 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems†
Robert W.
Bradley
,
Paolo
Bombelli
,
David J.
Lea-Smith
and
Christopher J.
Howe
*
Department of Biochemistry, University of Cambridge, Hopkins Building, Downing Site, CB2 1QW, UK. E-mail: ch26@cam.ac.uk
First published on 1st July 2013
Abstract
Biological photo-voltaic systems are a type of microbial fuel cell employing photosynthetic microbes at the anode, enabling the direct transduction of light energy to electrical power. Unlike the anaerobic bacteria found in conventional microbial fuel cells that use metals in the environment as terminal electron acceptors, oxygenic photosynthetic organisms are poorly adapted for electron transfer out of the cell. Mutant strains of the cyanobacterium Synechocystis sp. PCC 6803 were created in which all combinations of the three respiratory terminal oxidase complexes had been inactivated. These strains were screened for the ability to reduce the membrane-impermeable soluble electron acceptor ferricyanide, and the results were compared to the performance of the mutants in a biological photo-voltaic system. Deletion of the two thylakoid-localised terminal oxidases, the bd-quinol oxidase and cytochrome c oxidase resulted in a 16-fold increase in ferricyanide reduction rate in the dark compared to the wild-type. A further improvement to a 24-fold increase was seen upon deletion of the remaining “alternative respiratory terminal oxidase”. These increases were reflected in the peak power generated in the biological photo-voltaic systems. Inactivation of all three terminal oxidase complexes resulted in a substantial redirection of reducing power; in the dark the equivalent of 10% of the respiratory electron flux was channelled to ferricyanide, compared to less than 0.2% in the wild-type. Only minor improvements in ferricyanide reduction rates over the wild-type were seen in illuminated conditions, where carbon dioxide is preferentially used as an electron sink. This study demonstrates the potential for optimising photosynthetic microbes for direct electrical current production.
Introduction
The Earth has a rapidly growing and developing population which is projected to result in a 53% increase in global power consumption for the period 2008–2035 to an annual average of ∼26 TW.1 A heavy reliance on fossil fuels is likely to result in climate change with potentially devastating consequences for ecosystems around the world.2 Biological photo-voltaics is an emerging energy production technology which has the potential to use the planet's most abundant source of energy – sunlight – to produce electrical power directly with lower carbon emissions than with fossil fuels. A biological photo-voltaic (BPV) system is analogous to a fuel cell, employing photosynthetic microorganisms in the anodic half-cell to capture light energy, split water, and transmit the resulting electrons to the electrode.3,4 Unlike conventional photo-voltaic systems, the light harvesting material in a BPV device is self-assembling and self-repairing, and carbon is captured when this material is generated. The related microbial fuel cell (MFC) technology5 also exploits the benefits of using cheap biological catalysts at the anode – though organic matter, not light, is the ‘fuel’ in an MFC.The rapidly expanding field of MFC research has produced devices with impressive current densities,6 and near-perfect conversion efficiencies (from organic substrate to electrical current) have been reported.7 The anaerobic microbes commonly used in MFCs naturally make use of environmental minerals as terminal electron acceptors, and are therefore well adapted for electron transfer from the organism to the anode. Cyanobacteria are oxygenic photosynthetic microbes that are well adapted for light energy capture and charge separation, but their metabolism is directed towards using carbon dioxide as an electron sink; aerobic respiration is used for ATP production in the dark. The flow of electrons out of the organism is the limiting step in electrical current production in BPV devices by photosynthetic microbes, masking the latter's impressive light harvesting and charge-generation capabilities.8 Soluble redox-active compounds which mediate electron transfer from cell to anode are required to achieve reasonable current densities when using cells in suspension. Whilst lipid-soluble mediators (e.g. 1,4-benzoquinone)9 are able to access and extract the most current, in doing so they disrupt electron transfer processes within the organism, and this can eventually lead to cell death. The lipid-insoluble ferricyanide ion accepts electrons from the cell surface, and is not toxic, even over long periods.3 Although adding redox mediators would not be feasible in real-world applications of BPV technology, ferricyanide is a convenient experimental tool to indicate the amount of reducing power available at the cell surface, which will hopefully correlate with the performance of mediator-less BPV systems such as those based on biofilms.4 This study aims to alter the metabolism of the model cyanobacterium Synechocystis sp. PCC 6803 (Synechocystis) to improve the flow of electrons across the outer membranes.
Synechocystis has a typical Gram-negative arrangement of outer and inner membranes, plus multiple layers of thylakoid membranes within the cytoplasm. Protein complexes involved in both photosynthesis and respiration are present in the thylakoid membranes, with electrons from both processes sharing the plastoquinone (PQ) pool, as illustrated in Fig. 1. Some respiratory complexes are also found in the cytoplasmic membrane, forming a short respiratory chain of unknown function(s) which lacks the cytochrome b6f complex.10,11
 | ||
| Fig. 1 Schematic showing localisation of and electron transfer between respiratory/photosynthetic electron transport complexes. The cytoplasmic membrane contains no functional photosynthetic components. A simple respiratory electron transfer chain exists, formed of succinate (SDH) and/or NADH dehydrogenases (NDH-2) which reduce the plastoquinone (PQ) pool, which is oxidised in turn by the quinol oxidases ARTO and possibly Cyd. In the thylakoid membrane photosystem II (PSII) captures light energy and reduces the PQ pool; PSII is re-reduced by the oxygen evolving complex (OEC), and can also be oxidised by Flv2/4 to prevent photodamage – the terminal acceptor for the Flv2/4 pathway is yet to be identified. Photosynthetic electron transfer proceeds via the cytochrome b6f complex (cyt b6f), a soluble electron carrier (either cytochrome c6 (cyt-c6) or plastocyanin (PC)) to photosystem I (PSI), then to NADPH via ferredoxin (Fd) and ferredoxin-NADP+-reductase (FNR). Cyclic electron flow around PSI may also occur via FNR or the ferredoxin:quinone reductase (FQR) pathway. The terminal oxidases Cyd and COX help to regulate the redox balance of the thylakoid membrane during periods of illumination by oxidising the PQ pool and cyt-c6/PC respectively. In the dark they are the final electron acceptors for respiratory electrons entering the PQ pool from the NDH-1, NDH-2 and SDH complexes. This illustration represents the consensus opinion on the physiology of electron flow through the membranes in Synechocystis, and does not show cytosolic processes that consume reducing equivalents (e.g. nitrogen and carbon assimilation or oxygen reduction by Flv1/3). | ||
In the dark, oxygen is used as the terminal acceptor for respiratory electrons entering the PQ pool from the succinate or NAD(P)H dehydrogenase complexes – though it is likely that NDH-2 complexes are more involved in redox-sensing than respiration.12 The major terminal oxidase for respiratory electron flow is cytochrome c oxidase (COX).13 Electrons pass through the b6f complex, and then to COX via plastocyanin (PC) or cytochrome c6 (cyt-c6) resulting in the maximum number of protons being pumped for each electron. A smaller number of protons migrate across the membrane when the b6f complex is bypassed and plastoquinol is oxidised by the bd-quinol oxidase (Cyd) or the alternative respiratory terminal oxidase (ARTO), a bo-quinol oxidase. COX, like the b6f complex, is proposed to be located only in the thylakoid membrane of Synechocystis.10 In contrast, the ARTO appears to be located only in the cytoplasmic membrane,14 whilst Cyd has been confirmed in the thylakoid membrane15 and may be present in the cytoplasmic membrane.13 Hart et al. (2005)16 review the terminal oxidases present in cyanobacteria; for a more specific summary for Synechocystis, see Lea-Smith et al. (2013).17
Additional electron sources and sinks come into play in the light. Photosystem II (PSII) reduces the PQ pool with electrons produced by photolysis, and reducing equivalents can leave the membrane as NADPH via linear photosynthetic electron flow through the cytochrome b6f complex, PC/cyt-c6 and photosystem I (PSI). CO2 fixation by the Calvin cycle is the main sink for photosynthetic electrons, though NADPH is used for a variety of other cellular processes. As light intensity increases, the rate of electron production at PSII can overtake consumption by the Calvin cycle, resulting in the photosynthetic electron transfer chain becoming reduced. To avoid oxidative stress the thylakoid-localised terminal oxidases can be used to dissipate reducing equivalents,17 allowing PSII to be re-oxidised, although additional pathways exist to avoid the electron transfer chain becoming over-reduced. Cyd is primarily responsible for PQ pool oxidation and consequential PSII protection,15 though less effective electron dissipation could be performed by COX via the b6f complex.
We predicted that removing the terminal oxidases from Synechocystis in order to prevent the use of oxygen as a final electron acceptor would result in increased fluxes of electrons out of the cell. Unmarked mutant strains of Synechocystis were created which lacked one, two or all three terminal oxidases in all combinations. The rate of ferricyanide reduction by the mutant strains was assayed, and respiration and photosynthetic oxygen evolution rates were characterised. The mutants were then employed in a BPV device which used ferricyanide as an electron mediator in the anodic chamber, and had an open-air carbon–platinum cathode for water regeneration. The emphasis of this investigation is on the use of metabolic mutations to redirect reducing power out of the organism, not design of an optimised BPV device for maximum power output.
Materials and methods
Strains and culture conditions
Synechocystis sp. PCC 6803 was cultured photoautotrophically at 30 °C under continuous 40 μE m−2 s−1 white light, either with continuous shaking at 160 rpm in 100 ml flasks in BG11 minimal medium18 with 5 mM NaHCO3, or on BG11 with 1.5% agar plates with 1 mM TES-NaOH (pH 8.2) and 3 g l−1 Na2S2O3. Escherichia coli strain DH5α was cultured in LB medium at 37 °C, either in liquid medium with shaking at 220 rpm or on LB with 1.5% agar plates. Where appropriate, supplements were added at the following concentrations: ampicillin 100 μg ml−1, kanamycin 30 μg ml−1, sucrose 5% w/v.Mutant construction
Metabolic mutants of Synechocystis were created as previously described.17 Briefly, homologous recombination was used to replace the target genes ctaCIDIEI (COX subunits), ctaCII, (ARTO subunit II) and cydAB (Cyd subunits) with an aminoglycoside phosphotransferase (kanamycin resistance) plus sacB (levansucrase) cassette, followed by generation of unmarked mutants via a second homologous recombination step. The mutations do not result in polar effects on downstream genes.17 Mutant strain genotypes were confirmed by PCR. Standard molecular biology techniques were used.Ferricyanide reduction assay
Ferricyanide reduction assay experiments were conducted using four biological replicates, obtained by splitting a log phase (optical density at 750 nm = 0.4–0.6 Abs) starter culture. Cells were harvested in log phase by centrifugation at 1800 × g, and re-suspended in fresh BG11 medium to a chlorophyll (chl) concentration of 4.0 (nmol chl) ml−1. 3 ml of each biological replicate was taken for use in the Clark electrode, and the remainder was split equally between two conical flasks, one of which was covered with aluminium foil and tape to block all light. Potassium ferricyanide was added to 1 mM from a 200 mM stock in BG11, and the initial absorbance readings were taken before incubating the cells at 30 °C with constant shaking at 220 rpm and 14 or 40 μE m−2 s−1 red light (#106 filter, Lee Filters). A 1 ml sample of the suspension was removed, and the absorbance measured at 420, 680 and 750 nm (Helios Gamma UV-Vis spectrophotometer, Thermo Scientific), before and after spinning the sample for 5 minutes at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g to pellet cells. A total of four measurements were taken over a period of approximately 24 hours. Ferricyanide concentrations were calculated from the absorbance at 420 nm of the supernatant (1 Abs = 1 mM) after adjusting for residual cells, and the overall rate was the average of the three normalised rates between time points (weighted according to the period over which the rate was measured – see ESI,† Table S1 for formulae). When calculating the percentage of available electrons which were transferred to ferricyanide the proportions were calculated for each sample before being averaged.
000 g to pellet cells. A total of four measurements were taken over a period of approximately 24 hours. Ferricyanide concentrations were calculated from the absorbance at 420 nm of the supernatant (1 Abs = 1 mM) after adjusting for residual cells, and the overall rate was the average of the three normalised rates between time points (weighted according to the period over which the rate was measured – see ESI,† Table S1 for formulae). When calculating the percentage of available electrons which were transferred to ferricyanide the proportions were calculated for each sample before being averaged.
Oxygen electrode
Photosynthetic and respiratory oxygen exchange rates were measured using an oxygen electrode system (Rank Brothers Instruments Ltd.) maintained at 30 °C. Light was provided by red (625 nm emission peak) MR16 5 W LED lights (Deltech). Data were recorded with an ADC-20 datalogger (Pico Technology). Samples were subjected to an alternating dark/light regime, with light intensity in each illuminated period higher than the last; net photosynthesis for each light intensity was calculated by subtracting the respiration rate from the following dark period. Data points were interpolated by fitting a hyperbolic tangent function19 to the data.BPV construction
The four identical single-chamber BPV devices used in this study (ESI,† Fig. S1) were formed from 10 mm thick transparent Perspex, with a main anodic chamber of 25 mm height and 40 mm internal diameter. A 20 mm diameter carbon–platinum cathode impregnated on one side with Nafion (Ion Power, USA) formed the back wall of the anodic chamber. Transparent indium tin oxide (ITO, 60 Ω sq−1) on plastic PET (Sigma-Aldrich) formed the front of the anodic chamber. The junctions between Perspex layers were sealed with polydimethylsiloxane (PDMS) and held in place by front and back 10 mm transparent Perspex clamps which did not cover the electrode materials. The cyanobacteria-BG11-ferricyanide suspension was loaded into the device by way of 4 mm holes that fed into the top of the anodic chamber. The theoretical capacity of the chamber was 31.5 ml. The ITO-PET anode was replaced for each experiment; the other components were washed with deionised H2O before re-use. The electrodes were attached to the external circuit with a crocodile clip via a contact formed from a strip of stainless steel.BPV operation
Cells from four biological replicates were harvested in log phase by centrifugation for 10 min at 5000g and re-suspended to 2.5 (nmol chl) ml−1 in fresh BG11. Potassium ferricyanide was added to a final concentration of 1 mM, and the suspension was loaded into each device. The devices operated at 21 ± 1 °C. The channels were allowed to reach a stable voltage in the dark with an external resistance of 1 kΩ before light was provided. An MR16 red (625 nm emission peak) 5 W LED light illuminated the chamber at 40 μE m−2 s−1 through the transparent anode. The potential difference between anode and cathode was recorded using an ADC-20 datalogger (Pico Technology); current could be calculated via Ohm's law. Various external resistances ranging from 1 MΩ to 1 kΩ were placed in the external circuit in order to produce a power curve (power density against applied external resistance) for each strain. Each strain was characterised in quadruplicate, except where stated otherwise.Results
Ferricyanide reduction assay
Mutant strains of Synechocystis were assayed for the ability to reduce ferricyanide in continuous dark or light conditions – rates are presented in Fig. 2, normalised to chlorophyll as a proxy for cell density (previous investigations have found no difference in chlorophyll content among the mutants when cultured under continuous light17). In the dark assay (Fig. 2a) all single terminal oxidase mutants had a small (∼4-fold) increase in rate over the wild-type value of 2.0 ± 0.44 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 [average ± standard error of the mean]. The COX/ARTO double mutant had a slightly higher reduction rate of 12 ± 1.5 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 (6-fold increase), and a larger increase to 17 ± 5.2 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 was observed from the Cyd/ARTO strain. Inactivation of both thylakoid-localised terminal oxidases led to the largest rate increases. The COX/Cyd mutant reduced ferricyanide 19 times faster than the wild-type, a rate of 38 ± 3.4 pmol [Fe(CN)6]3− min−1 (nmol chl)−1. A 23-fold increase to 47 ± 5.7 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 was observed from the COX/Cyd/ARTO (triple) mutant.![Ferricyanide reduction rates. Ferricyanide reduction rates in (a), darkness, (b) 14 μE m−2 s−1 light, (c) 40 μE m−2 s−1 light. Rates are expressed in pmol [Fe(CN)6]3− min−1 (nmol chl)−1. The data are averages of at least three biological replicates; error bars show standard error of the mean. An asterisk indicates that an ANOVA test found the value differed significantly from the group of samples containing the wild-type with a p-value of less than or equal to 5%.](/image/article/2013/CP/c3cp52438h/c3cp52438h-f2.gif) | ||
| Fig. 2 Ferricyanide reduction rates. Ferricyanide reduction rates in (a), darkness, (b) 14 μE m−2 s−1 light, (c) 40 μE m−2 s−1 light. Rates are expressed in pmol [Fe(CN)6]3− min−1 (nmol chl)−1. The data are averages of at least three biological replicates; error bars show standard error of the mean. An asterisk indicates that an ANOVA test found the value differed significantly from the group of samples containing the wild-type with a p-value of less than or equal to 5%. | ||
In low (14 μE m−2 s−1) light conditions none of the mutants reduced ferricyanide much faster than the wild-type strain (Fig. 2b). The largest increase on the wild-type rate of 6.2 ± 1.2 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 was just 2.5-fold for the COX mutant, at 16 ± 1.9 pmol [Fe(CN)6]3− min−1 (nmol chl)−1.
The assay was repeated at 40 μE m−2 s−1 (Fig. 2c), though several-fold increases on the wild-type rate of 14 ± 1.4 pmol [Fe(CN)6]3− min−1 (nmol chl)−1 were again not seen. As observed in the dark condition, the Cyd/ARTO, COX/Cyd, and triple mutants had the largest increases in ferricyanide reduction rate – though the ranking of the three was reversed, with fold-increases of 3.2, 2.6 and 2.0 respectively.
In addition to the ferricyanide reduction assay, the respiration and photosynthetic oxygen evolution rates for each replicate were determined (ESI,† Fig. S2). The rate of oxygen consumption or evolution is a measure of the electron flow through the membrane-localised electron transport chains (four electrons per O2). For the replicates in the light, these data were used to calculate the proportion of electrons generated by water photolysis that was transferred to ferricyanide under the two different light intensities (Fig. 3a). The proportion was very low for all strains. Cyd/ARTO had the largest increase over wild type in both conditions: 0.82 ± 0.14% of photosynthetic electrons went to ferricyanide in 14 μE m−2 s−1 light compared to the wild-type proportion of 0.23 ± 0.067%, and 0.88 ± 0.18% compared to 0.33 ± 0.038% in 40 μE m−2 s−1 light. Compared to the wild-type, none of the mutants transferred a significantly smaller proportion of electrons to ferricyanide in the light. This indicates that any increases in the ferricyanide reduction rates of the mutants were due to a redirection of reducing power, rather than a change in the rate of photosynthetic generation of reducing equivalents (which would leave the proportion of electrons going to ferricyanide unchanged).
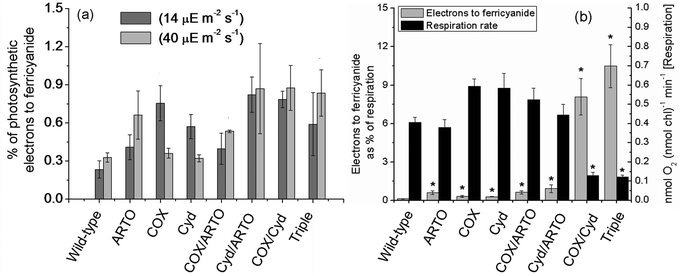 | ||
| Fig. 3 Redirection of reducing power in terminal oxidase mutants. (a) The percentage of photosynthetic electron flux, as determined by oxygen electrode assays, used to reduce ferricyanide. Dark grey bars are data from the 14 μE m−2 s−1 condition, light grey from the 40 μE m−2 s−1. (b) Electrons used to reduce ferricyanide in the dark (grey bars, primary y-axis), expressed as a percentage of the respiratory electron flux, and respiration rates (black bars, secondary y-axis). The data are averages of at least three biological replicates; error bars show standard error of the mean. Note the different scales on the y-axes. An asterisk indicates that an ANOVA test found the value differed significantly from the group of samples containing the wild-type with a p-value of less than or equal to 5%. | ||
Generation of reducing equivalents from stored carbohydrates in the dark does not require the consumption of oxygen per se, so the ratio of ferricyanide reduction rate to oxygen consumption rate does not necessarily equate to the proportion of electrons generated by carbohydrate oxidation which are transferred to ferricyanide. Therefore, expressing the amount of electrons transferred to ferricyanide in dark conditions as a percentage of the electrons used to reduce oxygen (Fig. 3b) encapsulates the redirection of reducing equivalents both to ferricyanide and away from oxygen. Most mutant strains show slight increases on the wild-type proportion of 0.13 ± 0.029%, with the exception of the two mutants lacking terminal oxidases in the thylakoid membrane, for which a much greater increase was observed. The amount of electrons used by the COX/Cyd strain to reduce ferricyanide is equivalent to 8.1 ± 1.4% of the electrons it uses to reduce oxygen, and the triple mutant uses 10.5 ± 1.7%. Respiration rates are included in Fig. 3b to emphasise that the increased proportion for the Cox/Cyd and triple mutants is partially due to decreased respiration rates. Data are listed in ESI,† Table S2.
Performance of mutants in a biological photo-voltaic device
In addition to measuring the reduction of ferricyanide spectroscopically, the peak power outputs were characterised from a BPV device operating with wild-type or mutant strains of Synechocystis and ferricyanide as the electron mediator (ESI,† Fig. S3). Power curves were generated in the dark at the end of a dark/40 μE m−2 s−1 light cycle; peak power is expressed relative to anode area. The power generated in the BPV system by each mutant system would be expected to depend on the rate of ferricyanide reduction, and a reasonable match between peak power (Fig. 4) and the ferricyanide reduction rate assay (Fig. 2) was indeed observed. As shown in Fig. 4, the peak power outputs for BPV systems operating with single mutants and the COX/ARTO double mutant were very similar to the peak power density of 0.52 ± 0.10 μW (nmol chl)−1 produced by wild-type cells. As observed in the ferricyanide reduction assay, BPV systems operating with the Cyd/ARTO, COX/Cyd and triple mutants performed best, with peak power outputs of 1.35 ± 0.21 μW (nmol chl)−1, 1.37 ± 0.46 μW (nmol chl)−1 and 2.33 ± 0.84 μW (nmol chl)−1 respectively, again matching the improvement on the wild-type seen in the ferricyanide reduction assay data. The peak power output from the triple mutant was established at an average current density of 33 ± 6.0 μA (nmol chl)−1 m−2, with this mutant producing a maximum current density of 64 ± 11 μA (nmol chl)−1 m−2. In contrast, the peak power output from the wild-type strain was established at an average current density of 27 ± 3.3 μA (nmol chl)−1 m−2, with a maximum current density of 49 ± 6.2 μA (nmol chl)−1 m−2. | ||
| Fig. 4 Peak power produced by terminal oxidase mutants in a BPV device. Data are averages of peak power produced at the end of a period of illumination from at least four biological replicates; error bars indicate standard error of the mean; power is normalised to anode area and cell density. An asterisk indicates that an ANOVA test found the value increased significantly from the group of samples containing the wild-type with a p-value of less than or equal to 5%. | ||
Discussion
Ferricyanide reduction in the dark
In the dark assay the single terminal oxidase mutants and the COX/ARTO double mutant showed only a minor increase in the rate of ferricyanide reduction compared to the wild-type. Assuming that Cyd is also present in the cytoplasmic membrane, these strains all possess at least one terminal oxidase to dissipate reducing power in both thylakoid and cytoplasmic membrane systems. The Cyd/ARTO mutant has both cytoplasmic terminal oxidases inactivated, and the result was an 8.5-fold increase in the rate of ferricyanide reduction. Inactivation of both thylakoid-localised terminal oxidases caused the COX/Cyd mutant to reduce ferricyanide 19 times faster than the wild-type. The improvement over the Cyd/ARTO strain is consistent with the thylakoid membrane being the major site of respiration – inactivation of terminal oxidases here causes a greater build-up of reducing equivalents compared to the cytoplasmic membrane. The highest rate of ferricyanide reduction was observed in the triple mutant; this strain is completely unable to use oxygen as a final electron acceptor from the membranes, resulting in the largest accumulation of reducing equivalents. The ferricyanide reduction rate of the triple mutant is not much greater than that of the COX/Cyd mutant, consistent with other observations that ARTO has a low oxygen reduction activity under most conditions.13Although the ratio of electrons used for ferricyanide reduction to the electrons used to reduce oxygen is a somewhat indirect measure of how electron flux is redirected in the dark, nevertheless, an increase in the ratio of ferricyanide reduction to oxygen consumption is desirable from the point of view of improving the performance of BPV devices – whether it is due to increased ferricyanide reduction rate or decreased ‘wastage’ of electrons through oxygen reduction. It has previously been shown that glycogen utilisation is lower in the COX/Cyd mutant, supporting the idea that the increased ferricyanide rates are due to re-direction of reducing equivalents, rather than an increase in the rate at which they are generated.17
Ferricyanide reduction in the light
When subjected to 14 μE m−2 s−1 light the ferricyanide reduction rate was similar in the double or triple mutants to the single mutants, suggesting that very little of the electron flux was used to reduce oxygen. Under low light conditions PSII is the rate-limiting step: carbon fixation happens fast enough to use all the electrons generated by photolysis, so the terminal oxidase “safety-valves” are not required and their inactivation produces no effect. The much higher ferricyanide reduction rates observed in the dark for the COX/Cyd and triple mutants compared to 14 μE m−2 s−1 light may be an effect of activating PSI (by illumination) as an alternative pathway for electrons to leave the thylakoid membrane.Increasing the light level to 40 μE m−2 s−1 produced a slight but not statistically significant increase in the ferricyanide reduction rates of the Cyd/ARTO, COX/Cyd, and triple mutants. The fact that inactivation of the terminal oxidases does not cause a substantial redirection of reducing equivalents towards ferricyanide under either 14 or 40 μE m−2 s−1 light conditions is not surprising, given that the organism can use alternative electron sinks, such as Flv2/4 or CO2. An additional observation is that the inactivation of COX/Cyd causes a significant decrease in photosynthetic oxygen evolution – see ESI,† Fig. S2. The lower rate of reducing equivalent generation counteracts any beneficial effect on raw ferricyanide reduction rate that deletion of these two respiratory terminal oxidases might have. The COX/Cyd and triple mutants also have significantly lower rates of aerobic respiration, but NAD(P)H production takes place upstream of the membrane-localised electron transport chains and would not necessarily be inhibited by a highly reduced PQ pool to the same degree as PSII.
Power generation by terminal oxidase mutants
Peak power production followed the pattern seen in the ferricyanide reduction assay, with the Cyd/ARTO, COX/Cyd, and triple mutants performing best. The light regime for the BPV devices was a dark/light cycle at moderate temperature, showing that these mutants perform better than wild-type cells under reasonably ‘natural’ conditions. Current production in the BPV device was not as high as observed in the ferricyanide reduction assay, which may well have been due to the lower operating temperature, cathodic inefficiency, or loss of current to oxygen at the anode, an unavoidable consequence of generating current from oxygenic photoautotrophs without having additional energy inputs such as sparging with N2 or an additional bias-potential.Insights into the mechanism of electron transfer
Whilst the molecular mechanism for ferricyanide reduction by Synechocystis remains to be defined, the results of this study fit with the conclusions of an earlier investigation which used inhibitors of the photosynthetic electron transport chain to probe the source of reducing equivalents used for ferricyanide reduction.3 That study concluded that NAD(P)H (rather than a quinol) was the likely substrate for ferricyanide reduction. The Cyd/ARTO, COX/Cyd and triple terminal oxidase mutant strains had the highest rates of ferricyanide reduction, and Gutthann et al.20 showed that these strains also had high hydrogen production rates – the substrate for the reduction of protons being NAD(P)H. We surmise that the increased NAD(P)H:NAD(P)+ ratio resulting from a more highly reduced PQ pool in the terminal oxidase mutants21 leads to an increased rate of ferricyanide reduction, as the reduced dinucleotide is the substrate for the uncharacterised ferricyanide reduction mechanism. It is also clear that ferricyanide reduction does not directly involve the terminal oxidases, as ferricyanide reduction rates were higher than the wild-type rate in all mutant strains.
Implications for future work
In order to translate the blockage of oxygen reduction more fully to an increase in ferricyanide (or other external acceptor) reduction rate, the capacity of the electron export mechanism needs to be increased. The potential for current production and the limitations which need to be overcome have been discussed elsewhere.8 That said, the fact that a simple combination of mutations can lead to a 4- to 5-fold increase in power output even in the absence of attempts to increase ‘ferricyanide reductase’ activity has exciting implications for the development of BPV systems for practical use.There are a number of other electron sinks or alternative pathways which could be removed or transiently suppressed in order to direct more reducing power towards electron export. Baebprasert et al.22 saw a positive effect on H2 production from deleting the nitrate and nitrite reductase enzymes – we would predict a similarly positive effect on ferricyanide reduction rates. Limiting the use of carbon fixation as an electron sink by transiently suppressing the Calvin cycle could potentially make a huge proportion of reducing power available for electron export. Photo-protection mechanisms, for example the Flv2/4 system,23 could also be considered wasteful routes of energy dissipation. The Synechocystis M55 mutant in which the ndhB gene is disrupted has a smaller but highly reduced NAD(P)H pool,21 and improved H2 production is observed in this mutant.24 Ferricyanide reduction rates for this mutant are dramatically higher than wild-type rates in dark and light conditions (ESI,† Table S3), but the M55 mutant grows very slowly without additional CO2.25 The Flv1/3 heterodimer also consumes NAD(P)H, reducing O2 in order to protect PSI from photodamage;26 and is another potential target for inactivation. Striking a balance between improving electrogenic activity and retaining enough metabolic diversity for the strain to thrive under a variety of conditions is an important consideration, especially when looking ahead to real-world applications. Without improving the capacity of electron export mechanisms, alterations to cyanobacterial metabolism will be deleterious. The next steps in optimising photosynthetic microbes for current production should focus on understanding and improving the electrogenic mechanism(s), in order for the organism to be able to cope with conditions that would otherwise lead to an excess of reducing power, and to reveal the full effect of redirecting metabolic electron flux.
Summary
Oxygenic photosynthetic organisms such as cyanobacteria have evolved to couple the capture of light energy to carbon dioxide fixation as efficiently as possible. During respiration or in times of excess electron production, reducing power can be dissipated to oxygen using terminal oxidases. This makes cyanobacteria poorly adapted for direct current extraction – especially compared to the (typically anaerobic) dissimilatory metal-reducing bacteria. The terminal oxidase complexes of the cyanobacterium Synechocystis sp. PCC 6803 were deleted to remove the possibility of using oxygen as an electron sink. This resulted in higher ferricyanide reduction rates as a consequence of creating an excess of reducing power. In illuminated conditions the ferricyanide reduction rates of mutant strains were only slightly increased compared to the wild-type, although higher light levels might exaggerate the difference. Larger gains were observed in dark conditions, especially from the COX/Cyd and triple deletion strains. The triple mutant transferred the equivalent of 10% of its aerobic respiratory electron flux to ferricyanide, causing a 23-fold increase in ferricyanide reduction rate compared to the wild-type. This substantial redirection shows it is possible to manipulate the metabolism of photosynthetic organisms rationally in order to improve their electrogenic capabilities, and is one of the first steps towards the creation of an organism designed to transduce light energy directly and efficiently to electrical power.Abbreviations
| ARTO | Alternative respiratory terminal oxidase |
| ATP | Adenosine triphosphate |
| BPV | Biological photo-voltaic |
| chl | Chlorophyll |
| cyt-c6 | Cytochrome c6 |
| COX | Cytochrome c oxidase |
| Cyd | bd-Quinol oxidase |
| MFC | Microbial fuel cell |
| PSI/II | Photosytem I/II |
| PC | Plastocyanin |
| PQ | Plastoquinone |
| Synechocystis | Synechocystis sp. PCC 6803. |
Acknowledgements
The authors are grateful for funding provided by the UK Engineering and Physical Sciences Research Council (EPSRC EP F047940).References
- U. S. Energy Information Administration, International Energy Outlook 2011, 2011.
- M. New, D. Liverman, H. Schroeder and K. Anderson, Philos. Trans. R. Soc., A, 2011, 369, 6–19 CrossRef.
- P. Bombelli, R. W. Bradley, A. M. Scott, A. J. Philips, A. J. McCormick, S. M. Cruz, A. Anderson, K. Yunus, D. S. Bendall, P. J. Cameron, J. M. Davies, A. G. Smith, C. J. Howe and A. C. Fisher, Energy Environ. Sci., 2011, 4, 4690–4698 CAS.
- A. J. McCormick, P. Bombelli, A. M. Scott, A. J. Philips, A. G. Smith, A. C. Fisher and C. J. Howe, Energy Environ. Sci., 2011, 4, 4699–4709 CAS.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS.
- H. Yi, K. P. Nevin, B.-C. Kim, A. E. Franks, A. Klimes, L. M. Tender and D. R. Lovley, Biosens. Bioelectron., 2009, 24, 3498–3503 CrossRef CAS.
- K. P. Nevin, H. Richter, S. F. Covalla, J. P. Johnson, T. L. Woodard, A. L. Orloff, H. Jia, M. Zhang and D. R. Lovley, Environ. Microbiol., 2008, 10, 2505–2514 CrossRef CAS.
- R. W. Bradley, P. Bombelli, S. J. L. Rowden and C. J. Howe, Biochem. Soc. Trans., 2012, 40, 1302–1307 CrossRef CAS.
- M. Torimura, A. Miki, A. Wadano, K. Kano and T. Ikeda, J. Electroanal. Chem., 2001, 496, 21–28 CrossRef CAS.
- M. Schultze, B. Forberich, S. Rexroth, N. G. Dyczmons, M. Roegner and J. Appel, Biochim. Biophys. Acta, 2009, 1787, 1479–1485 CrossRef CAS.
- T. Pisareva, J. Kwon, J. Oh, S. Kim, C. Ge, A. Wieslander, J.-S. Choi and B. Norling, J. Proteome Res., 2011, 10, 3617–3631 CrossRef CAS.
- C. A. Howitt, P. K. Udall and W. F. J. Vermaas, J. Bacteriol., 1999, 181, 3994–4003 CAS.
- C. A. Howitt and W. F. J. Vermaas, Biochemistry, 1998, 37, 17944–17951 CrossRef CAS.
- F. Huang, I. Parmryd, F. Nilsson, A. L. Persson, H. B. Pakrasi, B. Andersson and B. Norling, Mol. Cell. Proteomics, 2002, 1, 956–966 CAS.
- S. Berry, D. Schneider, W. F. J. Vermaas and M. Rögner, Biochemistry, 2002, 41, 3422–3429 CrossRef CAS.
- S. E. Hart, B. G. Schlarb-Ridley, D. S. Bendall and C. J. Howe, Biochem. Soc. Trans., 2005, 33, 832–835 CrossRef CAS.
- D. J. Lea-Smith, N. Ross, M. Zori, D. S. Bendall, J. S. Dennis, S. A. Scott, A. G. Smith and C. J. Howe, Plant Physiol., 2013, 162, 484–495 CrossRef CAS.
- R. Rippka, J. Deruelles, J. B. Waterbury, M. Herdman and R. Y. Stanier, J. Gen. Microbiol., 1979, 111, 1–61 CrossRef.
- B. E. Chalker, J. Theor. Biol., 1980, 84, 205–213 CrossRef CAS.
- F. Gutthann, M. Egert, A. Marques and J. Appel, Biochim. Biophys. Acta, 2007, 1767, 161–169 CrossRef CAS.
- J. W. Cooley and W. F. J. Vermaas, J. Bacteriol., 2001, 183, 4251–4258 CrossRef CAS.
- W. Baebprasert, S. Jantaro, W. Khetkorn, P. Lindblad and A. Incharoensakdi, Metab. Eng., 2011, 13, 610–616 CrossRef CAS.
- P. Zhang, M. Eisenhut, A.-M. Brandt, D. Carmel, H. M. Silén, I. Vass, Y. Allahverdiyeva, T. A. Salminen and E.-M. Aro, Plant Cell, 2012, 24, 1952–1971 CrossRef CAS.
- L. Cournac, G. Guedeney, G. Peltier and P. M. Vignais, J. Bacteriol., 2004, 186, 1737–1746 CrossRef CAS.
- T. Ogawa, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4275–4279 CrossRef CAS.
- Y. Allahverdiyeva, H. Mustila, M. Ermakova, L. Bersanini, P. Richaud, G. Ajlani, N. Battchikova, L. Cournac and E.-M. Aro, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4111–4116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp52438h |
| This journal is © the Owner Societies 2013 |