 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ab initio calculations of the optical absorption spectra of C60-conjugated polymer hybrids
Laura E.
Ratcliff
*ab and
Peter D.
Haynes
b
aLaboratoire de simulation atomistique (L_Sim), SP2M, UMR-E CEA/UJF-Grenoble 1, INAC, Grenoble, F-38054, France. E-mail: laura.ratcliff@cea.fr
bDepartments of Physics and Materials, Imperial College London, London, SW7 2AZ, UK
First published on 24th June 2013
Abstract
A recently developed linear-scaling density-functional theory (LS-DFT) formalism is used to calculate optical absorption spectra of hybrids of C60 and the conjugated polymers poly(para-phenylene) (PPP) and poly(para-phenylene vinylene) (PPV). The use of a LS formalism allows calculations on large systems with realistic proportions of C60, which has been of interest for the use of such materials in photovoltaics. Two different bonding structures are tested for the hybrid PPP and for both systems additional peaks are present in the absorption spectra below the original onset of absorption. By identifying the eigenstates involved in the relevant transitions, a weighted density difference is formed, demonstrating the transfer of charge between the polymer chain and the C60, in agreement with experiment. For the hybrid PPV, no additional peaks are observed in the absorption spectrum.
1 Introduction
Since the discovery in 1976 that conjugated polymers could be made conducting,1–3 they have been the subject of much research, both for reasons of fundamental theoretical interest and due to the promise of a wide range of exciting applications. They are highly attractive candidates for use in applications ranging from light-emitting diodes (LEDs) to photovoltaic cells.4–8 This is in part due to the ability to influence their properties via doping and modification of side chains, enabling for example the creation of materials with a wide range of energy gaps resulting in materials ranging from metallic to insulating.2,9 Furthermore, they benefit from useful mechanical properties such as the ability to be flexible, and from desirable processing methods, particularly the ability to process from solution, which also has the important effect of reducing the final cost of the materials.1,5,9 As a result, a great deal of effort has been extended into developing and studying such polymers with the hope of replacing inorganic semiconductors in some existing applications as well as creating new ones.For the purposes of this work, we are particularly interested in the application to photovoltaic cells, where polymer based devices could have a number of advantages over the standard silicon based solar cell, particularly in terms of cost. Indeed, conjugated polymers such as poly(para-phenylene) (PPP) and poly(para-phenylene vinylene) (PPV) (whose structures are depicted in Fig. 1) have been suggested as possible candidates for use as a donor when in combination with an acceptor such as the fullerene C60.5,8 For this use, certain criteria must be met, including the occurrence of charge transfer, which has been demonstrated in fullerene–polymer mixtures.9–12 Indeed, C60–PPV photovoltaic cells have been realized in a layered structure.13,14
 | ||
| Fig. 1 Schematics showing the structure of segments of PPP and PPV, with C atoms shown in white and H atoms in black. | ||
A useful way of studying these systems and comparing the behaviour of different conjugated polymers is through computational simulations, in particular first-principles calculations which allow the calculation of optical absorption spectra, among other useful properties. One very popular methodology is density-functional theory (DFT),15,16 which has been applied extensively to a large number of systems and shown to give remarkably accurate results for ground state properties, and can also be used as a good first approximation to excited state properties such as absorption spectra. The accuracy of these spectra will of course be limited by the approximations inherent in practical implementations of DFT, and arising from the fact that the Kohn–Sham eigenvalues have no rigorous correspondence with the true quasi-particle energies. Indeed, one can only achieve good agreement with experiment by applying the “scissor operator”, where the conduction states are rigidly shifted upwards in energy to correct for the underestimation of the band gap by DFT. However, previous calculations on PPP have shown good agreement with the experimental absorption spectra using the local density approximation (LDA)17 and so we expect the use of DFT to be a reasonable approximation for these systems. Furthermore, the systems of interest here involve many atoms, even when limiting ourselves to e.g. a single chain combined with a single molecule of C60, so that the cost of more accurate methods such as the GW approximation18–20 is prohibitively expensive for calculations on systems of this size. Indeed, even conventional DFT codes can become too expensive due to the cubic scaling bottleneck encountered for systems over a given size, and so it becomes useful to have a linear-scaling (LS) formulation of DFT, which takes advantage of the principle of nearsightedness and therefore allows one to access truly large systems.
To this end we have employed a recently developed methodology for calculating first the Kohn–Sham conduction states and then the imaginary component of the dielectric function in a LS-DFT framework.21,22 This new methodology combines the optimization of a second set of localized orbitals to describe the conductions states with the use of Fermi's golden rule and has been implemented in the LS-DFT code ONETEP.23–26 We have applied this methodology to the study of C60 hybrids of PPP and PPV.
In this paper we first outline the basic theory behind the ONETEP code, including the method used to calculate the Kohn–Sham conduction states. We then describe the method applied to the calculation of optical absorption spectra. Finally we will present results for the absorption spectra of the hybrid PPP and PPV, before summarizing our conclusions.
2 Methodology
ONETEP is a linear-scaling DFT code which takes advantage of the principle of near-sightedness, and in particular the exponential decay of the density matrix (DM) in systems with a band gap.27–29 This is achieved using a DM formulation, within a strictly localized representation of non-orthogonal generalized Wannier functions (NGWFs).30 The NGWFs are themselves represented in terms of a periodic cardinal sine (psinc) basis set,31 which allows them to be optimized during the calculation. The DM is written in separable form32,33 as: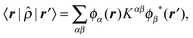 | (1) |
During the calculation the eigenstates are not directly referenced, however they can be recovered via a single diagonalization at the end of the calculation. This is an O(N3) process, however the prefactor is small as we optimize a minimal set of NGWFs that is much smaller than the number of psincs, so that the cost is negligible compared to the overall simulation time. This enables for example the calculation of densities of states (DOS) and band structures. However, as the NGWFs are optimized to minimize the total ground state energy, they are not expected to accurately represent the Kohn–Sham conduction states, and in some cases certain conduction states are found to be completely absent.21,22,34 In order to be able to calculate optical absorption spectra one therefore needs a method of optimizing a new set of NGWFs to accurately represent the unoccupied states. This is achieved by defining a projected Hamiltonian:
Ĥ − ![[small rho, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b7.gif) (Ĥ − σ), (Ĥ − σ), | (2) |
is the density operator and σ is an energy shift. The lowest eigenstates of this projected Hamiltonian are the lowest conduction states of interest and so it can be used in a similar procedure to ground state ONETEP calculations, optimizing a second set of NGWFs for a fixed number of conduction states, omitting the self-consistency process and instead re-using the ground state density and potential. Following the conduction NGWF optimization, the valence and conduction NGWFs are combined to form a joint valence–conduction basis set, which is capable of representing both the occupied and unoccupied Kohn–Sham states. Further details have been given elsewhere,21,22 including a demonstration of the linear-scaling behaviour of the method.
Once one has a process for calculating the Kohn–Sham conduction states as well as valence states, Fermi's golden rule can be applied to the calculation of optical absorption spectra. The methodology35 used here is the same as that applied in the cubic-scaling plane-wave pseudopotential (PWPP) code CASTEP.36 Using the dipole approximation, whereby only first order terms are included, one can write the imaginary component of the dielectric function as:
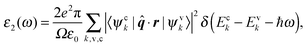 | (3) |
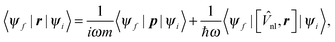 | (4) |
One could envisage constructing polymer–C60 devices in a variety of ways with either covalent or non-covalent bonds connecting the polymer to the C60, or by creating thin film devices. Indeed there are a wide variety of structures which have already been realized for C60–polymer hybrids, which include a number of structures with different bonding arrangements wherein a fullerene is covalently bonded to a polymer.38,39 We have chosen simple structures for hybrids of C60 with PPP and PPV with concentrations of approximately 25% by weight, in agreement with that used by Lane et al.12 in their experiment on C60-doped PPP. We have calculated the optical absorption spectra for these hybrid structures, and compared the results to those of isolated chains of PPP and PPV and an isolated molecule of C60.
Before generating structures for the hybrid polymers, individual calculations were performed for the two polymers and the fullerene, for validation. All calculations have been performed using the LDA exchange–correlation functional, with Γ point sampling only, norm-conserving pseudopotentials and periodic boundary conditions. The total energies were converged with respect to the psinc cut-off energy and NGWF radii – for all calculations the psinc grid spacing was set to be equivalent to a kinetic energy cut-off of 1115 eV,34 valence NGWF radii of 5.29 Å and conduction NGWF radii of 7.41 Å were used for all atoms, with four valence and conduction NGWFs per C atom and one valence and four conduction NGWFs per H atom. No truncation was applied to the density kernel.
3 Results and discussion
In order to test the accuracy of the method compared to traditional plane-wave DFT, we have calculated the density of states and the imaginary component of the dielectric function for a small unit cell of PPV containing 56 atoms, using ONETEP both with and without the optimization of a set of conduction NGWFs. These results are compared to PWPP results calculated using CASTEP in Fig. 2 and 3 respectively. The same pseudopotentials were used in both codes and the kinetic energy cut-off of the psinc and plane-wave basis sets were chosen to be approximately equivalent. As can be clearly seen from these results, the optimization of a set of conduction NGWFs is vital for good agreement with traditional DFT results. Further examples demonstrating the accuracy of the method are given elsewhere, together with a discussion on some of the limitations of the method.21,22 | ||
| Fig. 2 Density of states of PPV calculated with both ONETEP and a PWPP code. In the upper panel, the PWPP result is plotted with the ONETEP result in the valence NGWF basis only, in the lower panel the PWPP result is plotted with the ONETEP result in the joint valence–conduction NGWF basis. For each result, a Gaussian smearing width of 0.1 eV is used and only fifteen conduction states are included, which corresponds to the number which were optimized for the ONETEP conduction calculation. Reproduced with permission from ref. 22. | ||
 | ||
| Fig. 3 The imaginary component of the dielectric function of PPV calculated using the PWPP method and ONETEP both with (lower panel) and without (upper panel) a conduction calculation. A Gaussian smearing width of 0.1 eV is used. Reproduced with permission from ref. 22. | ||
Two different bonding structures were used for C60–PPP, which are depicted in Fig. 4(a) and (b). The C60–PPV was constructed using the same attachment mechanism as structure A. Unit cells of 676 (672) atoms were constructed in periodic boundary conditions for the hybrid polymers in structure A (B), with two molecules of C60 evenly spaced across the polymers. Large simulation cells of 239 Å × 28 Å × 40 Å and 259 Å × 26 Å × 26 Å for the doped PPP and PPV respectively were required to reduce the interaction between periodic copies of the polymers, thus the use of a linear-scaling methodology was further justified due to the large proportion of empty space in the simulation cell. Ground state and conduction calculations were then performed in ONETEP, followed by the calculation of the imaginary component of the dielectric function. The calculations on pure PPP and PPV were calculated in unit cells containing 280 and 560 atoms respectively, equivalent to samplings of 14 and 20 k-points with respect to the primitive unit cell, such that the spectra were converged.
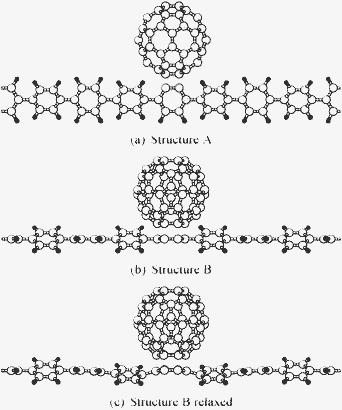 | ||
| Fig. 4 Schematics showing segments of the two different structures of C60–PPP, including both the unrelaxed and relaxed forms of structure B. | ||
The structures of both PPP and PPV were optimized using CASTEP and the structure of C60 was relaxed in the same manner. To reduce the computational cost, the hybrid structures were initially not optimized. The forces on the structures were calculated in ONETEP;26 the root mean squared (rms) forces for structures A and B of the hybrid PPP were 0.4 and 0.5 eV Å−1 respectively and the maximum forces were and 4.1 and 4.7 eV Å−1 respectively. For the hybrid PPV they were 0.4 and 5.1 eV Å−1. For the separated structures the forces were less than 0.1 eV Å−1. As these forces for the hybrid structures are rather high, a test was done for structure B of C60–PPP, by optimising the structure using ONETEP to give a rms force of 0.02 eV Å−1, and a maximum force of 0.1 eV Å−1. The changes in the structure are essentially localized to the section of the chain closest to the fullerene, with the polymer being bent away from the C60, as shown in Fig. 4(c). The resulting spectrum was then compared to that of the unoptimized structure, giving an estimate of the accuracy of the results for the other structures (see Fig. 5). This relaxation was of course subject to the constraint of periodic boundary conditions and thus while this gives an indication of a more optimal structure, it is entirely possible that the changes would be more significant if this constraint were removed; for example one might find that the polymer backbone becomes curved. The removal of the periodic boundary conditions might therefore provide an interesting avenue for future exploration of C60-doped polymers.
 | ||
| Fig. 5 The imaginary component of the dielectric function of C60–PPP in structures A and B, with both the unoptimized and optimized structure of B, compared to the sum of the spectra of pure PPP and C60. The spectra of pure PPP and C60 are also compared with the total, as is that of C60 and PPP in the same cell separated by a large distance (denoted C60 with PPP in the key). The three insets show a closer view of the spectra between 0.5 and 2.5 eV, highlighting the additional peaks. The results are plotted with a Gaussian smearing width of 0.05 eV. | ||
Results for the different hybrid PPP structures are plotted in Fig. 5, with the spectra of the PPP and C60 shown for comparison. The sum of the separate polymer and C60 results is also plotted. The results for both structures of C60–PPP show a number of small additional peaks below the original onset of absorption of PPP (approximately 2.4 eV), which are not present in either PPP or C60 on their own. These peaks are stronger for structure B than structure A. The relaxation of structure B has had a noticeable impact on the spectra, with changes both in position and relative height of the observed peaks. In particular, those peaks which are below the original onset of absorption have significantly decreased in height, which is likely due to the bending of the polymer away from the fullerene. However, the overall picture of additional peaks below this onset is still clearly preserved.
The imaginary component of the dielectric function has also been calculated for C60 and PPP in the same unit cell with the same cell dimensions as for the hybrid structures, but separated by a large distance (greater than 14 Å) so that they are not chemically bonded, the result for which is also shown in Fig. 5. In this case, the relative heights of the peaks have been affected, and an additional peak is present at around 3.5 eV, however no additional peaks are present below the original onset of absorption. This confirms that for those peaks below 2.4 eV to occur, the C60 and PPP must be in close proximity.
One of the advantages of computational spectroscopy is that it can be used to determine which transitions are responsible for a given peak. In this way insight can be gained into the origins of the additional peaks in the hybrid PPP system. For example, for the unoptimized structure B we can identify that there is a transition at 2.05 eV between the HOMO − 1 (highest occupied molecular orbital) and the LUMO + 12 (lowest occupied molecular orbital), for which the wavefunctions are plotted in Fig. 7(a) and (b). We can see that the electron density is primarily located on the polymer for the HOMO − 1, whereas for the LUMO + 12 it is primarily located on the fullerene. However, the three additional peaks below 2.4 eV result from a large number of transitions that are close together in energy. Therefore, rather than attempting to visualize all of the individual transitions involved, as we have done for this example, it is more useful to generate an average density change, weighted by the strength of the transition involved. To this end, we have used the following expression to calculate a weighted density difference nweighted:
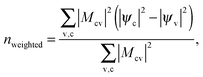 | (5) |
This analysis of the low energy peaks suggests that the charge transfer process is stronger for structure B of the hybrid polymer. This is possibly due to the fact that there is more overlap between the C60 and the chain, with six C atoms in proximity to the fullerene, rather than two. More importantly, we can see both from the bending of the chain during the relaxation of structure B and by visual inspection of the eigenstates close to the band gap that the pi-bonds on the phenyl ring adjacent to the fullerene have been disrupted, allowing for a strong mixing between states – an example of this can be seen in Fig. 7(a). For structure A, on the other hand, the angle between the fullerene and the polymer is such that no similar disruption occurs, as shown for the HOMO − 4 in Fig. 7(d). As a result, the additional peaks in the absorption spectrum for structure A are much less pronounced than those of structure B.
The spectrum for the hybrid C60–PPV is shown in Fig. 6, again plotted with those of PPV and C60. Unlike the PPP hybrids, the spectrum does not show any significant additional peaks, although some peaks have increased in intensity. The lack of additional peaks in the spectrum for C60–PPV could indicate that DFT alone is insufficient for capturing such behaviour in this particular system – it has been previously demonstrated that the inclusion of excitonic effects has a strong impact on the absorption spectrum of PPV.41 However, an alternate study by Pedersen using density-functional-based tight-binding (DFTB) indicates that excitonic effects are important for both PPP and PPV42 and so one also might not expect to see such peaks in C60–PPP without their inclusion, which we can see is not the case. Alternatively, it could be that the structure considered here is not ideal for the transfer of charge; in a further study by Pedersen43 charge transfer states are calculated in C60–PPV with an arrangement similar to structure B, but with a finite chain length, a range of separation distances between the C60 and the PPV greater than that used here and again using DFTB with the inclusion of excitonic effects. Given that both the structure and formalism are different from those used in our calculation it is difficult to say which might have the bigger effect, however as the low energy peaks in structure A of C60–PPP are already very small it seems more likely that it is the structure that is unfavourable, with too large an angle between the fullerene and the chain. We can also see from the spectra that the states on C60 are separated in energy from those of PPV, whereas with PPP there is a significant overlap in energy, particularly around 2.6 eV. This should make it easy for these states to mix in the hybrid PPP structure to create a lower-lying state, which would not be the case for PPV. The creation of such a state seems to be confirmed by the significant increase in the absorption spectra just below this energy seen for both PPP hybrids and for PPP and C60 in the same simulation cell. Finally, it could also be possible that there is some transfer of charge occurring between the PPV and the C60, but it is not visible in the absorption spectrum either because it is too weak, or because it is masked by other peaks which are already present.
 | ||
| Fig. 6 The imaginary component of the dielectric function of the C60–PPV hybrid, compared to the sum of the spectra of pure PPV and C60. The spectra of pure PPV and C60 are also compared with the hybrid spectrum in the lower half of the plot. The results are plotted with a Gaussian smearing width of 0.05 eV. | ||
 | ||
| Fig. 7 Two different eigenstates for a segment of unrelaxed structure B of C60–PPP, one occupied and one unoccupied, a weighted density difference for transitions below 2.4 eV for structure B and an eigenstate of structure A. For the weighted density, red indicates an increase in electron density and blue indicates a decrease. These figures were plotted using XCrySDen.40 | ||
We also observe here that it has long been known that the LDA is limited in its ability to treat long-range charge transfer states; in particular, the excitation energies of these states are chronically underestimated. This problem can be attributed to the so-called self-interaction error of DFT, which can result in excessive charge delocalization. Efforts to overcome this problem include the development of new exchange–correlation functionals which include non-local components e.g.ref. 44, and methods which aim to minimize this delocalization error such as constrained DFT.45,46 The results obtained in this work for charge transfer states should therefore be treated with caution and future calculations using for example an alternative functional would be recommended to confirm the above observations.
In practice, it is likely that chemically substituted PPP and PPV or other related conjugated polymers, such as poly[2-methoxy, 5-(2-ethylhexoxy)-1,4-phenylene vinylene] (MEH–PPV) or poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-p-phenylene vinylene] (MDMO–PPV), might be used instead of pure PPP and PPV, due to the increased solubility that can be achieved in organic solvents, which is useful for easy fabrication of electronic devices. Similarly, a lot of interest has been generated by fullerene derivatives such as [6,6]-phenyl-C60 butyric acid methyl ester (PCBM) as an alternative to pure C60 – there have, for example, been a number of studies on MDMO–PPV in conjunction with PCBM, including the development of new techniques for experimental characterization.47–50 Furthermore, it would not necessarily be possible to easily manufacture the structures investigated in this work. The above simplifications are justified as a first approximation in allowing us to model the absorption spectra of fullerene-conjugated polymer hybrids whilst keeping the computational costs low. However, it would be interesting to extend this work in future to the consideration of other hybrids such as those mentioned above, which, despite the larger unit cells involved, will still be fully accessible within a LS-DFT framework.
4 Conclusions
In conclusion, we have investigated the effect on the optical absorption spectra of the attachment of molecules of C60 to the two conjugated polymers PPP and PPV using a LS-DFT formalism, which has allowed us to simulate long chains with a realistic proportion of C60 with respect to the polymer chain length. For PPP, we considered two different bonding structures between the C60 and the chain. For both structures of the C60–PPP hybrid additional peaks were present in the absorption spectra below the original onset of absorption. By identifying the transitions between Kohn–Sham eigenstates which were responsible for these peaks and using the optical matrix elements to form a weighted density difference, we were able to demonstrate the transfer of electrons from the length of the chain to the C60 molecule, agreeing with experimental observations of charge transfer. For the PPV we did not find any additional peaks below the original onset of absorption, which is likely an indication that the bonding structure selected here is not optimal. Further calculations could be done in future on different bonding structures of C60–PPV such as structure B, in order to confirm this theory.A number of approximations and simplifications were made, including the use of a purely DFT based formalism, which was justified based on the impracticality of going beyond DFT for such large systems. In addition, relaxation of the hybrid structure was shown to affect the details of the absorption spectrum but not the overall picture and thus the use of unoptimized structures was also justified. Furthermore, whilst the structures used were not necessarily those directly achievable in experiment or likely to be the precise systems of interest for photovoltaic systems given that interest has shifted from pure PPP and PPV to various derivatives, this work provides an ideal starting point for the study of large fullerene–polymer complexes. Finally, this work could be easily extended in future to consider more complex polymers and fullerene–polymer combinations; indeed the linear-scaling formalism used here is ideal for the computational study of such materials and should prove to be useful in gaining further insights into systems which are of great interest for organic photovoltaics.
Acknowledgements
This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC). Calculations were performed on CX1 (Imperial College London High Performance Computing Service). P.D.H. acknowledges support from the Royal Society in the form of a University Research Fellowship.References
- A. J. Heeger, Rev. Mod. Phys., 2001, 73, 681–700 CrossRef CAS.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- H. Shirakawa, E. Louis, A. MacDiarmid, C. Chiang and A. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- G. Yu, Synth. Met., 1996, 80, 143–150 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- R. Friend, Pure Appl. Chem., 2001, 73, 425–430 CrossRef CAS.
- A. Moliton and R. C. Hiorns, Polym. Int., 2004, 53, 1397–1412 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS.
- Y. Wang, Nature, 1992, 356, 585–587 CrossRef CAS.
- L. Smilowitz, N. Sariciftci, R. Wu, C. Gettinger, A. Heeger and F. Wudl, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 13835 CrossRef CAS.
- P. Lane, M. Liess, Z. Vardeny, M. Hamaguchi, M. Ozaki and K. Yoshino, Synth. Met., 1997, 84, 641–642 CrossRef CAS.
- J. Halls, K. Pichler, R. Friend, S. Moratti and A. Holmes, Appl. Phys. Lett., 1996, 68, 3120–3122 CrossRef CAS.
- J. Halls, K. Pichler, R. Friend, S. Moratti and A. Holmes, Synth. Met., 1996, 77, 277–280 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- C. Ambrosch-Draxl, J. A. Majewski, P. Vogl and G. Leising, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 9668–9676 CrossRef CAS.
- L. Hedin, Phys. Rev., 1965, 139, A796–A823 CrossRef.
- R. W. Godby and R. J. Needs, Phys. Scr., 1990, T31, 227–231 CrossRef CAS.
- R. Del Sole, L. Reining and R. W. Godby, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 8024–8028 CrossRef CAS.
- L. E. Ratcliff, N. D. M. Hine and P. D. Haynes, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 165131 CrossRef.
- L. Ratcliff, Optical Absorption Spectra Calculated Using Linear-Scaling Density-Functional Theory, Springer, Heidelberg, 2013 Search PubMed.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Chem. Phys., 2005, 122, 084119 CrossRef.
- P. D. Haynes, C.-K. Skylaris, A. A. Mostofi and M. C. Payne, Phys. Status Solidi B, 2006, 243, 2489–2499 CrossRef CAS.
- N. D. M. Hine, P. D. Haynes, A. A. Mostofi, C.-K. Skylaris and M. C. Payne, Comput. Phys. Commun., 2009, 180, 1041–1053 CrossRef CAS.
- N. D. M. Hine, M. Robinson, P. D. Haynes, C.-K. Skylaris, M. C. Payne and A. A. Mostofi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195102 CrossRef.
- S. Ismail-Beigi and T. A. Arias, Phys. Rev. Lett., 1999, 82, 2127–2130 CrossRef CAS.
- W. Kohn, Int. J. Quantum Chem., 1995, 56, 229–232 CrossRef CAS.
- L. He and D. Vanderbilt, Phys. Rev. Lett., 2001, 86, 5341–5344 CrossRef CAS.
- C.-K. Skylaris, A. A. Mostofi, P. D. Haynes, O. Diéguez and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 035119 CrossRef.
- A. A. Mostofi, P. D. Haynes, C.-K. Skylaris and M. C. Payne, J. Chem. Phys., 2003, 119, 8842–8848 CrossRef CAS.
- R. McWeeny, Rev. Mod. Phys., 1960, 32, 335–369 CrossRef.
- E. Hernández and M. J. Gillan, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 10157–10160 CrossRef.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Phys.: Condens. Matter, 2005, 17, 5757 CrossRef CAS.
- C. J. Pickard, PhD thesis, University of Cambridge, 1997.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- A. J. Read and R. J. Needs, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13071–13073 CrossRef.
- E. Badamshina and M. Gafurova, Polym. Sci., Ser. B, 2008, 50, 215–225 CrossRef.
- C. Wang, Z.-X. Guo, S. Fu, W. Wu and D. Zhu, Prog. Polym. Sci., 2004, 29, 1079–1141 CrossRef CAS.
- A. Kokalj, Comput. Mater. Sci., 2003, 28, 155–168 CrossRef CAS.
- M. Rohlfing, M. Tiago and S. G. Louie, Synth. Met., 2001, 116, 101–105 CrossRef CAS.
- T. G. Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075207 CrossRef.
- T. G. Pedersen, J. Non-Cryst. Solids, 2006, 352, 2488–2491 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, J. Chem. Phys., 2007, 126, 191109 CrossRef.
- P. H. Dederichs, S. Blügel, R. Zeller and H. Akai, Phys. Rev. Lett., 1984, 53, 2512–2515 CrossRef CAS.
- Q. Wu and T. Van Voorhis, Phys. Rev. A, 2005, 72, 024502 CrossRef.
- L. Goris, K. Haenen, M. Nesládek, P. Wagner, D. Vanderzande, L. Schepper, J. Dhaen, L. Lutsen and J. Manca, J. Mater. Sci., 2005, 40, 1413–1418 CrossRef CAS.
- L. Goris, A. Poruba, L. Hodakova, M. Vanecek, K. Haenen, M. Nesladek, P. Wagner, D. Vanderzande, L. De Schepper and J. Manca, Appl. Phys. Lett., 2006, 88, 052113 CrossRef.
- W. Gong, M. A. Faist, N. J. Ekins-Daukes, Z. Xu, D. D. C. Bradley, J. Nelson and T. Kirchartz, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 024201 CrossRef.
- M. Faist, T. Kirchartz, W. Gong, R. Ashraf, I. McCulloch, J. de Mello, N. Ekins-Daukes, D. Bradley and J. Nelson, J. Am. Chem. Soc., 2011, 134, 685–692 CrossRef.
| This journal is © the Owner Societies 2013 |