
trans effect and trans influence: importance of metal mediated ligand–ligand repulsion†
Abstract
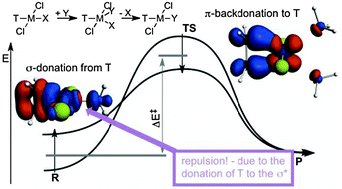
The trans effect and trans influence were investigated and rationalized in the aminolysis, a typical nucleophilic substitution reaction, of trans-TPtCl2NH3 complexes (T = NH3, PH3, CO and C2H4) using energy decomposition analysis, both along the reaction paths and on the stationary points, and Natural Orbital for Chemical Valence analysis. In order to scrutinize the underlying principles and the origin of the kinetic trans effect, plausible structural constraints were introduced in the decomposition analysis, which allowed eliminating the distance dependence of the interaction energy components. It was established that the trans effect can be rationalized with the interaction of the TPtCl2 and NH3 fragments in the reactant state and TPtCl2 and (NH3)2 fragments in the transition state. It was evinced quantitatively that the σ-donor ability of T indeed controls the stability of the reactant, whereas in the case of π-acids, backdonation stabilizes the transition state, for which conceptually two mechanisms are available: intrinsic and induced π-backdonation. In the destabilization of the reactant and also in the labilization of the leaving group (trans influence) repulsion plays a more important role than orbital sharing effects, which are the cornerstones of the widely accepted interpretations of the trans influence, such as competition for donation or limitation of the donation of the leaving group by the trans ligand T. This repulsive interaction was rationalized both in terms of donated electron density and also in the molecular orbital framework. NOCV orbitals indeed clearly show that the σ-trans effect can be envisioned as a donation from the trans ligand not only to the metal but also to the σ* orbital of the metal-leaving group bond, which manifests as a repulsion between the metal and the leaving group.
 Please wait while we load your content...
Please wait while we load your content...

