 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear magnetic resonance study of ion adsorption on microporous carbide-derived carbon†
Alexander C.
Forse
a,
John M.
Griffin
a,
Hao
Wang
ab,
Nicole M.
Trease
b,
Volker
Presser
c,
Yury
Gogotsi
d,
Patrice
Simon
e and
Clare P.
Grey
*ab
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: cpg27@cam.ac.uk
bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA
cINM – Leibniz-Institute for New Materials, Energy Materials Group, Campus D2 2, D-66123 Saarbrücken, Germany
dDepartment of Materials Science and Engineering and A.J. Drexel Nanotechnology Institute, Drexel University, Philadelphia, PA 19104, USA
eUniversité Paul Sabatier, CIRIMAT UMR CNRS 5085 Toulouse, 31062, France and Réseau sur le Stockage Electrochimique de l'Energie (RS2E), FR CNRS 3459, France
First published on 26th March 2013
Abstract
A detailed understanding of ion adsorption within porous carbon is key to the design and improvement of electric double-layer capacitors, more commonly known as supercapacitors. In this work nuclear magnetic resonance (NMR) spectroscopy is used to study ion adsorption in porous carbide-derived carbons. These predominantly microporous materials have a tuneable pore size which enables a systematic study of the effect of pore size on ion adsorption. Multinuclear NMR experiments performed on the electrolyte anions and cations reveal two main environments inside the carbon. In-pore ions (observed at low frequencies) are adsorbed inside the pores, whilst ex-pore ions (observed at higher frequencies) are not adsorbed and are in large reservoirs of electrolyte between carbon particles. All our experiments were carried out in the absence of an applied electrical potential in order to assess the mechanisms related to ion adsorption without the contribution of electrosorption. Our results indicate similar adsorption behaviour for anions and cations. Furthermore, we probe the effect of sample orientation, which is shown to have a marked effect on the NMR spectra. Finally, we show that a 13C → 1H cross polarisation experiment enables magnetisation transfer from the carbon architecture to the adsorbed species, allowing selective observation of the adsorbed ions and confirming our spectral assignments.
Introduction
Climate change and fossil fuel depletion are changing the world's attitude towards energy production and consumption. More renewable energy generation is required and technologies such as electric vehicles are being developed, both requiring the development of electrical energy storage devices such as batteries and electrical double-layer capacitors. Whilst batteries can store large amounts of energy for a given volume or mass, they suffer from limited cycle lives, restricted power handling capabilities and further limitations regarding safety and temperature requirements.1 Consequently, alternative energy storage devices such as electrical double-layer capacitors, also known as ultracapacitors or supercapacitors, have attracted significant attention. Supercapacitors store charge by ion electrosorption, with charge in the electrode balanced by a layer of ions of opposite polarity in the electrolyte.2 This non-faradaic storage mechanism allows rapid rates of charge and discharge and essentially unlimited cycle lives, making supercapacitors well suited to high power applications.3 Supercapacitors typically employ porous carbon electrodes as they have large surface areas for ion electrosorption, good electronic conductivities and relatively low production cost.4 In particular, activated carbons, derived from carbon-rich organic precursors such as coconut shells or wood, are the materials of choice in commercial devices.5 Such activated carbons exhibit pores with different diameters, often ranging from micropores (diameters less than 2 nm), and mesopores (diameters between 2 and 50 nm), to macropores (diameters greater than 50 nm) according to the IUPAC classification.6While a vast number of different carbons have been studied as supercapacitor electrode materials,4,6,7 carbide-derived carbons (CDCs), produced by chlorine treatment of metal carbides, have recently gained particular attention.8 CDCs have microporous structures with narrow pore size distributions.9,10 Control of the chlorination temperature used in the synthesis allows fine-tuning of the average pore size in the range 0.6 to 1.1 nm, with higher temperatures resulting in a larger average pore size.8 Chmiola et al. reported an increase in the capacitance of titanium carbide-derived carbon (TiC-CDC) at pore sizes of less than 1 nanometre, using an electrolyte comprising tetraethylammonium tetrafluoroborate (NEt4BF4) in acetonitrile.11 These results challenged the previous view that pores smaller than the solvated electrolyte ions do not contribute significantly to the capacitance. It was hypothesised that ion desolvation allowed a closer approach of charge centres at the electrode–electrolyte interface.12 Modelling and simulation studies have progressed the understanding of charge storage in microporous carbons,13–19 whilst new experimental methods have also been developed.20–24 Despite recent advances, further experimental techniques must be developed to directly probe the nature of ion adsorption and electrosorption in micropores.
In contrast to other techniques, nuclear magnetic resonance (NMR) spectroscopy allows the comprehensive investigation of the local structure and dynamics of electrode–electrolyte interfaces in a non-invasive and element selective way.25 This latter feature uniquely allows the separate observation of cations and anions in electrochemical systems. NMR has been used to study the adsorption of molecules on activated carbons,25–28 and the same techniques have been used to study adsorption inside carbon nanotubes (CNTs).29–32 It has been well established that resonances corresponding to molecules adsorbed on carbon surfaces show a shift to low frequencies (relative to the corresponding free species), making NMR a very useful tool to study ion adsorption. This approach has been extended to study the effect of an applied potential during the operation of supercapacitors with activated carbon electrodes. Ex situ experiments33,34 (NMR spectra acquired on a dismantled supercapacitor after electrochemical cycling) can be combined with magic-angle spinning (MAS) techniques to improve spectral resolution. However, in situ experiments35 (NMR spectra acquired during cycling) are, in principle, preferable since they give a realistic picture of working devices.
Whilst previous experimental NMR studies have focused on adsorption on a variety of activated carbons and CNTs, a more systematic approach is desirable to obtain detailed insight into ion adsorption within carbon micropores. To study the pore-size effect, here we focus our attention on CDCs with controlled pore sizes and narrow pore size distributions. NMR experiments allow the identification of in- and ex-pore ion environments. We carry out multinuclear NMR experiments to probe the adsorption of electrolyte cations and anions, shown to display very similar adsorption behaviour. We also explore some of the practical considerations relevant to envisaged in situ studies of supercapacitors, with sample orientation shown to be important for both interpreting spectra and improving resolution of features. Finally, a through-space magnetisation transfer experiment (from 13C enriched carbon to the 1H spins of the electrolyte cations) enables identification of species in close proximity to the carbon surface, providing conclusive evidence for our peak assignments.
Experimental
1. Carbon materials
TiC-CDC powder was synthesised by chlorine treatment of titanium carbide as described elsewhere in more detail.9 Commercial TiC (Alfa Aesar) with an average particle size of around 2 μm was used as the precursor material. TiC powder was placed in a quartz glass boat and transferred to a quartz glass tube furnace. The sample was heated to the desired temperature (600, 800, and 1000 °C) in an atmosphere of high purity argon gas, and the gas switched to dry chlorine for 3 h. Subsequent to the chlorine treatment, the gas was changed to pure argon, and then to pure hydrogen, with the sample held at 600 °C for 2 h to remove chlorine and chloride residues. Finally, the sample was cooled to room temperature in high purity argon. Samples treated in chlorine gas at a temperature X °C (e.g. 600 °C) are referred to as TiC-CDC-X (e.g. TiC-CDC-600).Isotopic Ti13C-CDC-1000 was synthesised as follows. A stoichiometric mixture of 13C powder (0.4 g, 99 at%, Sigma Aldrich) was mixed with crystalline titanium (1.7 g, ≥99.99 at%, Sigma Aldrich; particle size: 5–10 μm) in an agate mortar and then transferred to a graphite-plated alumina crucible. The resulting powder material was heated (10 K min−1) to 1550 °C in flowing argon under atmospheric pressure and held at that temperature for 12 h. Finally, the sample was cooled down to room temperature (10 K min−1). The resulting monolithic material was ground by hand with an agate mortar to an average particle size between 2 and 50 μm (see ESI,† Fig. S1). X-ray diffraction confirms that the material consisted predominantly of microcrystalline TiC, while a small fraction (<1 wt%) graphitic carbon (i.e., non-reacted precursor material) was found via Rietveld refinement. The titanium was extracted by treatment in dry chlorine. For this, the sample material was placed in a quartz glass boat, inserted in a tube furnace and flushed in Argon for 30 min. The sample was heated (10 K min−1) to 1000 °C and then kept, for 3 h in flowing chlorine. The sample was then flushed with argon gas and treated in flowing hydrogen at 600 °C for 3 h to remove residual chlorine and chloride species. After a final argon flushing, the resulting CDC powder was cooled to room temperature.
Carbon films were fabricated using the standard method for preparing film electrodes, with a mixture of carbon powder (95 wt%) and polytetrafluoroethylene (PTFE) binder (5 wt%). More details on the electrode preparation can be found in, for example, ref. 11.
2. Electrolytes
Tetraethylammonium tetrafluoroborate, NEt4BF4, (≥99.0%, Sigma Aldrich) 1.5 M in acetonitrile, ACN, (99.8%, Sigma Aldrich) was used in the majority of experiments, referred to as NEt4BF4/ACN. In cases where 1H NMR spectra were acquired, an electrolyte of the same salt and concentration was prepared using deuterated acetonitrile, D3CCN (99.80%, Eurisotop). We refer to this electrolyte as NEt4BF4/dACN.3. NMR sample preparation
4. NMR
NMR experiments were performed using Bruker Avance II spectrometers operating at magnetic field strengths of 7.05 and 9.4 T, corresponding to 1H Larmor frequencies of 300.2 and 400.4 MHz, respectively. 11B NMR spectra of samples in bags were recorded using a Bruker single-channel static NMR probe fitted with a 6 mm diameter solenoid coil. 19F NMR spectra of samples in bags were recorded using a Chemagnetics single-channel probe fitted with a 7 mm diameter solenoid coil. NMR spectra of samples contained within MAS rotors were obtained under static conditions using a Bruker 2.5 mm double resonance probe. Adsorption experiments were performed using a spin-echo pulse sequence (90°−τ–180°−τ – acquire) to avoid baseline distortions and to remove background signals associated with either the plastic bag or probe. Spin-echo τ delays of 50 μs (11B), and 100 μs (19F and 1H) were used, except for 19F NMR spectra of samples in bags, where a slightly longer τ delay of 200 μs was necessary to remove the stronger background signal associated with the PTFE resonance. Radiofrequency strengths of between 50–100 kHz were used for all nuclei studied. For the 13C → 1H CP NMR experiment, transverse magnetisation on 1H was obtained by cross-polarisation from 13C using a contact pulse duration of 8 ms (ramped for 1H). A spin-echo with a τ delay of 100 μs was incorporated prior to acquisition of the FID, during which no heteronuclear decoupling was applied. 11B NMR spectra were referenced relative to boric acid solution (0.1 M) at 19.6 ppm, 19F NMR spectra were referenced relative to hexafluorobenzene liquid at −164.9 ppm and 1H NMR spectra were referenced relative to tetramethylsilane using the CH3 resonance of liquid ethanol at 1.2 ppm as a secondary reference. All liquid reference samples were contained within MAS rotors, which were either inserted directly into the MAS probe, or wrapped in PTFE tape for use in static NMR probes. Sufficiently long recycle delays were used to ensure that none of the 1H, 11B and 19F signals were saturated, allowing for quantification of the different environments. Additional experimental details are given in the relevant figure caption.Results and discussion
1. 11B NMR study of anion adsorption on TiC-CDC-1000
To investigate the adsorption of the BF4 anions, 11B NMR experiments were performed. Experiments were performed on samples in bags, which represent a model for supercapacitor electrodes. Spectra were acquired with samples in the horizontal orientation (Fig. 1a) as in our previous work.35Fig. 1b shows 11B NMR spectra of samples in bags containing pieces of TiC-CDC-1000 film (6.0 mg) soaked with different volumes of NEt4BF4/ACN electrolyte. At a low loading volume of 2.0 μL a single broad peak is observed at −4.0 ppm. When the loading volume is increased to 5.0 μL, this first peak grows in intensity and shifts to higher frequency, whilst a second peak appears at 1.3 ppm. Addition of further electrolyte causes the peak at higher frequency to grow in intensity considerably, whilst the low frequency peak shows only small gains in intensity. For the highest loading volume of 10.0 μL, an additional small feature is observed at ∼0.1 ppm.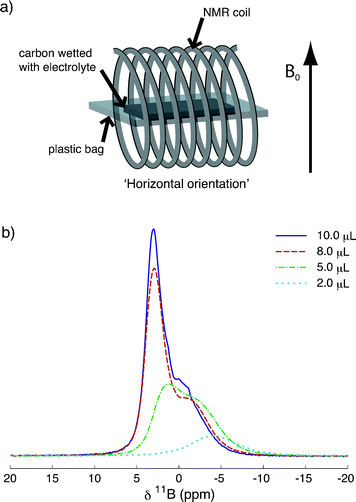 | ||
| Fig. 1 (a) The experimental set up used to acquire NMR spectra of samples in bags at the horizontal orientation. The applied magnetic field, B0, is shown. (b) Static 11B (9.4 T) NMR spectra of pieces of TiC-CDC-1000 film soaked with different volumes of NEt4BF4/ACN electrolyte (see legend), acquired in the horizontal orientation. Each spectrum is the result of coadding 1024 transients, separated by a recycle interval of 10 s. | ||
The different relative growths of the two peaks as a function of loading volume provides an indication of their origin. The approximately constant intensity of the low frequency feature above loading volumes of 5.0 μL suggests that this environment corresponds to anions in adsorption sites that are becoming saturated. Thus, the peak at low frequencies (−4.0 ppm for the 2.0 μL sample) is assigned to BF4 anions that are adsorbed to the carbon surface.35 Since TiC-CDC is predominantly microporous (see ESI,† Table S1 and Fig. S2) this peak can be more specifically assigned to anions adsorbed to the carbon inside the micropores, and is referred to as ‘in-pore’ in our study. As the in-pore sites begin to saturate, anions are forced to occupy positions in large reservoirs between the carbon particles in the film (see ESI,† Fig. S3), giving rise to the feature at higher frequencies (1.3 ppm for the 5.0 μL sample), referred to as ‘ex-pore’. Our previous work on an activated carbon35 proposed that this feature was due to anions either in the diffuse outer layer of the double layers or in larger pores. In TiC-CDC-1000, there is not space for diffuse layer formation in the pores considering that the diameter of fully-solvated BF4− is 1.16 nm36 and the average pore size in TiC-CDC-1000 is 0.93 nm (see ESI,† Table S1). The relatively broad line width of this resonance suggests that it does not correspond to a completely free liquid, but rather to a confined liquid. Thus, this peak must arise from anions confined in spaces between the primary particles. Finally, as the ex-pore sites become saturated, the carbon film can hold no further electrolyte. Therefore, the small feature at ∼0.1 ppm in the 10.0 μL sample is assigned to BF4 anions in pockets of liquid between the carbon film and the plastic bag, referred to as ‘free-electrolyte’. Indeed we observed this feature previously.35 The in-pore peak is seen to shift to higher frequencies on the addition of more electrolyte, this observation is discussed later.
Our assignments of the features in Fig. 1b are similar to those in other NMR work on the adsorption of molecules on activated carbons.25–28 In particular, one work on the adsorption of hydrogen gas on activated carbons assigned low and high frequency features to hydrogen adsorbed in the micropores and hydrogen in large intergranular pores.28 Parallels can also be drawn with work on the adsorption of molecules inside carbon nanotubes, which have assigned similar low and high frequency features to endohedral sites (inside the CNTs) and exohedral sites (outside the CNTs), respectively.29–32 Regardless of the nucleus studied, and whether the adsorbate is in the liquid or gaseous state, species adsorbed on carbon show a shift to low frequencies (of between 3 and 12 ppm for different carbons) from the corresponding non-adsorbed resonance. This suggests the chemical shift of adsorbed species is dominated by the influence of the carbon. The shift of the in-pore signal to low frequencies can be attributed to aromatic ring current effects associated with the graphene-like layers in the carbon.37 A molecule situated above an aromatic ring or a graphene-like layer is expected to exhibit a shift to low frequencies. Calculations of the chemical shift of molecules adsorbed inside CNTs have previously explored this phenomenon.38,39
2. The effect of carbon pore size on ion adsorption
Further studies were carried out on TiC-CDC-800 and TiC-CDC-600 to investigate the effect of the average pore size on the adsorption of electrolyte species. Fig. 2 shows the static 11B NMR spectra as well as porosity data for CDC samples studied in this work. Spectra were acquired in the same way as for TiC-CDC-1000 (Fig. 1b) to allow comparison. The spectra for TiC-CDC-800 (Fig. 2a) have the same qualitative form as for TiC-CDC-1000 (Fig. 1b). At the lowest loading volume of 2.0 μL the in-pore peak is observed at −4.0 ppm. As the loading level is increased to 5.0 μL the ex-pore peak is observed at −0.4 ppm. At the highest loading volume of 10.0 μL an additional peak assigned to free electrolyte is observed at 0.0 ppm. Interestingly, the addition of just 2.0 μL of electrolyte appears to effectively saturate the pores. This is in contrast to TiC-CDC-1000 (Fig. 1b) where the in-pore peak grows considerably on the addition of further electrolyte. For TiC-CDC-600 (Fig. 2b) the in-pore peak is visible at −5.3 ppm for the 2.0 μL sample. For higher loading volumes the ex-pore peak dominates the spectrum (−1.2 ppm for the 5.0 μL sample) and the in-pore feature is visible only as a broad shoulder. At the highest loading volume (15.0 μL) sharp free electrolyte features are observed at 1.4, 1.0 and 0.6 ppm. | ||
| Fig. 2 Static 11B (9.4 T) NMR spectra of pieces of TiC-CDC film soaked with different volumes of NEt4BF4/ACN electrolyte. Spectra are shown for (a) TiC-CDC-800 and (b) TiC-CDC-600. Each spectrum is the result of coadding 1024 transients, separated by a recycle interval of 10 s. (c) TiC-CDC pores sizes for carbons considered in this work as well as solvated and desolvated anion sizes36 are shown. dn is the pore size where n% of the pore volume is below that size such that d50 is the average pore size (d85 is referred to as the maximum pore size). | ||
The differences in the adsorption spectra for TiC-CDC-1000, -800 and -600 can be rationalised by considering the porosities of the different carbons and the size of the BF4 anion. Fig. 2c compares the sizes of solvated and desolvated BF4− to the average and maximum pore sizes for the different carbons studied in this work. For TiC-CDC-1000, the average pore size (d50) is quite similar to the solvated anion size and the maximum pore size is significantly larger, therefore many pores are accessible for adsorption. For TiC-CDC-800, the maximum pore size is approximately equal to the solvated ion size, and the average pore size is smaller, therefore fewer pores are accessible to the anions. This is reflected by the minimal increase in intensity of the in-pore peak on the addition of increasing volumes of electrolyte. Finally, for TiC-CDC-600, both the maximum and average pore sizes are significantly smaller than the solvated anion size and so few pores are accessible to the anions when there is no electrical potential applied to the sample. Consistent with this, the in-pore feature is hard to discern at most loading volumes. These results provide further evidence for our in- and ex-pore peak assignments, identified for TiC-CDC-1000 previously (Fig. 1b). It is interesting to note that in the absence of an applied voltage, few anions are adsorbed on TiC-CDC-600, yet electrochemical studies show this carbon to display the highest capacitance of the three carbons studied here.11,12 This illustrates that the electrosorption of ions must be related to (partial) desolvation once an electrical potential is applied. Indeed, the results suggest that ion desolvation does not play a significant role in the adsorption process in the absence of an applied voltage; if it did, essentially all of the carbon pores would be accessible to the anions for all three carbons studied.
3. The effect of sample orientation on NMR spectra
Recent NMR studies of systems contained within plastic bags40 and between flat plates41,42 have noted significant effects on the observed shift associated with the orientation of the sample with respect to the applied magnetic field. These effects have been attributed to bulk magnetic susceptibility (BMS) effects arising from the highly inhomogeneous shape of the sample. To investigate this effect, a TiC-CDC-1000 sample (6.0 mg) loaded with NEt4BF4/ACN electrolyte (10.0 μL) was studied in two different orientations, horizontal and vertical (Fig. 3a and b).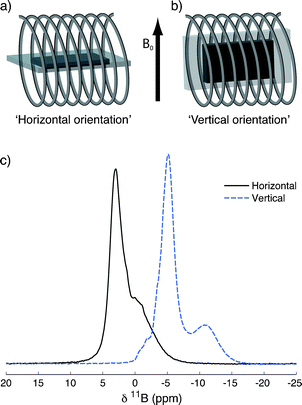 | ||
| Fig. 3 The horizontal (a) and vertical (b) orientations used to acquire NMR spectra of samples in bags. (c) Static (9.4 T) 11B NMR spectra of a piece of TiC-CDC-1000 film soaked with 10.0 μL of NEt4BF4/ACN electrolyte, in the horizontal and vertical orientations. Each spectrum is the result of coadding 1024 transients, separated by a recycle interval of 10 s. | ||
11B static NMR spectra are shown in Fig. 3c. As anticipated, the orientation of the sample relative to the applied magnetic field has a marked effect on the NMR spectrum. In the vertical orientation peaks are shifted to lower frequencies and better resolution between the in- and ex-pore peak is observed. On changing the sample orientation from horizontal to vertical, the in-pore peak position changes more than the ex-pore peak, with absolute changes of ∼10 and 8 ppm respectively. The small feature attributed to free-electrolyte appears to shift by a noticeably smaller amount of ∼2 ppm. This indicates that the in-pore, ex-pore and free electrolyte features experience different BMS effects. Crucially, future experiments should carefully consider the effects of sample orientation in the NMR coil. Indeed, this has been shown to be an important consideration in in situ studies of lithium-ion batteries.40
In the vertical orientation, increased resolution of the in- and ex-pore features allows for more accurate fitting of the spectra to enable an estimation of the fraction of anions that are adsorbed. A deconvolution was performed assuming a Gaussian line shape for the in-pore environment since a distribution of adsorption sites is expected in the micropores. The line shape corresponding to the ex-pore environment is expected to be close to a Lorentzian owing to its liquid-like form. However, confinement may result in reduced mobility and therefore some Gaussian broadening. Therefore the Gaussian/Lorentzian ratio in the fit was allowed to vary to encapsulate this behaviour. Fitting (see ESI,† Fig. S4) reveals that ∼21% of the total number of anions in the sample are adsorbed in the pores. Taking the specific surface area for TiC-CDC-1000 as 1723 m2 g−1 (see ESI,† Table S1) and assuming a monolayer of adsorbed anions on a planar surface, the area containing one BF4 anion is calculated to be 5.4 nm2 (see ESI† for details). This is comparable to the theoretical lower limit of 3.0 nm2 per anion, calculated for a close packed square lattice (see ESI,† Fig. S5) of solvated anions and cations with sizes36 of 1.16 and 1.30 nm respectively, the larger effective footprint of the anion being expected due to the curvature of the internal carbon surfaces.
4. 19F NMR – a further probe of anion adsorption
A 19F NMR adsorption study was carried out on TiC-CDC-1000 to probe the adsorption of the BF4 anions in more detail. A preliminary experiment showed the NMR spectra to have a very similar orientation dependence to that shown in Fig. 3, with better resolution of peaks in the vertical orientation (see ESI,† Fig. S6). Fig. 4 shows the static 19F spectra of samples in bags containing pieces of TiC-CDC-1000 film (6.0 mg) soaked with different volumes of NEt4BF4/ACN electrolyte, acquired in the vertical orientation. The 19F spectra reveal in- and ex-pore environments for BF4−, consistent with the 11B spectra (Fig. 1b). Moreover, the relative growth of these features as a function of loading volume is very similar to that seen in the 11B experiment. At a low loading volume of 2.0 μL the in-pore peak is observed at −156.4 ppm (Fig. 4). When the loading volume is increased to 5.0 μL the ex-pore peak begins to become visible at higher frequency. Addition of further electrolyte causes the in-pore environment to shift to lower frequencies and to saturate, whilst the ex-pore environment shows a considerable increase in intensity. For the highest loading volume of 15.0 μL a shoulder attributed to free electrolyte (between the carbon film and the plastic bag) is observed at −150.6 ppm. Fitting of the 10.0 μL spectrum (see ESI,† Fig. S4) reveals that ∼26% of the anions in the sample are adsorbed in the pores. Assuming a monolayer of adsorbed anions on a planar surface, the area containing one BF4 anion is calculated to be 4.4 nm2. This is in good agreement with the value of 5.4 nm2 obtained above for the corresponding 11B spectrum. The small difference between these two values likely arises from the experimental errors associated with weighing the carbon and syringing the electrolyte in the fabrication of the two different samples used in these measurements. We note an apparent shift for the in-pore peak to lower frequencies on loading with increased volumes of electrolyte. The opposite trend is observed for spectra of the same samples acquired in the horizontal orientation (see ESI,† Fig. S7), suggesting that this shift is due to a BMS effect. | ||
| Fig. 4 Static (7.05 T) 19F NMR spectra of TiC-CDC-1000 film pieces (6.0 mg) soaked with different volumes of NEt4BF4/ACN electrolyte, acquired in the vertical orientation. Each spectrum is the result of coadding 64 transients, separated by a recycle interval of 10 s. | ||
The 19F and 11B spectra lead us to the same conclusion, namely, that there are two main environments for the anions inside the carbon film. This is consistent with the assertion that the chemical shift of the in-pore peak arises from the carbon, and that adsorption is physical and not chemical in nature. The separation of the in-pore and ex-pore environments is very similar for 11B and 19F, measured as 5.7 ppm and 5.5 ppm respectively for the 10.0 μL samples in the vertical orientation. This suggests that the fluorine and boron atoms experience similar shielding effects from the carbon.
5. Comparison between anion and cation adsorption
To probe the NEt4 cation environments, 1H NMR experiments were performed on TiC-CDC-1000 in plastic bags (see ESI,† Fig. S8). Whilst spectra show qualitative agreement with studies of the BF4 anions, with both in- and ex-pore features, they are complicated by the 1H signal from the plastic bag used to contain the sample. To circumvent this issue associated with samples in bags and to allow direct comparison of the anion and cation environments, 19F and 1H experiments were performed on samples in MAS rotors. A MAS probe was used for the experiments and the spectra were acquired in static mode, the sample orientation corresponding to the magic-angle (MA; 54.74°). Fig. 5 shows 19F and 1H NMR spectra of TiC-CDC-1000 (3.0 mg) samples in MAS rotors soaked with different volumes of NEt4BF4/dACN electrolyte. Static 19F (Fig. 5a) and 1H spectra (Fig. 5b) are acquired on the same samples, and spectra of the neat electrolyte are also overlaid. | ||
| Fig. 5 (a) 19F and (b) 1H NMR (7.05 T) spectra of TiC-CDC-1000 (3.0 mg) soaked with different volumes of NEt4BF4/dACN electrolyte in MAS rotors, acquired in static mode at the magic-angle. The same carbon mass to electrolyte volume ratios were used as for the experiments on samples in bags. 19F and 1H spectra are shown for the same samples, allowing for direct comparison of the anion and cation environments. The spectrum of free electrolyte is also shown for comparison; the –CH2– and –CH3 resonances are labelled in the 1H spectrum. Each 19F spectrum is the result of coadding 16 transients with a recycle interval of 15 s, whilst for each 1H spectrum 32 transients and a recycle interval of 8 s were used. | ||
Whilst some resolution is lost at the MA sample orientation due to negligible BMS effects (compared to Fig. 4), the same general trends seen in the previous spectra (Fig. 1b and 4) are reproduced. In both 19F and 1H spectra, only the in-pore peak is seen at a loading volume of 1.0 μL, and the ex-pore environment appears for the 2.5 μL sample. Further addition of electrolyte causes only small increases in intensity of the in-pore environment as it begins to saturate, whilst the ex-pore peak increases in intensity considerably. Since the NEt4 cations have a –CH2– and a –CH3 group, two resonances are seen for the ex-pore environment (and neat electrolyte), the –CH2– resonance appearing at higher frequency. Interestingly, the ex-pore environments show very similar resonant frequencies to the neat electrolyte peaks.
Careful NMR studies of planar lithium-ion battery electrodes have shown that BMS effects are minimised when samples are orientated at the MA,40 signals for species within the electrodes giving rise to resonances with shifts close to their isotropic values obtained under MAS. This phenomenon could be rationalised by considering the (long-range) dipolar interactions that occur between nuclear spins and the magnetic moments of the nearby particles within the electrode films, the orientation dependent shifts being significant when paramagnetic and conducting (carbon and metal) particles were present. The overlap of the neat electrolyte and ex-pore peaks in our system at the MA (Fig. 5) indicates that the shift of the latter feature is largely dominated by BMS (long-range) interactions. This overlap adds further support to our assignment of the ex-pore species being located between carbon particles without any significant adsorption occurring. While the resonant frequency of the in-pore feature is also affected by BMS effects, a substantial contribution to its shift must arise from a local field interaction, that is from the nearby ring currents of the carbon.27,28,37 We note that the –CH2– and –CH3 resonances are not resolvable for the in-pore environment due to their broad shape.
The key observation from Fig. 5 is that the 19F and 1H spectra have an almost identical form. This indicates that the adsorption behaviour of anions and cations is the same in the absence of an applied voltage. For systems with a point of zero charge close to pH 7.0, this behaviour is expected if charge neutrality is to be maintained in the solution and the carbon. Indeed this has been observed in molecular dynamics simulations of an ionic liquid.16 It is noted that spectra of the 5.0 and 7.5 μL samples have a very similar form, the resonances from the 7.5 μL sample being only marginally more intense. This behaviour is attributed to loss of electrolyte during micro-syringing of the larger volume. We also note that the 4.0 μL sample has a larger in-pore occupation than the other samples, though we do not offer an explanation for this deviation.
6. Through-space magnetisation transfer experiment
Our NMR experiments on CDCs soaked with different volumes of electrolyte provide insight into the nature of the observed resonances and strongly indicate the presence of two distinct ion environments. However the resolution of these environments remains limited. For future experiments, it would be advantageous to observe just the in-pore environment; that is ions adsorbed on the carbon surface in the micropores. Cross polarisation (CP) is routinely used in NMR for magnetisation transfer over short distances.43 It is typically used to achieve a signal enhancement, but can also be used for spectral editing so as to favour species close to carbon (or other nuclei).44–47 In view of this, CDCs enriched with the NMR-active 13C isotope (99% enrichment) were prepared.Fig. 6 shows the static direct excitation and 13C → 1H CP spectra of a Ti13C-CDC-1000 (3.0 mg) sample in a MAS rotor with 7.5 μL NEt4BF4/dACN electrolyte. This loading volume was chosen since the direct excitation spectrum is expected to show both in- and ex-pore peaks (Fig. 5b). Indeed, direct excitation gives a very similar spectrum to that for natural abundance TiC-CDC-1000. In contrast, the 13C → 1H CP experiment reveals solely the in-pore environment. This confirms that these cations are spatially close to the carbon, since magnetisation transfer is by dipolar coupling, a short range through-space interaction. This observation confirms our peak assignments. Indeed the absence of ex-pore peaks confirms that these cations are further from the carbon than the in-pore cations. We note that in the 13C→1H CP spectrum it is still not possible to resolve the in-pore –CH2– and –CH3 resonances, as the overall line width is much bigger than the expected separation of these resonances (∼2 ppm). Whilst other CP contact times (the time during which magnetisation is transferred) were studied, we only present the spectrum for the longest time (8 ms) here, since it gave the greatest signal intensity.
 | ||
| Fig. 6 Direct excitation and 13C → 1H CP NMR spectra (7.05 T) of TiC-CDC-1000 (3.0 mg) soaked with 7.5 μL of NEt4BF4/dACN electrolyte in a MAS rotor, acquired in static mode at the magic-angle. CP only shows protons (in the cations) that are spatially close to the carbon, confirming that these (in-pore) cations are adsorbed to the surface. The direct excitation and CP spectra are the result of coadding 32 and 4608 transients respectively, transients separated by a recycle interval of 3 s. Spectra are scaled for ease of comparison. The pulse sequence for the CP experiment is given in the ESI† (Fig. S9). | ||
The 13C → 1H CP experiment could be applied to study adsorption on other carbon materials, both to provide more definitive peak assignments as well as to resolve peaks from adsorbed and non-adsorbed species. For example, it could confirm whether resonances assigned to molecules located in the exohedral sites of carbon nanotubes29,30 are adsorbed to the exterior of the CNTs or not. The technique could also be used to confirm the presence of layered interfaces, such as that proposed for methanol gas inside large CNTs where one layer is adsorbed to the carbon and the other is in the centre of the nanotube.29 The 13C → 1H CP experiment could help resolve the in-pore peak when features overlap, as in 1H NMR spectra of hydrogen gas adsorbed on activated carbons with large pore sizes.28
Conclusions
This work has presented a systematic NMR study of electrolyte adsorption on TiC-CDCs, a model carbon morphology used as a supercapacitor electrode material. The experiments have provided fundamental insight into the different electrolyte ion environments present in the carbon. In-pore features appear at lower NMR frequencies and arise from ions adsorbed in the micropores of the carbon, while ex-pore features arise from free ions in the spaces between carbon particles. These environments have been studied as a function of pore size, the results corroborating our spectral assignments. For TiC-CDC-600, the carbon with the smallest pore size studied, few anions were adsorbed in the micropores as a large fraction of the total pore volume is virtually inaccessible to solvated anions. Multinuclear NMR experiments point towards identical adsorption behaviour for anions and cations.Our work highlights some of the experimental considerations that will be key to in situ studies of supercapacitors. The effects of sample orientation have been explored and 13C → 1H CP NMR experiments, with 13C-enriched carbons, have been developed to select the ions adsorbed in the pores. While careful choice of sample orientation can improve spectral resolution, CP experiments allow selection of just the adsorbed species in the NMR spectrum. This is highly desirable since it is these ions that contribute to the charge storage in supercapacitors. A combination of the techniques presented here will be of great use in envisaged in situ experiments as well as experiments in the broader field of adsorption on porous carbons.
Acknowledgements
ACF, JMG, HW and CPG acknowledge the Sims Scholarship (ACF), EPSRC and the EU ERC for funding. ACF and JMG thank the NanoDTC (Cambridge) for travel funding. NMT was supported by Northeastern Center for Chemical Energy Storage, an Energy Frontier Research Center funded by U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award #DE-SC0001294. VP acknowledges funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. YG was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, under Award No. DE-FG02-07ER46473. PS acknowledges funding from the European Research Council (ERC, Advanced Grant, ERC-2011-AdG, Project 291543 – IONACES). The authors are grateful to Mohamed Shamma for help with the synthesis of isotopic TiC, Boris Dyatkin for his help with gas sorption analysis and synthesis of isotopic CDC, and Min Heon for his support with scanning electron microscopy (all at Drexel University). Dr Mesut Aslan (INM) is thanked for his help with gas sorption analysis. VP thanks Prof. Eduard Arzt for his continuing support.Notes and references
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS.
- M. Winter and R. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- J. R. Miller and A. Burke, Electrochem. Soc. Interface, 2008, 17, 53–57 CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- A. Burke, Electrochim. Acta, 2007, 53, 1083–1091 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810–833 CrossRef CAS.
- G. Laudisio, R. K. Dash, J. P. Singer, G. Yushin, Y. Gogotsi and J. E. Fischer, Langmuir, 2006, 22, 8945–8950 CrossRef CAS.
- R. Dash, J. Chmiola, G. Yushin, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer and S. Kucheyev, Carbon, 2006, 44, 2489–2497 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2012 DOI:10.1021/ar200306b.
- M. V. Fedorov and A. A. Kornyshev, Electrochim. Acta, 2008, 53, 6835–6840 CrossRef CAS.
- J. C. Palmer, A. Llobet, S.-H. Yeon, J. E. Fischer, Y. Shi, Y. Gogotsi and K. E. Gubbins, Carbon, 2010, 48, 1116–1123 CrossRef CAS.
- C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS.
- S. Kondrat and A. Kornyshev, J. Phys.: Condens. Matter, 2011, 23, 022201 CrossRef CAS.
- S. Kondrat, N. Georgi, M. V Fedorov and A. A. Kornyshev, Phys. Chem. Chem. Phys., 2011, 13, 11359–11366 RSC.
- S. Kondrat, C. R. Pérez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 CAS.
- R. Lin, P. L. Taberna, J. Chmiola, D. Guay, Y. Gogotsi and P. Simon, J. Electrochem. Soc., 2009, 156, A7–A12 CrossRef CAS.
- T. M. Arruda, M. Heon, V. Presser, P. C. Hillesheim, S. Dai, Y. Gogotsi, S. V. Kalinin and N. Balke, Energy Environ. Sci., 2013, 6, 225–231 CAS.
- M. D. Levi, G. Salitra, N. Levy, D. Aurbach and J. Maier, Nat. Mater., 2009, 8, 872–875 CrossRef CAS.
- S. Boukhalfa, L. He, Y. B. Melnichenko and G. Yushin, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef.
- M. D. Levi, S. Sigalov, G. Salitra, R. Elazari, D. Aurbach, L. Daikhin and V. Presser, J. Phys. Chem. C, 2013, 117, 1247–1256 CAS.
- R. K. Harris, T. V. Thompson, P. R. Norman, C. Pottage and A. N. Trethewey, J. Chem. Soc., Faraday Trans., 1995, 91, 1795–1799 RSC.
- R. K. Harris, T. V. Thompson, P. R. Norman and C. Pottage, Carbon, 1999, 37, 1425–1430 CrossRef CAS.
- L. M. Dickinson, R. K. Harris, J. A. Shaw, M. Chinn and P. R. Norman, Magn. Reson. Chem., 2000, 38, 918–924 CrossRef CAS.
- R. J. Anderson, T. P. McNicholas, A. Kleinhammes, A. Wang, J. Liu and Y. Wu, J. Am. Chem. Soc., 2010, 132, 8618–8626 CrossRef CAS.
- X. Liu, X. Pan, W. Shen, P. Ren, X. Han and X. Bao, J. Phys. Chem. C, 2012, 116, 7803–7809 CAS.
- W. Sekhaneh, M. Kotecha, U. Dettlaff-Weglikowska and W. S. Veeman, Chem. Phys. Lett., 2006, 428, 143–147 CrossRef CAS.
- Q. Chen, J. L. Herberg, G. Mogilevsky, H.-J. Wang, M. Stadermann, J. K. Holt and Y. Wu, Nano Lett., 2008, 8, 1902–1905 CrossRef CAS.
- K. Shen and T. Pietrass, J. Phys. Chem. B, 2004, 108, 9937–9942 CrossRef CAS.
- S.-I. Lee, K. Saito, K. Kanehashi, M. Hatakeyama, S. Mitani, S.-H. Yoon, Y. Korai and I. Mochida, Carbon, 2006, 44, 2578–2586 CrossRef CAS.
- M. Deschamps, E. Gilbert, P. Azais, E. Raymundo-Piñero, M. R. Ammar, P. Simon, D. Massiot and F. Béguin, Nat. Mater., 2013, 12, 351–358 CrossRef CAS.
- H. Wang, T. K.-J. Köster, N. M. Trease, J. Ségalini, P.-L. Taberna, P. Simon, Y. Gogotsi and C. P. Grey, J. Am. Chem. Soc., 2011, 133, 19270–19273 CrossRef CAS.
- Y.-J. Kim, Y. Masutzawa, S. Ozaki, M. Endo and M. S. Dresselhaus, J. Electrochem. Soc., 2004, 151, E199–E205 CrossRef CAS.
- P. Lazzeretti, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS.
- N. A. Besley and A. Noble, J. Chem. Phys., 2008, 128, 101102 CrossRef.
- M. Kibalchenko, M. C. Payne and J. R. Yates, ACS Nano, 2011, 5, 537–545 CrossRef CAS.
- N. M. Trease, L. Zhou, H. J. Chang, B. Y. Zhu and C. P. Grey, Solid State Nucl. Magn. Reson., 2012, 42, 62–70 CrossRef CAS.
- R. Ulrich, R. W. Glaser and A. S. Ulrich, J. Magn. Reson., 2003, 164, 115–127 CrossRef CAS.
- R. W. Glaser and A. S. Ulrich, J. Magn. Reson., 2003, 164, 104–114 CrossRef CAS.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid-state NMR Basic Principles and Practice, Momentum Press, 2012 Search PubMed.
- J. Kolmas and W. Kolodziejski, Chem. Phys. Lett., 2012, 554, 128–132 CrossRef CAS.
- J. Kolmas and W. Kolodziejski, Chem. Commun., 2007, 4390–4392 RSC.
- W. Kolodziejski, A. Corma, K. Wozniak and J. Klinowski, J. Phys. Chem., 1996, 100, 7345–7351 CrossRef CAS.
- J. L. White, L. W. Beck and J. F. Haw, J. Am. Chem. Soc., 1992, 114, 6182–6189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images and porosity analysis of carbons, spectral fits, derivation of formulae used to calculate area per anion, supporting NMR spectra and pulse sequences. See DOI: 10.1039/c3cp51210j |
| This journal is © the Owner Societies 2013 |