 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the correlation between dye coverage and photoelectrochemical performance in dye-sensitized solar cells†
Viktor
Johansson
a,
Lilian
Ellis-Gibbings
b,
Trevor
Clarke
b,
Mikhail
Gorlov
a,
Gunther G.
Andersson
*b and
Lars
Kloo
*a
aApplied Physical Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: Larsa@kth.se
bFlinders Centre for NanoScale Science and Technology, Flinders University, PO Box 2100, Adelaide, SA 5001, Australia. E-mail: Gunther.Andersson@flinders.edu.au
First published on 21st October 2013
Abstract
Concentration depth profiles of the ruthenium based dyes Z907 and N719 adsorbed onto titania are measured directly and used for determining the adsorption isotherm of the dyes. Dye layers formed by both grow in islands on the titania which do not cover the entire titania surface even at the maximum coverage. Impedance spectroscopy in conjunction with the adsorption isotherms shows that recombination losses mainly appear between the dye and the electrolyte solution. The short circuit current and the efficiency increase linearly with the dye coverage. The open circuit voltage slightly increases with increasing dye coverage which is interpreted as most likely to be a consequence of the higher charge in the particles upon higher dye loading on the TiO2 surface.
1. Introduction
From the perspective of global usage of finite fossil energy resources and the anthropogenic perturbation of the atmospheric as well as hydrospheric carbon dioxide balance alternative sources of energy are urgently required. The essentially infinite and high influx of solar irradiation makes the conversion of solar light into either electricity (solar cells) or energy-rich substances (solar fuels) the most promising alternative for future global energy systems.The dye-sensitized solar cell (DSC) was in one large step transferred from a purely academic interest to a realistic alternative for large-scale energy production by the pioneering work of Grätzel and O'Regan in 1991.1 Today, the area of DSCs is a very active field of research and early commercialization efforts with at least 50–60 papers published in refereed journals and 5–10 patents are made public every week. The vast majority of scientific articles published concern new components or new materials for DSCs, most commonly new sensitizing dyes or new electrode materials or morphologies.2 Only a small part of the published work is devoted to fundamental understanding. In terms of patent applications, researchers in China and Korea are currently the most active;3 again, the main focus is new materials and components.
A DSC is essentially a typical electrochemical cell, consisting of three components only; two electrodes and a redox-active electrolyte. The difference from other types of electrochemical cells, such as a battery, is that (at least) one of the electrodes is made photosensitive, allowing the conversion of light into energy-rich electrons, photoelectrons. This makes the photoelectrode the most essential component of the DSC, and it is therefore no surprise that the main focus in research and patent activities concerns the photoelectrode. The light absorption takes place at the photoelectrode interface, where typically a sensitizer, either an organic or organometallic dye molecule, or a quantum dot, is attached to a semiconductor substrate. The major improvement made by Grätzel and O'Regan in 1991 was to employ a nanostructured, wide bandgap semiconductor material (TiO2) as a substrate for the sensitizer. If the sensitizer and semiconductor substrate are chosen in a suitable way, the photoexcited sensitizer will efficiently inject the excited electron into the semiconductor whereafter it diffuses to the back contact and can be utilized before its return to the DSC via the second component, the counter electrode. The final and the third component, the electrolyte, then has the task of returning the electron to the oxidized sensitizer to complete the electrochemical circuit.
Extensive work is devoted to optimizing the performance of the sensitizing systems. Typically, the nanostructured photoelectrode semiconductor material is sensitized, dyed, through immersion into a dilute solution of the sensitizing dye. Using solution depletion techniques, the dye adsorption tends to follow a typical Langmuir isotherm4–6 and also deviations from that behaviour have been reported.7,8 According to presumption, the dye will then adsorb to the semiconductor surface through an anchoring group, most commonly a carboxylate group.9–13 Also, it is presumed that the dye molecules organize into a self-assembled monolayer (SAM).11,14,15 The main argument for the formation of a well-ordered SAM is the Langmuir type of adsorption together with simple injection kinetics of photoelectrons from the adsorbed and excited dye molecules. However, recent studies based on AFM and neutral impact collision ion scattering spectroscopy (NICISS) have started to question these presumptions:16,17 firstly, it is clear that the dye molecules aggregate extensively both in solution and upon adsorption. Secondly, the dye molecules so far studied do not form SAMs. Instead, they tend to form islands of multiple dye layer thickness. Thirdly, the increase in dye coverage upon increasing dye bath concentration follows a Langmuir isotherm model although the unit of growth is not a single dye molecule, but rather a dye island. A recent AFM study indicates that the dye molecule islands are typically of a size around 50 nm and even thicker than indicated by the previous NICISS studies.18 In addition, a recent study by O'Regan and co-workers indicates that the photoelectron injection process may be more complex than previously thought.19
In this work, we will study two of the most commonly used organometallic dyes, the N71920 and Z90721 dyes. The photovoltaic performance and electron kinetics will be followed as a function of the dye coverage level, and the results obtained will be correlated with results from NICISS on the dye organization at the different coverage levels.
2. Experimental section
2.1. Reagents and electrolytes
Guanidinium thiocyanate (GNCS) was purchased from Alfa Aesar. N-Methylbenzimidazole (NMBI), iodine, KI, ethanol, tert-butanol, acetonitrile and propionitrile were purchased from Aldrich. 1-Butyl-3-methylimidazolium iodide (BMII) was purchased from Alfa Aesar. The dye N-719 cis-RuL2(SCN)2 (L = 2,2′-bipyridyl-4,4′-dicarboxylic acid) was purchased from Dyesol and the dye Z907 cis-RuLL′(SCN)2 (L = 2,2′-bipyridyl-4,4′-dicarboxylic acid; L′ = 4,4′-di-nonyl-2′-bipyridyl) was bought from Solaronix. The electrolyte used in this work was composed of 0.8 M BMII, 0.3 M NMBI, 0.5 M GNCS, 0.1 M iodine, and 0.05 M KI in a solvent consisting of an 85/15 vol% mixture of acetonitrile and propionitrile.2.2. Device fabrication
The fabrication of the solar cell devices is described as follows. Fluorine-doped SnO2 (FTO) glass plates (Pilkington-TEC8) were cleaned in water and ethanol. The conducting glass substrates were immersed in 40 mM aqueous TiCl4 solution at 60 °C for 30 minutes and then washed with water and ethanol. Two layers of TiO2 paste containing ∼25 nm sized TiO2 particles were first printed on the conducting glass substrates (∼7 μm thickness), followed by one layer containing scattering particles (∼3 μm, PST-400C, JGC Catalysts and Chemicals LTD), and a final thickness of 10 ± 1 μm was obtained. The screen-printed TiO2 electrodes were gradually heated to 500 °C in an oven (Nabertherm Controller P320) in an air atmosphere for 30 min. After sintering the electrodes received a TiCl4 treatment as described above, and a second and final sintering at 500 °C for 30 min was performed. The prepared TiO2 electrodes were heated again at 300 °C for 10 min before use. After cooling to 80 °C, the TiO2 electrodes were immersed in the solutions of N719 and Z907, respectively, for 24 h rendering the desired dye surface coverage.After washing with ethanol and drying under air flow, the sensitized titania electrodes were assembled with thermally platinized counter electrodes (CEs) using a 25 μm thin thermoplastic frame (Surlyn). The electrolyte was introduced under vacuum through a prefabricated drilled hole in the CEs. The hole was sealed afterwards with a 50 μm thermoplastic frame and a glass plate. A minimum of three cells per dye surface coverage was prepared and investigated. The active area was obtained through masking giving 0.25 cm2.
2.3. Current vs. potential characteristics
A solar simulator (Newport 91160) was used to give an irradiation of 100 mW cm−2 [the equivalent of 1 sun at air mass (AM) 1.5G] at the surface of solar cells. The overall light-to-electricity conversion efficiency (η), fill factor (FF), open-circuit voltage (Voc), and short-circuit current density (Jsc) were obtained through the current–voltage characteristics of a solar cell at room temperature. These characteristics and I–V diagrams were monitored and recorded using a computerized Keithley model 2400 source unit.2.4. Photoelectrochemical measurements
Electron lifetime, transport time and accumulation for the solar cells were determined using a white-light-emitting diode (Luxeon Star 1 W) as the light source.22 Voltage and current traces were recorded using a 16-bit resolution data acquisition board (DAQ National Instruments) in combination with a current amplifier (Stanford Research SR570). Lifetimes were determined by monitoring photovoltage transient at different light intensities applying a small square-wave modulation (2 Hz modulation frequency, 10 mV modulation amplitude, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz sampling frequency) to the baseline intensity. First-order kinetic models were used to extract the time constants.
000 Hz sampling frequency) to the baseline intensity. First-order kinetic models were used to extract the time constants.
2.5. Electrochemical impedance measurements
Measurements were performed using a Autolab PGstat12 potentiostat with an impedance module. Impedance measurements were done in the frequency range of 10 kHz to 10 mHz, using 20 mV AC amplitude, with illumination provided by a 5 W white Luxeon LED. Impedance was measured under dark and light conditions. The cell was illuminated with a set intensity and impedance was measured under open-circuit conditions. In the dark a bias potential was applied. The response was analyzed by the equivalent circuit models previously described by Bisquert and Fabregat.23 The recombination resistance (Rrec) was simply extracted from Nyquist plots of the DSCs. The data used in the figures for electrochemical impedance measurements were obtained from cells identical to those used for photoelectrochemical measurements.2.6. Solution depletion experiments
A colloidal paste of TiO2 nanoparticles (Dyesol, DSL 18NR-T) was screen-printed onto glass slides and sintered at 500 °C for 1 h to give nanoporous TiO2 films with a surface area of 0.25 cm2 and a consistent average thickness of 10 μm (measured using a profilometer).N719 dye was purchased from Dyesol and used as received to make solutions of various concentrations in ethanol. Each solution was separated into two sample vials, into one a TiO2 film substrate was immersed, and the other was kept as reference. After 24 h the amount of adsorbed dye was determined by comparing the difference in absorbance at the 530 nm peak using the linear absorption coefficient (1.36 × 104 M−1 cm−1) obtained from the reference solutions of known concentration. The absorbance of the solutions was recorded using a Cary 300 Bio UV-vis spectrophotometer.
Z907 was purchased from Solaronix and used as received to make solutions of various concentrations in a 1:1 mixture by initial volume of tert-butanol–acetonitrile. It can be noted here that the adsorption isotherm for N719 in the same solvent mixture is highly similar to that obtained in ethanol. The same procedure as previously described for the N719 system was used with the difference that a peak at 290 nm was used for the measurements based on a linear absorption coefficient of 3.80 × 104 M−1 cm−1.
2.7. Neutral impact collision ion scattering spectroscopy (NICISS)
The details of the NICISS technique are described in ref. 24. A brief summary is given here. NICISS is an established technique for determining the concentration depth profiles of the elements at soft matter surfaces. The probing depth is about 20 nm and the depth resolution is a few Å in the near-surface area.24 In a NICISS experiment the target is bombarded with a pulsed beam of inert gas ions with a kinetic energy of several keV.24 Mostly helium ions are used as projectiles. The energy of the neutral projectiles backscattered from the atoms forming the target is determined by their time-of-flight (TOF) from the target to the detector. The projectiles lose energy during the backscattering process. This energy loss depends on the mass of the target atom and is used to identify the element from which a projectile is backscattered. A second type of energy loss appears on the trajectory of the projectile through the bulk due to small-angle scattering and electronic excitations of the molecules constituting the target (stopping power).25 This second type of energy loss is used to determine the depth of the atom from which a projectile is backscattered. In combination, these two types of energy losses are used to determine the concentration depth profiles of the specific elements. Because helium is lighter than hydrogen, helium cannot be backscattered from hydrogen, and no energy loss profiles of hydrogen appear in NICIS spectra. Instead a broad background of sputtered hydrogen is present. The data evaluation procedure for obtaining the concentration depth profiles is described in detail in ref. 26. NICISS has been used before to analyze dye/electrode interfaces in DSCs.16 From the NICISS measurements the concentration depth profiles of the dye can be derived through the ruthenium signal.2.8. Sample preparation for NICISS experiments
Mesoporous layers of TiO2 were prepared using the DSL 18NR-T paste from Dyesol. The paste was used as received (batch 278) containing 19.4% TiO2 loading with particle size averaging at 20 nm. These titania nanoparticles are prevalently of anatase structure.27 The surfaces were prepared using the doctor blade method on a clean indium tin oxide (ITO) coated glass substrate. The thickness of the layer is determined by the thickness of the tape frame used. Subsequently, the ITO/titania substrate was heated in an oven for 30 minutes at 450 ± 20 °C. The temperature was increased and decreased slowly to avoid cracking of the titania layer. The paste contains organic fillers, an organic plasticiser, and terpineol as solvent. These are burnt off during the sintering process and help to control the porosity of the layer.28The ITO/titania substrates were heated to 80 °C for 15 minutes prior to immersion into the dye solution in order to remove surface contaminants. Z907 solutions were made by dissolving the appropriate mass of dye in a 1:1 acetonitrile–tert-butanol mixture followed by consecutive dilutions. The samples were dyed for 24 hours in approximately 4 ml solution and rinsed thoroughly with ethanol prior to analysis. Samples were kept in sealed containers protected from light. The typical sample size was approximately 1.5 cm2.
The dye adsorption was investigated using NICISS for concentrations ranging from 0.0015 mmol L−1 to 0.045 mmol L−1. The latter concentration is the maximum solubility of Z907 in a 1:1 acetonitrile–tert-butanol mixture. The concentrations shown are the initial solution concentrations. For low concentrations, the concentration of the dye solution changes significantly during the adsorption process and the effect associated with this will be discussed later on.
3. Results and discussion
3.1. Photoelectrochemical characterization
Working electrodes with dye surface coverage levels of 5%, 25%, 50% and 100% were manufactured for both the N719 and the Z907 dye and assembled into fully operating dye-sensitized solar cells. The coverage is estimated from solution depletion measurements. Apart from the cell with the highest loading, all cells have to be considered as being inhomogeneously loaded with dye across the thickness of the titania layer (higher dye coverage at the top and lower the further down). Jena et al. found that a soaking time of 50 minutes is sufficient for the diffusion of the dye similar to those in the present work up to a depth to which light can penetrate into a dye loaded titania layer.29,30 Dürr et al. have shown that Fick diffusion describes the diffusion of dyes into porous titania layers.31 Based on this knowledge only the sample with the highest loading can be considered as being homogenously loaded up to the depth relevant for a cell under illumination. In this context, it should be carefully highlighted that the percentages given refer to the maximum coverage level, and that they do not presume that the surface is fully covered by the dye at the 100% coverage level (see discussion below). The total coverage of the surface with the dye was determined with NICISS and will be described below. Apart from the difference in dye and dye coverage, all cells contain the same components and have the same composition. Any difference in photoelectrochemical performance should therefore be attributable to the difference in dye coverage only.The photovoltaic performance of the DSCs is given in Table 1 and Fig. 1. Looking at the I–V data a few conclusions can be drawn. Firstly, the cells based on N719 and Z907 behave conceptually the same way. Fill factors (ff) are fairly high (about 0.7) and remain constant with a tendency to lower values at 100% coverage. Given the fact that the titania layer is inhomogenously covered with the dye, the performance of the samples with less than 100% coverage has to be considered as an average across the varying coverage of the titania film. The productive processes of electron conduction in the cell under irradiation thus appear to be similar for similar dye surface coverage levels.
| Dye | Coverage [% of max. coverage] | η [%] | V oc [V] | I sc [mA cm−2] | ff [−] |
|---|---|---|---|---|---|
| N719 | 5 | 2.1 ± 0.3 | 0.62 ± 0.01 | 4.9 ± 0.4 | 0.70 ± 0.02 |
| 25 | 3.0 ± 0.3 | 0.63 ± 0.01 | 7.0 ± 0.4 | 0.70 ± 0.02 | |
| 50 | 4.1 ± 0.4 | 0.66 ± 0.01 | 9.0 ± 0.6 | 0.69 ± 0.02 | |
| 100 | 7.4 ± 0.1 | 0.72 ± 0.01 | 16.0 ± 0.2 | 0.65 ± 0.01 | |
| Z907 | 5 | 1.4 ± 0.3 | 0.58 ± 0.03 | 3.3 ± 0.7 | 0.72 ± 0.01 |
| 25 | 3.2 ± 0.2 | 0.62 ± 0.01 | 7.2 ± 0.5 | 0.72 ± 0.01 | |
| 50 | 4.4 ± 0.5 | 0.65 ± 0.01 | 10.0 ± 0.6 | 0.66 ± 0.05 | |
| 100 | 7.6 ± 0.2 | 0.69 ± 0.01 | 15.7 ± 0.5 | 0.69 ± 0.01 | |
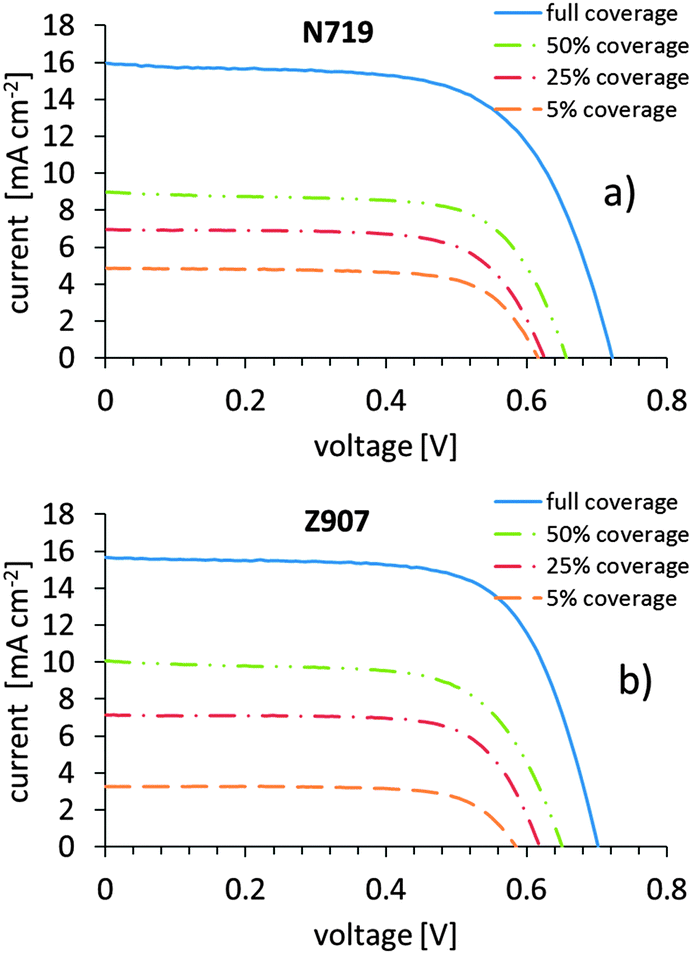 | ||
| Fig. 1 The average I–V-curves obtained for different dye surface coverages of the (a) N719 dye and (b) the Z907 dye. | ||
There is on the other side a distinct increase in open-circuit voltage (Voc) with increasing dye surface coverage. Presuming the electrolyte redox potential to be constant, this is often taken as an indication that the pseudo-Fermi level is higher at higher coverage, and that may simply be a consequence of the higher charge in the particles upon higher dye loading on the TiO2 surface. Sometimes, higher Voc values are taken as an indication of lower recombination losses from the dyed titania film to the electrolyte. However, as we will soon experience from electrochemical impedance spectroscopy (EIS), such an interpretation is not obvious.
The current increases with increasing dye surface coverage, as expected. A higher dye loading is expected to yield more injected photoelectrons as the photo current (Isc) is higher. The dependence of the current as well as the open circuit voltage, the fill factor and the efficiency on dye surface coverage will be discussed below. In this context it should be noted that cells with 0% coverage (bare titania) give low voltages of about 0.3 V and very low currents and consequently marginal conversion efficiencies. The non-sensitized semiconductor is thus a very bad photoelectrode.
EIS data for the corresponding DSCs are given in Fig. 2 as Nyquist plots. Typically, semicircles from three different processes can be extracted from such data corresponding to charge-transfer processes taking place at characteristic ac frequencies in the range studied here. Detailed analyses can yield electrolyte diffusion resistance (at low ac frequencies), conducting glass substrate sheet resistance and counter electrode charge-transfer resistance (at high ac frequencies), all of which are important for the overall DSC performance. All of these are expected to be analogous with the DSCs studied, and all results indicate that so is the case. Therefore, these processes are not further analyzed nor discussed. However, a fourth process can typically be monitored in EIS of DSCs, the loss of photoelectrons from the dyed titania photoelectrode to the oxidized component of the electrolyte redox system. This process normally appears as a rather large semicircle in the middle of an EIS Nyquist plot.
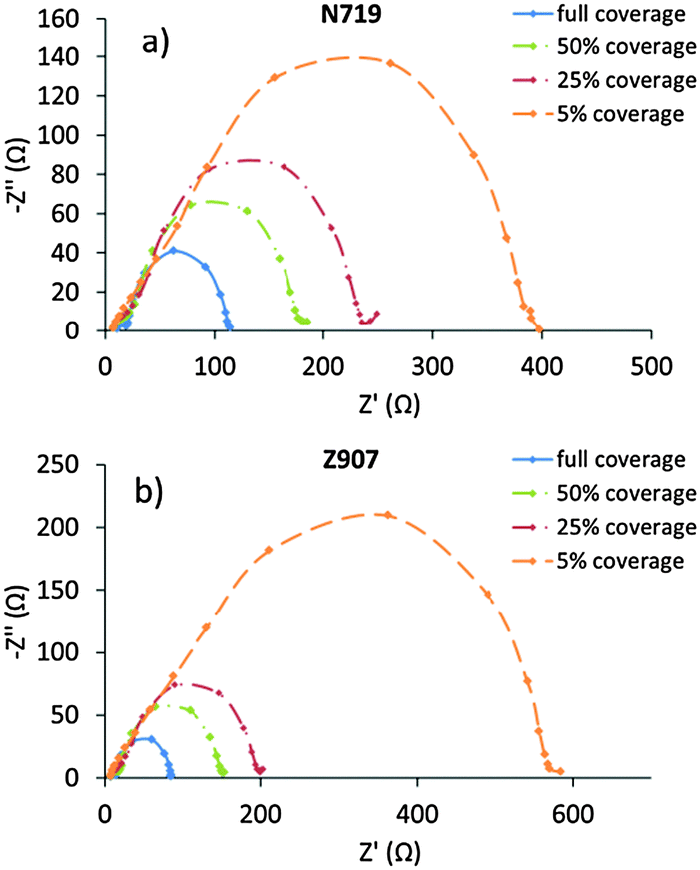 | ||
| Fig. 2 The EIS Nyquist plots at different dye surface coverages of DSCs containing the (a) N719 dye and (b) the Z907 dye. | ||
Again, the EIS data of DSCs based on N719 and Z907 behave in an analogous way. It is notable here that the recombination resistance (Rrec) increases as the dye surface coverage decreases. The direct interpretation of these results is that recombination losses increase as the dye loading increases, and, as a consequence, the major loss pathway goes through the dye rather than from the ‘bare’ titania surface; at least in the presence of an electrolyte additive such as NMBI. However, since the dye coverage is changed, also the amount of protons released upon adsorption differs between different coverage levels. Although the electrolytes contain classical bases, such as the NMBI, part of the protons are expected to adsorb onto the titania surface and may thus also affect its capacitance. A crude estimate of the difference between 5% and 100% of maximum coverage suggests that the band shifts could amount to several hundreds of millivolts. At the same time, of course the anionic character of the adsorbed dye molecules counteracts this effect. In conclusion, we cannot avoid noting that the change in photovoltage with coverage levels is in the range of 100 mV for both sensitizing dyes from low to high coverage levels, and this suggests that the simple interpretation of the recombination resistance above is somewhat dubious. Lifetimes estimated from frequencies of the recombination semicircles are very similar and amount to about (0.2 ± 0.1) s for all systems studied. These values are in agreement with the lifetime estimates derived from other methods at lower light intensities (vide infra). It is notable here that the electrolyte used in all DSCs contains the acclaimed surface-blocking additive NMBI. The exact effect of the NMBI is not known, and it may be direct, in terms of surface blocking of recombination losses, or it may be indirect, for instance shifting the acid–base equilibria in the electrolyte. Nevertheless, control experiments with analogous DSCs without NMBI give substantially lower efficiencies mainly attributable to recombination losses. The Rrec for both dyes at 100% of maximum dye surface coverage is about 100 Ω (in the presence of NMBI in the electrolyte), and at lower coverage the resistance is just under 400 Ω for N719 and 600 Ω for Z907.
The photoelectrochemical measurements offer some insights into the electron dynamics of the DSCs under illumination. Electron lifetimes are plotted against light intensity in Fig. 3. However, such data are analogous to lifetimes vs. photocurrent, and offer information on how electron density in the titania particles affects the lifetime. It is notable that differences between different dye surface coverage levels are small. Possibly, one can acclaim reverse behavior for DSCs containing the two dyes; slightly longer electron lifetimes at higher dye surface coverage for DSCs containing N719 and vice versa for DSCs containing Z907. However, differences are small, and it may be equally adequate to claim essentially no difference between either of the dye surface coverages nor between the two sensitizing dyes. Also transport times vs. light intensity (Fig. S1, ESI†) and accumulated charge vs. open-circuit voltage (Fig. S2, ESI†) show no significant difference between dye coverage levels nor between sensitizing dyes.
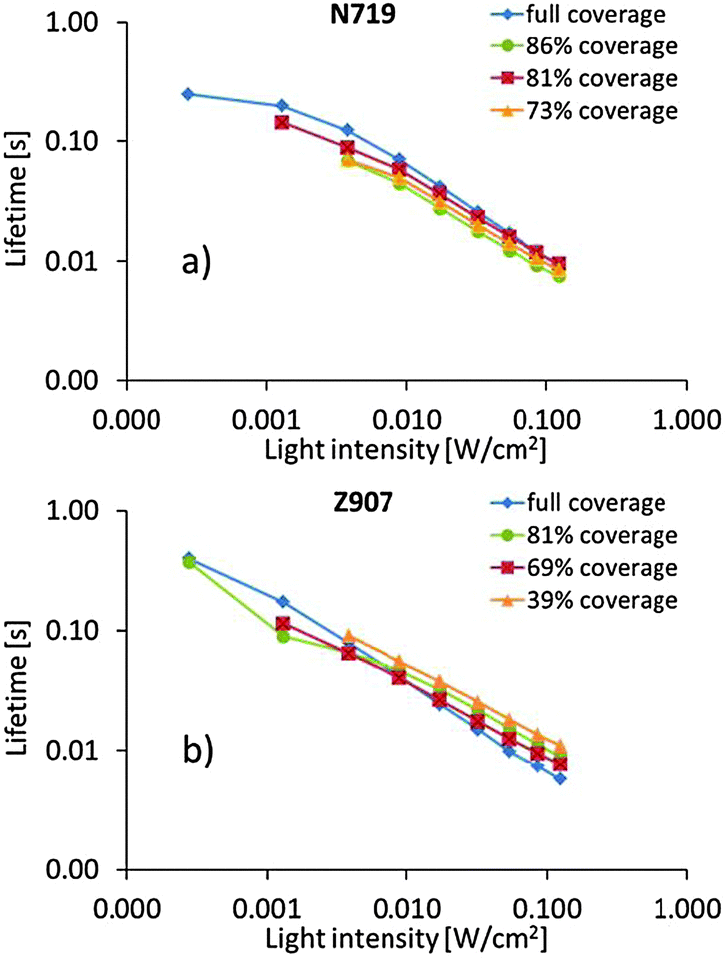 | ||
| Fig. 3 Plots of electron lifetime vs. light intensity of DSCs containing the (a) N719 dye and (b) the Z907 dye. | ||
3.2. Concentration depth profiles and adsorption isotherms
In Fig. 4, the concentration depth profiles of Z907 on porous titania are shown. The concentration depth profiles are deconvoluted and corrected for the spherical shape of the particles forming the substrate as described in detail in ref. 16. We describe here briefly the main steps of this procedure. The measured concentration depth profiles are corrected for the shape of the particles forming the substrate because the path length of the trajectory of the helium projectiles through the dye layer adsorbed on the titania depends on where the projectile hits the titania particle. If the projectile hits the sample at the top of a titania particle, the path length of the projectile through the dye layer is the same as the thickness of the dye layer as illustrated in ref. 16. However, if the projectile hits the sample not at the top of the titania particle, the path length is longer than the thickness of the dye layer as illustrated in ref. 16. A measured spectrum will consist of an average of signals caused by projectiles having hit the titania sphere at various positions relative to the center. The influence on the spectrum does not depend on the size of the particles but only on the shape of the particles which can be assumed to be spherical. The size of the particle does not play a role as the time of flight distance does not change: the size of a particle is about 8 orders of magnitude smaller than the time of flight length. Subsequently the spectrum corrected for the shape of the titania particles is deconvoluted for the energy resolution of the spectrum. It must be emphasized that in this procedure of evaluating the NICISS measurement the roughness of the surface is taken into account.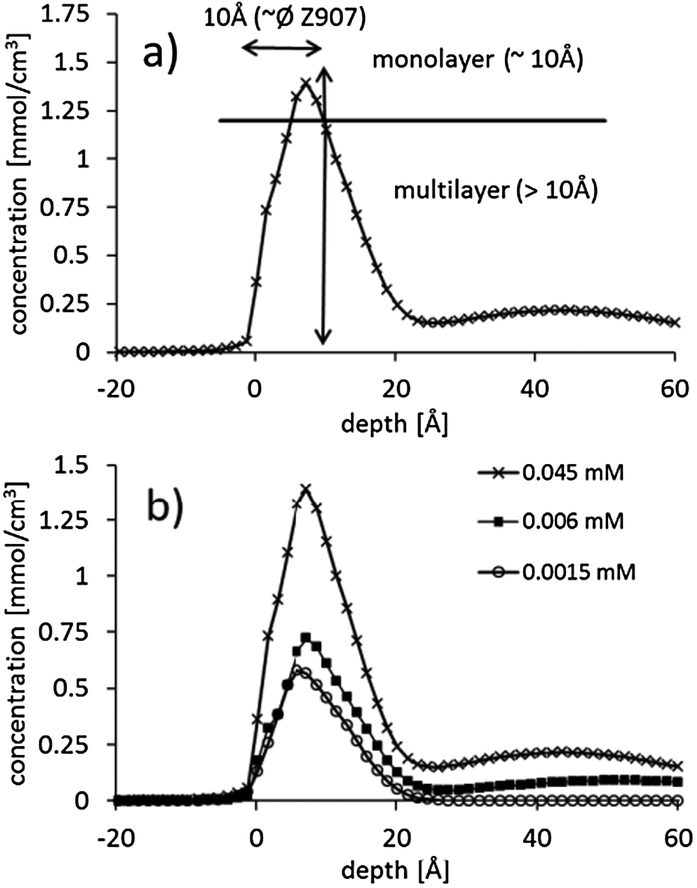 | ||
| Fig. 4 Concentration depth profiles determined from the NICIS spectra: (a) 0.045 mM, (b) 0.006 mM and (c) 0.0015 mM. | ||
The concentration depth profiles are measured only at the surface of the titania electrode. However, because the entire electrode is formed by the same titania material, the adsorption of the dye onto the titania surface can be considered as representing the adsorption onto the titania through the entire electrode.
By integrating the concentration depth profiles corrected for the spherical shape of the particles the total amount of dye adsorbed on the surface is determined. In Fig. 5, the adsorption on samples fabricated with various dye concentrations is shown as the adsorption isotherm. The maximum concentration of the dye solution investigated was 0.045 mM as this is the maximum solubility of Z907 in the tert-butanol–acetonitrile mixture. The maximum surface coverage is (2.6 ± 0.3) × 10−10 mol cm−2 and is slightly higher than that found on N719 on nanoporous titania.16 It has to be noted here the surface coverage means coverage of the titania surface and not of the entire electrode. The only difference is that the maximum coverage of Z907 is reached at a lower concentration compared with N719. For the depletion adsorption isotherm the maximum was gauged with the help of the adsorption isotherm measured with NICISS. The highest concentration of the solution depletion isotherm is 0.04 mM. At this concentration the titania surface in the porous titania layer is homogeneously covered and the coverage determined with NICISS corresponds to the coverage of the titania surface throughout the titania layer. The depletion adsorption isotherm shows a different slope than the NICISS adsorption isotherm. The reason for the discrepancy is most likely that the titania layer is not homogenously soaked with dye due to the decreasing dye concentration. The uptake of the dye from the solution is thus not only determined by the decrease in adsorption of the titania when decreasing the concentration of the dye solution, but also by the decrease in the rate of diffusion into the titania layer. This effect is also the likely reason for the discrepancy between the isotherms determined by the solution depletion method and NICISS, as found in ref. 16.
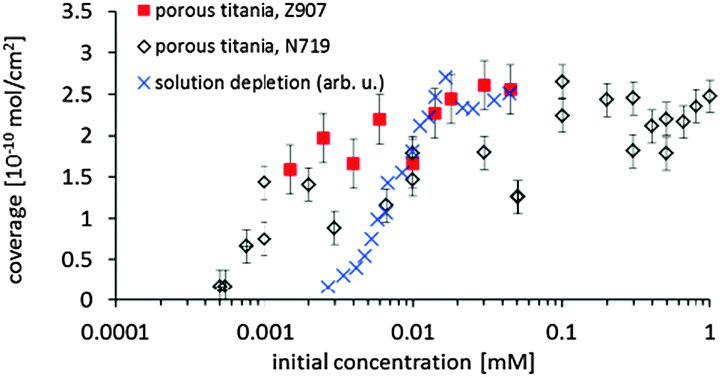 | ||
| Fig. 5 Adsorption isotherm of Z907 and N719 on porous titania. The values for the solution depletion method are shown in arbitrary units and scaled such that the values at the maximum adsorption overlap with those measured with NICISS. | ||
In the single concentration depth profiles shown in Fig. 4 it can be seen that the dye does adsorb in multilayers. In the case that the dye would adsorb as a monolayer, the total width of the profile should be not more than 10 Å, i.e. the average diameter of the Z907 molecule, because the position of the ruthenium within the dye layer would have some distribution but centered somewhere in the middle of the layer. The total width of the main contribution of the profile is about 20 Å which is about twice of that of a monolayer. This finding is similar to that found for N719.16 However, the shape of the Z907 concentration depth profiles is slightly different to that of N719. The Z907 profiles can rather be interpreted as double layers, a structure similar to that suggested by Ni et al.,18 while the profiles of N719 show a more gradual change from the maximum concentration to the uncovered substrate. The shape of the concentration depth profiles does not change with the Z907 coverage, which allows for the conclusion that the Z907 grows in islands, similarly to the N719.
The meaning of maximum (100%) coverage needs to be discussed. Our interpretation is in percentage of maximum coverage. The maximum coverage may suggest that the titania surface is fully covered. However, that is most likely not the case. Taking into account the surface concentration obtained from the NICISS study (N719: 2.2 × 10−10 mol cm−2 corresponding to 1.4 × 1014 molecules per cm2; Z907: 2.6 × 10−10 mol cm−2 corresponding to 1.6 × 1014 molecules per cm2), together with the typical area of 1 nm2 covered by a single dye molecule, we obtain 1.3 monolayers on average for N719 and 1.6 monolayers on average for Z907. The average thickness of the dye layer can be obtained from comparing the amount of dye in each of the sections with the amount of the dye in the interval 0 to 10 Å by calculating
 | (1) |
In Fig. 6, the short-circuit current, open-circuit voltage, efficiency and fill factor for various coverage levels of N719 and Z907 on nanoporous titania are shown. It can be seen that the fill factor stays rather constant with increasing dye coverage. The open-circuit voltage increases slightly, which is most likely due to the change in the pseudo-Fermi level of the bare titania as compared to the titania surface covered with the dye. The open-circuit voltages and in particular the short-circuit current increase linearly with dye coverage. Given the growth of the dye layer a co-operative effect could be expected. With increasing dye coverage the number of pathways for transport of the electrons from the dye to the electrolyte (recombination losses) decreases, as the contact area between the dye islands and the electrolyte solution decreases in favour of the contact area between the dye islands. Thus, instead of losing the electron to the electrolyte the electron moves via another dye molecule to the titania. Such a co-operative effect is not seen in Fig. 6a and b. However, it must be noted that the actual coverage of the titania surface inside the dye layer is inhomogeneous with respect to the depth and might blur such an effect in graphs, such as those in Fig. 6.
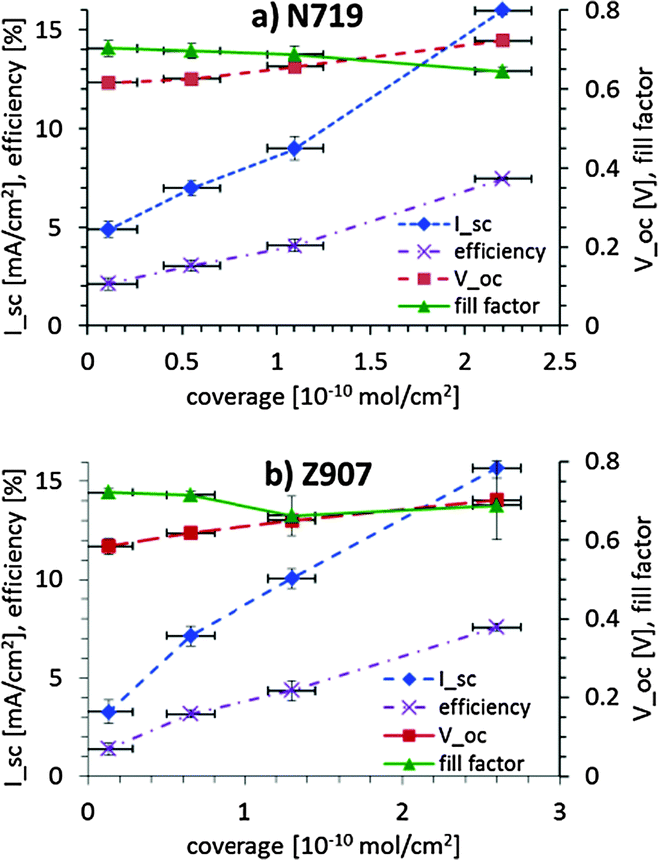 | ||
| Fig. 6 Short-circuit current, open-circuit voltage, efficiency and fill factor for various coverage levels of (a) N719 and (b) Z907 on nanoporous titania. | ||
4. Conclusions
In this work, it is noted that DSCs containing the two different sensitizing organometallic dyes N719 and Z907 display very similar photovoltaic performance in spite of the fact that they exhibit rather different organization patterns. N719 forms islands of multilayer thickness already from low coverage levels, whereas Z907 forms islands with a bilayer formation (also from low coverage levels).From the impedance spectra it was found that recombination losses increase with increasing dye coverage, indicating that the dye molecules represent a major recombination pathway. However, it is also clear that electrolyte additives, such as NMBI, play a very important role in preventing direct and probably even more efficient recombination directly from the photoelectrode surface.
The current, as well as the efficiency, increase linearly while the open-circuit voltage slightly increases and the fill factor slightly decreases. The increase in current with the coverage of titania with dye molecules means that the performance of the DSCs follows the number of dye molecules adsorbed irrespective of the dye organization. The increase in open-circuit voltage with increasing dye coverage is attributed to the fact that the pseudo-Fermi level is higher at higher coverage, which may simply be a consequence of the higher charge in the particles upon higher dye load on the TiO2 surface. Also, the surface concentration of cations, such as protons, may affect the conduction band energy.
Finally, from the formation of islands, their shape and thickness as well as the maximum coverage it can be concluded that the titania surface is incompletely covered with dye molecules. This means that even at the highest coverage a fraction of the titania surface is exposed to the electrolyte unless this fraction of the titania surface is covered with a co-adsorbate.
Acknowledgements
We acknowledge the Swedish National Research Council, Swedish Energy Agency, the Knut and Alice Wallenberg Foundation as well as Flinders University for financial support.Notes and references
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–739 CrossRef CAS.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- H. Pettersson, K. Nonomura, L. Kloo and A. Hagfeldt, Energy Environ. Sci., 2012, 5, 7376–7380 CAS.
- A. Fattori, L. Peter, K. McCall, N. Robertson and F. Marken, J. Solid State Electrochem., 2010, 14, 1929–1936 CrossRef CAS PubMed.
- R. Katoh, K. Yaguchi and A. Furube, Chem. Phys. Lett., 2011, 511, 336–339 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1916, 38, 2221–2295 CrossRef CAS.
- K. Kilså, E. I. Mayo, B. S. Brunschwig, H. B. Gray, N. S. Lewis and J. R. Winkler, J. Phys. Chem. B, 2004, 108, 15640–15651 CrossRef.
- S. Ardo, Y. Sun, A. Staniszewski, F. N. Castellano and G. J. Meyer, J. Am. Chem. Soc., 2010, 132, 6696–6709 CrossRef CAS PubMed.
- F. De Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Graetzel, J. Phys. Chem. C, 2010, 114, 6054–6061 CAS.
- K. E. Lee, M. A. Gomez, T. Regier, Y. Hu and G. P. Demopoulos, J. Phys. Chem. C, 2011, 115, 5692–5707 CAS.
- J. Lim, Y. S. Kwon, S.-H. Park, I. Y. Song, J. Choi and T. Park, Langmuir, 2011, 27, 14647–14653 CrossRef CAS PubMed.
- F. Risplendi, G. Cicero, G. Mallia and N. M. Harrison, Phys. Chem. Chem. Phys., 2013, 15, 235–243 RSC.
- V. Shklover, M. K. Nazeeruddin, S. M. Zakeeruddin, C. Barbé, A. Kay, T. Haibach, W. Steurer, R. Hermann, H. U. Nissen and M. Grätzel, Chem. Mater., 1997, 9, 430–439 CrossRef.
- P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef PubMed.
- D. Robert and J. V. Weber, Adsorption, 2000, 6, 175–178 CrossRef CAS.
- L. Ellis-Gibbings, V. Johansson, R. B. Walsh, L. Kloo, J. S. Quinton and G. G. Andersson, Langmuir, 2012, 28, 9431–9439 CrossRef CAS PubMed.
- P. Marquet, G. Andersson, A. Snedden, L. Kloo and R. Atkin, Langmuir, 2010, 26, 9612–9616 CrossRef CAS PubMed.
- J.-S. Ni, C.-Y. Hung, K.-Y. Liu, Y.-H. Chang, K.-C. Ho and K.-F. Lin, J. Colloid Interface Sci., 2012, 386, 359–365 CrossRef CAS PubMed.
- M. Juozapavicius, M. Kaucikas, J. J. van Thor and B. C. O'Regan, J. Phys. Chem. C, 2013, 117, 116–123 CAS.
- K. Kalyanasundaram and M. K. Nazeeruddin, Chem. Phys. Lett., 1992, 193, 292–297 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, I. Exnar and M. Gratzel, Chem. Commun., 2002, 2972–2973 RSC.
- G. Boschloo, L. Häggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS PubMed.
- F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117–131 CrossRef CAS PubMed.
- G. Andersson and H. Morgner, Surf. Sci., 1998, 405, 138–151 CrossRef CAS.
- G. Andersson, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 75, 032901 CrossRef.
- G. Andersson, T. Krebs and H. Morgner, Phys. Chem. Chem. Phys., 2005, 7, 136–142 RSC.
- A. Zaban, S. T. Aruna, S. Tirosh, B. A. Gregg and Y. Mastai, J. Phys. Chem. B, 2000, 104, 4130–4133 CrossRef CAS.
- M. Grätzel and J. R. Durrant, in Nanostructured and Photoelectrochemical Systems for Solar Photon Conversion, ed. M. D. Archer and A. J. Nozik, Imperial College Press, 2007, pp. 503–536 Search PubMed.
- A. K. Jena and P. Bhargava, RSC Adv., 2013, 3, 2655–2661 RSC.
- B. O'Regan, L. Xiaoe and T. Ghaddar, Energy Environ. Sci., 2012, 5, 7203–7215 CAS.
- M. Dürr, A. Schmid, M. Obermaier, A. Yasuda and G. Nelles, J. Phys. Chem. A, 2005, 109, 3967–3970 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp52486h |
| This journal is © the Owner Societies 2014 |