 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CH2OO Criegee biradical yields following photolysis of CH2I2 in O2†
Daniel Stonea, Mark Blitz*ab, Laura Daubneya, Trevor Inghamab and Paul Seakinsab
aSchool of Chemistry, University of Leeds, UK. E-mail: m.blitz@leeds.ac.uk
bNational Centre for Atmospheric Science, University of Leeds, UK
First published on 12th September 2013
Abstract
Yields of CH2OO and CH2IO2 from the reaction of CH2I radicals with O2 are reported as a function of total pressure, [N2] and [O2] at T = 295 K using three complementary methods. Results from the three methods are similar, with no observed additional dependence on [O2]. The CH2I + O2 reaction has a yield of ∼18% CH2OO at atmospheric pressure.
Criegee biradicals (CR2OO) are key reaction intermediates in the ozonolysis of unsaturated organic compounds,1 and their involvement in the atmospheric oxidation of alkenes has long been postulated.2,3 Despite much effort, direct observations of Criegee biradicals have only recently been reported for CH2OO4–8 and CH3CHOO.9 Photolysis of CH2I2 in the presence of O2 has been shown to produce CH2OO at low pressures through the reactions:5,10
| CH2I2 + hν → CH2I + I | (R1) |
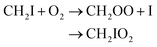 | (R2) |
| CH2I + O2 → CH2IOO# |
| CH2IOO# → CH2OO + I | (R2a) |
| CH2IOO# + M → CH2IO2 + M | (R2b) |
![Chemical activation scheme to describe the reaction between CH2I and O2(R2), where the initially formed excited species CH2IO2# either proceeds to produce CH2OO + I (k2a) or is collisionally stabilised to produce the CH2IO2 peroxy radical or (k2b[M]).](/image/article/2013/CP/c3cp52466c/c3cp52466c-s1.gif) | ||
| Scheme 1 Chemical activation scheme to describe the reaction between CH2I and O2(R2), where the initially formed excited species CH2IO2# either proceeds to produce CH2OO + I (k2a) or is collisionally stabilised to produce the CH2IO2 peroxy radical or (k2b[M]). | ||
The yields of CH2OO and CH2IO2 from CH2I + O2 were recently measured by Huang et al.10 as a function of [N2], [O2] and [He] by monitoring the I atoms produced in (R1) and (R2a)via their infrared absorption owing to F′′ = 4 → F′ = 3 of the 2P3/2 → 2P1/2 spin–orbit transition at 7603.138 cm−1. Given the stoichiometry between CH2I and I in (R1), it is possible to infer the fraction of CH2I radicals producing CH2OO through comparison of the I atom yields from the instant photolytic production in (R1) and the slower production via(R2a). While there is potential for multi-photon dissociation of CH2I2 to produce CH2 + 2I, it is expected that this is relatively minor compared to production of CH2I + I.16–19 Huang et al. showed that the yield of CH2OO decreases with total pressure, consistent with collisional stabilisation of the CH2IOO# intermediate to CH2IO2.
However, Huang et al. also reported significant differences in the I atom yields from (R2a) (and thus in CH2OO yields) between experiments performed in N2 buffer gas and those performed in O2, indicating a much greater efficiency of O2 for stabilisation of CH2IOO# to CH2IO2 compared to N2, and an unusual interaction between CH2IOO# and O2.
In this work we report observations of the yields of CH2OO and CH2IO2 from CH2I + O2 following laser flash photolysis of CH2I2–N2–O2 gas mixtures as a function of [N2], [O2] and total pressure using several complementary methods at total pressures between 25 and 450 Torr. Experiments were initially performed to monitor I atom fluorescence, thus enabling inference of the yields of CH2OO and CH2IO2 in the manner described by Huang et al.10 Subsequent experiments monitored the yields of HCHO from reactions of CH2OO–CH2IO2 in the presence of excess SO2 or NO by laser-induced fluorescence (LIF) of HCHO at λ ∼ 353.1 nm. Full experimental details are given in the ESI.† All experiments were performed at T = 295 K unless stated otherwise.
Fig. 1 shows the typical I atom signal following photolysis of CH2I2–O2–N2. The instantaneous photolytic production of iodine atoms through (R1) can be clearly distinguished from the subsequent growth in (R2a). The I atom signals were analysed using eqn (1):
 | (1) |
![Iodine atom signal following photolysis of CH2I2 in the presence of O2. For this plot total pressure (N/V) = 3.27 × 1017 cm−3 (∼10 Torr, predominantly N2); [O2] = 4.02 × 1015 cm−3; [CH2I2] = 5.03 × 1012 cm−3. The time resolution is such that iodine production from photolysis and reaction can be identified. The fit to eqn (1) is shown by the solid line.](/image/article/2013/CP/c3cp52466c/c3cp52466c-f1.gif) | ||
| Fig. 1 Iodine atom signal following photolysis of CH2I2 in the presence of O2. For this plot total pressure (N/V) = 3.27 × 1017 cm−3 (∼10 Torr, predominantly N2); [O2] = 4.02 × 1015 cm−3; [CH2I2] = 5.03 × 1012 cm−3. The time resolution is such that iodine production from photolysis and reaction can be identified. The fit to eqn (1) is shown by the solid line. | ||
The absolute iodine atom yield from reaction (R2a) is given by the ratio S1/S0, and was observed to decrease with increasing total pressure of N2, consistent with production of the CH2OO Criegee biradical at low pressures and stabilisation of the chemically activated CH2IO2# species to the CH2IO2 peroxy radical at higher pressures. Solution of the I atom yield from (R2) (ΦI), and thus the CH2OO yield, is given by the Stern–Volmer relationship in eqn (2):
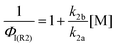 | (2) |
Fig. 2 shows the Stern–Volmer plot for reaction (R2). The intercept of the iodine atom Stern–Volmer plot is 1.08 ± 0.12, consistent with channel 2a being the dominant bimolecular process. The slope of the Stern–Volmer plot gives the Stern–Volmer quenching coefficient (k2b/k2a), and is equal to (2.28 ± 0.11) × 10−19 cm3 for these experiments, similar to the value of k2b/k2a = (3.1 ± 0.2) × 10−19 cm3 reported by Huang et al.10 for experiments in N2 buffer gas. For stabilisation of CH2IO2# by O2, Huang et al. report a value of k2b/k2a = (4.09 ± 0.32) × 10−18 cm3. The iodine atom experiments in this work were conducted at low [O2] (∼4 × 1015 cm−3) to ensure (R2a) was sufficiently slow to provide confidence in the resolution of the photolytic I atom production from the chemical I atom production. The effects of CH2IO2# stabilisation by O2 were thus investigated in the HCHO yield experiments.
![Stern–Volmer analyses for CH2OO yields from CH2I + O2 as a function of total pressure. Main panel shows results from this work monitoring iodine atom production (squares; intercept = 1.08 ± 0.12; slope = (2.28 ± 0.11) × 10−19 cm3), and HCHO production in the presence of SO2 (triangles; intercept = 1.46 ± 0.25; slope = (0.95 ± 0.24) × 10−19 cm3) and NO (circles; intercept = 1.41 ± 0.30; slope = (1.33 ± 0.31) × 10−19 cm3). Constraining the intercepts to unity for fits to SO2 and NO data gives slopes of (1.37 ± 0.10) × 10−19 cm3 and (1.71 ± 0.16) × 10−19 cm3, respectively. Data shown for SO2 and NO were taken over a range of [O2] ((0.1–7.8) × 1018 cm−3). A fit to all data reported in this work gives an intercept of 1.10 ± 0.23 and a slope of (1.90 ± 0.22) × 10−19 cm3 (shown by the solid line). Error bars shown on the plot and those given for the fits are 1σ, with fits weighted to the experimental errors. Separate lines of best fit for results from the different methods are not shown for clarity but are given in the ESI. The inset plot shows results from this work together with parameterisations given by Huang et al. for N2 (solid light grey line), O2 (broken black line) and air (broken dark grey line).](/image/article/2013/CP/c3cp52466c/c3cp52466c-f2.gif) | ||
| Fig. 2 Stern–Volmer analyses for CH2OO yields from CH2I + O2 as a function of total pressure. Main panel shows results from this work monitoring iodine atom production (squares; intercept = 1.08 ± 0.12; slope = (2.28 ± 0.11) × 10−19 cm3), and HCHO production in the presence of SO2 (triangles; intercept = 1.46 ± 0.25; slope = (0.95 ± 0.24) × 10−19 cm3) and NO (circles; intercept = 1.41 ± 0.30; slope = (1.33 ± 0.31) × 10−19 cm3). Constraining the intercepts to unity for fits to SO2 and NO data gives slopes of (1.37 ± 0.10) × 10−19 cm3 and (1.71 ± 0.16) × 10−19 cm3, respectively. Data shown for SO2 and NO were taken over a range of [O2] ((0.1–7.8) × 1018 cm−3). A fit to all data reported in this work gives an intercept of 1.10 ± 0.23 and a slope of (1.90 ± 0.22) × 10−19 cm3 (shown by the solid line). Error bars shown on the plot and those given for the fits are 1σ, with fits weighted to the experimental errors. Separate lines of best fit for results from the different methods are not shown for clarity but are given in the ESI.† The inset plot shows results from this work together with parameterisations given by Huang et al. for N2 (solid light grey line), O2 (broken black line) and air (broken dark grey line). | ||
Fig. 3a shows a typical kinetic trace for HCHO following photolysis of CH2I2–O2–N2 in the absence of any additional co-reagent (i.e. SO2 or NO), in which HCHO is produced in the system by reactions (R3–R6):
| CH2OO + I → HCHO + IO | (R3) |
| CH2IO2 + I → CH2IO + IO | (R4) |
| CH2IO2 + CH2IO2 → 2CH2IO + O2 | (R5) |
| CH2IO → HCHO + I | (R6) |
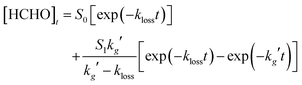 | (3) |
 | ||
| Fig. 3 HCHO fluorescence signals following photolysis of CH2I2 in the presence of O2. Panel (a) shows HCHO signals at 150 Torr in the absence of any co-reagent with the fit to eqn (3). Panel (b) shows HCHO signals at 250 Torr in the presence of SO2, with the fit to eqn (4). Panel (c) shows HCHO signals at 250 Torr in the presence of NO, with the fit to eqn (4). The inset plots in (b) and (c) show the evolution of the signals to longer times. | ||
Experiments conducted in excess SO2 or NO did not result in a decrease in the HCHO yield on addition of the co-reagent, indicating complete titration of both CH2OO and CH2IO2 to HCHO. In both cases biexponential growth of HCHO was observed, as shown in Fig. 3b and c, with the observed HCHO signal in both cases described by eqn (4):
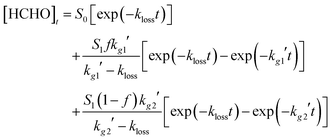 | (4) |
For both SO2 and NO experiments, the rate of the initial fast HCHO growth displayed a linear dependence on [SO2] or [NO], respectively, with kg1′ determined over the range 5000–60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1. The rate of the slower secondary growth was independent of [SO2] or [NO], and occurred at a similar rate to the HCHO growth observed in the absence of any additional co-reagent, and thus attributed to HCHO production viareaction (R3) or (R4–R6). The fact that kg1′ ≫ kg2′ means that f is reliably determined, and that kg1′ is determined without any significant influence from the more complicated kinetics associated with the slower kinetics, kg2′.
000 s−1. The rate of the slower secondary growth was independent of [SO2] or [NO], and occurred at a similar rate to the HCHO growth observed in the absence of any additional co-reagent, and thus attributed to HCHO production viareaction (R3) or (R4–R6). The fact that kg1′ ≫ kg2′ means that f is reliably determined, and that kg1′ is determined without any significant influence from the more complicated kinetics associated with the slower kinetics, kg2′.
The fast HCHO in the presence of SO2 is consistent with production from CH2OO + SO2:
| CH2OO + SO2 → HCHO + SO3 | (R7) |
Reactions of peroxy radicals with NO are typically fast (for example, kCH3O2+NO = 7.7 × 10−12 cm3 s−1 at 298 K25), while Welz et al.5 reported an upper limit for the rate coefficient for reaction of CH2OO with NO of <6 × 10−14 cm3 s−1. Therefore, we propose that the fast HCHO growth in experiments with NO results from the reaction of CH2IO2 with NO (R8) followed by the rapid decomposition of CH2IO to HCHO and I in (R6),15 with the slower growth resulting from (R3):
| CH2IO2 + NO → CH2IO + NO2 | (R8) |
| CH2IO → HCHO + I | (R6) |
While the slower growth of HCHO is not strictly pseudo-first-order, but is treated as such by eqn (4), simulations (described in the ESI†) show that the yields of HCHO from the two growth processes are well described by eqn (4) and the yields from the two processes (i.e. S1 and f) are faithfully determined by fitting to eqn (4). In both systems, the rate of the fast growth process (6000–60000 for SO2; 5000–20000 s−1 for NO) is significantly faster than that of the slower growth process (∼300–500 s−1), ensuring that the two growth processes are essentially decoupled and the HCHO yields from the two growth processes can be distinguished, and that the rate coefficient describing the fast growth is equal to that for the pseudo-first-order reactions, (R7) or (R8). We assign no kinetic information to kg2′ for either system, and as shown in the ESI,† the approximation of the slower growth process to pseudo-first-order kinetics leads to uncertainties in the yields of only 2–3%.
As noted above, there was no change in the total HCHO yield in the system upon the addition of either SO2 or NO, and this was observed to be the case at all total pressures. At low pressures where the CH2OO yield is close to unity, the addition of SO2 leads to reaction (R7) and formation of HCHO with close to 100% yield.23 In the ESI,† Fig. S3 compares the HCHO signal in the system with and without the addition of SO2. The fact that both traces observe the same amount of HCHO in the system adds validity to the assumption that in the absence of reagents, reactions (R3–R6), lead to 100% HCHO formation. At higher total pressures where CH2IO2 formation is significant, the reason the HCHO yield is still 100% is because there is no reaction between the peroxy radical and SO2, which is in agreement with the literature,24 and the peroxy radical is titrated to HCHO via(R4–R6). In the case of NO, it is the peroxy radical that reacts rapidly with the NO (R8) to form HCHO, but there is no significant reaction between the CH2OO and NO, in accord with the results from Welz et al.,5 and therefore HCHO is formed on a slow timescale viareaction (R3). This again leads to 100% yield of HCHO in the system independent of total pressure, in accord with the data.
Thus, the fractional contributions of the fast and slow growth processes to the total HCHO yields in the presence of NO and SO2 can be used to identify the yields of CH2OO and CH2IO2 from the reaction of CH2I with O2. The fractional contribution of the fast growth process to the total HCHO yield in the presence of NO thus reflects the yield of CH2IO2 from (R2) (i.e. YCH2IO2 = k2b[M]/(k2a + k2b[M]) = fNO and YCH2OO = k2a/(k2a + k2b[M]) = 1 − fNO), while the fractional contribution of the fast growth process to the total HCHO yield in the presence of SO2 reflects the yield of CH2OO from (R2) (i.e. YCH2OO = k2a/(k2a + k2b[M]) = fSO2 and YCH2IO2 = k2b[M]/(k2a + k2b[M]) = 1 − fSO2).
Fig. 2 also shows the Stern–Volmer analysis for CH2OO yields determined by the SO2 and NO experiments (i.e. Stern–Volmer plots for 1/fSO2 and 1/(1−fNO), respectively). Experiments with SO2 (triangles) give k2b/k2a = (0.95 ± 0.24) × 10−19 cm3, while those with NO (circles) give k2b/k2a = (1.33 ± 0.31) × 10−19 cm3, with intercepts of 1.46 ± 0.25 and 1.41 ± 0.30, respectively. Constraining the intercepts to unity in the fits to data from the SO2 and NO experiments gives k2b/k2a = (1.37 ± 0.10) × 10−19 cm3 and k2b/k2a = (1.71 ± 0.16) × 10−19 cm3, respectively. Further details can be found in the ESI.† The relative errors in the SO2 and NO experiments are typically larger than those for the iodine atom experiments owing to the need to fit a greater number of parameters in eqn (4) compared to eqn (1), and the smaller range of pressures in the Stern–Volmer plot for which the yields can be determined by the SO2 or NO method (at low and high pressures, where one of CH2OO or CH2IO2 dominates the HCHO growth it is difficult to resolve the two growth components, and thus to retrieve the relative yields, in the fit to eqn (4)). From Fig. 2, the fact that within error there is reasonable agreement in the CH2OO yields from the HCHO and the iodine atom experiments is further indication that all the sources of HCHO in each of the systems are understood and defined.
Experiments in both SO2 and NO were performed over a range of O2 concentrations, with measurements taken using 100% O2 buffer gas in both cases (see Fig. 4), in order to test if O2 has a significant effect on the CH2OO yield. In contrast to the work of Huang et al.,10 no dependence of k2b/k2a on [O2] was observed in any of our measurements. A fit to all our data reported here gives k2b/k2a = (1.90 ± 0.22) × 10−19 cm−3, with an intercept of (1.10 ± 0.23). Huang et al. noted that their observed difference in CH2IO2# stabilisation efficiency by N2 and O2 was an unusual result, with N2 and O2 often displaying similar collisional stabilisation efficiencies. It was proposed that O2 may not be acting as a simple collision partner to remove excess energy in CH2IO2#, but that there may be a reactive process occurring between O2 and CH2IO2#, potentially resulting in production of HCHO, IO and O2. However, an investigation of CH2I + O2 by Gravestock et al.15 could not identify IO as a product of the reaction even when more than 10% of O2 was present at 30 Torr total pressure, and our measurements of HCHO yields in this work are not consistent with the production of HCHO from this reaction.
![Inverse of CH2OO yields from CH2I + O2 as a function of [O2] for experiments with (a) SO2 at 150 Torr (circles) and 350 Torr (squares); (b) NO at 50 Torr (open squares), 150 Torr (circles), 250 Torr (triangles), 350 Torr (filled squares) and 450 Torr (inverted triangles). Horizontal lines show the average inverse CH2OO yield at each pressure for all experiments SO2 (panel a) and NO (panel b). Error bars are 1σ in the fits to eqn (4).](/image/article/2013/CP/c3cp52466c/c3cp52466c-f4.gif) | ||
| Fig. 4 Inverse of CH2OO yields from CH2I + O2 as a function of [O2] for experiments with (a) SO2 at 150 Torr (circles) and 350 Torr (squares); (b) NO at 50 Torr (open squares), 150 Torr (circles), 250 Torr (triangles), 350 Torr (filled squares) and 450 Torr (inverted triangles). Horizontal lines show the average inverse CH2OO yield at each pressure for all experiments SO2 (panel a) and NO (panel b). Error bars are 1σ in the fits to eqn (4). | ||
At present, there does not appear to be any simple explanation as to the differences between this work and the work of Huang et al. in the apparent yields of CH2OO and CH2IO2 from CH2I + O2 as a function of pressure. While the work of Huang et al. indicates a CH2OO yield of only ∼4% in air at 760 Torr, our results indicate a yield of ∼18%, with potentially significant implications for the oxidation chemistry of halogen containing organic compounds and for our understanding of atmospheric chemistry in marine regions with high concentrations of species such as CH2I2.26–32
In conclusion, we have measured the yields of CH2OO and CH2IO2 from the reaction of CH2I radicals with O2 as a function of total pressure and as a function of [N2] and [O2] using three complementary methods. Results from the three methods are similar, with no observed dependence of the CH2OO yield on [O2]. We estimate that the reaction between CH2I and O2 reaction has a yield of ∼18% of the CH2OO Criegee biradical at atmospheric pressure.
Acknowledgements
The authors are grateful to the National Centre for Atmospheric Science (NCAS) and the Engineering and Physical Sciences Research Council (EPSRC, grant reference EP/J010871/1) for funding.References
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- R. Criegee and G. Wenner, Justus Liebigs Ann. Chem., 1949, 564, 9–15 CrossRef CAS.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- C. A. Taatjes, G. Meloni, T. M. Selby, A. J. Trevitt, D. L. Osborn, C. J. Percival and D. E. Shallcross, J. Am. Chem. Soc., 2008, 130, 11883–11885 CrossRef CAS PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204–207 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2012, 14, 10391–10400 RSC.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Am. Chem. Soc., 2012, 134, 20045–20048 CrossRef CAS PubMed.
- Y.-T. Su, Y.-H. Huang, H. A. Witek and Y.-P. Lee, Science, 2013, 340, 174–176 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- H. Huang, A. J. Eskola and C. A. Taatjes, J. Phys. Chem. Lett., 2012, 3, 3399–3403 CrossRef CAS.
- S. Enami, T. Yamanaka, S. Hashimoto, M. Kawasaki, K. Tonokura and H. Tachikawa, Chem. Phys. Lett., 2007, 445, 152–156 CrossRef CAS PubMed.
- S. Enami, Y. Sakamoto, T. Yamanaka, S. Hashimoto, M. Kawasaki, K. Tonokura and H. Tachikawa, Bull. Chem. Soc. Jpn., 2008, 81, 1250–1257 CrossRef CAS.
- S. Enami, J. Ueda, M. Goto, Y. Nakano, S. Aloisio, S. Hashimoto and M. Kawasaki, J. Phys. Chem. A, 2004, 108, 6347–6350 CrossRef CAS.
- J. Sehested, T. Ellermann and O. J. Nielsen, Int. J. Chem. Kinet., 1994, 26, 259–272 CrossRef CAS.
- T. J. Gravestock, M. A. Blitz, W. J. Bloss and D. E. Heard, ChemPhysChem, 2010, 11, 3928–3941 CrossRef CAS PubMed.
- G. Hancock and V. Haverd, Chem. Phys. Lett., 2003, 372, 288–294 CrossRef CAS.
- H. M. Su, W. T. Mao and F. N. Kong, Chem. Phys. Lett., 2000, 322, 21–26 CrossRef CAS.
- U. Bley, F. Temps, H. G. Wagner and M. Wolf, Ber. Bunsen-Ges. Phys. Chem., 1992, 96, 1043–1048 CAS.
- R. A. Alvarez and C. B. Moore, J. Phys. Chem., 1994, 98, 174–183 CrossRef CAS.
- A. J. Eskola, D. Wojcik-Pastuszka, E. Ratajczak and R. S. Timonen, Phys. Chem. Chem. Phys., 2006, 8, 1416–1424 RSC.
- A. Masaki, S. Tsunashima and N. Washida, J. Phys. Chem., 1995, 99, 13126–13131 CrossRef CAS.
- J. C. Ianni, Kintecus, Windows Version 2.80, 2002, www.kintecus.com Search PubMed.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1961 CrossRef.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- C. M. Roehl, J. B. Burkholder, G. K. Moortgat, A. R. Ravishankara and P. J. Crutzen, J. Geophys. Res., Atmos., 1997, 102, 12819–12829 CrossRef CAS.
- L. J. Carpenter, W. T. Sturges, S. A. Penkett, P. S. Liss, B. Alicke, K. Hebestreit and U. Platt, J. Geophys. Res., [Atmos.], 1999, 104, 1679–1689 CrossRef CAS.
- L. J. Carpenter, Chem. Rev., 2003, 103, 4953–4962 CrossRef CAS PubMed.
- C. Ordonez, J. F. Lamarque, S. Tilmes, D. E. Kinnison, E. L. Atlas, D. R. Blake, G. S. Santos, G. Brasseur and A. Saiz-Lopez, Atmos. Chem. Phys., 2012, 12, 1423–1447 CrossRef CAS.
- A. Saiz-Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. von Glasow, J. C. G. Martin, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773–1804 CrossRef CAS PubMed.
- A. Saiz-Lopez and R. von Glasow, Chem. Soc. Rev., 2012, 41, 6448–6472 RSC.
- L. J. Carpenter, S. D. Archer and R. Beale, Chem. Soc. Rev., 2012, 41, 6473–6506 RSC.
- T. J. Dillon, M. E. Tucceri, R. Sander and J. N. Crowley, Phys. Chem. Chem. Phys., 2008, 10, 1540–1554 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp52466c |
| This journal is © the Owner Societies 2013 |