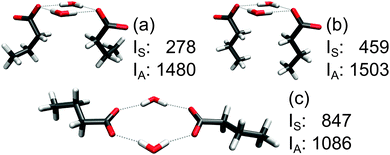 | ||
| Fig. 7 Intensities IS and IA (km mol−1) of two microsolvated butanoic acid anions. The geometries of three local energy minima (no constraints were applied) were chosen to realize different hydrogen-bonding situations and chain conformations that may occur in complex systems and demonstrate the tunability of the intensity ratio. | ||
4.9 On the folding mechanism
The transition from a linear to a folded chain configuration causes an increase of the Coulomb repulsion between the carboxylate groups, which depends on the chain length m. Table 2 shows the energy differences between linear and folded configurations for the lowest-energy structure of each motif. In the case of the succinate (m = 2), the folded-1 structure is compared with the bridged-3 one. Considering the bare Coulomb interaction between the carboxylate fragments, an increase with m is evident (ECoul in Table 2), which essentially originates from their larger separation in the linear configuration (dlinear). In the folded states, the separation dfolded varies only little due to the constraints of the water cluster. As a consequence, the number of water molecules required to stabilize the folded state increases with m.| m | n | d linear | d folded | E Coul | Dianion | Water | Dianion–water | ZPVE | E f |
|---|---|---|---|---|---|---|---|---|---|
| a Energy difference between the folded-1 and the linear-1 (bridged-3) structure that is shown in Fig. 2 (3); CEPA/1 calculations (aug-cc-pVDZ basis set) with BSSE correction, geometries and ZPVE from PBE/aug-cc-pVDZ. b Data from ref. 4. c C–C separation of the carboxylate carbons (Å). d Bare Coulomb interaction of the COO− fragments, calculated from HF atomic partial charges (natural population analysis). | |||||||||
| 2 | 10 | 3.83 | 3.09 | 15.3 | 9.5 | 14.0 | −27.6 | 0.1 | −4.0 |
| 4 | 12 | 6.47 | 4.84 | 18.9 | 13.9 | 2.8 | −20.4 | 1.9 | −1.8 |
| 14 | 6.51 | 4.76 | 19.2 | 14.5 | 5.3 | −27.0 | 2.0 | −5.2 | |
| 6b | 12 | 8.92 | 4.95 | 26.5 | 23.0 | 5.6 | −33.7 | 2.7 | −2.4 |
| 14 | 8.95 | 4.48 | 38.7 | 31.1 | 24.0 | −63.0 | 1.8 | −6.1 | |
| 16 | 8.98 | 5.38 | 23.2 | 18.6 | 3.5 | −31.5 | 2.6 | −6.8 | |
In all three dianions, the enhanced Coulomb repulsion in the folded state is compensated by an increased dianion–water interaction, whereas the energy of the water cluster without the dianion is slightly increased (Table 2). This means that the water–water interaction does not contribute at all to the stabilization of the folded state. Interestingly, Demireva et al. proposed a different mechanism for the stabilization of the folded configuration in a recent microhydration study of diaminoheptane dications.3 Their measured IRPD spectra indicate an increase in the intensity ratio of the water OH-stretching bands of bonded vs. free water molecules, i.e., from water molecules that donate hydrogen bonds. This increase was observed when a critical number of water molecules is exceeded and is missing in the spectrum of the monocation solvated by n/2 water molecules and was therefore interpreted to result from a folding transition. As the aminoheptane can only donate hydrogen bonds, this points towards a stabilization due to newly formed water–water bonds upon folding, although this scenario certainly does not exclude a stabilizing contribution from the water–dication interaction.
5 Conclusions
In this work, we have studied the effects of microhydration on the IR spectra of succinate, adipate, and sebacate dianions using IRPD experiments and quantum chemical calculations. Like for the suberate dianion, we observe a clear transition from linear to folded structure for the adipate and sebacate dianions, when reaching a critical number of water molecules. The latter increases monotonically with the chain length and is 13 in the case of the adipate, in agreement with a previous PES study,12 and 18–19 for the sebacate. In the case of the smaller succinate dianion, we showed for the first time that a folded configuration is assumed with 12 water molecules, which is not evident from the PES experiment.12 The transition between the linear and folded motif is less well defined for the succinate dianion for two reasons: (1) the possibility of bridging water molecules that bind to both carboxylates simultaneously, without chain isomerization. These bridged structures feature a normalized νS intensity IS/(IS + IA) that varies between the values for the folded structures and those of linear structures with separated water clusters and (2) the intensity IS of the symmetric band is already reduced in the linear structure, when compared with IS for longer chains, due to the strong ion–ion interaction. Hence, the assignment of the succinate–water clusters to one of the three motifs is uncertain for 4–10 water molecules, the transition from linear to folded chain configuration occurs between 8 and 12 water molecules. Moreover, the succinate dianion is the only system where we find the νS band intensity recovering for large cluster sizes (n > 16). This is consistent with the assumption that the linear configuration dominates in bulk solution of the succinate, whereas adipate and longer carboxylate dianions prefer surface solvation.12For all three dianions and the previously studied suberate,4 our calculations surprisingly show that the energetic mechanism behind the folding is the increased water–solute interaction, whereas the water–water interactions do not stabilize the folded state over the linear one.
We identified the mechanisms behind the intensity quenching. In addition to the resonant interaction between the induced dipoles discussed by Wende et al.,4 we found two important factors that strongly modulate the intensities IS and IA. The isomerization of the outermost C–C–C–C dihedral, which is present in all folded structures of the investigated dianions, reduces IS. Hydrogen bonds between water and the carboxylate group affect both IS and IA and can either enhance or quench the intensity, depending on their orientation to the carboxylate group. Hydrogen bonds in the direction of the C–COO bond enhance IS while reducing IA, whereas the opposite applies to hydrogen bonds with a lateral or out-of-plane orientation, which are formed by bridging water molecules upon folding. These two factors can tune the normalized νS band intensity in the range 0.16–0.49 (intensity ratio IS/IA of 0.19–0.94). These findings will prove useful to interpret IR data of deprotonated carboxylic acids, such as FTIR difference spectra, which are sensitive to changes in the hydrogen-bonded networks that solvate these anions in a biological environment.
Acknowledgements
We acknowledge financial support from the Spanish Juan de la Cierva, the European Research Council Advanced Grant DYNamo (ERC-2010-AdG-267374), Spanish Grants (FIS2010-21282-C02-01 and PIB2010US-00652), Grupos Consolidados UPV/EHU del Gobierno Vasco (IT578-13), Ikerbasque, the European Commission projects CRONOS (Grant number 280879-2 CRONOS CP-FP7), and a German Humboldt fellowship.References
- N. S. Nagornova, T. R. Rizzo and O. V. Boyarkin, Science, 2012, 336, 320–323 CrossRef CAS PubMed.
- T. M. Chang, J. S. Prell, E. R. Warrick and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 15805–15813 CrossRef CAS PubMed.
- M. Demireva, J. T. O'Brien and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 11216–11224 CrossRef CAS PubMed.
- T. Wende, M. Wanko, L. Jiang, G. Meijer, K. R. Asmis and A. Rubio, Angew. Chem., Int. Ed., 2011, 50, 3807–3810 CrossRef CAS PubMed.
- J. A. Silveira, K. A. Servage, C. M. Gamage and D. H. Russell, J. Phys. Chem. A, 2013, 117, 953–961 CrossRef CAS PubMed.
- K. R. Asmis, T. Wende, M. Brummer, O. Gause, G. Santambrogio, E. C. Stanca-Kaposta, J. Dobler, A. Niedziela and J. Sauer, Phys. Chem. Chem. Phys., 2012, 14, 9377–9388 RSC.
- K. Mizuse and A. Fujii, J. Phys. Chem. A, 2012, 116, 4868–4877 CrossRef CAS PubMed.
- X. B. Wang and L. S. Wang, Annu. Rev. Phys. Chem., 2009, 60, 105–126 CrossRef CAS PubMed.
- X. B. Wang, J. Yang and L. S. Wang, J. Phys. Chem. A, 2008, 112, 172–175 CrossRef CAS PubMed.
- B. Minofar, L. Vrbka, M. Mucha, P. Jungwirth, X. Yang, X. B. Wang, Y. J. Fu and L. S. Wang, J. Phys. Chem. A, 2005, 109, 5042–5049 CrossRef CAS PubMed.
- X. Yang, Y. J. Fu, X. B. Wang, P. Slavicek, M. Mucha, P. Jungwirth and L. S. Wang, J. Am. Chem. Soc., 2004, 126, 876–883 CrossRef CAS PubMed.
- B. Minofar, M. Mucha, P. Jungwirth, X. Yang, Y.-J. Fu, X.-B. Wang and L.-S. Wang, J. Am. Chem. Soc., 2004, 126, 11691–11698 CrossRef CAS PubMed.
- K. R. Asmis and D. M. Neumark, Acc. Chem. Res., 2012, 45, 43–52 CrossRef CAS PubMed.
- G. Murdachaew, M. Valiev, S. M. Kathmann and X.-B. Wang, J. Phys. Chem. A, 2012, 116, 2055–2061 CrossRef CAS PubMed.
- N. O. B. Lüttschwager, T. N. Wassermann, R. A. Mata and M. A. Suhm, Angew. Chem., Int. Ed., 2013, 52, 463–466 CrossRef PubMed.
- D. J. Goebbert, T. Wende, R. Bergmann, G. Meijer and K. R. Asmis, J. Phys. Chem. A, 2009, 113, 5874–5880 CrossRef CAS PubMed.
- D. J. Goebbert, G. Meijer and K. R. Asmis, AIP Conf. Proc., 2009, 1104, 22–29 CrossRef CAS.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- TURBOMOLE V6.1 2009, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Neese, A. Hansen, F. Wennmohs and S. Grimme, Acc. Chem. Res., 2009, 42, 641–648 CrossRef CAS PubMed.
- S. E. Cabaniss, J. A. Leenheer and I. F. McVey, Spectrochim. Acta, Part A, 1998, 54, 449–458 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the MD simulations, a detailed comparison of theoretical and experimental spectra, energetics and IR data of the minimum-energy structures, and benchmark calculations. See DOI: 10.1039/c3cp52824c |
| ‡ Current address: Physical and Theoretical Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QZ, United Kingdom. |
| § Current address: State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, P. R. China. |
| This journal is © the Owner Societies 2013 |