 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst example of a high-level correlated calculation of the indirect spin–spin coupling constants involving tellurium: tellurophene and divinyl telluride†
Yury Yu.
Rusakov
a,
Leonid B.
Krivdin
*a,
Freja F.
Østerstrøm
b,
Stephan P. A.
Sauer
b,
Vladimir A.
Potapov
a and
Svetlana V.
Amosova
a
aA.E. Favorsky Irkutsk Institute of Chemistry, Russian Academy of Sciences, Siberian Branch, 1 Favorsky St., 664033 Irkutsk, Russia. E-mail: krivdin_office@irioch.irk.ru
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark
First published on 3rd July 2013
Abstract
This paper documents the very first example of a high-level correlated calculation of spin–spin coupling constants involving tellurium taking into account relativistic effects, vibrational corrections and solvent effects for medium sized organotellurium molecules. The 125Te–1H spin–spin coupling constants of tellurophene and divinyl telluride were calculated at the SOPPA and DFT levels, in good agreement with experimental data. A new full-electron basis set, av3z-J, for tellurium derived from the “relativistic” Dyall's basis set, dyall.av3z, and specifically optimized for the correlated calculations of spin–spin coupling constants involving tellurium was developed. The SOPPA method shows a much better performance compared to DFT, if relativistic effects calculated within the ZORA scheme are taken into account. Vibrational and solvent corrections are next to negligible, while conformational averaging is of prime importance in the calculation of 125Te–1H spin–spin couplings. Based on the performed calculations at the SOPPA(CCSD) level, a marked stereospecificity of geminal and vicinal 125Te–1H spin–spin coupling constants originating in the orientational lone pair effect of tellurium has been established, which opens a new guideline in organotellurium stereochemistry.
Introduction
Recent advances in the implementation of different correlated methods, such as SOPPA, SOPPA(CC2), SOPPA(CCSD), EOM-CCSD, CCSD and various density functional methods, for the calculation of spin–spin coupling constants1–6 has resulted in a vast amount of papers dealing with the calculation of spin–spin coupling constants of different types involving the most popular magnetic isotopes – 1H, 13C, 15N, 19F, 29Si, 31P and even 77Se, at the modern high-level non-empirical and DFT levels (for references, see the reviews in ref. 2,3,7,8). To the best of our knowledge, no correlated non-empirical calculations have been attempted for the spin–spin couplings involving “heavy” 125Te, which is also a popular isotope among the NMR community worldwide. There are two commonplace reasons for this gap: firstly, the absence of a full-electron tellurium basis set of sufficient quality in most popular program codes, and secondly, the traditional “scare” of relativistic effects which, for certain, should be of particular importance for this type of spin–spin coupling constants and which are not implemented for the calculation of spin–spin couplings in most quantum chemical programs. In the present paper, we report the first example of such a calculation performed for 125Te–1H coupling constants in tellurophene (1) and divinyl telluride (2) using our newly developed full-electron tellurium basis set, specifically optimized for the correlated calculations of spin–spin coupling constants involving tellurium.
Apart from being of purely theoretical interest, tellurophene and divinyl telluride are important starting materials for organic synthesis and the preparation of new organotellurium compounds.9–15 As an example of the first type (i.e., dealing with organic synthesis), diaryl tellurophenes are involved in the tellurium–lithium exchange reaction giving 1,4-dilithio-1,3-butadienes, which can be further applied for the preparation of siloles.15 As an example of the second type (i.e., preparing organotellurium compounds), a number of important substituted divinyl tellurides, prospective monomers in polymer synthesis, were obtained recently from tellurium tetrahalides and acetylene.16–18
For the conformationally rigid tellurophene, we performed calculations of geminal and vicinal 125Te–1H coupling constants at different levels of theory, taking into account relativistic effects, vibrational corrections and solvent effects, while for the conformationally labile divinyl telluride, such calculations were carried out with the emphasis on the conformational effects. As a result, a marked stereospecificity of geminal and vicinal tellurium–hydrogen spin–spin coupling constants across double bonds originating in the orientational lone pair effect of tellurium (amounting to more than 100 Hz!) has been established, which provides a new guideline in organotellurium stereochemistry.
Experimental details
General synthetic procedure
Tellurophene (1) was obtained from elemental tellurium and diacetylene,19 and divinyl telluride (2) was prepared from elemental tellurium and acetylene.13,14 Efficient syntheses of tellurophene and divinyl telluride in high yields were achieved via the generation of a telluride anion from elemental tellurium by the action of KOH and reducing reagents (tin dichloride, hydrazine hydrate) followed by the addition of the telluride anion to the triple bond of acetylene or diacetylene.13,14,19 Compounds 1 and 2 were identified based on their 1H, 13C and 125Te NMRspectra.NMR measurements
Experimental values of J(Te,H) were obtained in chloroform-d from the proton-coupled 125Te NMR spectra, as shown in Fig. 1 for tellurophene. 1H, 13C and 125Te NMRspectra were recorded on a Bruker AVANCE-400 spectrometer (1H, 400.13 MHz; 13C, 100.62 MHz; 125Te, 126.24 MHz) in a 5 mm broadband probe at 25 °C in CDCl3 with hexamethyldisiloxane (1H, 13C) and dimethyltelluride (125Te) as internal standards. Experimental measurements of 125Te–1H spin–spin coupling constants were carried out from the proton-coupled 125Te spectra accumulated overnight using a spectral width of 4 kHz with 16 K data points. | ||
| Fig. 1 Proton-coupled 125Te NMRspectrum of tellurophene (1) in CDCl3 at 25 °C (126.24 MHz). The chemical shift scale (δ, ppm) is referenced to Me2Te used as an internal standard. | ||
Computational strategy
Quantum chemical methods
First of all, we used the conformationally rigid tellurophene (1) to perform benchmark calculations of its two possible spin–spin coupling constants involving tellurium and the protons of the tellurophene ring, geminal 2J(Te,H) and vicinal 3J(Te,H), at two different levels of theory – the second order polarization propagator approach (SOPPA) and, on the other hand, density functional theory (DFT). For the former (SOPPA), we applied the known methods – namely, the parent SOPPA20–23 itself in combination with CC2, known as SOPPA(CC2),24,25 and with CCSD, referred to as SOPPA(CCSD).23,26 In the general SOPPA formalism, the ground-state wavefunction used in the calculation of spin–spin coupling constants as linear-response property is approximated with the MP2 wavefunction, while in the SOPPA(CC2) and SOPPA(CCSD) methods, the MP2 correlation coefficients are replaced accordingly with CC2 and CCSD single and double amplitudes, which is regarded24,26 to generally improve the description of the electron correlation at the MP2 level. For the latter (DFT), we used the most common three-parameter hybrid functional of Becke27 in combination with the correlation functional of Lee, Yang and Parr,28 the so-called B3LYP, and the parameter-free generalized gradient functional of Perdew, Burke and Ernzerhof (PBE)29 with a predetermined amount of exact exchange, known as PBE0.30,31 Both functionals, B3LYP and PBE0, demonstrated a rather good performance in our recent calculations of 29Si–1H spin–spin couplings across the double bonds in the structurally related alkenylsilanes,32,33 so it was believed that they would be appropriate for the present case as well.Scalar relativistic corrections were calculated with the zero order regular approximation34–39 (known as ZORA) at the DFT-B3LYP level.
Both vibrational and solvent corrections were evaluated at the DFT-B3LYP level, the former using the zero-point vibrational energy (ZPVE) approach, as described by Ruud et al.,40 while the latter within the PCM scheme for chloroform.
Basis sets
In the non-relativistic calculations of the coupling constants, we employed Sauer's standard contracted basis set, aug-cc-pVTZ-J,41 for the hydrogens involved in spin–spin coupling with tellurium while standard Dunning's cc-pVDZ basis sets42–44 were used throughout for all uncoupled atoms. For tellurium, we used a full-electron basis set derived in this paper (and referred to further on as av3z-J) from the “relativistic” Dyall's basis set, dyall.av3z,45,46 motivated by the common practice of combining Dyall's and Dunning's basis sets42–44 in calculations of spectral parameters of molecules containing heavy elements, e.g. Hg(CH3)2.47–50 Here, we use different basis sets for the coupled and uncoupled atoms in the calculations of tellurium–hydrogen coupling constants based on the general idea of locally dense basis sets employing large sets of functions on a particular atom or small molecular region and smaller or attenuated sets elsewhere.51–53 This approach has been shown to be very effective in calculating spin–spin coupling constants of many different types at the highly computationally demanding SOPPA, SOPPA(CC2) and SOPPA(CCSD) levels at lower computational costs and reliable accuracy when coupled atoms are specified within the triple zeta basis sets augmented with tight s-functions, while uncoupled atoms are determined with the standard basis sets of double zeta quality (for references, see the review in ref. 6).The av3z-J basis set was specifically optimized for the correlated calculations of spin–spin coupling constants involving tellurium using our original technique applied earlier for the creation of the aug-cc-pVTZ-J family of basis sets,23,41,54–58 which is essentially to extend the basis set until convergence of the coupling constants is achieved. As a test molecule, we employed the simplest hydride of tellurium, H2Te, whose geometry had been optimized at the MP2 level with the av3z/aug-cc-pVTZ basis set. In a series of SOPPA calculations on H2Te with Dyall's completely uncontracted av3z basis set for Te and the aug-cc-pVTZ-J basis set for H, it was determined that the 1J(125Te,1H) coupling constant in H2Te is not significantly changed by extending the dyall.av3z Te basis set with additional s-, p-, d- or f-type functions with larger exponents. In a second series of calculations, an optimal contraction scheme for the dyall.av3z Te basis set consisting of 29 s-, 22 p-, 16 d- and 2 f-type sets of functions was then searched, which would change the 1J(125Te,1H) coupling constant by less than 1 Hz. Generally in the aug-cc-pVTZ-J family of basis sets, the molecular orbital coefficients of the dyall.av3z/aug-cc-pVTZ-J Hartree–Fock calculation on H2Te were employed as contraction coefficients. First, a contraction scheme for the s-type functions was determined followed by the p- and d-type functions. The final, contracted av3z-J basis set for tellurium then consists of 19 s-, 14 p-, 8 d- and 2 f-type sets of functions, as shown in the ESI.†
In the calculation of the relativistic corrections at the ZORA-DFT-B3LYP level, a Slater-type TZ2P basis set59 was used.
Software
All the geometry optimizations were performed with the GAMESS code60 at the MP2/aug-cc-pVTZ-pp level without symmetry constraints, taking into account solvent effects (chloroform) within the PCM scheme with the exception of the geometry optimization of H2Te during the generation of the av3z-J basis set, which was carried out with the DALTON package61 at the MP2/av3z/aug-cc-pVTZ level. Theoretical values of J(Te,H) were calculated as a sum of the four non-relativistic coupling contributions to the total coupling, J, namely, the Fermi contact, JFC, spin–dipolar, JSD, diamagnetic spin–orbital, JDSO, and paramagnetic spin–orbital, JPSO, terms. The SOPPA, SOPPA(CC2), SOPPA(CCSD) and DFT calculations of the 125Te–1H spin–spin coupling constants together with their solvent and vibrational corrections have been carried out with the DALTON package.23,61–63 Relativistic corrections to the 125Te–1H spin–spin coupling constants were calculated at the ZORA-DFT-B3LYP/TZ2P level within the ADF program code.64 The NBO analysis of divinyl telluride was performed at the HF level using the GAUSSIAN 09 suite of programs.65Results and discussion
The non-relativistic values of the 125Te–1H coupling constants of tellurophene calculated at different SOPPA and DFT levels in comparison with the experimental data are compiled in Table 1. It follows that for both couplings, 2J(Te,H) and 3J(Te,H), the Fermi contact contribution dominates. However, for the vicinal coupling, 3J(Te,H), the overall contribution of the non-contact terms is almost negligible, while for the geminal coupling constant, 2J(Te,H), the contribution of the paramagnetic spin–orbital term is about 11 Hz (at the SOPPA level) or even 14 Hz (at the DFT level), which is essentially more than 10% of its total value and is thus, far from negligible.| Spin–spin coupling constant | Method | J DSO | J PSO | J SD | J FC | J |
|---|---|---|---|---|---|---|
| All couplings and coupling contributions are in Hz.a Measured in CDCl3 at 25 °C. | ||||||
| 2 J(Te,H) | DFT-B3LYP | 0.3 | 14.2 | −0.3 | −114.5 | −100.3 |
| DFT-PBE0 | 0.3 | 14.0 | −0.2 | −108.5 | −94.4 | |
| SOPPA | 0.3 | 11.4 | −0.6 | −101.9 | −90.8 | |
| SOPPA(CC2) | 0.3 | 11.1 | −0.5 | −100.8 | −89.9 | |
| SOPPA(CCSD) | 0.3 | 11.4 | −0.6 | −98.5 | −87.4 | |
| Experimenta | (−)101.7 | |||||
| 3 J(Te,H) | DFT-B3LYP | 0.5 | −0.4 | −1.2 | −20.4 | −21.5 |
| DFT-PBE0 | 0.5 | −0.6 | −1.3 | −17.9 | −19.3 | |
| SOPPA | 0.5 | −0.7 | −0.9 | −27.8 | −28.9 | |
| SOPPA(CC2) | 0.5 | −0.6 | −0.9 | −28.1 | −29.1 | |
| SOPPA(CCSD) | 0.5 | −0.7 | −0.9 | −26.3 | −27.4 | |
| Experimenta | (−)20.2 | |||||
The most interesting conclusion which can be reached from the analysis of the data presented in Table 1 is that at the non-relativistic level, both DFT methods give better results in comparison with the experimental data than any of the SOPPA methods.
However, in our opinion, the better performance of DFT in this particular case is nothing more than a fortuitous cancellation of errors due to not reaching the complete basis set limit,66–70 the well-known phenomenon in computational quantum chemistry. Indeed, taking into account the relativistic effects calculated with the zero order regular approximation together with vibrational and solvent effects drastically changes the situation, as can be seen from the data given in Table 2.
| Spin–spin coupling constant | Method | J nrel | ΔJrel | ΔJvib | ΔJsol | J |
|---|---|---|---|---|---|---|
| All couplings and coupling contributions are in Hz. All relativistic, vibrational and solvent corrections are calculated at the DFT-B3LYP level.a Measured in CDCl3 at 25 °C. | ||||||
| 2 J(Te,H) | DFT-B3LYP | −100.3 | −17.6 | −0.1 | −0.1 | −118.1 |
| DFT-PBE0 | −94.4 | −17.6 | −0.1 | −0.1 | −112.2 | |
| SOPPA | −90.8 | −17.6 | −0.1 | −0.1 | −108.6 | |
| SOPPA(CC2) | −89.9 | −17.6 | −0.1 | −0.1 | −107.7 | |
| SOPPA(CCSD) | −87.4 | −17.6 | −0.1 | −0.1 | −105.2 | |
| Experimenta | (−)101.7 | |||||
| 3 J(Te,H) | DFT-B3LYP | −21.5 | 5.1 | 0.6 | −0.6 | −16.4 |
| DFT-PBE0 | −19.3 | 5.1 | 0.6 | −0.6 | −14.2 | |
| SOPPA | −28.9 | 5.1 | 0.6 | −0.6 | −23.8 | |
| SOPPA(CC2) | −29.1 | 5.1 | 0.6 | −0.6 | −24.0 | |
| SOPPA(CCSD) | −27.4 | 5.1 | 0.6 | −0.6 | −22.3 | |
| Experimenta | (−)20.2 | |||||
It follows that ZPVE and PCM corrections (ΔJvib and ΔJsol) are of minor importance for both 2J(Te,H) and 3J(Te,H) couplings while scalar relativistic effects (ΔJrel) contribute to the calculated values of these couplings by up to ca. −18 and +5 Hz, respectively, which totals to accordingly, 17 and 23% of their non-relativistic values, see Table 2. It can now clearly be seen that taking into account relativistic corrections essentially worsens the DFT results and dramatically improves the performance of the SOPPA methods, the latter results being in very good agreement with the experimental results.
For the conformationally labile divinyl telluride, prior to the calculations of the 125Te–1H spin–spin coupling constants, we performed a theoretical conformational analysis at the MP2/aug-cc-pVTZ-pp level taking into account solvent effects (chloroform) within the PCM scheme, which revealed the existence of three true-minimum rotational conformers with a ratio of A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B:C = 65:20:15 at 300 K (within the Boltzmann distribution scheme) confirmed by the vibrational harmonic analysis (Fig. 2). In the most favorable conformer A, the vinyl groups are in the skewed s-cis and s-trans orientations with out-of-plane deviations of accordingly 16 and 51°. On the other hand, the B and C conformers are characterized by the s-trans orientation of both vinyl groups in each conformer, with out-of-plane deviations of 46 and 36°, respectively.
:B:C = 65:20:15 at 300 K (within the Boltzmann distribution scheme) confirmed by the vibrational harmonic analysis (Fig. 2). In the most favorable conformer A, the vinyl groups are in the skewed s-cis and s-trans orientations with out-of-plane deviations of accordingly 16 and 51°. On the other hand, the B and C conformers are characterized by the s-trans orientation of both vinyl groups in each conformer, with out-of-plane deviations of 46 and 36°, respectively.
 | ||
| Fig. 2 Rotational conformers of divinyl telluride localized at the MP2/aug-cc-pVTZ-pp level taking into account solvent effects (CDCl3) within the PCM scheme. Given in the parenthesis are their relative energies (kJ mol−1). Element colors: tellurium – tawny, carbon – grey, hydrogen – light grey. | ||
Given in Table 3 are the conformationally averaged 125Te–1H spin–spin coupling constants in divinyl telluride calculated at the SOPPA(CCSD) level including relativistic corrections, which are in a very good agreement with their experimental values. This is a very encouraging result and it is even more encouraging in view of the fact that the total “relativistic” values of 2J(Te,HX) differ dramatically in different conformers of 2 (e.g., −57 Hz in A and −10 Hz in C), which means that conformational averaging together with relativistic corrections are of crucial importance for the high-level correlated calculations of 125Te–1H spin–spin coupling constants.
|
|
||||||
|---|---|---|---|---|---|---|
| Spin–spin coupling constant | Conformer | J nrel | ΔJrel | J | ΔJavera | Experimentb |
| All couplings and coupling contributions are in Hz.a Conformationally averaged.b Measured in CDCl3 at 25 °C. | ||||||
| 2 J(Te,HX) | A | −57.4 | −12.9 | −70.3 | −50.5 | (−)48.7 |
| B | −13.6 | −1.1 | −14.7 | |||
| C | −10.3 | −0.9 | −11.2 | |||
| 3 J(Te,HA) | A | −41.2 | 8.6 | −32.6 | −39.1 | (−)43.0 |
| B | −56.1 | 6.2 | −49.9 | |||
| C | −57.8 | 4.4 | −53.4 | |||
| 3 J(Te,HB) | A | −22.6 | 3.9 | −18.7 | −20.5 | (−)20.7 |
| B | −26.8 | 3.0 | 23.8 | |||
| C | −26.9 | 2.9 | 24.0 | |||
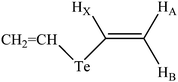
Another interesting consequence which follows from the dramatic difference of 2J(Te,HX) in the different rotational conformers of 2 is that this coupling should demonstrate a marked stereospecificity with respect to the internal rotation around the Te–C bond. In our previous publications, we have reported similar effects for J(P,HX) and J(Se,HX) in the structurally related trivinyl phosphine71 and divinyl selenide72 and explained their stereochemical behavior in terms of the lone pair effect of phosphorous and selenium. It now appears that the lone pair of tellurium provides the same effect on the 125Te–1H spin–spin coupling constants. In this respect, it should be outlined that according to the NBO analysis performed in this study, the tellurium atom possesses two different lone pairs, one of almost pure p-character (p-type) and another one providing a considerable amount of s-character (σ-type) and being in the orthogonal position to the former. Shown in Fig. 3 are the naturally localized molecular orbitals describing both types of lone pairs in 1 and 2: the p-type (Fig. 3a and c) and σ-type (Fig. 3b and d), and it is noteworthy that based on our previous findings,73 it should be the lone pair of a σ-type which provides the so-called orientational lone pair effect upon the values of the 125Te–1H coupling constants under consideration.
 | ||
| Fig. 3 The p- and σ-type tellurium lone pairs in tellurophene (a and b) and divinyl telluride (c and d), as follows from the NBO analysis. | ||
To verify all these arguments which deal with the tellurium orientational lone pair effect, we calculated the dihedral angle dependences of all three 125Te–1H couplings, 2J(Te,HX), 3J(Te,HA) and 3J(Te,HB), in divinyl telluride (2) with respect to the internal rotation around the Te–C bond, see Fig. 4. Negative geminal coupling, 2J(Te,HX), provides the most striking behavior, decreasing (increasing in absolute value) by more than 100 Hz (!) on going from an s-trans (φ = 0°) to an s-cis (φ = 180°) arrangement of the vinyl group rotating around the Te–C bond, which corresponds to transoidal and cisoidal orientations of the tellurium lone pair towards the geminal 125Te–1H coupling pathway. The same effect but of smaller value and of opposite sign were found earlier for the positive geminal couplings, 2J(P,HX) and 2J(Se,HX), in trivinyl phosphine71 and divinyl selenide.72 Both negative transoidal, 3J(Te,HA), and cisoidal, 3J(Te,HB), vicinal couplings markedly increase (decrease in absolute value) on going from a transoidal to a cisoidal orientation of the tellurium lone pair towards the vicinal 125Te–1H coupling pathway. The same effect was also reported for the 3J(P,H) and 3J(Se,H) couplings due to the orientational lone pair effect of phosphorous and selenium.71,72
 | ||
| Fig. 4 Dihedral angle dependences of the 2J(Te,H) and 3J(Te,H) coupling constants in divinyl telluride (2) calculated at the SOPPA(CCSD) level. The value of φ = 0° is assigned to the s-trans orientation of the vinyl group rotating around the Te–C bond, as shown. | ||
Thus, it follows that geminal and vicinal 125Te–1H coupling constants show marked stereospecificity originating in the orientational lone pair effect of tellurium, in line with the same effects observed for phosphorous71,74,75 and selenium,56,72,73,76–81 which is of prime importance for the stereochemical studies of organotellurium compounds.
Concluding remarks
The present paper documents the first example of a high-level correlated calculation of the 125Te–1H spin–spin coupling constants in tellurophene and divinyl telluride using a newly developed full-electron basis set, av3z-J, for tellurium, which was derived from the “relativistic” Dyall's basis set, dyall.av3z, and specifically optimized for the correlated calculations of spin–spin coupling constants involving tellurium. This new basis set can and will be used for further high-level calculations of spin–spin coupling constants involving tellurium in a wide series of organotellurium compounds. Very good agreement with the experimental results is achieved at the SOPPA, SOPPA(CC2) and SOPPA(CCSD) levels provided that the relativistic effects calculated within the ZORA scheme are taken into account. This is a very encouraging result in view of the prospective calculations of 125Te–1H spin–spin coupling constants in a wide variety of organotellurium compounds. It is noteworthy that for the conformationally labile organotellurium molecules, the conformational averaging of the calculated 125Te–1H spin–spin couplings is a crucial point, while the vibrational and solvent corrections are of only minor importance. The second distinguished conclusion reached in this study is the established marked stereospecificity of geminal and vicinal 125Te–1H spin–spin coupling constants originating in the orientational lone pair effect of tellurium, which provides a new guideline to the stereochemical structure of organotellurium compounds.Acknowledgements
Financial support from the Russian Foundation for Basic Research (Grant No. 11-03-00022a) and the Danish Center for Scientific Computing is greatly acknowledged.Notes and references
- T. Helgaker, M. Jaszuński and K. Ruud, Chem. Rev., 1999, 99, 293 CrossRef CAS
.
- J. Vaara, Phys. Chem. Chem. Phys., 2007, 9, 5399 RSC
- L. B. Krivdin and R. H. Contreras, Annu. Rep. NMR Spectrosc., 2007, 61, 133 CAS
- T. Helgaker, M. Jaszuński and M. Pecul, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 249 CrossRef CAS
- T. Helgaker, S. Coriani, P. Jørgensen, K. Kristensen, J. Olsen and K. Ruud, Chem. Rev., 2012, 112, 543 CrossRef CAS
- Y. Y. Rusakov and L. B. Krivdin, Russ. Chem. Rev., 2013, 82, 99 Search PubMed
- R. H. Contreras, J. E. Peralta, C. G. Giribet, M. C. Ruiz de Azua and J. C. Facelli, Annu. Rep. NMR Spectrosc., 2000, 41, 55 CrossRef CAS
- R. H. Contreras, V. Barone, J. C. Facelli and J. E. Peralta, Annu. Rep. NMR Spectrosc., 2003, 51, 167 CAS
- G. Zeni, D. S. Ludtke, R. B. Panatieri and A. L. Braga, Chem. Rev., 2006, 106, 1032 CrossRef CAS
- V. I. Minkin and I. D. Sadekov, Compr. Heterocycl. Chem. III, 2008, 3, 1007 Search PubMed
- N. K. Gusarova, A. A. Tatarinova and L. M. Sinegovskaya, Sulfur Rep., 1991, 11, 1 Search PubMed
- F. Fringuelli, G. Marino and A. Taticchi, Adv. Heterocycl. Chem., 1977, 21, 119 CAS
- B. A. Trofimov, S. V. Amosova, N. K. Gusarova, V. A. Potapov and A. A. Tatarinova, Sulfur Lett., 1983, 1, 151 Search PubMed
- N. K. Gusarova, B. A. Trofimov, A. A. Tatarinova, V. A. Potapov, A. V. Gusarov, S. V. Amosova and M. G. Voronkov, Zh. Org. Khim., 1989, 25, 39 (
Chem. Abstr.
, 1989
, 111
, 114711
) Search PubMed
- M. Katkevics, S. Yamaguchi, A. Toshimitsu and K. Tamao, Organometallics, 1998, 17, 5796 CrossRef CAS
- M. V. Musalova, S. V. Amosova and V. A. Potapov, Molecules, 2012, 17, 5770 Search PubMed
- V. A. Potapov, M. V. Musalova and S. V. Amosova, Russ. Chem. Bull., 2012, 61, 204 Search PubMed
- V. A. Potapov, M. V. Musalov, M. V. Musalova and S. V. Amosova, Russ. Chem. Bull., 2009, 58, 2404 Search PubMed
- V. A. Potapov and S. V. Amosova, Metalloorg. Khim., 1990, 3, 1197 (
Chem. Abstr.
, 1991
, 114
, 121949
) Search PubMed
- E. S. Nielsen, P. Jørgensen and J. Oddershede, J. Chem. Phys., 1980, 73, 6238 CrossRef CAS
- M. J. Packer, E. K. Dalskov, T. Enevoldsen, H. J. Aa. Jensen and J. Oddershede, J. Chem. Phys., 1996, 105, 5886 CrossRef CAS
- K. L. Bak, H. Koch, J. Oddershede, O. Christiansen and S. P. A. Sauer, J. Chem. Phys., 2000, 112, 4173 CrossRef CAS
- T. Enevoldsen, J. Oddershede and S. P. A. Sauer, Theor. Chem. Acc., 1998, 100, 275 CrossRef CAS
- H. Kjær, S. P. A. Sauer and J. Kongsted, J. Chem. Phys., 2010, 133, 144106 Search PubMed
- H. Kjær, S. P. A. Sauer, J. Kongsted, Y. Yu. Rusakov and L. B. Krivdin, Chem. Phys., 2011, 381, 35 Search PubMed
- S. P. A. Sauer, J. Phys. B: At., Mol. Opt. Phys., 1997, 30, 3773 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS
- Y. Y. Rusakov, L. B. Krivdin, V. M. Nosova and A. V. Kisin, Magn. Reson. Chem., 2012, 50, 278 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, V. M. Nosova, A. V. Kisin and V. G. Lakhtin, Magn. Reson. Chem., 2012, 50, 665 Search PubMed
- C. Chang, M. Pelissier and P. Durand, Phys. Scr., 1986, 34, 394 CAS
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505 CrossRef CAS
- S. K. Wolff, T. Ziegler, E. van Lenthe and E. J. Baerends, J. Chem. Phys., 1999, 110, 7689 CrossRef CAS
- J. Autschbach and T. Ziegler, J. Chem. Phys., 2000, 113, 936 CrossRef CAS
- K. Ruud, P.-O. Åstrand and P. R. Taylor, J. Chem. Phys., 2000, 112, 2668 CrossRef CAS
- P. F. Provasi, G. A. Aucar and S. P. A. Sauer, J. Chem. Phys., 2001, 115, 1324 CrossRef CAS
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358 CrossRef CAS
- K. G. Dyall, Theor. Chem. Acc., 2002, 108, 335 CrossRef CAS
- K. G. Dyall, Theor. Chem. Acc., 2006, 115, 441 CrossRef CAS
- V. Arcisauskaite, J. I. Melo, L. Hemmingsen and S. P. A. Sauer, J. Chem. Phys., 2011, 135, 044306 CrossRef
- V. Arcisauskaite, S. Knecht, S. P. A. Sauer and L. Hemmingsen, Phys. Chem. Chem. Phys., 2012, 14, 2651 RSC
- V. Arcisauskaite, S. Knecht, S. P. A. Sauer and L. Hemmingsen, Phys. Chem. Chem. Phys., 2012, 14, 16775 RSC
- V. Arcisauskaite, S. Knecht, S. P. A. Sauer and L. Hemmingsen, Phys. Chem. Chem. Phys., 2012, 14, 16070 RSC
- D. B. Chesnut and E. F. C. Byrd, Chem. Phys., 1996, 213, 153 CrossRef CAS
- P. F. Provasi, G. A. Aucar and S. P. A. Sauer, J. Chem. Phys., 2000, 112, 6201 Search PubMed
- M. Sanchez, P. F. Provasi, G. A. Aucar and S. P. A. Sauer, Adv. Quantum Chem., 2005, 48, 161 Search PubMed
- S. P. A. Sauer and W. T. Raynes, J. Chem. Phys., 2000, 113, 3121 CrossRef CAS
- V. Barone, P. F. Provasi, J. E. Peralta, J. P. Snyder, S. P. A. Sauer and R. H. Contreras, J. Phys. Chem. A, 2003, 107, 4748 CrossRef CAS
- Y. Yu. Rusakov, L. B. Krivdin, S. P. A. Sauer, E. P. Levanova and G. G. Levkovskaya, Magn. Reson. Chem., 2010, 48, 44 CAS
- P. F. Provasi and S. P. A. Sauer, J. Chem. Phys., 2010, 133, 054308 CrossRef
- E. D. Hedegård, J. Kongsted and S. P. A. Sauer, J. Chem. Theory Comput., 2011, 7, 4077 CrossRef
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS
- DALTON, A Molecular Electronic Structure Program, Release Dalton2011 (2011), see http://daltonprogram.org/.
- O. Vahtras, H. Ågren, P. Jørgensen, H. J. A. Jensen, S. B. Padkjær and T. Helgaker, J. Chem. Phys., 1992, 96, 6120 CrossRef CAS
- T. Helgaker, M. Watson and N. C. Handy, J. Chem. Phys., 2000, 113, 9402 CrossRef CAS
- ADF2009.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands (2009), see http://www.scm.com.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
- T. Kupka, Magn. Reson. Chem., 2008, 46, 851 Search PubMed
- T. Kupka, Magn. Reson. Chem., 2009, 47, 959 Search PubMed
- T. Kupka, Magn. Reson. Chem., 2009, 47, 210 Search PubMed
- T. Kupka, Magn. Reson. Chem., 2009, 47, 674 CrossRef CAS
- T. Kupka, M. Stachów, M. Nieradka, J. Kaminsky, T. Pluta and S. P. A. Sauer, Magn. Reson. Chem., 2011, 49, 231 Search PubMed
- S. V. Fedorov, L. B. Krivdin, Y. Y. Rusakov, N. A. Chernysheva and V. L. Mikhailenko, Magn. Reson. Chem., 2010, 48, 548 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, N. V. Istomina, V. A. Potapov and S. V. Amosova, Magn. Reson. Chem., 2008, 46, 979 Search PubMed
- Y. Y. Rusakov and L. B. Krivdin, Magn. Reson. Chem., 2012, 50, 557 Search PubMed
- S. V. Fedorov, L. B. Krivdin, Y. Y. Rusakov, I. A. Ushakov, N. V. Istomina, N. A. Belogorlova, S. F. Malysheva, N. K. Gusarova and B. A. Trofimov, Magn. Reson. Chem., 2009, 47, 288 Search PubMed
- W. H. Hersh, S. T. Lam, D. J. Moskovic and A. J. Panagiotakis, J. Org. Chem., 2012, 77, 4968 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, N. V. Orlov and V. P. Ananikov, Magn. Reson. Chem., 2011, 49, 570 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, N. V. Istomina, E. P. Levanova and G. G. Levkovskaya, Aust. J. Chem., 2009, 62, 734 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, L. K. Papernaya and A. A. Shatrova, Magn. Reson. Chem., 2012, 50, 169 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, V. A. Potapov, M. V. Penzik and S. V. Amosova, Magn. Reson. Chem., 2011, 49, 389 Search PubMed
- K. E. Kövér, A. A. Kumar, Y. Y. Rusakov, L. B. Krivdin, T.-Z. Illyés and L. Szilágyi, Magn. Reson. Chem., 2011, 49, 190 Search PubMed
- Y. Y. Rusakov, L. B. Krivdin, A. A. Kumar, L. Szilágyi and K. E. Kövér, Magn. Reson. Chem., 2012, 50, 488 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Exponents and contraction coefficients of the av3z-J basis set for Te. See DOI: 10.1039/c3cp51462e |
| This journal is © the Owner Societies 2013 |